
Synthesis of Cyclobutane Analogue 4: Preparation of Purine and Pyrimidine Carbocyclic Nucleoside Derivatives

 ,
, 
Abstract
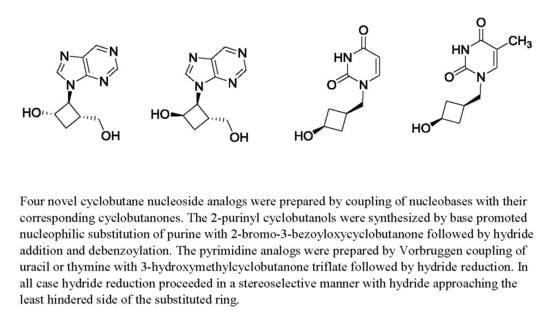
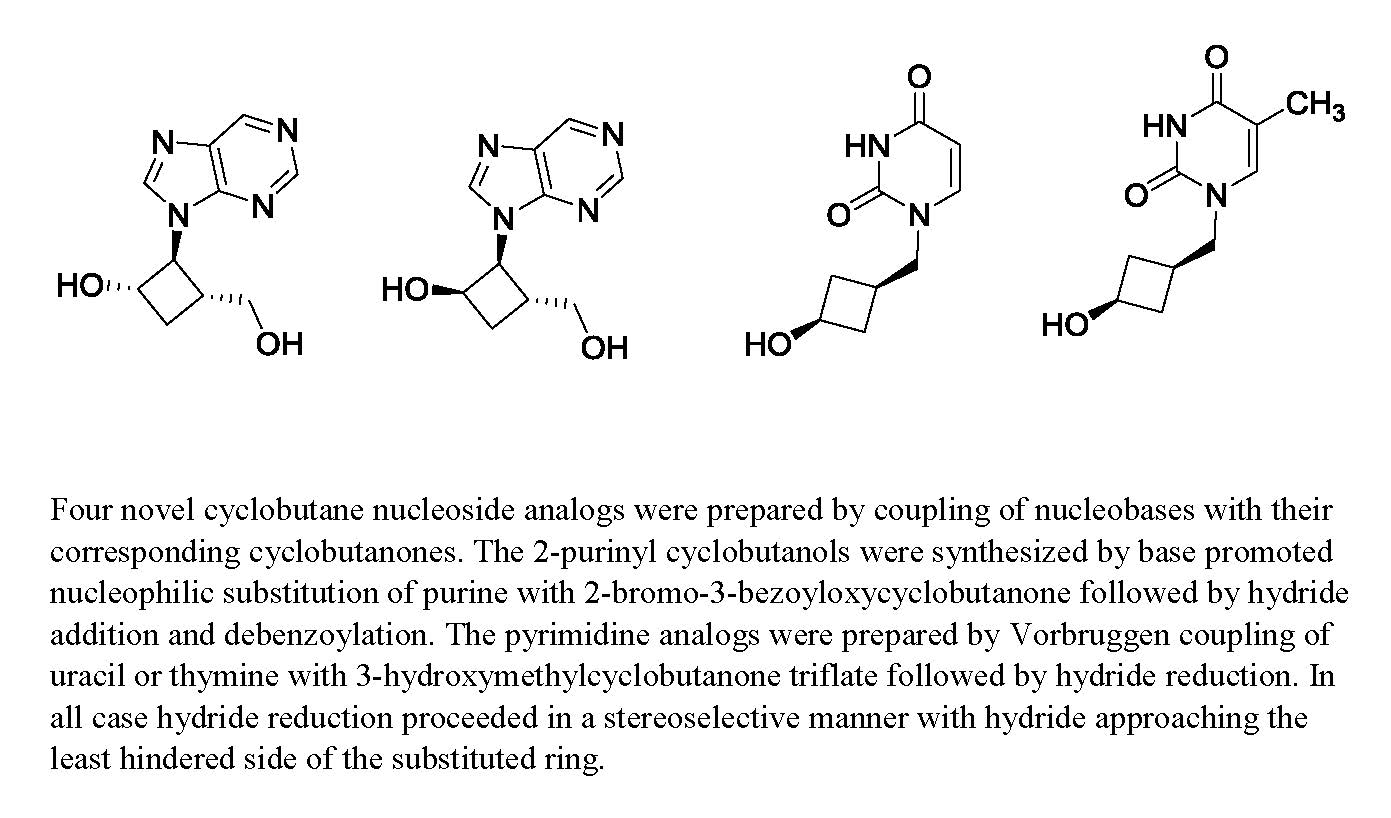
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of Purine Carbocyclic Analogues
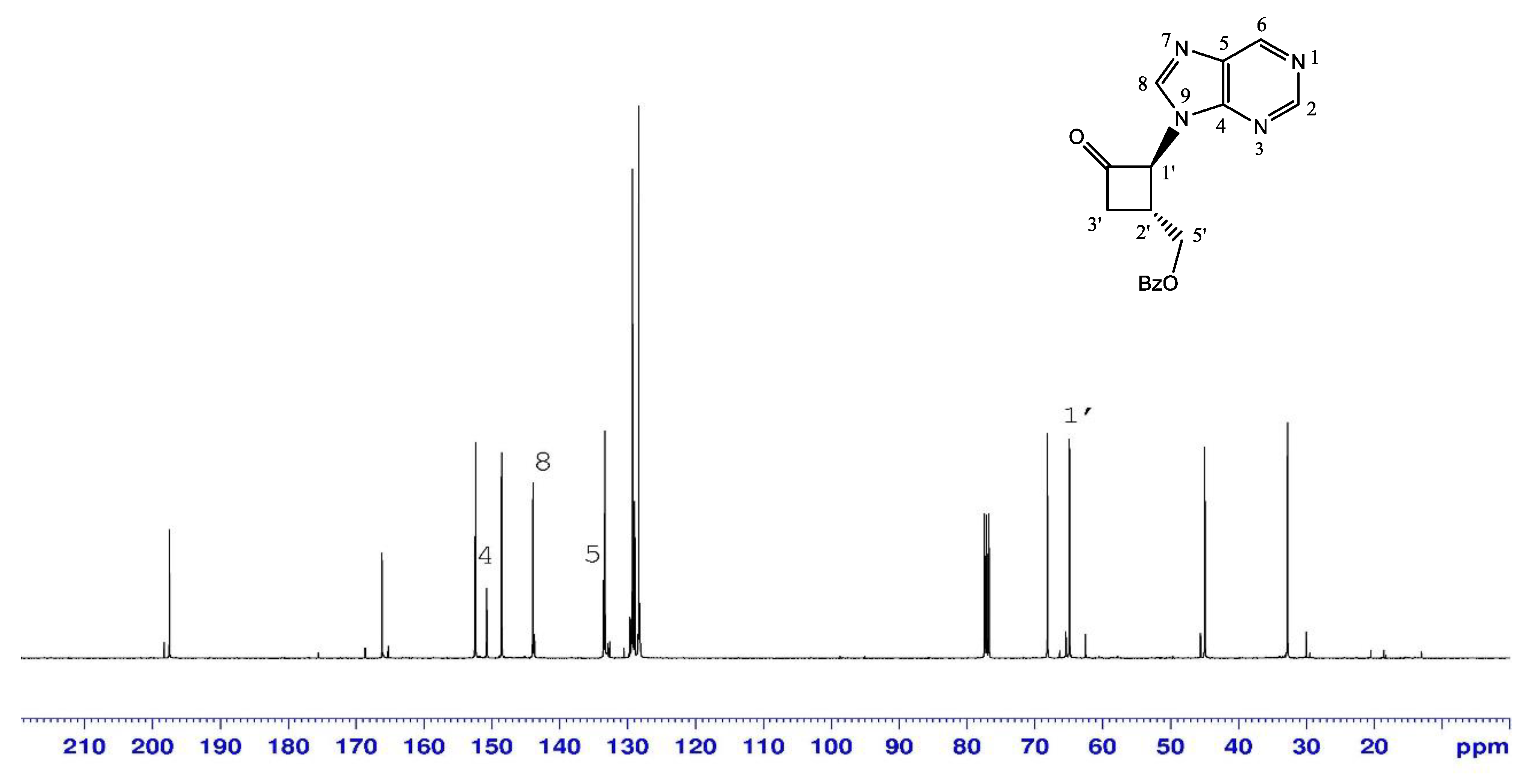
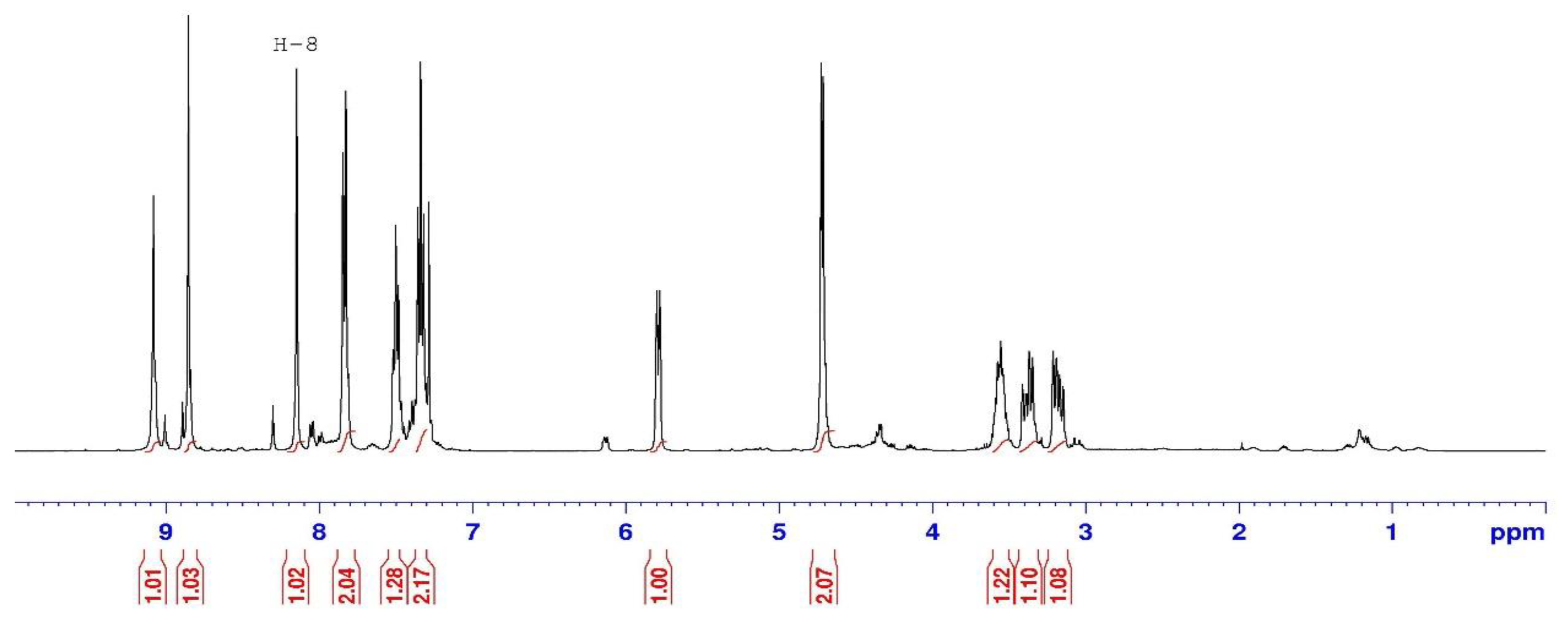
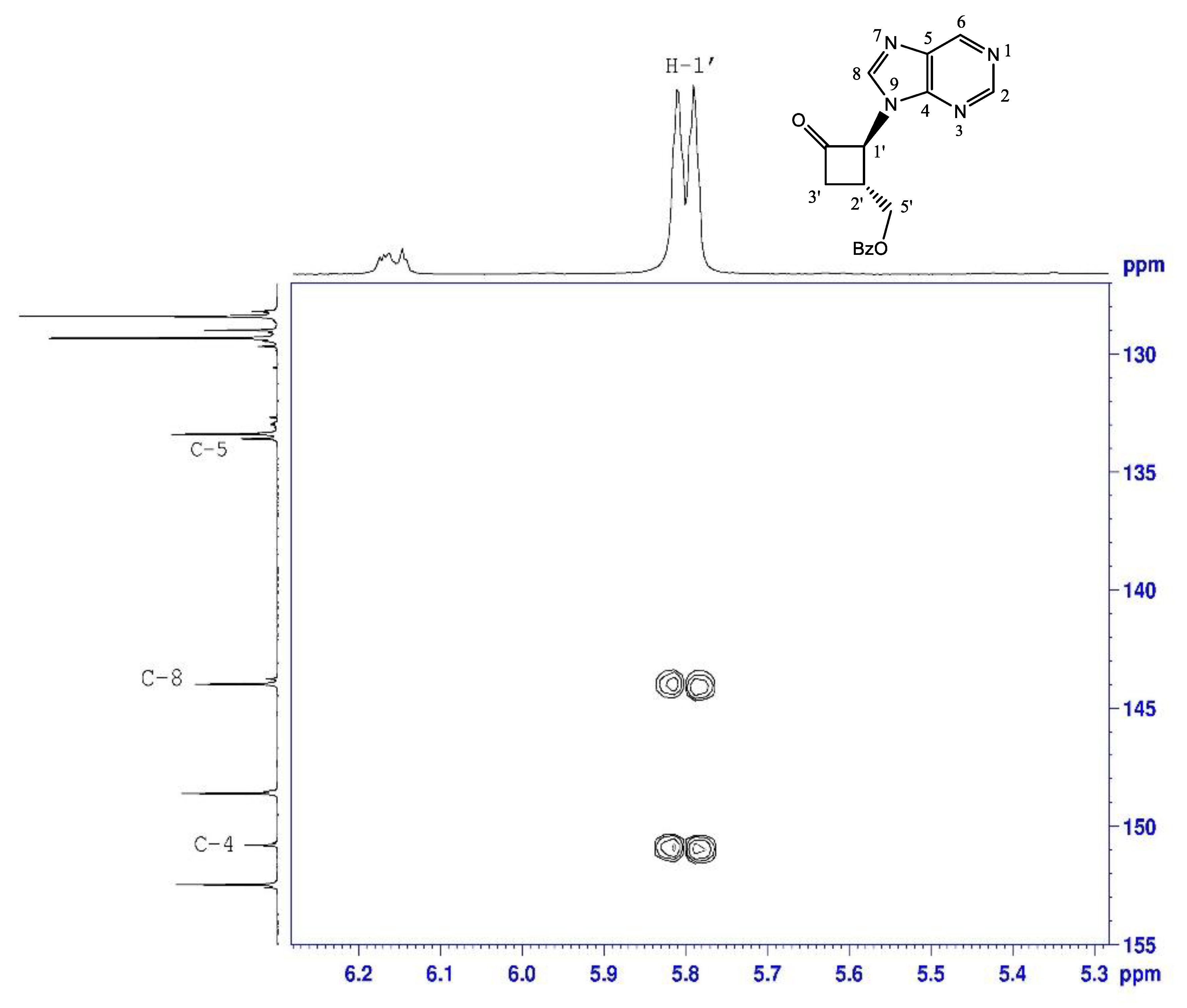
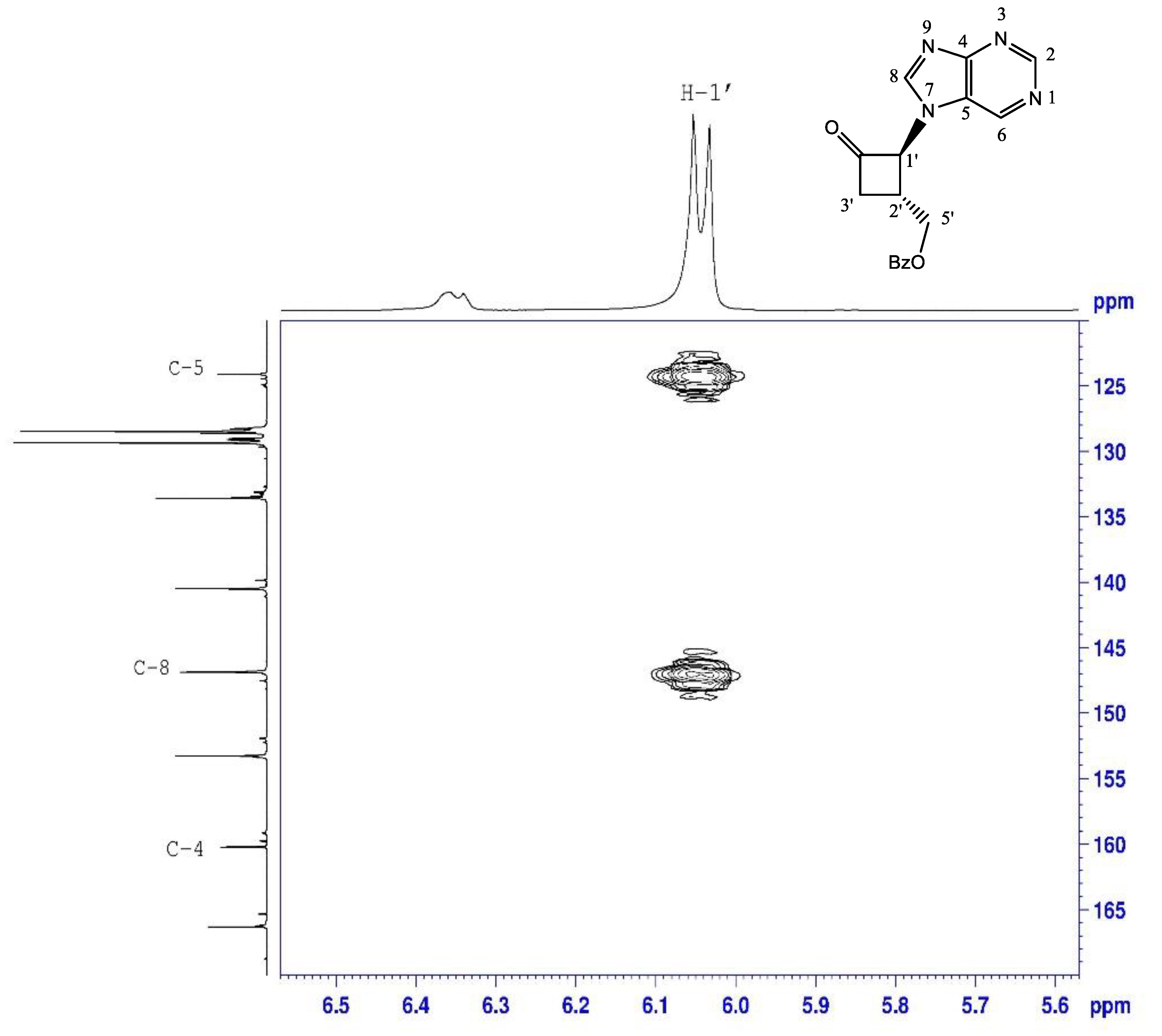
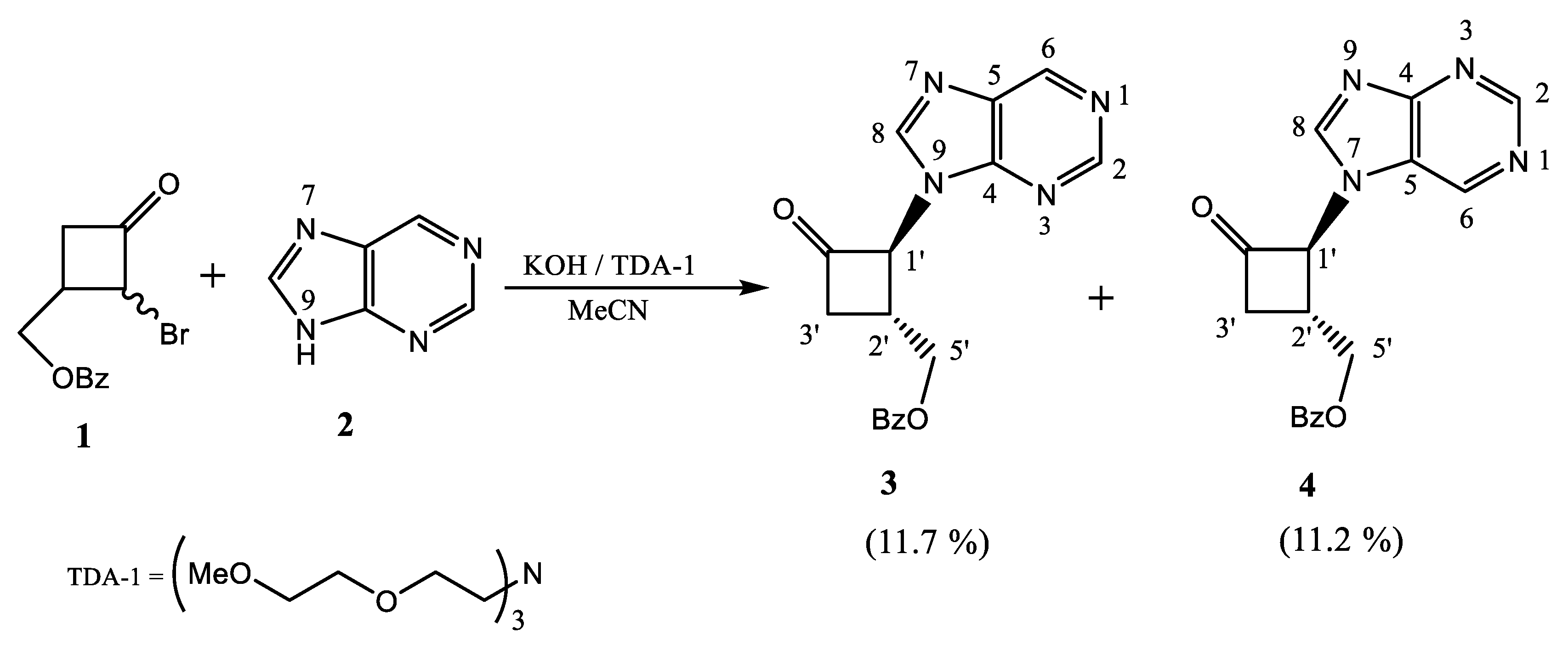
2.1.1. N-Alkylation of Purine to Bromocyclobutanone 1
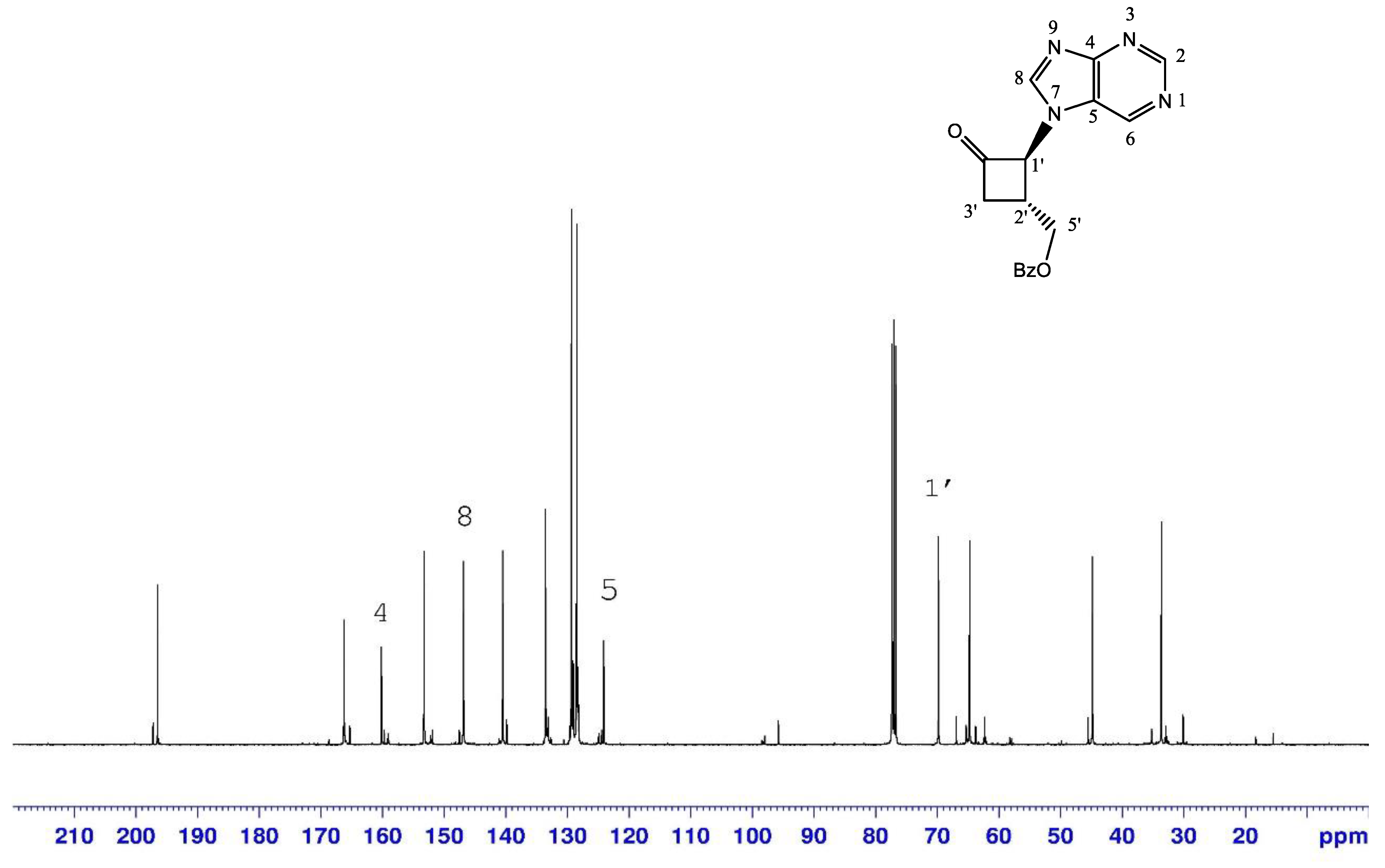
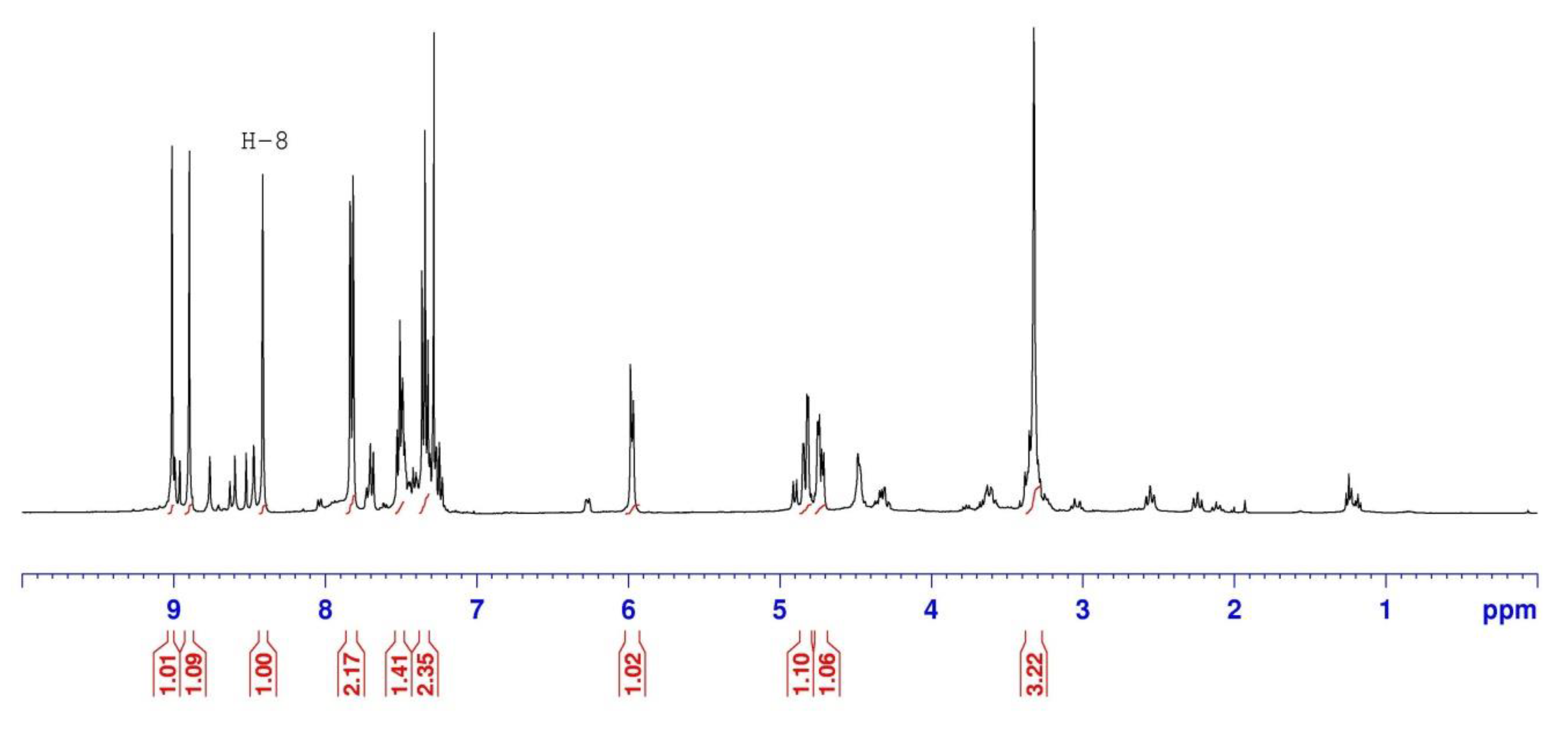
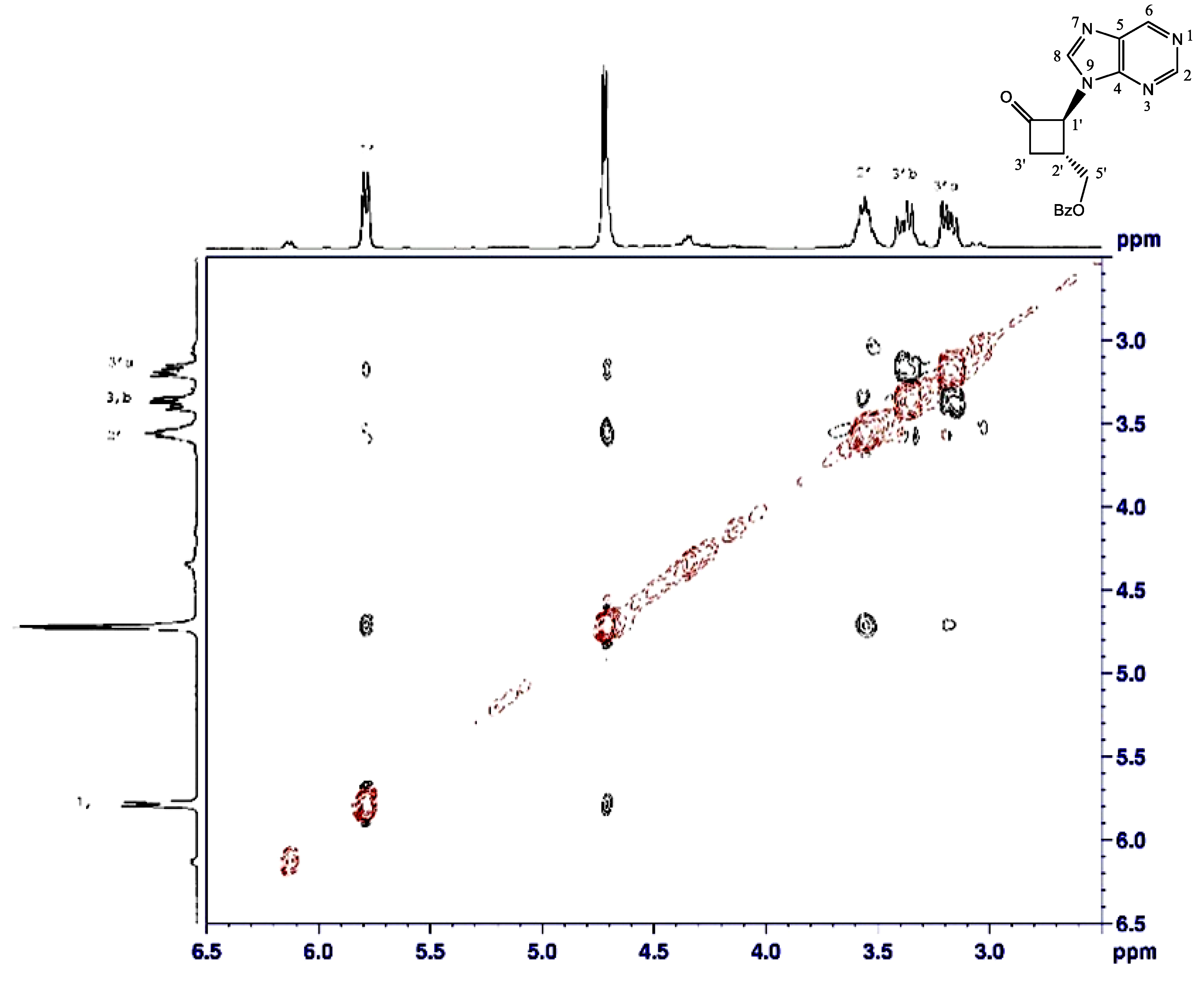
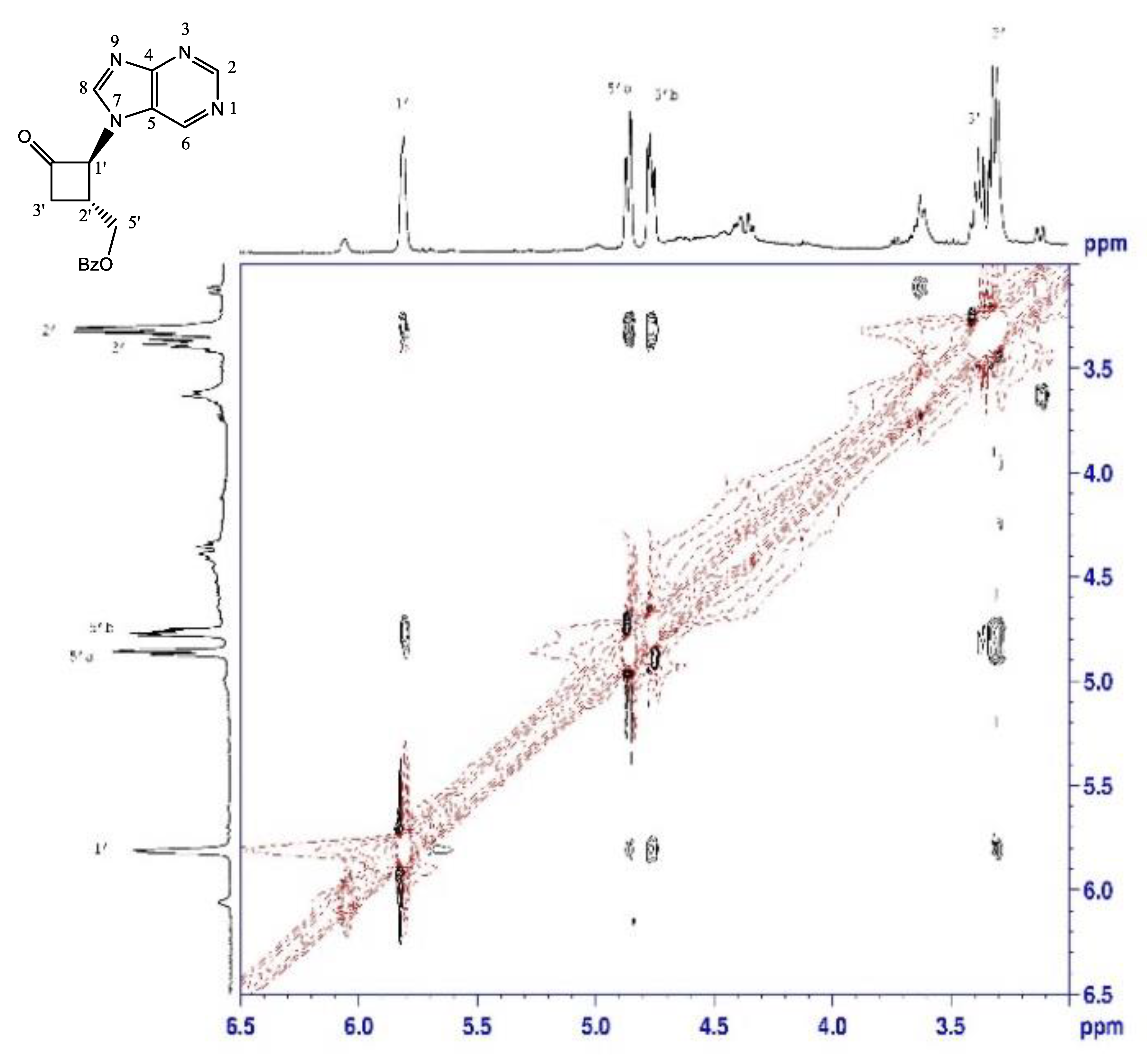
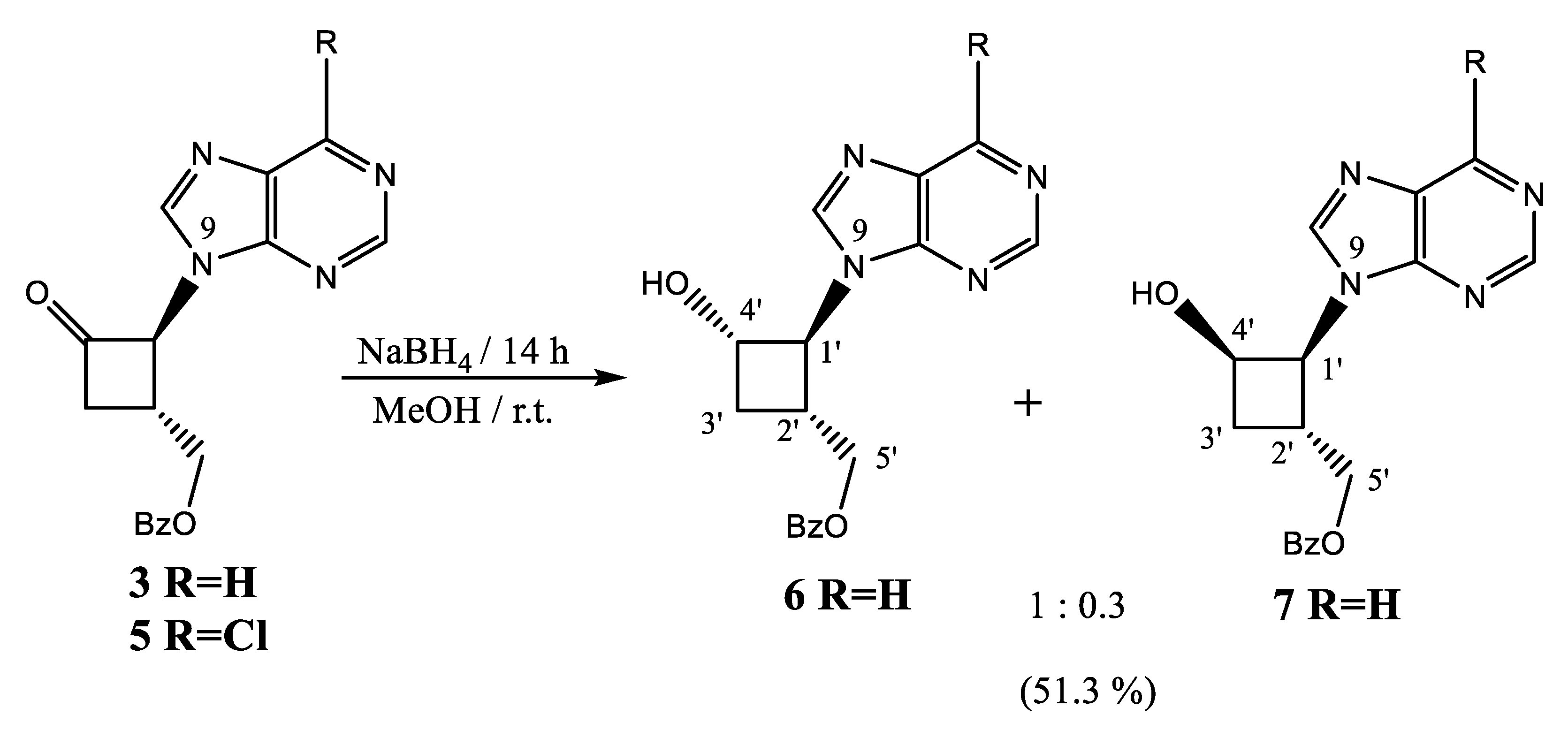
2.1.2. Reduction of Ketone Derivatives
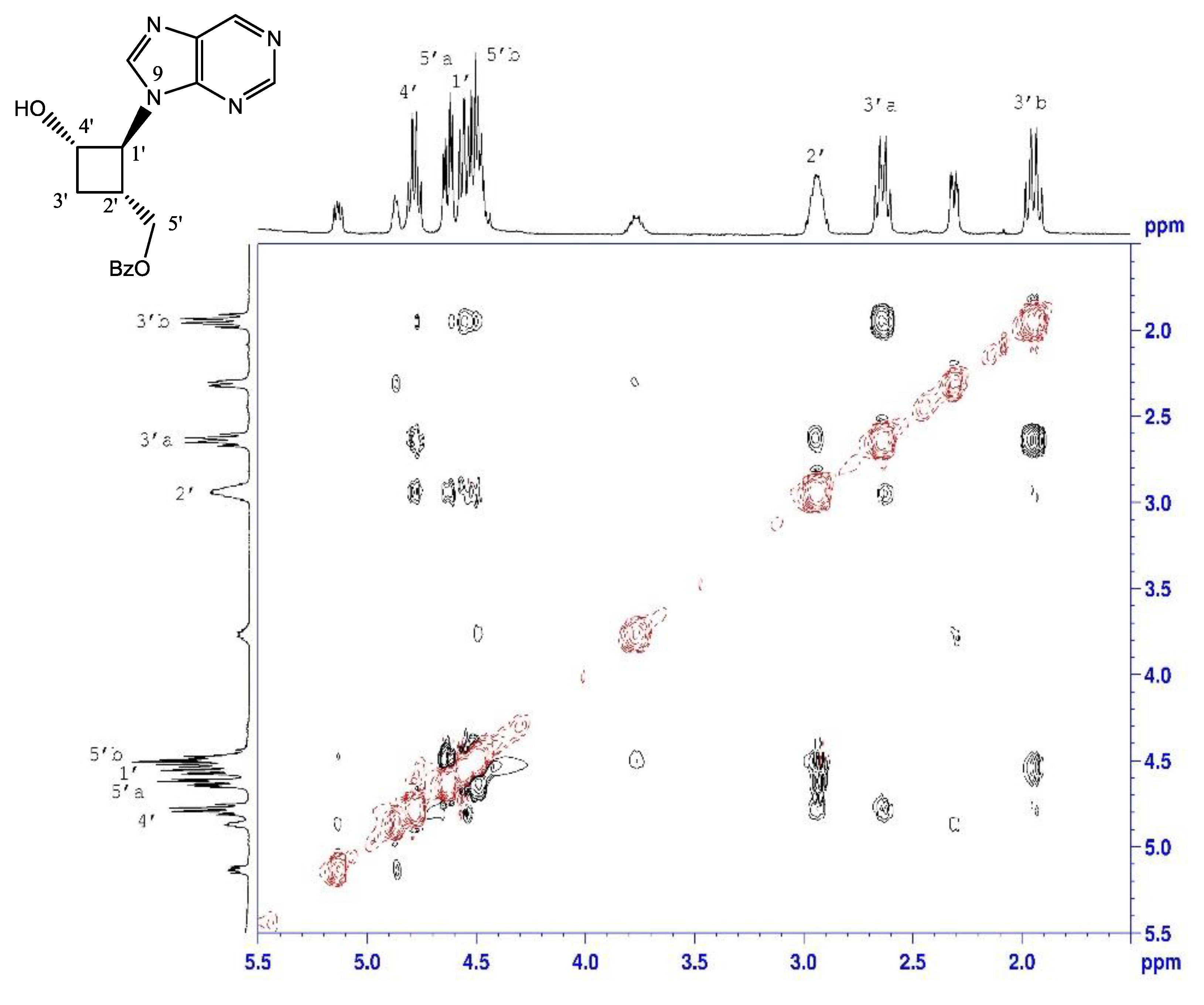
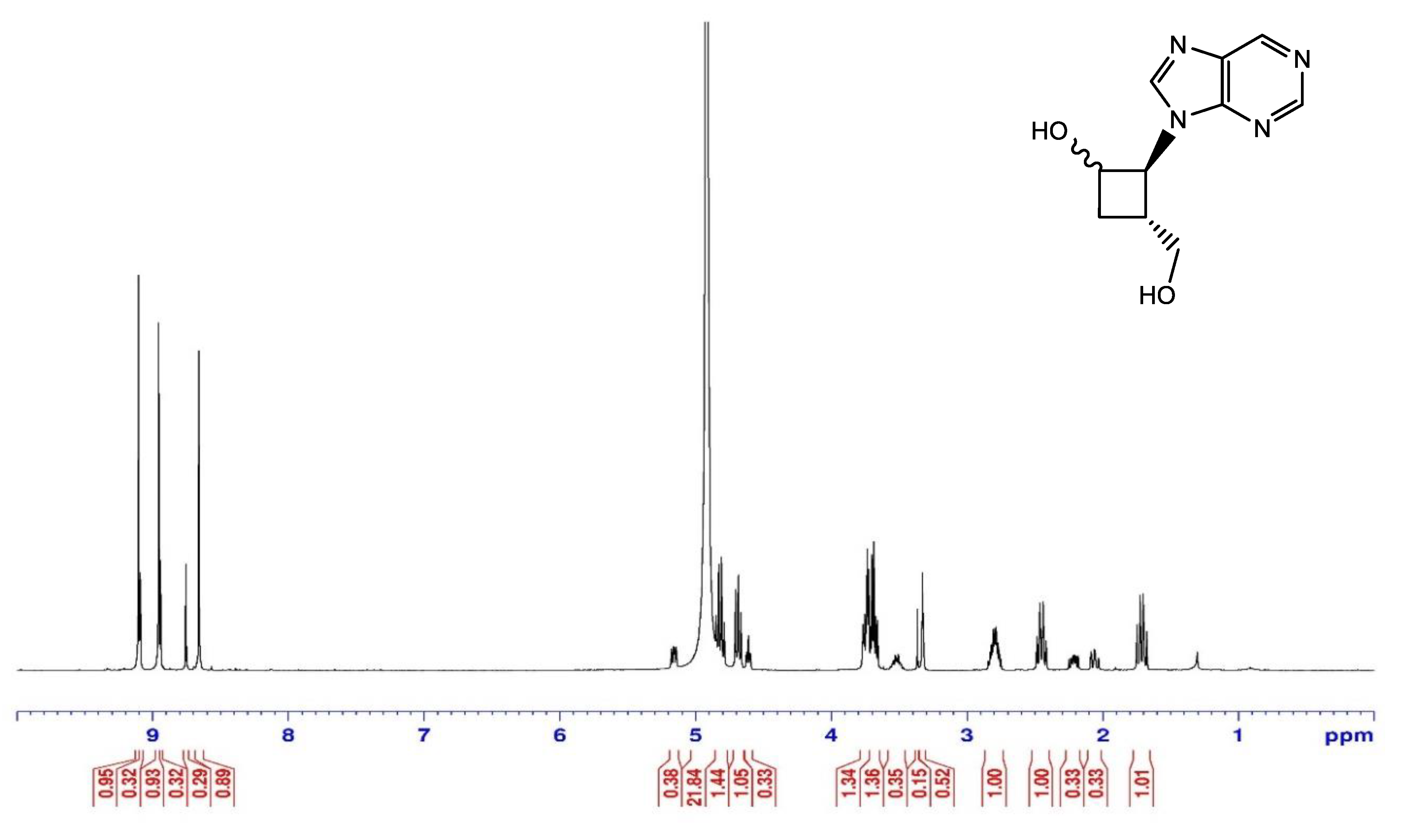
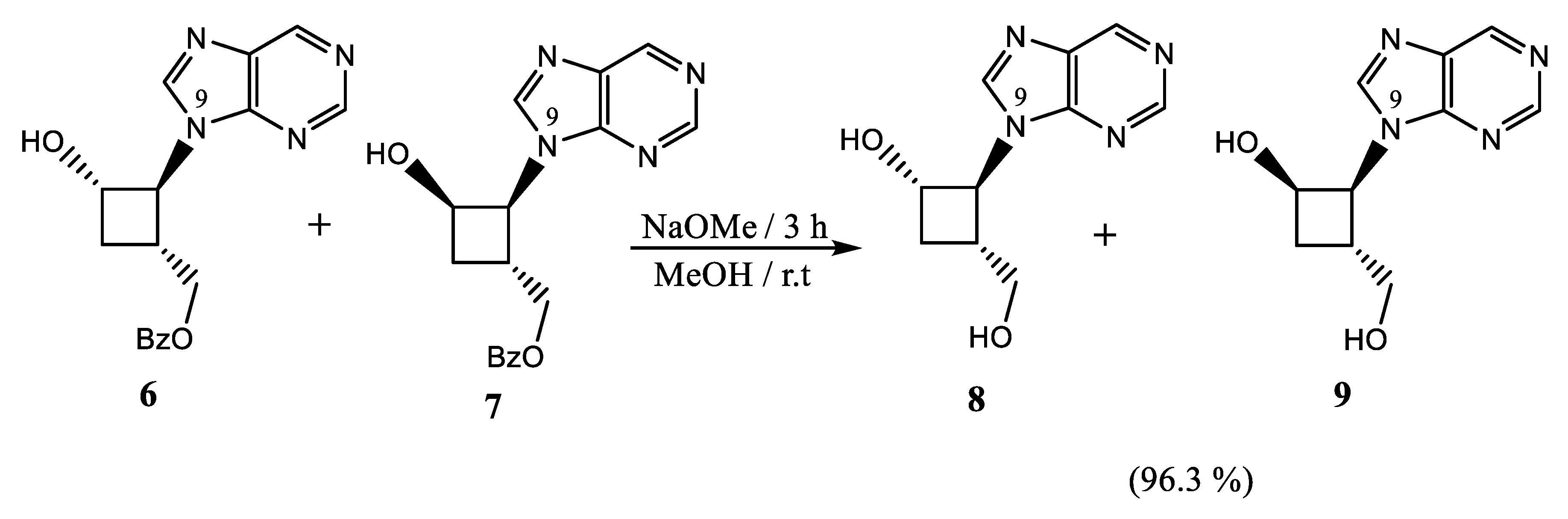
2.1.3. Debenzoylation of Purine Alcohols
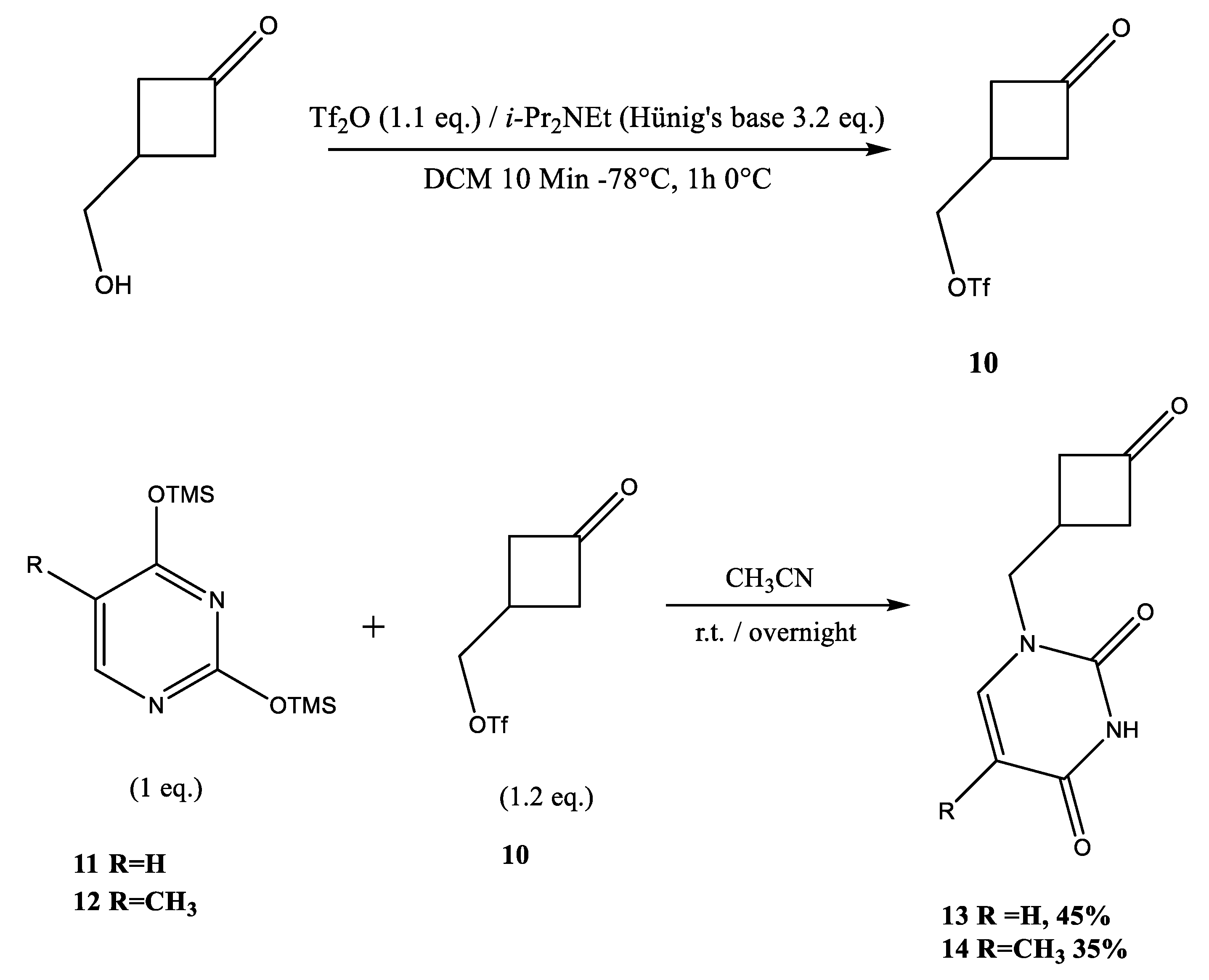
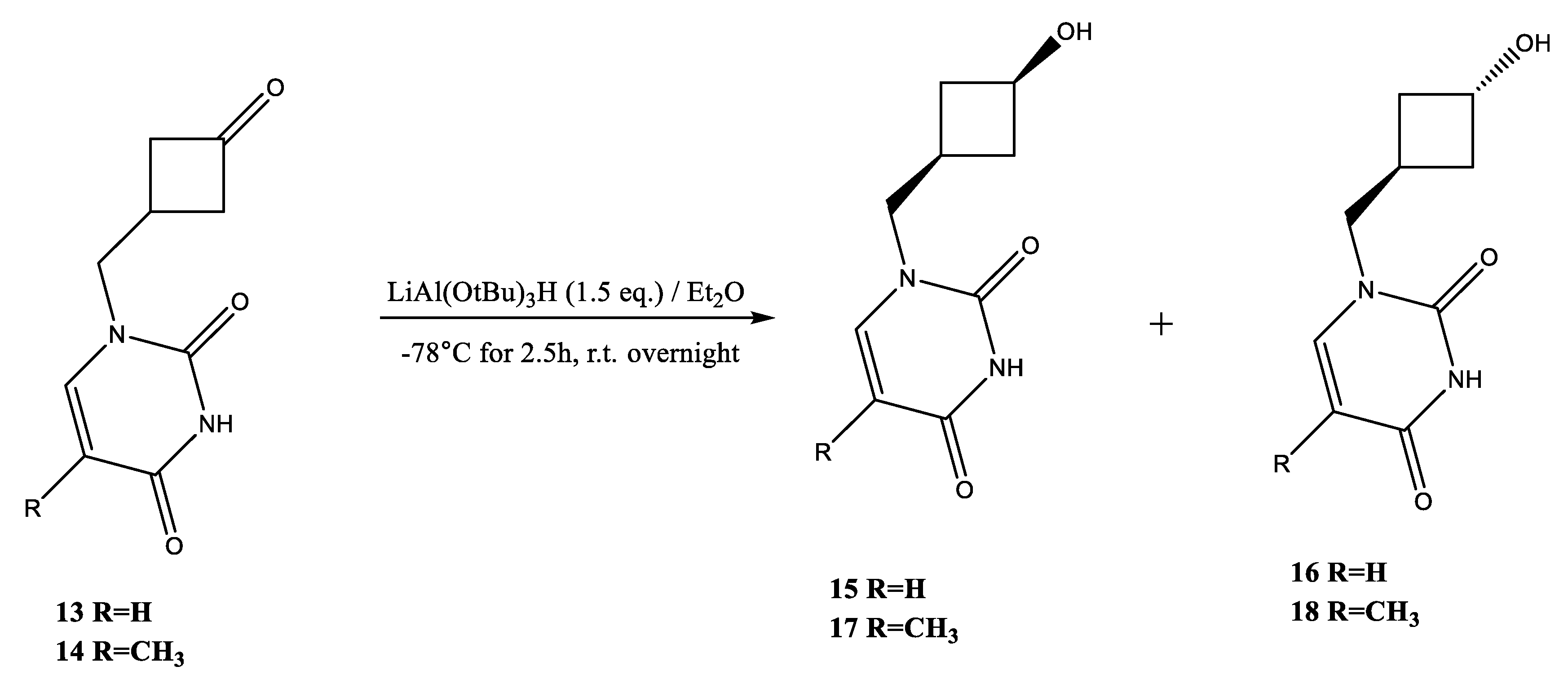
2.2. Synthesis of Pyrimidine Carbocyclic Nucleoside Analogues
3. Summary and Conclusions
4. Experimental
4.1. N-Alkylation of Purine
4.2. Reduction of N-9-Purine Ketone 3
4.3. Major Alcohol
4.4. Minor Alcohol
4.5. Deprotection of Purine Alcohols 6 and 7
4.6. Preparation of Ketone 13
4.7. Preparation of Ketone 14
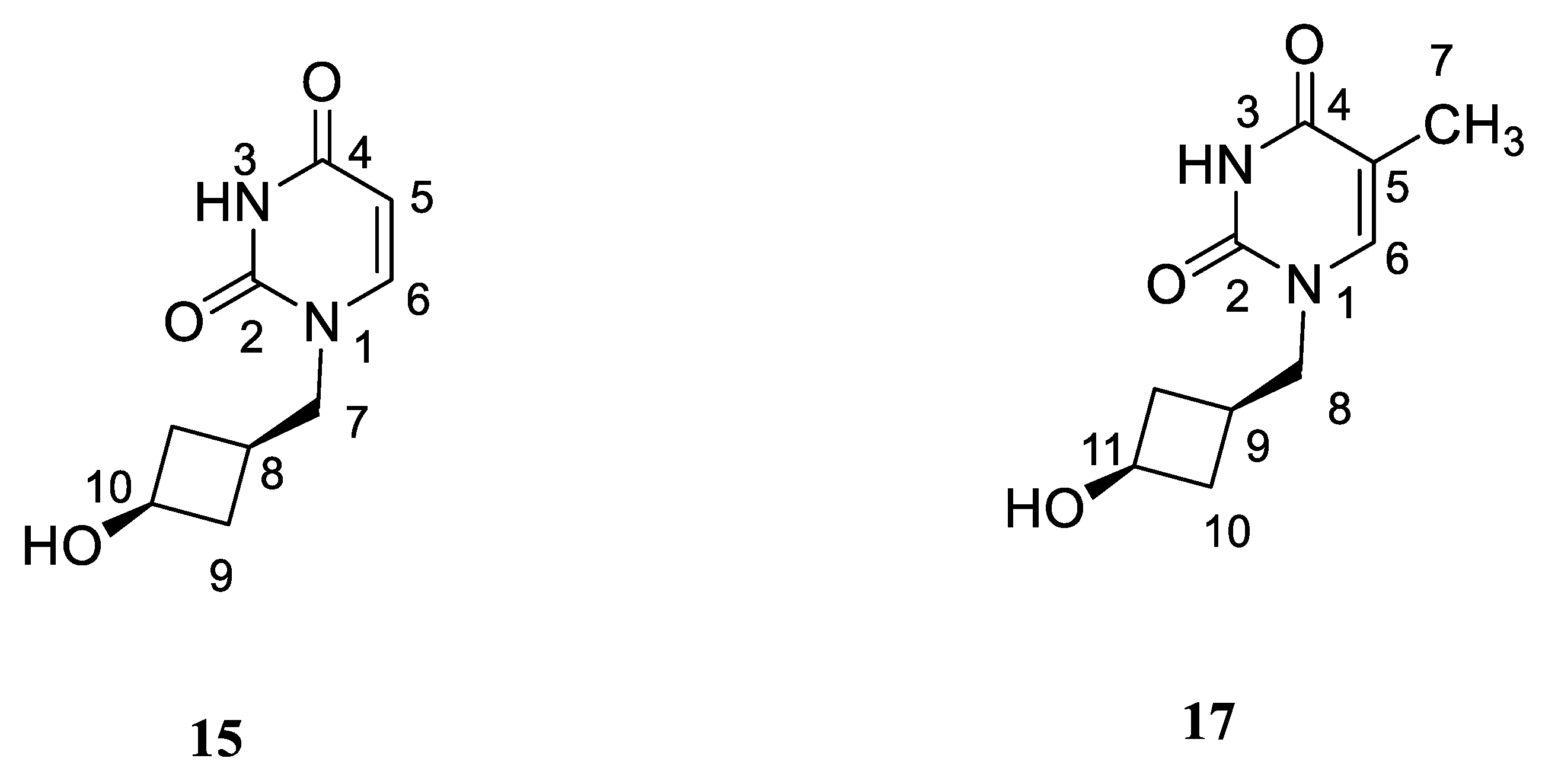
4.8. Preparation of Alcohol 15
4.9. Preparation of Alcohol 17
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Clercq, E. Strategies in the design of antiviral drugs. Nat. Rev. Drug Discov. 2002, 1, 13–25. [Google Scholar] [CrossRef]
- De Clercq, E. AIDS in the Third World: How to stop the HIV infection? Verh. K. Acad. voor Geneeskd. van Belgi. 2007, 69, 81–104. [Google Scholar]
- Chen, Q.; Davidson, A. Synthesis, conformational study and antiviral activity of L-like neplanocin derivatives. Bioorganic Med. Chem. Lett. 2017, 27, 4436–4439. [Google Scholar] [CrossRef]
- Coates, J.A.V.; Inggall, H.J.; Pearson, B.A.; Penn, C.R.; Storer, R.; Williamson, C.; Cameron, J.M. Carbovir: The (–) enantiomer is a potent and selective antiviral agent against human immunodeficiency virus in vitro. Antivir. Res. 1991, 15, 161–168. [Google Scholar] [CrossRef]
- Faletto, M.B.; Miller, W.H.; Garvey, E.P.; St. Clair, M.H.; Daluge, S.M.; Good, S.S. Unique intracellular activation of the potent anti-human immunodeficiency virus agent 1592U89. Antimicrob. Agents Chemother. 1997, 41, 1099–1107. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, H.; Shimizu, N.; Shimada, N.; Takita, T.; Takeuchi, T. Inhibition of infectivity of human immunodeficiency virus by oxetanocin. J. Antibiot. 1987, 40, 1077–1078. [Google Scholar] [CrossRef]
- Hayashi, S.; Norbeck, D.W.; Rosenbrook, W.; Fine, R.L.; Matsukura, M.; Plattner, J.J.; Broder, S.; Mitsuya, H. Cyclobut-A and cyclobut-G, carbocyclic oxetanocin analogs that inhibit the replication of human immunodeficiency virus in T cells and monocytes and macrophages in vitro. Antimicrob. Agents Chemother. 1990, 34, 287–294. [Google Scholar] [CrossRef] [Green Version]
- Bisacchi, G.S.; Braitman, A.; Cianci, C.W.; Clark, J.M.; Field, A.K.; Hagen, M.E.; Hockstein, D.R.; Malley, M.F.; Mitt, T.; Slusarchyk, W.A.; et al. Synthesis and antiviral activity of enantiomeric forms of cyclobutyl nucleoside analogs. J. Med. Chem. 1991, 34, 1415–1421. [Google Scholar] [CrossRef]
- Bisacchi, G.S.; Singh, J.; Godfrey, J.D., Jr.; Kissick, T.P.; Mitt, T.; Malley, M.F.; Marco, J.D.; Gougoutas, J.Z.; Mueller, R.H.; Zahler, R. Regioselective coupling of tetraalkylammonium salts of 6-iodo-2-aminopurine to a cyclobutyltriflate: Efficient preparation of homochiral BMS-180,194, a potent antiviral carbocyclic nucleoside. J. Org. Chem. 1995, 60, 2902–2905. [Google Scholar] [CrossRef]
- Singh, J.; Bisacchi, G.S.; Ahmad, S.; Godfrey, J.D., Jr.; Kissick, T.P.; Mitt, T.; Kocy, O.; Vu, T.; Papaioannou, C.G.; Wong, M.K.; et al. A practical asymmetric synthesis of the antiviral agent lobucavir, BMS-180194. Org. Process Res. Dev. 1998, 2, 393–399. [Google Scholar] [CrossRef]
- Genovesi, E.V.; Lamb, L.; Medina, I.; Taylor, D.; Seifer, M.; Innaimo, S.; Colonno, R.J.; Clark, J.M. Antiviral efficacy of lobucavir (BMS-180194), a cyclobutyl-guanosine nucleoside analogue, in the woodchuck (Marmota monax) model of chronic hepatitis B virus (HBV) infection. Antivir. Viral Res. 2000, 48, 197–203. [Google Scholar] [CrossRef]
- Hanson, R.L.; Shi, Z.; Brzozowski, D.B.; Banerjee, A.; Kissick, T.P.; Singh, J.; Pullockaran, A.J.; North, J.T.; Fan, J.; Howell, J.; et al. Regioselective enzymatic aminoacylation of lobucavir to give an intermediatefor lobucavir prodrug. Bioorganic Med. Chem. 2000, 8, 2681–2687. [Google Scholar] [CrossRef]
- Darses, B.; Greene, A.E.; Coote, S.C.; Poisson, J.F. Expedient approach to chiral cyclobutanones: Asymmetric synthesis of cyclobut-G. Org. Lett. 2008, 10, 821–824. [Google Scholar] [CrossRef]
- Ebead, A.; Fournier, R.; Lee-Ruff, E. Synthesis of Cyclobutane Nucleosides. Nucleosides Nucleotides Nucleic Acids 2011, 30, 391–404. [Google Scholar] [CrossRef]
- Hassan, M.; Yaseen, A.; Ebead, A.; Audette, G.; Lee-Ruff, E. Synthesis of cyclobutane nucleoside analogues 3: Preparation of carbocyclic derivatives of oxetanocin. Nucleosides Nucleotides Nucleic Acids 2018, 37, 518–531. [Google Scholar] [CrossRef]
- Montgomery, J.; Temple, C.J. Synthesis of Potential Anticancer Agents. XXVI. The Alkylation of 6-Chloropurine2. Am. Chem. Soc. 1961, 83, 630. [Google Scholar] [CrossRef]
- Zorbach, W.W.; Tipson, R.S. Synthetic Procedures in Nucleic Acid Chemistry, Volume I; Zorbach, W.W.; Tipson, R.S. John Wiley and Sons: New York, NY, USA, 1968. [Google Scholar]
- Kazimierczuk, Z.; Cottam, H.; Revankar, G.; Robins, R.J. Synthesis of 2’-deoxytubercidin, 2’-deoxyadenosine, and related 2’-deoxynucleosides via a novel direct stereospecific sodium salt glycosylation procedure. Am. Chem. Soc. 1984, 106, 6379. [Google Scholar] [CrossRef]
- Seela, F.; Bourgeois, W. Synthesis of Indole 2’,3’-Dideoxyribonucleosides. Synthesis 1990, 10, 945–950. [Google Scholar] [CrossRef]
- Hockova, D.; Budesinsky, M.; Marek, R.; Marek, J.; Holy, A. Regioselective Preparation of N7- and N9-Alkyl Derivatives of N6-[ (Dimethylamino) methylene]adenine Bearing an Active Methylene Group and Their Further Derivatization Leading to α-Branched Acyclic Nucleoside Analogues. Eur. J. Org. Chem. 1999, 1999, 2675–2682. [Google Scholar] [CrossRef]
- Rustullet, A.; Alibes, R.; March, P.; Figueredo, M.; Font, J. Stereoselective Route to Oxetanocin Carbocyclic Analogues Based on a [2 + 2] Photocycloaddition to a Chiral 2(5H)-Furanone. Org. Lett. 2007, 9, 2827. [Google Scholar] [CrossRef]
- Kjellberg, J.; Johansson, N. Characterization of N7 and N9 alkylated purine analogues by 1H and 13C nmr. Tetrahedron 1986, 42, 6541. [Google Scholar] [CrossRef]
- Choi, W.B.; Wilson, L.J.; Yeola, S.; Liotta, D.C.; Schinazi, R.F.J. In situ complexation directs the stereochemistry of N-glycosylation in the synthesis of thialanyl and dioxolanyl nucleoside analogs. Am. Chem. Soc. 1991, 113, 9377. [Google Scholar] [CrossRef]
- Vorbrüggen, H.; Ruh-Pohlenz, C. Synthesis of Nucleosides. Org. React. 2000, 97, 489. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 13 and 14 are available from the authors. |


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N-9-ketone 3 | N-7-ketone 4 | |
|---|---|---|
| H-8 | 8.12 | 8.41 |
| C-1’ | 68.2 | 69.8 |
| C-4 | 150.8 | 160.2 |
| C-5 | 133.6 | 124.1 |
| C-8 | 144 | 146.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasaneen, N.; Ebead, A.; Hassan, M.M.; Afifi, H.; Hunter, H.; Lee-Ruff, E.; El-Gohary, N.S.; Maarouf, A.R.; El-Emam, A.A. Synthesis of Cyclobutane Analogue 4: Preparation of Purine and Pyrimidine Carbocyclic Nucleoside Derivatives. Molecules 2019, 24, 3235. https://doi.org/10.3390/molecules24183235
Hasaneen N, Ebead A, Hassan MM, Afifi H, Hunter H, Lee-Ruff E, El-Gohary NS, Maarouf AR, El-Emam AA. Synthesis of Cyclobutane Analogue 4: Preparation of Purine and Pyrimidine Carbocyclic Nucleoside Derivatives. Molecules. 2019; 24(18):3235. https://doi.org/10.3390/molecules24183235
Chicago/Turabian StyleHasaneen, Noha, Abdelaziz Ebead, Muhammad Murtaza Hassan, Hanan Afifi, Howard Hunter, Edward Lee-Ruff, Nadia S. El-Gohary, Azza R. Maarouf, and Ali A. El-Emam. 2019. "Synthesis of Cyclobutane Analogue 4: Preparation of Purine and Pyrimidine Carbocyclic Nucleoside Derivatives" Molecules 24, no. 18: 3235. https://doi.org/10.3390/molecules24183235
APA StyleHasaneen, N., Ebead, A., Hassan, M. M., Afifi, H., Hunter, H., Lee-Ruff, E., El-Gohary, N. S., Maarouf, A. R., & El-Emam, A. A. (2019). Synthesis of Cyclobutane Analogue 4: Preparation of Purine and Pyrimidine Carbocyclic Nucleoside Derivatives. Molecules, 24(18), 3235. https://doi.org/10.3390/molecules24183235