PSTP-3,5-Me Inhibits Osteoclast Differentiation and Bone Resorption



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
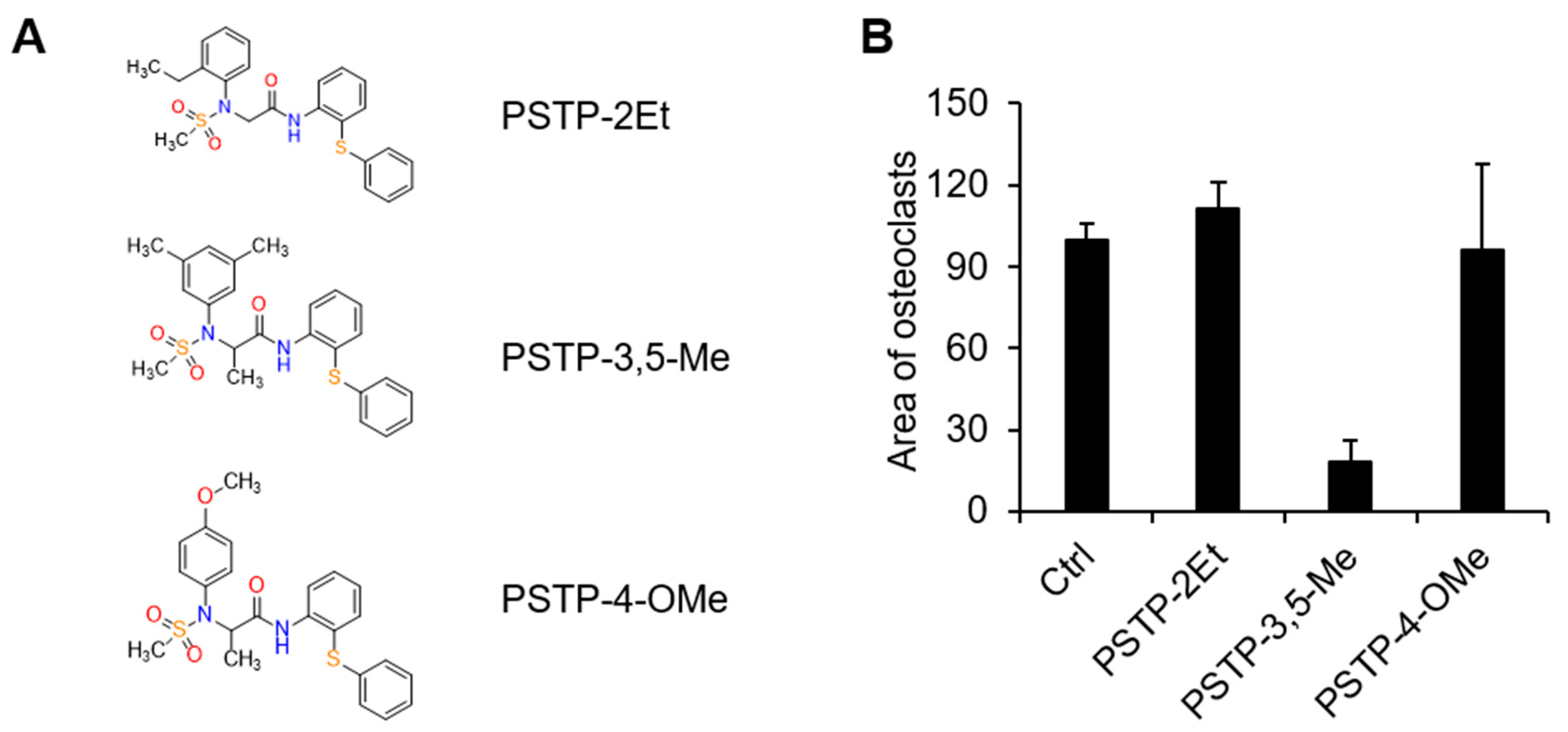
2.1. PSTP-3,5-Me Inhibits Osteoclast Differentiation
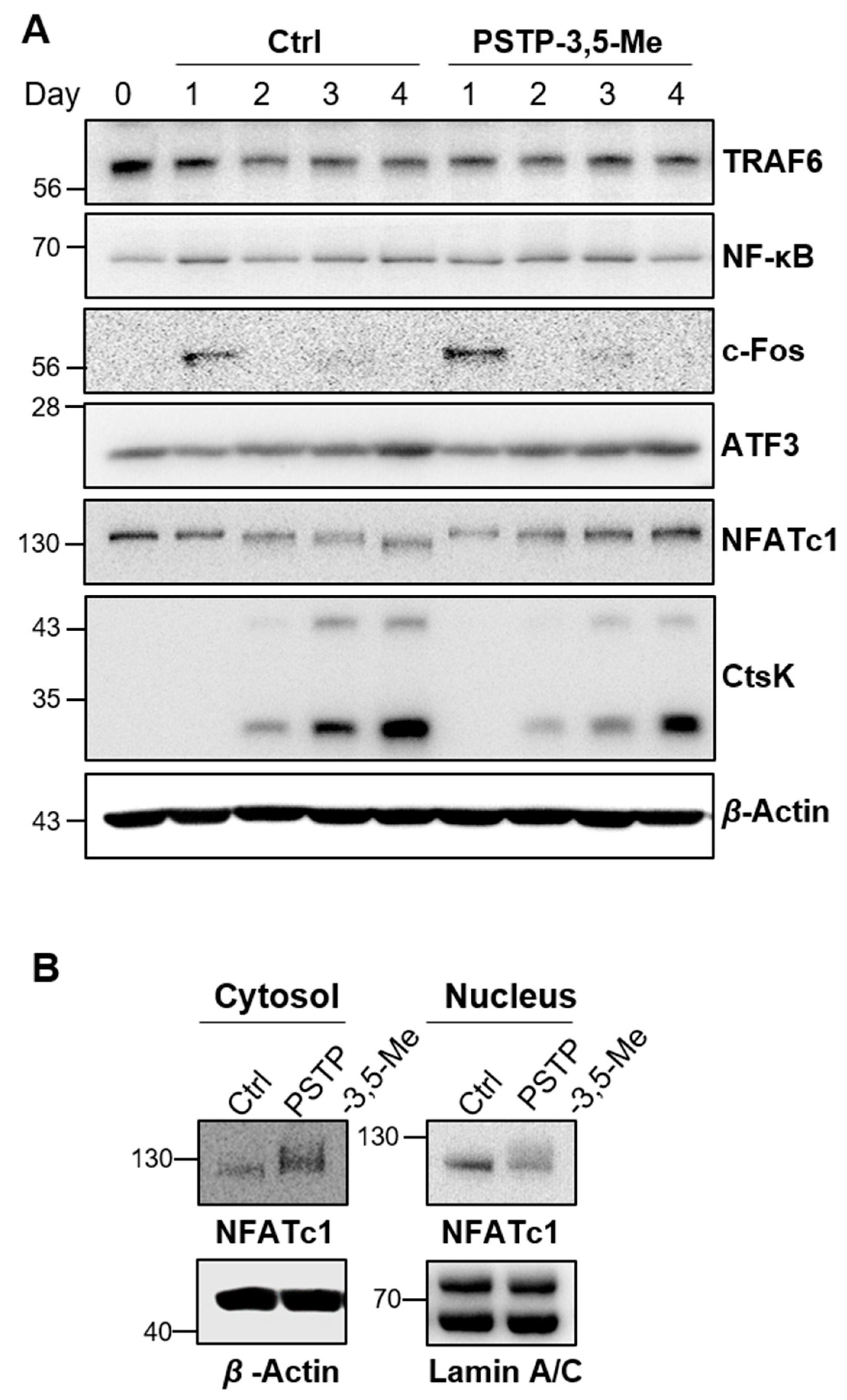
2.2. PSTP-3,5-Me Inhibits Osteoclast Differentiation Mediated by Reduced CtsK and NFATc1 Expressions
2.3. PSTP-3,5-Me Suppresses Nuclear Translocation of NFATc1
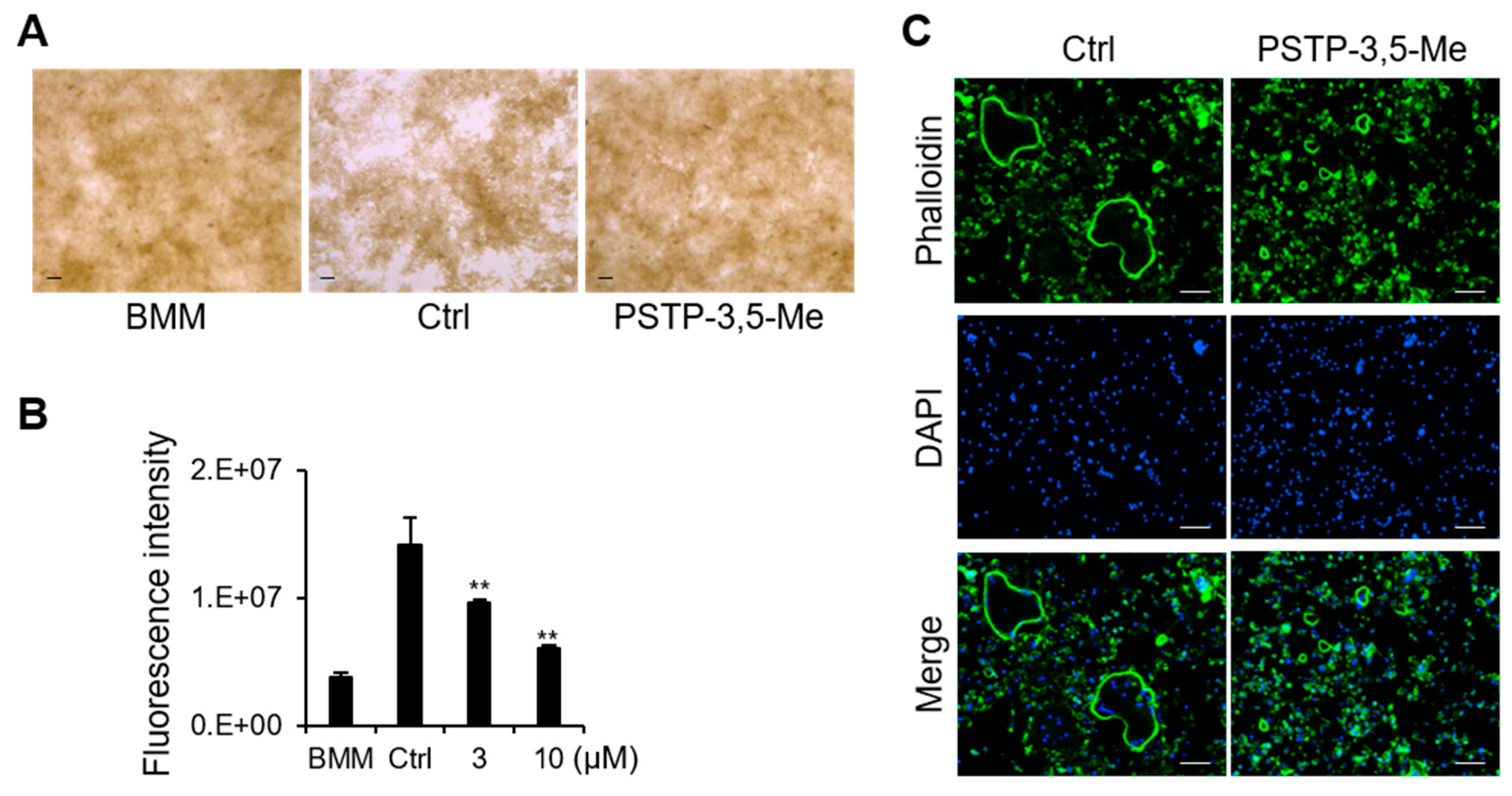
2.4. PSTP-3,5-Me Inhibits Actin-Ring Formation and Bone-Resorption Activity
2.5. PSTP-3,5-Me Does Not Affect Osteoblast Differentiation
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.2. Osteoclast Differentiation and TRAP Staining
4.3. Cell Viability Assay
4.4. Quantitative Real-Time PCR (qRT-PCR)
4.5. Western Blotting
4.6. Osteoblast Differentiation and ALP Staining
4.7. Bone Resorption Assay
4.8. Actin Ring Formation
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nagy, V.; Penninger, J.M. The RANKL-RANK story. Gerontology 2015, 61, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Ducy, P.; Schinke, T.; Karsenty, G. The osteoblast: A sophisticated fibroblast under central surveillance. Science 2000, 289, 1501–1504. [Google Scholar] [CrossRef] [PubMed]
- Teitelbaum, S.L. Bone resorption by osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, K.; Kim, I.; Seong, S.; Kim, S.W.; Kim, N. Role of anoctamin 5, a gene associated with gnathodiaphyseal dysplasia, in osteoblast and osteoclast differentiation. Bone 2019, 120, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; McDonald, J.M. Disorders of bone remodeling. Annu. Rev. Pathol. 2011, 6, 121–145. [Google Scholar] [CrossRef] [PubMed]
- Palagano, E.; Menale, C.; Sobacchi, C.; Villa, A. Genetics of osteopetrosis. Curr. Osteoporos. Rep. 2018, 16, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Motiur Rahman, M.; Takeshita, S.; Matsuoka, K.; Kaneko, K.; Naoe, Y.; Sakaue-Sawano, A.; Miyawaki, A.; Ikeda, K. Proliferation-coupled osteoclast differentiation by RANKL: Cell density as a determinant of osteoclast formation. Bone 2015, 81, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, N.K.; Lee, S.Y. Current understanding of RANK signaling in osteoclast differentiation and maturation. Mol. Cells 2017, 40, 706–713. [Google Scholar]
- Narahara, S.; Sakai, E.; Kadowaki, T.; Yamaguchi, Y.; Narahara, H.; Okamoto, K.; Asahina, I.; Tsukuba, T. KBTBD11, a novel BTB-Kelch protein, is a negative regulator of osteoclastogenesis through controlling Cullin3-mediated ubiquitination of NFATc1. Sci. Rep. 2019, 9, 3523. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, N. Regulation of NFATc1 in osteoclast differentiation. J. Bone. Metab. 2014, 21, 233–241. [Google Scholar] [CrossRef]
- Cicek, M.; Vrabel, A.; Sturchio, C.; Pederson, L.; Hawse, J.R.; Subramaniam, M.; Spelsberg, T.C.; Oursler, M.J. TGF-beta inducible early gene 1 regulates osteoclast differentiation and survival by mediating the NFATc1, AKT, and MEK/ERK signaling pathways. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Winslow, M.M.; Pan, M.; Starbuck, M.; Gallo, E.M.; Deng, L.; Karsenty, G.; Crabtree, G.R. Calcineurin/NFAT signaling in osteoblasts regulates bone mass. Dev. Cell 2006, 10, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Aliprantis, A.O.; Ueki, Y.; Sulyanto, R.; Park, A.; Sigrist, K.S.; Sharma, S.M.; Ostrowski, M.C.; Olsen, B.R.; Glimcher, L.H. NFATc1 in mice represses osteoprotegerin during osteoclastogenesis and dissociates systemic osteopenia from inflammation in cherubism. J. Clin. Investig. 2008, 118, 3775–3789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef]
- Song, I.; Kim, J.H.; Kim, K.; Jin, H.M.; Youn, B.U.; Kim, N. Regulatory mechanism of NFATc1 in RANKL-induced osteoclast activation. Febs. Lett. 2009, 583, 2435–2440. [Google Scholar] [CrossRef] [Green Version]
- Kolluri, S.K.; Kopparapu, P.R.; Pearce, M. Small Molecule Bcl-2 Functional Converters as Cancer Therapeutics by Inducing Growth Inhibition or Apoptosis. Pct. Int. Appl. WO 2018102766 A2, 7 June 2018. [Google Scholar]
- Kim, N.D.; Kim, H.G.; Kang, Y.M.; Sa, G.H.; Sung, S.J.; Lee, T.H. Pharmaceutical Composition Containing Integrin Inhibitor as Active Ingredient for Prevention or Treatment of Inflammatory Diseases. Republic Korean Kongkae Taeho Kongbo. KR 2017026798 A, 16 February 2017. [Google Scholar]
- Abbas, Y.A. Synthesis of some new sulfonamides derivatives of toluidine as possible antimicrobial agents. Egypt. J. Pharm. Sci. 1995, 36, 187–195. [Google Scholar]
- Greig, I.R.; Coste, E.; Ralston, S.H.; van′t Hof, R.J. Development of triarylsulfonamides as novel anti-inflammatory agents. Bioorg. Med. Chem. Lett. 2013, 23, 816–820. [Google Scholar] [CrossRef]
- Yang, P.; Wang, L.; Feng, R.; Almehizia, A.A.; Tong, Q.; Myint, K.Z.; Ouyang, Q.; Alqarni, M.H.; Wang, L.; Xie, X.Q. Novel triaryl sulfonamide derivatives as selective cannabinoid receptor 2 inverse agonists and osteoclast inhibitors: Discovery, optimization, and biological evaluation. J. Med. Chem. 2013, 56, 2045–2058. [Google Scholar] [CrossRef]
- Goldfarb, D.S. Method Using Lifespan-altering Compounds for Altering the Lifespan of Eukaryotic Organisms, and Screening for Such Compounds. U.S. Patent US 20090163545 A1, 25 June 2009. [Google Scholar]
- Yagi, M.; Miyamoto, T.; Sawatani, Y.; Iwamoto, K.; Hosogane, N.; Fujita, N.; Morita, K.; Ninomiya, K.; Suzuki, T.; Miyamoto, K.; et al. DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J. Exp. Med. 2005, 202, 345–351. [Google Scholar] [CrossRef]
- Miyamoto, H.; Suzuki, T.; Miyauchi, Y.; Iwasaki, R.; Kobayashi, T.; Sato, Y.; Miyamoto, K.; Hoshi, H.; Hashimoto, K.; Yoshida, S.; et al. Osteoclast stimulatory transmembrane protein and dendritic cell-specific transmembrane protein cooperatively modulate cell-cell fusion to form osteoclasts and foreign body giant cells. J. Bone Min. Res. 2012, 27, 1289–1297. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, K.; Jin, H.M.; Song, I.; Youn, B.U.; Lee, S.H.; Choi, Y.; Kim, N. Negative feedback control of osteoclast formation through ubiquitin-mediated down-regulation of NFATc1. J. Biol. Chem. 2010, 285, 5224–5231. [Google Scholar] [CrossRef]
- Li, J.; Zeng, L.; Xie, J.; Yue, Z.; Deng, H.; Ma, X.; Zheng, C.; Wu, X.; Luo, J.; Liu, M. Inhibition of osteoclastogenesis and bone resorption in vitro and in vivo by a prenylflavonoid xanthohumol from hops. Sci. Rep. 2015, 5, 17605. [Google Scholar] [CrossRef]
- Trajanoska, K.; Rivadeneira, F. The genetic architecture of osteoporosis and fracture risk. Bone 2019, 126, 2–10. [Google Scholar] [CrossRef]
- Zanker, J.; Duque, G. Osteoporosis in older persons: Old and new players. J. Am. Geriatr. Soc. 2019, 67, 831–840. [Google Scholar] [CrossRef]
- Raisz, L.G. Pathogenesis of osteoporosis: Concepts, conflicts, and prospects. J. Clin. Investig. 2005, 115, 3318–3325. [Google Scholar] [CrossRef]
- Ukon, Y.; Makino, T.; Kodama, J.; Tsukazaki, H.; Tateiwa, D.; Yoshikawa, H.; Kaito, T. Molecular-based treatment strategies for osteoporosis: A literature review. Int. J. Mol. Sci. 2019, 20, 2557. [Google Scholar] [CrossRef]
- Milat, F.; Ebeling, P.R. Osteoporosis treatment: A missed opportunity. Med. J. Aust. 2016, 205, 185–190. [Google Scholar] [CrossRef]
- Tabatabaei-Malazy, O.; Salari, P.; Khashayar, P.; Larijani, B. New horizons in treatment of osteoporosis. Daru 2017, 25, 2. [Google Scholar] [CrossRef]
- Peichl, P.; Marteau, R.; Griesmacher, A.; Kumpan, W.; Schedl, R.; Prosquil, E.; Fasol, P.; Broll, H. Salmon calcitonin nasal spray treatment for postmenopausal women after hip fracture with total hip arthroplasty. J. Bone Min. Metab. 2005, 23, 243–252. [Google Scholar] [CrossRef]
- Vahle, J.L.; Long, G.G.; Sandusky, G.; Westmore, M.; Ma, Y.L.; Sato, M. Bone neoplasms in F344 rats given teriparatide [rhPTH(1-34)] are dependent on duration of treatment and dose. Toxicol. Pathol. 2004, 32, 426–438. [Google Scholar] [CrossRef]
- Takayanagi, H. The role of NFAT in osteoclast formation. Ann. N. Y. Acad. Sci. 2007, 1116, 227–237. [Google Scholar] [CrossRef]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef] [Green Version]
- Huynh, H.; Wan, Y. mTORC1 impedes osteoclast differentiation via calcineurin and NFATc1. Commun. Biol. 2018, 1, 29. [Google Scholar] [CrossRef]
- Kiani, A.; Habermann, I.; Haase, M.; Feldmann, S.; Boxberger, S.; Sanchez-Fernandez, M.A.; Thiede, C.; Bornhauser, M.; Ehninger, G. Expression and regulation of NFAT (nuclear factors of activated T cells) in human CD34+ cells: Down-regulation upon myeloid differentiation. J. Leukoc. Biol. 2004, 76, 1057–1065. [Google Scholar] [CrossRef]
- Crabtree, G.R.; Olson, E.N. NFAT signaling: Choreographing the social lives of cells. Cell 2002, 109, S67–S79. [Google Scholar] [CrossRef]
- Graef, I.A.; Chen, F.; Crabtree, G.R. NFAT signaling in vertebrate development. Curr. Opin. Genet. Dev. 2001, 11, 505–512. [Google Scholar] [CrossRef]
- Horsley, V.; Pavlath, G.K. NFAT: Ubiquitous regulator of cell differentiation and adaptation. J. Cell. Biol. 2002, 156, 771–774. [Google Scholar] [CrossRef]
- Kim, K.; Lee, S.H.; Ha Kim, J.; Choi, Y.; Kim, N. NFATc1 induces osteoclast fusion via up-regulation of Atp6v0d2 and the dendritic cell-specific transmembrane protein (DC-STAMP). Mol. Endocrinol. 2008, 22, 176–185. [Google Scholar] [CrossRef]
- Yagi, M.; Ninomiya, K.; Fujita, N.; Suzuki, T.; Iwasaki, R.; Morita, K.; Hosogane, N.; Matsuo, K.; Toyama, Y.; Suda, T.; et al. Induction of DC-STAMP by alternative activation and downstream signaling mechanisms. J. Bone Min. Res. 2007, 22, 992–1001. [Google Scholar] [CrossRef]
- Miyamoto, T. Regulators of osteoclast differentiation and cell-cell fusion. Keio J. Med. 2011, 60, 101–105. [Google Scholar] [CrossRef]
- Chiu, Y.H.; Schwarz, E.; Li, D.; Xu, Y.; Sheu, T.R.; Li, J.; de Mesy Bentley, K.L.; Feng, C.; Wang, B.; Wang, J.C.; et al. Dendritic cell-specific transmembrane protein (DC-STAMP) regulates osteoclast differentiation via the Ca2+/NFATc1 axis. J. Cell. Physiol. 2017, 232, 2538–2549. [Google Scholar] [CrossRef]
- Ahn, S.H.; Chen, Z.; Lee, J.; Lee, S.W.; Min, S.H.; Kim, N.D.; Lee, T.H. Inhibitory effects of 2N1HIA (2-(3-(2-fluoro-4-methoxyphenyl)-6-oxo-1(6H)-pyridazinyl)-N-1H-indol-5-ylacetamide) on osteoclast differentiation via suppressing cathepsin K expression. Molecules 2018, 23, 3139. [Google Scholar] [CrossRef]
- Cho, E.; Lee, J.K.; Lee, J.Y.; Chen, Z.; Ahn, S.H.; Kim, N.D.; Kook, M.S.; Min, S.H.; Park, B.J.; Lee, T.H. BCPA {N,N′-1,4-butanediylbis[3-(2-chlorophenyl)acrylamide]} inhibits osteoclast differentiation through increased retention of peptidyl-prolyl cis-trans isomerase never in mitosis a-interacting 1. Int. J. Mol. Sci. 2018, 19, 3463. [Google Scholar] [CrossRef]
Sample Availability: PSTP compounds are commercially available (Chembridge Corp., Chicago, IL, USA). |




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, E.; Chen, Z.; Lee, J.; Lee, S.; Lee, T.-H. PSTP-3,5-Me Inhibits Osteoclast Differentiation and Bone Resorption. Molecules 2019, 24, 3346. https://doi.org/10.3390/molecules24183346
Cho E, Chen Z, Lee J, Lee S, Lee T-H. PSTP-3,5-Me Inhibits Osteoclast Differentiation and Bone Resorption. Molecules. 2019; 24(18):3346. https://doi.org/10.3390/molecules24183346
Chicago/Turabian StyleCho, Eunjin, Zhihao Chen, Jinkyung Lee, Sunwoo Lee, and Tae-Hoon Lee. 2019. "PSTP-3,5-Me Inhibits Osteoclast Differentiation and Bone Resorption" Molecules 24, no. 18: 3346. https://doi.org/10.3390/molecules24183346
APA StyleCho, E., Chen, Z., Lee, J., Lee, S., & Lee, T.-H. (2019). PSTP-3,5-Me Inhibits Osteoclast Differentiation and Bone Resorption. Molecules, 24(18), 3346. https://doi.org/10.3390/molecules24183346