Benzoxaboroles—Novel Autotaxin Inhibitors
by
Kristina Kraljić
, 
Dubravko Jelić
,
Dinko Žiher
,
Adam Cvrtila
,
Snježana Dragojević
,
Verona Sinković
and
Milan Mesić
* Fidelta Ltd., Prilaz baruna Filipovića 29, 10000 Zagreb, Croatia
*
Author to whom correspondence should be addressed.
Molecules 2019, 24(19), 3419; https://doi.org/10.3390/molecules24193419
Submission received: 18 August 2019
/
Revised: 12 September 2019
/
Accepted: 17 September 2019
/
Published: 20 September 2019
(This article belongs to the Collection Efficient Pharmaceutical and Chemical Approaches for Anticancer Therapy: Design, Preliminary Evaluations, and Further Developments)
Abstract
:Autotaxin (ATX) is an extracellular enzyme that hydrolyses lysophosphatidylcholine (LPC) to lysophosphatidic acid (LPA), which has a role in the mediation of inflammation, fibrosis and cancer. ATX is a drug target that has been the focus of many research groups during the last ten years. To date, only one molecule, Ziritaxestat (GLPG1690) has entered the clinic; it is currently in Phase 3 clinical trials for idiopathic pulmonary fibrosis. Other small molecules, with different binding modes, have been investigated as ATX inhibitors for cancer including compounds possessing a boronic acid motif such as HA155. In this work, we targeted new, improved inhibitors of ATX that mimic the important interactions of boronic acid using a benzoxaborole motif as the acidic warhead. Furthermore, we aimed to improve the plasma stability of the new compounds by using a more stable core spacer than that embedded in HA155. Compounds were synthesized, evaluated for their ATX inhibitory activity and ADME properties in vitro, culminating in a new benzoxaborole compound, 37, which retains the ATX inhibition activity of HA155 but has improved ADME properties (plasma protein binding, good kinetic solubility and rat/human plasma stability).
1. Introduction
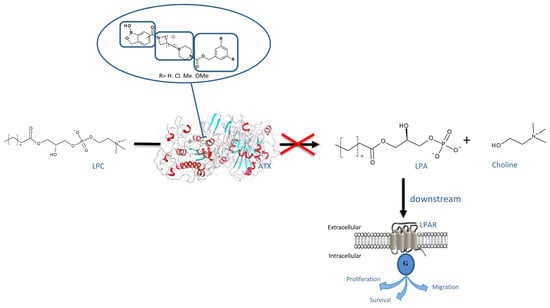
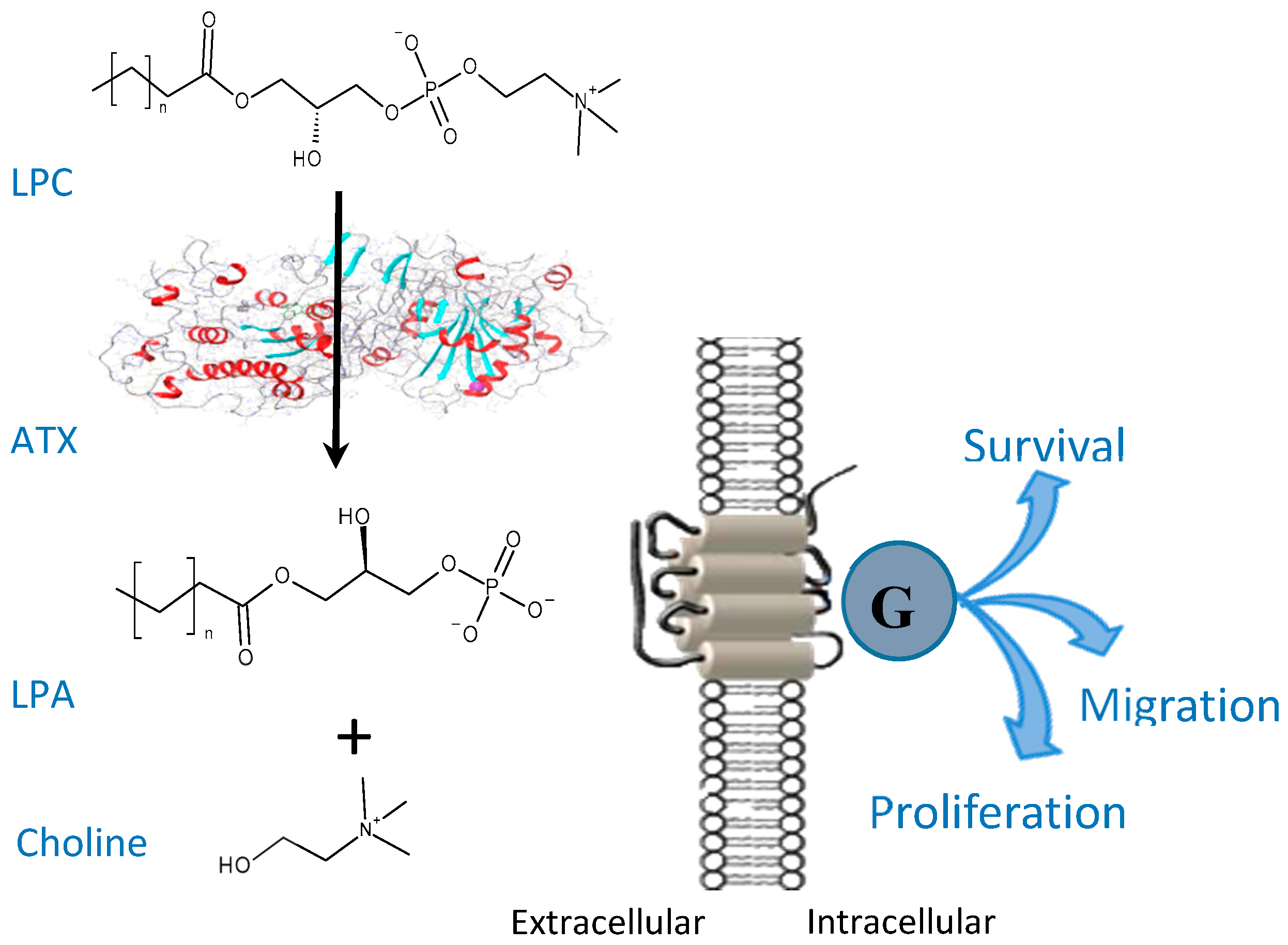
Autotaxin (ATX) is a lysophospholipase D enzyme that hydrolyses the bioactive lipid molecule lysophosphatidylcholine (LPC) to form lysophosphatidic acid (LPA) and choline (Figure 1) [1]. ATX is present in cerebrospinal fluid, blood, peritoneal fluid and synovial fluid [2].
LPA is a mediator of several pathophysiological processes, such as inflammation, fibrosis and cancer [3]. LPA modulates six different G protein-coupled receptors (GPCRs) in the cell membrane. It has been reported that LPA has pro-tumorigenic effects on ovarian, breast and prostate cancer cells [4].
High levels of LPA have also been found in the peritoneal liquids (ascites) of women suffering from ovarian cancer [5]. High levels of LPA (up to 10 times higher than normal) have generally been observed in cancer patients compared to healthy subjects [6]. In mice, Nagano reported that a 4 mg/kg i.v. injection of an ATX inhibitor (such as 3BoA) significantly decreases LPA plasma levels (almost to zero), indicating that ATX is the major enzyme responsible for LPA production [7].
In the search for non-lipid like ATX inhibitors, a thiazolidinedione carboxylic acid analog HA51 (Figure 2) has been reported [8]. Inspired by the proteasome inhibitor bortezomib, in which the boronic acid group interacts with a N-terminal threonine oxygen nucleophile in the proteasome, the carboxylic group in HA51 (Figure 2) was similarly replaced by the boronic acid isostere. The derived compound HA130 has reported activity of 28 nM in an in vitro LPC choline release assay. Crystallography studies published by Hausman [9] further supported the hypothesis that HA130 is engaged in interactions with Thr209 residue and that the 4-fluorobenzyl moiety reaches deep in a hydrophobic pocket.
The molecule HA130 is a potent inhibitor of ATX. Furthermore, in vivo proof of principle studies have demonstrated a rapid decrease of LPA levels in the plasma of mice after an intravenous administration (1 nmol/g), with no overt toxicity issues observed [8]. HA130, in a dose-dependent manner, inhibited the ATX-mediated migration of A2058 human melanoma cells. However, the stability of HA130 in vivo was very poor (half-life less than five minutes) [10]. A later publication from the same authors [11] reported on HA155, a boronic acid regioisomer of HA130, which has a better inhibition of ATX. The same was observed for HA130, as the presence of a double bond attached to the thiazolidinedione heterocycle may cause HA155 to act as a Michael acceptor and may cause in vivo instability. Therefore, in an effort to improve stability, a reduction of the double bond in HA155, and the introduction of the rigid bicyclic tetrahydroisoquinoline as a central ring core have been performed [11].
More recent developments around boronic acid-based ATX inhibitors were published in 2013 by Nagano’s group [7], in which thiazolinone analogues bearing benzylboronic acid groups were investigated. The boronic acid warhead, as in HA155, was designed to bind to Zn2+ ions and to Thr209. Changes designed to optimize lipophilic interactions with the active site of ATX, afforded derivative 3BoA (Figure 2) with good inhibitory properties (IC50 = 13 nM). Compound 3BoA was further tested in vitro and in vivo, showing several very promising pharmacologically related features. In brief 3BoA is more potent than HA155 as an LPA production inhibitor in incubated plasma and cell-motility assays, and it exerts a more favorable metabolic stability and decreases rapidly plasma LPA levels in mice after a 4 mg/kg intravenous administration [10].
A recent (2017) publication by Lanier et al. [12] reported a fragment based approach used to explore phenylboronic acids as warheads towards ATX inhibitors. They tested more than 650 boronic acid fragments. combining in silico computational chemistry filters and crystallography, allowing them to identify fragments that were consistent to known SAR against ATX [12].
The non-boronic acid compound, PF-8380, is one of the most potent in vitro inhibitors of ATX (Figure 2) with an IC50 = of 2.8 nM in a human enzyme assay. PF-8380 was developed by Pfizer in the context of identifying new anti-inflammatory drugs indicating promising in vivo efficacy. Notably, in a separate study, PF-8380 produced a significant anti-invasive effect in glioblastoma cell lines, delaying simultaneously glioma tumor growth progression in vivo [8].
The Ovaa group has speculated that replacing carboxylic acid warheads with less acidic boronic acid warheads may be beneficial for ATX inhibition. Benzoxaboroles, (cyclic boronic acid esters that have lower pKa values that are 1–2 units lower than boronic acids [13]) may have improved binding characteristics towards the ATX enzyme. Crystallographic studies by Hausman have demonstrated further insight for the boronic acid binding mode, in which Thr209 and Zinc atoms are in a complex with boronic acids [9]. This was illustrated by the ring strain generated in the five-membered oxaborole ring where the boron atom was in a neutral, trigonal-planar form [13]. Benzoxaboroles have physicochemical properties which are a consequence of the relatively strong Lewis acidic center on the boron atom and the presence of a free hydroxyl group [14]. The sp2 hybridized boron atom possesses an empty p-orbital which accepts electrons from the hydroxyl group of threonine 209 (Thr209) that explains adduct formation of boronic acids, as in the recent modelling of Lanier [12]. According to Lanier et al. [12], a boronic acid motif with this type of interaction may enhance binding affinity up to 1000 fold. Similarly, we assumed that the binding mode and the mechanism of adduct formation in the ATX active site would resemble phenylboronic acids (Figure 3).
Our group was interested in new ATX inhibitors for cancer treatment. Based on the previous work [9] where boronic acids were used as warheads, we designed and synthesized novel ATX inhibitors with a benzoxaborole as an isostere aiming for the same binding pattern (like HA155) to the active pocket but with better overall drug properties. Working around the potential stability issue due to presence of the double bond, we designed different linkers, introducing more rigidity. We also used the 3,5-substituted benzylic group as a hydrophobic lipid binding motif.
Our design and synthesis of novel ATX inhibitors focused on inhibitors with the benzoxaborole head moiety as the acidic headgroup, the substituted benzyl carbamate moiety as the lipophilic portion, and a rigid core spacer constituted of two saturated heterocyclic rings (Figure 4).
The structures of the warheads and spacer groups to be combined together are shown in Figure 5.
The design of the novel ATX inhibitors was also based on the available protein-ligand x-ray crystal structures in the PDB (Protein Data Bank) (Figure 6).
ATX inhibitors [16] (Figure 7) target the hydrophobic lipid-binding pocket in the central catalytic phosphodiesterase (PDE) domain of ATX (Figure 8A). This is located underneath a shallow groove that accommodates lysophospholipids (Figure 8B). In the active site, the first Zn2+ ion is in the tetrahedral arrangement [17] and is coordinated by the catalytic Thr209 as well as the side chains of Asp171, Asp358, and His359. The second Zn2+ ion coordinates with Asp311, His315 and His474, and, usually, a solvent molecule or counter-ion. Furthermore, an open tunnel (or ‘channel’; Figure 6C), which is partially hydrophobic in nature, is located in close proximity where a variety of molecules can be accommodated, forming a ‘T-intersection’ with the shallow groove [18,19].
In our hands, a standard docking protocol within Glide (with and without H-bond/metal constraints) failed to dock HA155 in the active site of ATX in the pose observed in the x-ray structure (PDB ID: 2XRG [9]) This pose has the characteristic interaction of Thr209 with the boron atom. Based on natural bond orbital (NBO) calculations [20], the existence of polar character of Thr209 and boron atom of HA155 hybridized to sp3 character, we applied a covalent docking approach. Using covalent docking, HA155 was successfully docked in the conformation observed in the crystal structure (RMSD (heavy atoms) = 0.68 Å, Figure 6). This same approach was therefore applied to our compounds in this paper. (Figure 7).
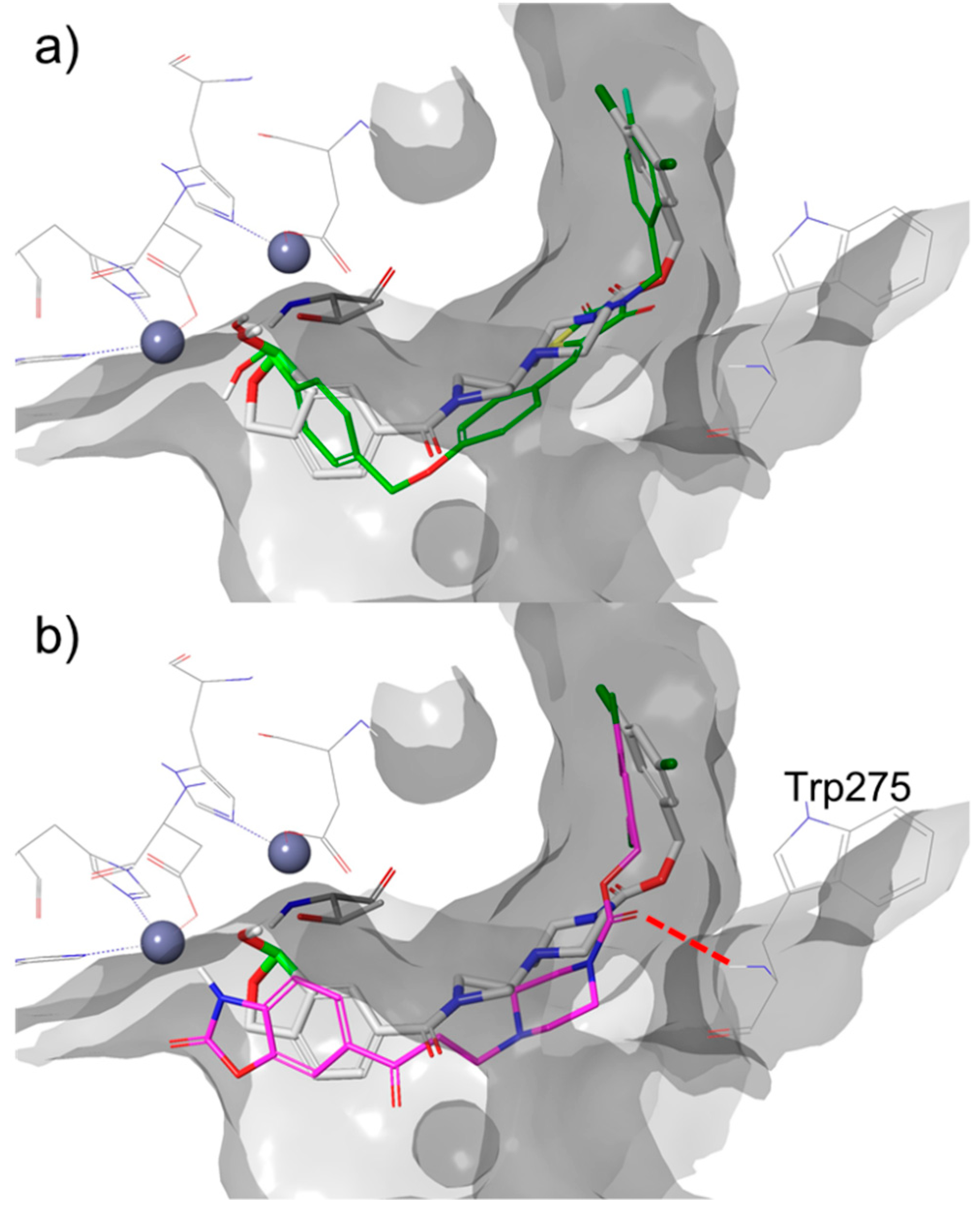
The dative covalent bond between the boron atom and Thr209 is required for these inhibitors [11], and docking poses thus generated are in line with the previously reported co-crystalized structures of ATX (Figure 8a). In addition, this reversible covalent bond places the hydroxyl moiety of the boronate between both zinc ions in the ATX active site (Figure 7). The docking studies also showed that the other oxygen atom of the boronate is involved in an interactions with the amides of Thr209 (d(O…H–NThr209) = 2.6 Å) and/or Asn230 Thr209 (d(O…H–NAsn230) = 2.3 Å), which are in close proximity. The binding of the rest of the molecule is mainly driven by hydrophobic interactions in similar way as other ligands with similar ‘tails’—such as PF-8380 (PDB ID: 5L0K [21], Figure 8b). The aromatic ring at the end of the ‘tail,’ a hydrophobic 3,5-dichlorobenzene, points directly into a deep hydrophobic pocket. The hydrogen bond between the carbonyl group pf the carbamate and the Trp 275 amide has been observed in the generated docking poses for some compounds described in this paper, similar to what is revealed in the co-crystalized structure with PF-8380 (d (O…H–NTrp275) = 3.1 Å, Figure 8b) and ATX.
As previously suggested for SAR transfer between series of active compounds, targeting this hydrogen bond can be useful in the modification of this class of benzoxaboroles [22]. The lipophilic tail of the molecule was designed according to data published for the PF-8380 molecule. This is where the carbonyl group creates a hydrogen bond with the acceptor of a Trp275 [9]. To reduce the degrees of conformational freedom, the rigidity of the molecule increased by the introduction of several acyclic rings. These rings would bridge the between warhead benzoxaborole and carbamate moiety (Figure 8).
During the final preparation of the manuscript, the Kang group analyzed the topological water network in the binding pocket of ATX. Using this pharmacophoric insight, new molecules were synthesized and tested for their ATX inhibitory activities [23].
2. Results
2.1. ATX Inhibition
All 21 new benzoxaboroles derivatives prepared herein were evaluated for ATX inhibition in vitro using a biochemical choline detection assay, combining elements of assays already described in the literature [24]. ATX activity was measured using Lyso PC (16:1) as a substrate and HA155 was used as the standard, control, compound.
clogP values were calculated for all compounds using the Percepta software [25]. Chrom logD was determined experimentally using a procedure described in the supplementary data. Furthermore, the correlation between calculated clogP values and experimentally determined Chrom log D values were explored for molecules containing boron functional groups such as boronic acids and benzoxaboroles. The results are presented in Table 1.
2.2. ADME Properties
Eight of the most active compounds, together with HA155, were selected for characterization of in vitro ADME properties such as solubility, microsomal metabolic stability, plasma stability and plasma protein binding (PPB). Results are presented in Table 2.
3. Discussion
The modification of several regions had dramatic effects on the inhibition of ATX. Thus, the hydrophobic part of the inhibitors showed sensitivity to phenyl substitution—the unsubstituted phenyl compound (41) being almost inactive (23 µM). The 3,5 dimethoxyphenyl analogue (49) was a slightly better (11 µM). Ten times better potency was achieved when comparing to 41 with 3,5-dimethyl substitution on phenyl ring for compound 45. The original 3,5-dichlorosubstitution (already described for PF-8380) remained the most favorable substitution pattern for the majority of the compounds prepared. SAR around the core spacer showed that the piperazine–azetidine bicycle was the best spacer. The position of the boron atom on the aromatic ring (acidic headgroup) was also very important and differed to that observed with HA155. In our case, the most active compound was the 6-amido substituted benzoxaborole (compound 37), whilst 4-amido or 7-amido substitution was not tolerated. Among 21 tested compounds, four compounds showed ATX inhibition in the nanomolar range of activity. The IC50 for the most active compound (37) was 130 nM. In our LPC choline released assay, HA155 also showed nanomolar activity (88 nM) but was less potent then reported in published data (5.7 nM) [11]. Keeping in mind that the analogue of HA155 with tetrahydroisoquinoline as a rigid core spacer was significantly less potent (ten times) then HA155, our molecule 36 with space core rigidity was only two times less potent that HA155.
The kinetic solubility in plasma bovine serum (PBS) at pH 7.4, determined by turbidimetric method, was >100 µM for the majority of tested compounds. The most active compound 36 had a solubility of 30–100 µM, while HA155 suffered from very poor solubility (1–3 µM). Results regarding metabolic stability in rat and human liver microsomes suggest that the majority of tested compounds would be classified a high-predicted in vivo hepatic clearance (expressed as percentage of liver blood flow, %LBF). Two exceptions include compounds 37 and 45, which had a moderate predicted in vivo clearance (62% and 45%, respectively) in rats. However, in human liver microsomes, all compounds showed high-predicted in vivo clearance, whereas the comparator compound showed a moderate predicted in vivo clearance. The results of plasma stability which were measured at four hours indicated a good stability of compound 37 that remained 71.2% of its initial concentration in rat and 79.1% of its initial concentration in human plasma. However, HA155 was less stable with only 65.7% of its initial concentration in rat and 42.9% of its initial concentration in rat and human plasma, respectively, remaining after four hours. The binding to plasma proteins (PPB) of newly synthesized compounds was significantly lower when compared with binding of standard compounds that showed a very high PPB of >99.9% in both tested species. That would impact efficacy of 37 in comparison to HA155 since their free fractions are different more than 20 times assuming that both molecules have the same binding kinetics kon/koff.
4. Experimental Section
4.1. Materials and Instruments
All chemicals, solvents, and chemical and biochemical reagents were of analytical grade and purchased from commercial sources (Merck, Merck KGaA, Darmstadt, Germany, Fluka Sigma-Aldrich Laborchemikalien GmbH, Hannover, Germany, Alfa Aesar, Karlsruhe, Germany and Sigma, St. Louis, MO, USA, Combi-blocks, Combi-blocks Inc.San Diego, USA, TCI, TCI-Europe, Zwijndrecht, Belgium, Kemika, Kemika d.d., Zagreb, Croatia). All starting materials were obtained from commercial sources (Merck, Merck KGaA, Darmstadt, Germany, Fluka Sigma-Aldrich Laborchemikalien GmbH, Hannover, Germany, Alfa Aesar, Karlsruhe, Germany, and Sigma, St. Louis, MO, USA) and used without further purification.
The progress of all reactions was checked by UPLC-MS/UV Waters system (Waters, Waters Corporation, Milford, USA) and thin layer chromatography (TLC) using precoated Silica Gel 60F254 sheets (Merck, Darmstadt, Germany). The spots on plates were visualized under UV light (254 nm).
Column chromatography was performed by an Interchim Puriflash 450 system or by the Waters Mass Directed AutoPurification system.
The 1H and 13C nucleic magnetic resonance (NMR) spectra were recorded at 400 MHz on a Bruker 400 spectrometer (Bruker Analytische Messtechnik GmbH, Rheinstetten, Germany). Chemical shifts were determined relatively to the signals of residual protons of the deuterated solvent (DMSO). Chemical shifts are reported in delta (δ) units in parts per million (ppm), and splitting patterns are designated as s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet and br, broad. Coupling constants were recorded in Hertz (Hz). High resolution mass spectra were determined on an Agilent 1100 series.
4.2. Synthesis
4.2.1. General Procedure for the Preparation of Benzoxaborolic Acids
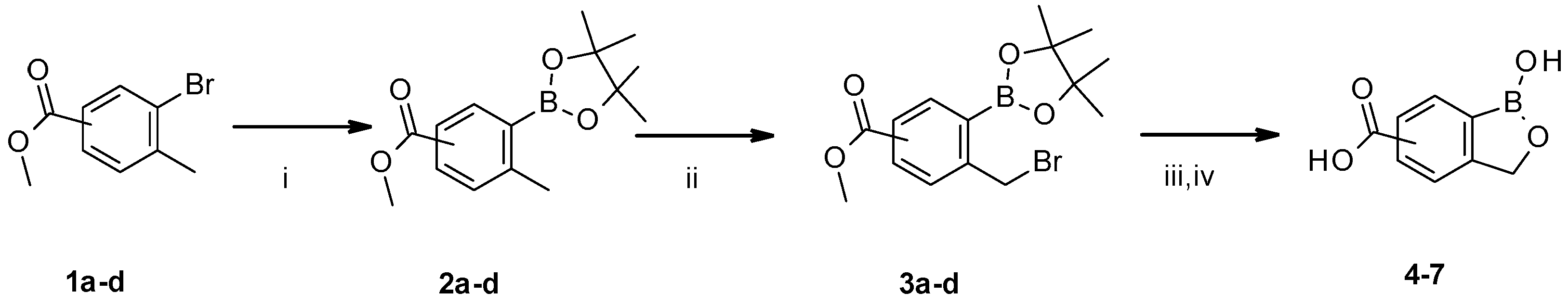
Reactions were performed under argon atmosphere using syringe septa technique. Tolyl bromide 1a–d (8.731 mmol), pinacol diboron (12.223 mmol, 1.4 eq) and KOAc (26.193 mmol, 3 eq) were dissolved in dioxane (180 mL) at rt, and the resulting mixture was bubbled with argon. PdCl2(dppf) (0.873 mol, 0.1 eq) was added and stirring was continued for 16 h at 90 °C. The reaction mixture was cooled to rt, quenched with NH4Cl (70 mL), and extracted with Et2O (3 × 70 mL). The organic layers were combined, dried over Na2SO4/MgSO4, filtered off end evaporated in vacuo to give raw material which was been purified by Interchim Puriflash 450 in the gradient of MeOH in dichloromethane (DCM): 0%–5% in 20 column volumes. The appropriate fractions were combined and evaporated in vacuo to give corresponding intermediates 2a–d. [27] To a solution of corresponding intermediates 2a–d (7.243 mmol) in benzene (40 mL), NBS (7.605 mmol, 1.05 eq) and AIBN (0.362 mmol, 0.05 eq) were added and the reaction mixture stirred at reflux. four hours later solvent had evaporated in vacuo, and Et2O was added to the residue (40 mL). The resulting precipitate was filtered off, and the filtrate got extracted with 20% KOH (3 × 30 mL). The aqueous layers were combined and stirred at rt 90 min. The solution was cooled to 0 °C, and pH was adjusted to 2 with 6N aqueous HCl solution. The white precipitate was collected by filtration to afford corresponding benzoxaborole acids 4–7 (Scheme 1).
methyl 4-methyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate 2a [28]
3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoic acid 2b [29]
methyl 2-methyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate 2c [22]
methyl 3-methyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate 2d, Yield: 53%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 7.84–7.88 (m, 1H), 7.49–7.57 (m, 2H), 3.97 (s, 3H), 2.49 (s, 3H), 1.47 (s, 12H) 13C NMR (DMSO-d6, 100 MHz, δ ppm): 167.7, 140.8, 133.6, 132.6, 128.8, 125.7, 83.4, 52.3, 25.0, 21.0 HRMS: m/z calculated for C15H21BO4 [M + H]+ 276.1642; found 276.1639.
1-hydroxy-3H-2,1-benzoxaborole-6-carboxylic acid 4 [30]
1-hydroxy-3H-2,1-benzoxaborole-5-carboxylic acid 5 [30]
1-hydroxy-3H-2,1-benzoxaborole-7-carboxylic acid 6 [31]
1-hydroxy-3H-2,1-benzoxaborole-4-carboxylic acid 7, Yield: 58%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.28 (s, 1H), 8.02 (m, 1H), 7.96 (m, 1H), 7.50 (m, 1H), 5.21 (s, 2H) 13C NMR (DMSO-d6, 100 MHz, δ ppm): 167.2, 134.9, 132.1, 127.4, 71.3 HRMS: m/z calculated for C8H7BO4 [M − H]− 176.0401; found 176.0397.
4.2.2. General Procedure for the Preparation of Carbamate Intermediates
The first step in the synthetic pathway was the reduction of an appropriate carboxylic acid (8e–g) by LiAlH4. The entire reaction was performed under an argon atmosphere using a syringe septa technique. To a solution of lithium aluminum hydride (0.063 mol, 1.2 eq) in THF (15 mL) that was cooled in an ice bath, a solution of the substituted benzoic acid (8e–g) (0.052 mol, 1 eq) in THF (65 mL) was added dropwise during 30 minutes. The resulting mixture was stirred at ambient temperature for 2 h. To the reaction mixture were added: Water (3 mL), a 1M solution of NaOH (3 mL), and then water (10 mL) whilst stirring continued for a further 90 minutes at ambient temperature. The mixture was extracted with EtOAc (3 × 30 mL). The organic layers were combined, dried over Na2SO4/MgSO4, filtered and evaporated in vacuo to give a crude product which was purified by Interchim Puriflash 450 with a gradient of EtOAc in cyclohexane. The appropriate fractions were combined and evaporated in vacuo to give corresponding alcohols (9e–g). The resultant alcohols (0.039 mol, 1 eq) and CDI (0.039 mol, 1 eq) were dissolved in DMF (100 mL) at ambient temperature, and the resulting mixture was treated with the Boc protected piperazine (0.035 mol, 0.9 eq), and stirring was continued for 16 h at ambient temperature. Solvent was evaporated in vacuo. Water (50 mL) was added to the residue. Extraction with dichloromethane (DCM) (3 × 30 mL) followed. The organic layers were combined, dried over Na2SO4/MgSO4, filtered off, end evaporated in vacuo to give raw material which was purified by Interchim Puriflash 450 in the gradient of MeOH in DCM: 0%–3% in 20 CV. The appropriate fractions were combined and evaporated in vacuo to give corresponding Boc protected piperazine carbamates (10e–h). To a solution of Boc protected piperazine carbamate (10e–h) (0.035 mol, 1 eq) in DCM (200 mL), cooled in an ice bath, TFA (0.353 mol, 10 eq) was added. The resulting mixture was stirred at ambient temperature for 30 minutes. The pH of the reaction mixture was adjusted to 8 with 6N aqueous solution of NaOH and layers were separated. The aqueous layer was washed with DCM (2 × 100 mL). The organic layers were combined, dried over Na2SO4/MgSO4, filtered off and evaporated in vacuo to give corresponding piperazine carbamates intermediates (11e–h). The piperazine carbamate intermediates (11e–h) (0.010 mol, 1 eq) and corresponding N-Boc protected cyclic ketone (0.010 mol, 1 eq) were dissolved in DCM (45 mL) at rt, and to the resulting solution NaBH(OAc)3 (0.013 mol, 1.3 eq) was added in portions before stirring was continued for 16 h at rt. The reaction mixture was quenched with a saturated solution of NaHCO3 (30 mL). The layers were separated, and the aqueous layer was washed with DCM (2 × 20 mL). The organic layers were combined, dried over Na2SO4/MgSO4, filtered off and evaporated in vacuo to give crude product which was purified by Interchim Puriflash 450, in a gradient of MeOH in DCM: 0%–5% in 20 column volumes. The appropriate fractions were combined and evaporated in vacuo to give corresponding Boc protected intermediates (12–20). Finally, the Boc protecting group was cleaved with TFA to obtain carbamate intermediates (21–29) (Scheme 2).
O4-tert-butyl O1-[(3,5-dichlorophenyl)methyl] piperazine-1,4-dicarboxylate [32]
O4-tert-butyl O1-[(3,5-dimethylphenyl)methyl] piperazine-1,4-dicarboxylate 10f, Yield: 86%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 6.94 (s, 3H), 4.99 (s, 2H), 3.32–3.39 (m, 2H), 2.25 (s, 6H), 1.39 (s, 9H) 13C NMR (DMSO-d6, 100 MHz, δ ppm): 179.3, 154.0, 153.7, 137.4, 136.5, 129.5, 125.3, 79.1, 66.4, 43.2, 28.0, 20.8 HRMS: m/z calculated for C15H21BO4 [M + H-100]+ 276.1642; found 276.1641.
O4-tert-butyl O1-[(3,5-dimethoxyphenyl)methyl] piperazine-1,4-dicarboxylate 10g, Yield: 83%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 7.84–7.88 (m, 2H), 7.49–7.57 (m, 1H), 5.02 (s, 2H), 3.72 (s, 6H), 3.32–3.39 (m, 2H), 1.47 (s, 12H) 13C NMR (DMSO-d6, 100 MHz, δ ppm): 179.7, 179.5, 154.3, 153.7, 139.0, 105.3, 99.4, 79.1, 66.1, 55.1, 27.9 HRMS: m/z calculated for C19H28N2O6 [M + Na]+ 403.1840; found 403.1825.
O1-benzyl O4-tert-butyl piperazine-1,4-dicarboxylate 10h [33]
(3,5-dimethylphenyl)methyl piperazine-1-carboxylate 11f, Yield: 98%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 8.19 (br. s, 1H), 6.93 (m, 3H), 5.00 (s, 2H), 2.62–2.70 (m, 2H), 2.25 (s, 6H) 13C NMR (DMSO-d6, 100 MHz, δ ppm): 179.7, 179.5, 163.2, 161.0, 137.4, 129.3, 129.2, 125.5, 125.4, 70.0, 66.5, 66.2, 45.1, 20.8 HRMS: m/z calculated for C14H20N2O2 [M + H]+ 249.1598; found 249.1599.
(3,5-dimethoxyphenyl)methyl piperazine-1-carboxylate 11g, Yield: 97%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 6.39–6.45 (m, 3H), 4.99 (s, 2H), 3.72 (s, 6H), 2.58–2.68 (m, 2H) 13C NMR (DMSO-d6, 100 MHz, δ ppm): 179.2, 160.5, 160.4, 154.4, 139.3, 105.3, 105.1, 99.3, 65.8, 55.2, 55.1, 45.3, 44.5 HRMS: m/z calculated for C14H20N2O4 [M + H]+ 281.1496; found 281.1522.
(3,5-dichlorophenyl)methyl 4-(1-tert-butoxycarbonylazetidin-3-yl)piperazine-1-carboxylate 12, Yield: 72%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 7.74–7.85 (m, 1H), 7.5–7.72 (m, 2H), 5.30 (s, 2H), 3.99–4.12 (m., 2H), 3.80–3.94 (m, 2H), 3.52–3.71 (m, 4H), 1.60 (s, 9H) 13C NMR (DMSO-d6, 100 MHz, δ ppm): 155.6, 154.1, 141.2, 134.1, 127.7, 126.1, 78.6, 64.7, 53.0, 48.6, 43.2, 28.0 HRMS: m/z calculated for C20H27Cl2N3O4 [M + H]+ 444.1451; found 444.1450.
(3,5-dichlorophenyl)methyl 4-(1-tert-butoxycarbonylpyrrolidin-3-yl)piperazine-1-carboxylate 13, Yield: 60%, 1H NMR (DMSO-d6, 400 MHz, δ ppm) 7.52–7.60 (m, 1H), 7.36–7.47 (m, 2H), 5.03 (s, 2H), 3.37–3.56 (m, 4H), 3.09–3.21 (m, 1H), 3.00–3.07 (m, 1H), 2.84–2.99 (m, 1H), 2.64–2.87 (m, 1H), 2.37–2.45 (m, 1H), 1.89–1.92 (m., 1H), 1.37 (s, 9H) 13C NMR (DMSO-d6, 100 MHz, δ ppm): 153.9, 141.2, 134.1, 127.4, 126.3, 126.2, 115.3, 64.8, 64.7, 64.4, 64.7, 50.8, 49.2, 44.7, 28.9 HRMS: m/z calculated for C21H29Cl2N3O4 [M + H]+ 458.1608; found 458.1625.
(3,5-dichlorophenyl)methyl 4-(1-tert-butoxycarbonyl-4-piperidyl)piperazine-1-carboxylate 14, Yield: 77%, 1H NMR (DMSO-d6, 400 MHz δ ppm) 7.60 (s, 1H), 7.41–7.51 (m, 2H), 5.11 (s, 2H), 3.92–4.04 (m, 2H), 3.39–3.49 (m, 2H), 2.69–2.76 (m, 2H), 2.38–2.52 (m, 2H), 1.69–1.71 (m, 2H), 1.43 (s, 9H), 1.20–1.35 (m, 2H) 13C NMR (DMSO-d6, 100 MHz, δ ppm): 154.0, 153.8, 141.3, 134.0, 127.4, 126.1, 78.5, 64.6, 60.8, 48.3, 43.9, 28.0, 27.6 HRMS: m/z calculated for C22H31Cl2N3O4 [M + H]+ 472.1764; found 472.1774.
(3,5-dimethylphenyl)methyl 4-(1-tert-butoxycarbonylazetidin-3-yl)piperazine-1-carboxylate 15, Yield: 67%, 1H NMR (DMSO-d6, 400 MHz δ ppm) 7.89–7.96 (m, 3H), 4.97 (s, 2H), 3.75–3.83 (m, 2H), 3.59–3.68 (m, 2H), 3.35–3.39 (m, 2H), 2.97–3.06 (m,1H), 2.24 (s, 6H), 2.17–2.23 (m, 2H), 1.47 (s, 9H) 13C NMR (DMSO-d6, 100 MHz, δ ppm): 179.4, 155.6, 154.4, 137.4, 136.6, 129.2, 125.5, 125.4, 78.6, 66.3, 53.0, 48.7, 43.2, 28.0, 20.8 HRMS: m/z calculated for C22H33N3O4 [M + H]+ 404.2544; found 404.2561.
(3,5-dimethylphenyl)methyl 4-(1-tert-butoxycarbonylpyrrolidin-3-yl)piperazine-1-carboxylate 16, Yield: 59%, 1H NMR (DMSO-d6, 400 MHz, δ ppm) 7.01 (s, 3H), 5.08 (s, 2H), 3.38–3.44 (m, 4H), 3.11–3.26 (m, 1H), 2.91–3.03 (m, 1H), 2.71–2.87 (m, 1H), 2.39–2.49 (m, 1H), 2.30–2.39 (m, 1H), 2.29 (s, 6H), 1.61–1.75 (m, 1H), 1.42 (s, 9H) 13C NMR (DMSO-d6, 100 MHz, δ ppm): 179.0, 154.3, 153.4, 137.4, 136.6, 129.2, 125.4, 78.3, 66.2, 63.5, 62.7, 50.8, 49.3, 49.2, 44.6, 44.4, 43.3, 28.9, 28.1, 20.8 HRMS: m/z calculated for C23H36N3O4 [M + H]+ 418.2711; found 418.2718.
(3,5-dimethoxyphenyl)methyl 4-(1-tert-butoxycarbonylazetidin-3-yl)piperazine-1-carboxylate 17, Yield: 74%, 1H NMR (DMSO-d6, 400 MHz, δ ppm) 6.45–6.51 (m, 2H), 6.42 (s, 1H), 4.99 (s, 2H), 3.75–3.87 (m, 2H), 3.72 (s, 6H), 3.59–3.68 (m, 2H), 3.34–3.46 (m, 4H), 2.98–3.07 (m, 1H), 1.36 (s, 9H) 13C NMR (DMSO-d6, 100 MHz, δ ppm): 179.1, 160.5, 155.5, 154.3, 139.2, 105.2, 99.3, 78.6, 66.0, 55.2, 55.1, 53.0, 48.7, 43.2, 28.0 HRMS: m/z calculated for C22H33N3O6 [M + H]+ 436.2442; found 436.2428.
(3,5-dimethoxyphenyl)methyl 4-(1-tert-butoxycarbonylpyrrolidin-3-yl)piperazine-1-carboxylate 18, Yield: 55%, 1H NMR (DMSO-d6, 400 MHz, δ ppm) 6.46–6.49 (m, 2H), 6.40–6.45 (m, 1H), 4.99 (s, 2H), 3.72 (s, 6H), 3.45–3.53 (m, 1H), 3.35–3.42 (m, 4H), 2.87–2.98 (m, 1H), 2.61–2.69 (m, 1H), 2.34–2.43 (m, 1H), 2.27–2.35 (m, 1H), 1.94–2.06 (m, 1H), 1.37 (s, 9H) 13C NMR (DMSO-d6, 100 MHz, δ ppm): 179.5, 160.5, 105.2, 99.3, 65.9, 55.2 28.1 HRMS: m/z calculated for C22H35N3O6 [M + H]+ 450.2599 found 450.2641.
(3,5-dichlorophenyl)methyl 4-(azetidin-3-yl)piperazine-1-carboxylate 21, Yield: 72%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 7.51–7.62 (m, 1H), 7.38–7.42 (m, 2H), 5.06 (s, 2H), 4.07–4.16 (m., 2H), 3.78–3.14 (m, 2H), 3.52–3.71 (m, 2H), 13C NMR (DMSO-d6, 100 MHz, δ ppm): 179.1, 161.1, 154.1, 141.2, 134.1, 127.5, 126.2, 64.8, 54.8, 53.0, 50.5, 48.6, 43.2 HRMS: m/z calculated for C15H19Cl2N3O2 [M + H]+ 344.0922; found 344.0931.
3,5-dichlorophenyl)methyl 4-pyrrolidin-3-ylpiperazine-1-carboxylate 22, Yield: 99%, 1H NMR (DMSO-d6, 400 MHz, δ ppm) 7.52–7.60 (m, 1H), 7.36–7.47 (m, 2H), 5.03 (s, 2H), 3.00–3.07 (m, 1H), 2.84–2.99 (m, 1H), 2.75–2.83 (m, 1H), 2.64–2.87 (m, 1H), 2.59–2.64 (m, 1H), 1.87–1.89 (m, 1H), 1.55–1.68 (m, 1H) 13C NMR (DMSO-d6, 100 MHz, δ ppm): 154.0, 141.2, 134.1, 127.4, 126.2, 126.1, 64.7, 64.1, 51.0, 48.6, 44.7, 43.4, 28.5 HRMS: m/z calculated for C16H21Cl2N3O2 [M + H]+ 458.1608; found 458.1625.
(3,5-dichlorophenyl)methyl 4-(4-piperidyl)piperazine-1-carboxylate 23, Yield: 99%, 1H NMR (DMSO-d6, 400 MHz, δ ppm) 7.52–7.60 (m, 1H), 7.36–7.47 (m, 2H), 5.03 (s, 2H), 3.92–4.04 (m, 2H), 3.39–3.49 (m, 2H), 2.69–2.76 (m, 2H), 2.38–2.52 (m, 2H), 1.69–1.71 (m, 2H), 1.20–1.35 (m, 2H) 13C NMR (DMSO-d6, 100 MHz, δ ppm): 154.0, 153.8, 141.3, 134.0, 127.4, 126.1, 64.6, 48.3, 43.9 HRMS: m/z calculated for C17H23Cl2N3O2 [M + H]+ 372.124; found 372.1237.
(3,5-dimethylphenyl)methyl 4-(azetidin-3-yl)piperazine-1-carboxylate 24, Yield: 99%, 1H NMR (DMSO-d6, 400 MHz, δ ppm) 6.94–7.96 (m, 3H), 4.99 (s, 2H), 3.72–3.79 (m, 2H), 3.64–3.71 (m, 2H), 2.25 (s, 6H) 13C NMR (DMSO-d6, 100 MHz, δ ppm): 179.5, 165.4, 154.4, 137.4, 136.6, 129.3, 125.4, 66.3, 56.0, 54.7, 49.4, 48.4, 20.9, 20.8 HRMS: m/z calculated for C22H33N3O4 [M + H]+ 404.2544; found 404.2561.
(3,5-dimethylphenyl)methyl 4-pyrrolidin-3-ylpiperazine-1-carboxylate 25, Yield: 99%, 1H NMR (DMSO-d6, 400 MHz, δ ppm) 6.89–7.05 (m, 3H), 5.01 (s, 2H), 3.73 (m, 1H), 3.31–3.46 (m, 1H), 3.11–3.26 (m, 1H), 2.91–3.03 (m, 1H), 2.71–2.87 (m, 1H), 2.39–2.49 (m, 1H), 2.30–2.39 (m, 1H), 2.29 (s, 6H), 1.61–1.75 (m, 1H) 13C NMR (DMSO-d6, 100 MHz, δ ppm): 179.5, 154.4, 137.4, 136.6, 129.3, 125.4, 66.3, 62.8, 50.7, 47.2, 44.0, 43.3, 27.4, 20.9 HRMS: m/z calculated for C18H27N3O2 [M + H]+ 318.2181; found 318.2186.
(3,5-dimethoxyphenyl)methyl 4-(azetidin-3-yl)piperazine-1-carboxylate 26, Yield: 95%, 1H NMR (DMSO-d6, 400 MHz, δ ppm) 6.45–6.51 (m, 2H), 6.42 (s, 1H), 4.99 (s, 2H), 3.75–3.87 (m, 2H), 3.72 (s, 6H), 3.59–3.68 (m, 2H), 3.34–3.46 (m, 2H), 2.98–3.07 (m, 1H), 2.24 (m, 4H) 13C NMR (DMSO-d6, 100 MHz, δ ppm): 179.6, 160.5, 154.3, 139.2, 105.2, 99.3, 66.0, 56.0, 55.2, 49.2, 48.4, 43.1 HRMS: m/z calculated for C17H25N3O4 [M + H]+ 336.1918; found 336.1939.
(3,5-dimethoxycyclohexa-1,5-dien-1-yl)methyl 4-pyrrolidin-3-ylpiperazine-1-carboxylate 27, Yield: 93%, 1H NMR (DMSO-d6, 400 MHz, δ ppm) 6.46–6.49 (m, 2H), 6.40–6.45 (m, 1H), 4.99 (s, 2H), 3.72 (s, 6H), 3.00–3.18 (m, 2H), 2.87–2.98 (m, 1H), 2.70–2.83 (m, 1H), 2.59–2.63 (m, 1H), 2.37–2.47 (m, 2H), 1.87–1.96 (m, 1H), 1.54–1.67 (m, 1H) 13C NMR (DMSO-d6, 100 MHz, δ ppm): 179.5, 160.5, 154.2, 139.2, 150.2, 105.1, 99.3, 66.0, 64.3, 55.2, 41.1, 48.3, 28.5 HRMS: m/z calculated for C18H27N3O4 [M + H]+ 350.2074; found 350.2089.
4.2.3. General Procedure for the Preparation of Novel Autotaxin Inhibitors
To a solution of benzoxaborolic acid (4–7) (0.232 mmol, 1 eq), the carboxylate intermediate (21–29) (0.232 mmol) in DCM (1 mL), EDAC (0.372 mmol, 1.6 eq) and DMAP (0.009 mmol, 0.04 eq) were added at ambient temperature, and the resulting mixture was stirred for 16 h. The solvent was evaporated in vacuo, and the raw material was purified by a Waters Mass Directed AutoPurification system, giving corresponding novel autotaxin inhibitors (30–50) (Scheme 3).
(3,5-dichlorophenyl)methyl 4-[1-(1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)pyrrolidin-3-yl]piperazine-1-carboxylate30, Yield: 79%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.29 (br.s, 1H), 7.87 (d, J = 7.6 Hz, 1H), 7.61 (d, J = 7.5 Hz, 1H), 7.55 (s, 1H), 7.35–7.48 (m, 3H), 5.05 (s, 2H), 5.01 (s, 2H), 3.71–3.80 (m, 2H), 3.58–3.67 (m, 2H), 3.50–3.58 (m, 2H), 2.84–2.94 (m, 2H), 2.73–2.84 (m, 2H), 2.34–2.47 (m, 2H), 2.08–2.18 (m, 2H), 13C NMR (DMSO-d6, 400 MHz, δ ppm): 168.6, 141.2, 134.1, 129.7, 129.3, 127.5, 126.2, 121.3, 69.9, 67.9, 63.7, 62.4, 54.3, 25.9 HREI-MS: m/z calculated for C24H26BCl2N3O5 [M]+ 517.1452; found 517.1448.
(3,5-dichlorophenyl)methyl 4-[1-(1-hydroxy-3H-2,1-benzoxaborole-7-carbonyl)pyrrolidin-3-yl]piperazine-1-carboxylate31, Yield: 53%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.28 (br.s, 1H), 7.78 (d, J = 7.7 Hz, 1H), 7.69–7.73 (m, 1H),7.56 (s, 1H) 7.48–7.51 (m, 1H), 7.39–7.43 (m, 1H), 5.05 (s, 2H), 5.01 (s, 2H), 3.70–3.82 (m, 2H), 3.52–3.63 (m, 2H), 3.26–3.42 (m, 4H), 3.18–3.29 (m, 1H), 2.34–2.43 (m, 4H), 1.99–2.15 (m, 2H), 13C NMR (DMSO-d6, 400 MHz, δ ppm): 168.6, 141.2, 134.1, 129.7, 129.3, 127.5, 126.2, 121.3, 69.9, 67.9, 63.7, 62.4, 54.3, 25.9, HREI-MS: m/z calculated for C24H26BCl2N3O5 [M]+ 517.1452; found 517.1446.
(3,5-dichlorophenyl)methyl 4-[1-(1-hydroxy-3H-2,1-benzoxaborole-5-carbonyl)pyrrolidin-3-yl]piperazine-1-carboxylate 32, Yield: 68%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.33 (br.s, 1H), 7.66–7.88 (m, 1H), 7.47–7.59 (m, 2H), 7.35–7.46 (m, 3H), 5.04 (s, 2H), 5.00 (s, 2H), 3.70–3.76 (m, 2H), 3.48–3.62 (m, 2H), 2.85–2.92 (m, 4H), 2.06–2.17 (m, 4H), 1.95–2.06 (m, 2H) 13C NMR (DMSO-d6, 400 MHz, δ ppm): 168.4, 134.1, 131.9, 129.9, 127.4, 126.2, 125.8, 120.0, 99.8, 98.6, 62.0, 25.2, 19.7, 8.1, HREI-MS: m/z calculated for C24H26BCl2N3O5 [M]+ 517.1452; found 517.1451.
(3,5-dichlorophenyl)methyl 4-[1-(1-hydroxy-3H-2,1-benzoxaborole-4-carbonyl)pyrrolidin-3-yl]piperazine-1-carboxylate) 33, Yield: 59%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.32 (br.s, 1H), 7.82–7.86 (m, 1H), 7.71–7.74 (m, 1H), 7.62–7.58 (m, 1H), 7.34–7.42 (m, 2H),7.41–7.51 (m, 1H), 5.03 (s, 2H), 5.00 (s, 2H), 3.70–3.82 (m, 2H), 3.37–3.54 (m, 2H), 3.23–3.32 (m, 5H), 2.15–2.25 (m, 4H), 1.96–2.25 (m, 2H), 13C NMR (DMSO-d6, 400 MHz, δ ppm): 168.4, 134.1, 131.9, 129.9, 127.4, 126.2, 125.8, 120.0, 99.8, 98.6, 62.0, 25.2, 19.7, 8.1 HREI-MS: m/z calculated for C24H26BCl2N3O5 [M]+ 517.1452; found 517.1448.
3,5-dichlorophenyl)methyl 4-[1-(1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)-4-piperidyl]piperazine-1-carboxylate34, Yield: 81%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.31 (br.s., 1H), 7.71 (d, J = 7.6 Hz, 1H), 7.54 (s, 1H), 7.45–7.47 (m, 2H) 7.38–7.42 (m, 2H), 5.05 (s, 2H), 5.00 (s, 2H), 4.41–4.53 (m, 1H), 3.54–3.65 (m, 1H), 2.93–3.07 (m, 1H), 2.70–2.82 (m, 1H), 2.50–2.54 (m, 1H), 2.44–2.54 (m, 4H), 2.43–2.47 (m, 4H), 1.77–1.87 (m, 1H), 1.63–1.74 (m, 1H), 1.29–1.42 (m, 2H) 13C NMR (DMSO-d6, 400 MHz, δ ppm): 169.1, 154.9, 154.0, 141.2, 134.9, 134.0, 129.2, 128.7, 127.4, 126.1, 121.4, 69.9, 64.7, 60.8, 48.4, 43.9, HREI-MS: m/z calculated for C25H28BCl2N3O5 [M]+ 531.1608; found 531.1606.
(3,5-dichlorophenyl)methyl 4-[1-(1-hydroxy-3H-2,1-benzoxaborole-5-carbonyl)-4-piperidyl]piperazine-1-carboxylate35, Yield: 73%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.32 (br.s., 1H), 7.75 (d, J = 7.6 Hz, 1H), 7.54 (s, 1H), 7.40 (d, J = 7.4 Hz, 1H), 7.5–7.42 (m, 2H), 7.29–7.34 (m, 1H), 5.05 (s, 2H), 4.99 (s, 2H), 4.20–4.42 (m, 1H), 3.50–3.60 (m, 2H), 2.94–3.05 (m, 2H), 2.70–2.80 (m, 1H), 2.50–2.54 (m, 1H), 2.44–2.54 (m, 4H), 2.43–2.47 (m, 4H), 1.77–1.86 (m, 1H), 1.60–1.69 (m, 1H) 13C NMR (DMSO-d6, 400 MHz, δ ppm): 168.8, 154.0, 141.5, 138.4, 134.0, 130.5, 127.3, 126.3, 125.1, 119.5, 69.9, 64.8, 60.7, 48.3, 43.9, 28.4, HREI-MS: m/z calculated for C25H28BCl2N3O5 [M]+ 531.1608; found 531.1607.
(3,5-dichlorophenyl)methyl 4-[1-(1-hydroxy-3H-2,1-benzoxaborole-4-carbonyl)-4-piperidyl]piperazine-1-carboxylate 36, Yield: 59%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.93 (s, 1H), 7.52–7.62 (m, 2H), 7.41 (s, 1H), 7.20–7.34 (m, 2H), 7.15–7.18 (m, 1H), 5.06 (s, 4H), 4.30–4.41 (m, 1H), 3.87–4.00 (m, 2H), 2.91–3.05 (m, 2H), 2.67–2.80 (m, 1H), 2.57–2.66 (m, 4H), 2.50–2.54 (m, 1H), 1.62–1.83 (m, 1H) 13C NMR (DMSO-d6, 400 MHz, δ ppm): 168.8, 154.0, 141.5, 138.4, 134.0, 130.5, 127.3, 126.3, 125.1, 119.5, 69.9, 64.8, 60.7, 48.3, 43.9, 28.4 HREI-MS: m/z calculated for C25H28BCl2N3O5 [M]+ 531.1608; found 531.1605.
(3,5-dichlorophenyl)methyl 4-[1-(1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)azetidin-3-yl]piperazine-1-carboxylate 37, Yield: 83%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.29 (br.s., 1H), 8.00–8.02 (m, 1H), 7.73 (d, J = 7.7 Hz, 1H), 7.55 (s, 1H), 7.47 (d, J = 7.9 Hz, 1H), 7.40 (d, J = 1.9 Hz, 2H), 5.06 (s, 2H), 5.02 (s, 2H), 4.03–4.14 (m, 2H), 3.84–3.90 (m, 1H), 3.36–3.48 (m, 4H), 3.13–3.21 (m, 1H), 2.25–2.36 (m, 4H), 13C NMR (DMSO-d6, 400 MHz, δ ppm): 169.2, 156.7, 154.0, 141.3, 134.0, 131.7, 129.9, 127.5, 126.2, 121.6, 70.2, 64.8, 53.9, 48.9, 43.2, HREI-MS: m/z calculated for C23H24BCl2N3O5 [M]+ 503.1295; found 503.1299.
(3,5-dichlorophenyl)methyl 4-[1-(1-hydroxy-3H-2,1-benzoxaborole-5-carbonyl)azetidin-3-yl]piperazine-1-carboxylate 38, Yield: 77%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.33 (br.s., 1H), 7.78 (d, J = 7.8 Hz, 1H), 7.62–7.64 (m, 1H), 7.54–7.58 (m, 2H), 7.41 (d, J = 1.9 Hz, 2H), 5.06 (s, 2H), 5.00 (s, 2H), 4.20–4.28 (m,2H), 3.99–4.12 (m, 2H), 3.09–3.20 (m, 1H), 2.24–2.32 (m, 4H), 13C NMR (DMSO-d6, 400 MHz, δ ppm): 169.2, 156.7, 154.0, 141.3, 134.0, 131.7, 129.9, 127.5, 126.2, 121.6, 70.2, 64.8, 53.9, 48.9, 43.2, HREI-MS: m/z calculated for C23H24BCl2N3O5 [M]+ 503.1295; found 503.1301.
benzyl 4-[1-(1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)pyrrolidin-3-yl]piperazine-1-carboxylate 39, Yield: 82%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.26 (br.s, 1H), 7.70–7.80 (m, 1H), 7.54–7.70 (m, 1H), 7.41–7.49 (m, 1H), 7.24–7.41 (m, 5H), 5.06 (s, 2H), 5.01 (s, 2H), 3.71–3.79 (m, 2H), 3.58–3.67 (m, 2H), 3.49–3.57 (m, 2H), 2.84–2.92 (m, 2H), 2.74–2.83 (m, 2H), 2.34–2.43 (m, 2H), 2.07–2.17 (m, 2H), 13C NMR (DMSO-d6, 400 MHz, δ ppm): 168.5, 155.5, 154.2, 136.8, 135.4, 129.6, 129.2, 128.4, 127.9, 121.3, 69.9, 66.2, 63.7, 62.2, 52.5, 50.7, 47.9, 43.3, HREI-MS: m/z calculated for C24H28BN3O5 [M]+ 449.2231; found 449.2240.
benzyl 4-[1-(1-hydroxy-3H-2,1-benzoxaborole-5-carbonyl)pyrrolidin-3-yl]piperazine-1-carboxylate 40, Yield: 71%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.30 (br.s, 1H), 7.71–7.81 (m, 1H), 7.48–7.53 (m, 1H), 7.40–7.47 (m, 1H), 7.27–7.38 (m, 5H), 5.05 (s, 2H), 5.00 (s, 2H), 3.72–3.79 (m, 2H), 3.58–3.67 (m, 2H), 3.45–3.53 (m, 4H), 3.38–3.44 (m, 4H), 3.19–3.29 (m, 1H), 2.06–2.28 (m, 2H), 13C NMR (DMSO-d6, 400 MHz, δ ppm): 168.4, 154.5, 154.0, 138.6, 137.0, 132.0, 128.5, 127.8, 127.5, 125.6, 120.0, 69.9, 66.1, 63.6, 62.3, 50.7, 43.4, HREI-MS: m/z calculated for C24H28BN3O5 [M]+ 449.2231; found 449.2243.
benzyl 4-[1-(1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)azetidin-3-yl]piperazine-1-carboxylate 41, Yield: 87%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.31 (br.s., 1H), 7.78 (d, J = 7.9 Hz, 1H), 7.65 (s, 1H), 7.58 (d, J = 7.7 Hz, 1H), 7.29–7.39 (m, 5H), 5.07 (s, 2H), 5.03 (s, 2H), 4.03–4.15 (m, 2H), 3.85–3.91 (m, 1H), 3.39–3.46 (m, 4H), 3.14–3.21 (m, 1H), 2.25–2.34 (m, 4H), 13C NMR (DMSO-d6, 400 MHz, δ ppm): 169.5, 156.5, 154.5, 137.1 132.0, 130.2, 128.5, 127.8, 127.6, 121.5, 69.9, 66.1, 57.1, 53.5, 52.9, 48.7, 43.1, HREI-MS: m/z calculated for C23H26BN3O5 [M]+ 435.2075; found 435.2079.
benzyl 4-[1-(1-hydroxy-3H-2,1-benzoxaborole-5-carbonyl)azetidin-3-yl]piperazine-1-carboxylate 42, Yield: 53%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.33 (br.s., 1H), 7.78 (d, J = 7.6 Hz, 1H), 7.65 (s, 1H), 7.58 (d, J = 7.6 Hz, 1H), 7.29–7.39 (m, 5H), 5.07 (s, 2H), 5.03 (s, 2H), 4.04–4.14 (m, 2H), 3.85–3.90 (m, 2H), 3.37–3.45 (m, 4H), 3.12–3.19 (m, 1H), 2.24–2.35 (m, 4H), 13C NMR (DMSO-d6, 400 MHz, δ ppm): 169.1, 154.5, 153.9, 136.8, 135.0, 134.4, 130.5, 128.5, 127.9, 126.4, 120.4, 69.9, 66.2, 56.5, 53.6, 52.2, 48.7, 43.1, HREI-MS: m/z calculated for C23H26BN3O5 [M]+ 435.2075; found 435.2078.
(3,5-dimethylphenyl)methyl 4-[1-(1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)pyrrolidin-3-yl]piperazine-1-carboxylate 43, Yield: 79%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.32 (br.s, 1H), 7.79–7.92 (m, 1H), 7.57–7.68 (m, 1H), 7.45 (d, J = 7.8 Hz, 1H), 6.91–7.02 (m, 3H), 5.01 (s, 2H), 4.97 (s, 2H), 3.69–3.82 (m, 2H), 3.54–3.67 (m, 4H), 3.32–3.51 (m, 2H), 2.17–2.38 (m, 1H), 1.97–2.06 (m, 1H), 13C NMR (DMSO-d6, 400 MHz, δ ppm): 168.5, 155.4, 137.5, 136.6, 129.6, 129.3, 125.5, 121.3, 63.8, 62.3, HREI-MS: m/z calculated for C26H32BN3O5 [M]+ 477.2544; found 477.2546.
(3,5-dimethylphenyl)methyl 4-[1-(1-hydroxy-3H-2,1-benzoxaborole-5-carbonyl)pyrrolidin-3-yl]piperazine-1-carboxylate 44, Yield: 52%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.34 (br.s, 1H), 7.76–(d, J = 7.5 Hz, 1H), 7.56 (s, 1H), 7.47–7.55 (m, 1H), 6.88–6.97 (m, 3H), 4.92–5.03 (m, 4H), 3.70–3.82 (m, 2H), 3.37–3.45 (m, 2H), 3.23–3.32 (m, 5H), 2.24 (s, 6H), 2.15–2.25 (m, 4H), 1.96–2.17 (m, 2H), 13C NMR (DMSO-d6, 400 MHz, δ ppm): 168.4, 154.5, 153.7, 137.5, 136.6, 130.5, 129.3, 125.7, 125.4, 120.0, 63.7, 62.4, 20.8, HREI-MS: m/z calculated for C26H32BN3O5 [M]+ 477.2544; found 477.2547.
(3,5-dimethylphenyl)methyl 4-[1-(1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)azetidin-3-yl]piperazine-1-carboxylate 45, Yield: 78%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.29 (br.s., 1H), 8.01 (d, J = 1.6 Hz, 1H), 7.73 (d, J = 7.8 Hz,1H), 7.47 (d, J = 7.7 Hz, 1H), 6.88–6.98 (m, 3H), 5.02 (s, 2H), 4.97 (s, 2H), 4.03–4.23 (m, 2H), 3.82–3.90 (m, 2H), 3.36–3.44 (m, 4H), 3.12–3.20 (m, 1H), 2.26–2.34 (m, 4H), 2.24 (s, 6H), 13C NMR (DMSO-d6, 400 MHz, δ ppm): 169.3, 156.3, 154.5, 137.4, 136.5, 131.6, 130.1, 129.2, 128.0, 125.4, 124.2, 121.5, 70.0, 66.3, 62.9, 53.6, 48.7, 20.8, HREI-MS: m/z calculated for C25H30BN3O5 [M]+ 463.2388; found 463.2390.
(3,5-dimethylphenyl)methyl 4-[1-(1-hydroxy-3H-2,1-benzoxaborole-5-carbonyl)azetidin-3-yl]piperazine-1-carboxylate 46, Yield: 77%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.32 (br.s., 1H), 7.78 (d, J = 7.6 Hz, 1H), 7.63 (s, 1H), 7.56 (d, J = 7.6 Hz,1H), 6.88–6.98 (m, 3H), 5.00 (s, 2H), 4.97 (s, 2H), 4.26–4.38 (m, 2H), 3.83–3.4.02 (m, 2H), 3.35–3.42 (m, 4H), 3.11–3.18 (m, 1H), 2.25–2.33 (m, 4H), 2.24 (s, 6H), 13C NMR (DMSO-d6, 400 MHz, δ ppm): 169.3, 156.3, 154.5, 137.4, 136.5, 131.6, 130.1, 129.2, 128.0, 125.4, 124.2, 121.5, 70.0, 66.3, 62.9, 53.6, 48.7, 20.8 HREI-MS: m/z calculated for C25H30BN3O5 [M]+ 463.2388; found 463.2391.
(3,5-dimethoxyphenyl)methyl 4-[1-(1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)pyrrolidin-3-yl]piperazine-1-carboxylate 47, Yield: 71%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.29 (br.s, 1H), 7.78 (d, J = 7.7 Hz, 1H), 7.63 (s, 1H), 7.56 (d, J = 7.6 Hz, 1H), 6.38–6.54 (m, 3H), 4.93–5.06 (m, 4H), 4.26–4.40 (m, 4H), 3.83–4.02 (m, 2H),3.73 (s, 6H), 3.35–3.42 (m, 4H), 3.11–3.18 (m, 1H), 2.25–2.33 (m, 4H), 13C NMR (DMSO-d6, 400 MHz, δ ppm): 160.6, 129.4, 121.6, 105.6, 99.5, 69.9, 64.0, 55.4, 50.8, HREI-MS: m/z calculated for C26H32BN3O7 [M]+ 509.2442; found 509.2444.
(3,5-dimethoxyphenyl)methyl 4-[1-(1-hydroxy-3H-2,1-benzoxaborole-5-carbonyl)pyrrolidin-3-yl]piperazine-1-carboxylate 48, Yield: 66%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.27 (br.s, 1H), 7.78 (d, J = 7.4 Hz, 1H), 7.64 (s, 1H), 7.46–7.63 (m, 1H), 6.45–6.53 (m, 2H), 6.43 (s, 1H), 4.93–5.08 (m, 4H), 3.71–3.75 (m, 2H), 3.70 (s,6H), 3.46–3.68 (m, 6H), 3.18–3.29 (m, 1H), 2.33–2.46 (m, 4H), 1.97–2.18 (m, 2H), 13C NMR (DMSO-d6, 400 MHz, δ ppm): 160.3, 130.7, 130.3, 125.6, 125.4, 120.0, 106.8, 105.3, 99.3, 55.1, HREI-MS: m/z calculated for C26H32BN3O7 [M]+ 509.2442; found 509.2444.
(3,5-dimethoxyphenyl)methyl 4-[1-(1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)azetidin-3-yl]piperazine-1-carboxylate 49, Yield: 83%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.27 (br.s., 1H), 7.99–8.01 (m, 1H), 7.73 (d, J = 8.0 Hz, 1H), 7.47 (d, J = 8.0 Hz, 1H), 6.41–6.50 (m, 3H), 5.00 (s, 2H), 4.99 (s, 2H), 4.26–4.33 (m, 4H), 3.84–3.91 (m, 2H), 3.72 (s, 6H), 3.35–3.48 (m, 4H), 3.12–3.20 (m, 1H), 2.25–2.35 (m, 4H), 13C NMR (DMSO-d6, 400 MHz, δ ppm): 160.6, 156.4, 154.5, 139.3, 134.3, 131.8, 130.1, 130.0, 129.5, 129.1, 121.5, 105.2, 99.4, 55.2, 54.4, 53.5, HREI-MS: m/z calculated for C25H30BN3O7 [M]+ 495.2286; found 495.2285.
((3,5-dimethoxyphenyl)methyl 4-[1-(1-hydroxy-3H-2,1-benzoxaborole-5-carbonyl)azetidin-3-yl]piperazine-1-carboxylate 50, Yield: 73%, 1H NMR (DMSO-d6, 400 MHz, δ ppm): 9.34 (br.s., 1H), 7.79 (d, J = 7.4 Hz, 1H), 7.65 (s, 1H), 7.59 (d, J = 7.4 Hz, 1H), 6.42–6.51 (m, 3H), 5.00 (s, 2H), 4.99 (s, 2H), 4.23–4.31 (m, 2H), 3.83–3.91 (m, 2H), 3.72 (s, 6H), 3.34–3.46 (m, 4H), 3.14–3.20 (m, 1H), 2.23–2.36 (m, 4H), 13C NMR (DMSO-d6, 400 MHz, δ ppm): 169.2, 160.5, 154.3, 139.4, 134.9, 130.6, 126.2, 120.7, 106.7, 105.8, 99.3, 69.9, 66.0, 55.1, 53.6, 48.7, 43.3, HREI-MS: m/z calculated for C25H30BN3O7 [M]+ 495.2286; found 495.2286.
4.3. Molecular Modelling Studies
All visualizations and molecular modelling studies were done using Schrodinger Suite 2018-1 [19]. The co-crystal structure of ATX in complex with the HA155 boronic acid inhibitor (PDB ID: 2XRG) [9] was used as a template for the docking studies due to the similarity between HA155 and examined molecules. The protein was prepared (modelled) using Protein Preparation Wizard (with Epik v4.3, Impact v7.8, and Prime v5.1) using default settings. Missing hydrogen atoms and sidechains were added, the hydrogen bond network was optimized, and the structure was minimized using the OPLS-2005 force field [38]. The initial neutral 3D structures of ligands were generated by LigPrep and OPLS2005 force-field was used.
Water molecules were removed from the protein structure, and zinc ions of the active site were retained within the structure for docking studies. Docking was performed using Covalent Docking v1.3 [15] within Glide v7.8, where Thr209 of ATX was selected as a reactive residue in the formation of covalent bond in the reaction of boron acid addition [20]. Default values were used for the receptor grid generation around HA155 ligand in the binding site. Performance of the covalent docking was tested by re-docking of HA155 into ATX (Figure 5). This verified protocol was used for the docking of boronate compounds in this paper.
4.4. Biochemical Enzyme Assay
Benzoxaborole derivatives were tested in the choline detection assay for ATX inhibition measuring ATX activity using LysoPC (16:1) as a substrate (Figure 9). Mother plates with serial dilutions of compounds in pure DMSO were prepared from 10 mM DMSO stock solutions. Storplate-384-deep-well-V plates (Perkin Elmer) were also used for mother plate preparation. Compounds were diluted in a DMSO in a ratio of 1:3. An aliquot of 200 nL of compound solution was transferred from the mother plate to the test plate with a Mosquito nanoliter liquid handling system (TTP labtech). The assay was performed in Spectra-Plate-384 TC, transparent, (Perkin Elmer, Cat#6007658) using a 20 µL assay volume. The total final percentage of DMSO in the test media was 1%. Compounds were tested at 11 consecutive three-fold dilutions starting from 50 µM (final concentrations) in duplicate, covering range of 0.0008–50 μM to determine IC50 values. The control compound (HA 155) was tested on every test plate. After dispensing of compounds directly to the dry plate, we added 10 μL of recombinant ATX (2 ng/mL) in a Tris–HCl buffer (140 mM NaCl, 1mM MgCl2, 5 mM KCl, 3 mM CaCl2, 1 mg/ml bovin serum albumin (BSA, fatty acid free) and 100 mM Tris–HCl, pH 9). Finally, 10 μL of 200 μM LysoPC (16:1) in a Tris–HCl buffer was added to each well, while the plate was incubated 90 min at 37 °C. The above-described solution with dimethyl sulfoxide but without the test compound was used as a control. LysoPC without ATX was used as a control for the autohydrolysis of LysoPC. After 1.5 h of incubation, 20 μL of stop solution and developer was added {Choline oxidase (1.2 U/mL), Peroxidase (1.2 U/mL), TOOS (2.7 mM), 4-Aminoantipyrine (2.7 mM), and EDTA (50 mM)} to the reaction mixture, and absorbance was measured after 30 min of incubation. The absorbance was measured by using Perkin-Elmer EnVision plate reader (λ = 555 nm). Data were analyzed using GraphPad Prism software. The calculation of IC50 data, curves and quality control (QC) analysis was made by using Excel tools and GraphPadPrism software. Briefly, individual concentration–effect curves were generated by plotting the logarithm of the tested concentration of tested compounds (X) vs. corresponding percentage inhibition values (Y) using a least squares (ordinary) fit. Best fit IC50 values were calculated using the Log (inhibitor) vs. response – variable slope (four parameters) equation, where Y = 100/(1 + 10^((LogIC50–X)*HillSlope)). QC criteria parameters (Z,’ S:B, ΔmP) were checked for every plate.
4.5. Lipophilicity Determination
Chrom logD values were determined from the following equation: Chrom logD = 0.0857 × CHI − 2 [39]. Chromatographic hydrophobic index (CHI) values were determined from the gradient retention times (RT). These values approximately corresponded to the volume percentage of organic component in the mobile phase when the compound elutes. CHI values were determined at pH 7.4. Chromatograms were obtained using on Agilent 1100 Series liquid chromatography system with an HPLC diode-array detector (DAD) coupled with a Micromass Quattro micro atmosphere pressure ionization (API) mass spectrometer. The column used for analyses was Phenomenex Luna C18 (50 × 3 mm, 5 µm, 100 Å). The column was maintained at 25 °C. The flow rate was 1.0 mL/min. The mobile phase was composed of 50 mM ammonium acetate in water at pH 7.4 (solvent A) and acetonitrile (solvent B). The solvent gradient condition was initially 0.5% B, and then it increased linearly to 100% B over 3.0 min, remaining at 100% B for 0.5 min, then decreasing linearly to 0% B over 0.2 min followed by an equilibration period of 1.3 min prior to the next injection. The total experimental duration was 5 min. Injection volume was 2.0 µl. Samples temperature was maintained at 15 °C in the autosampler chamber. A group of ten standard compounds having different CHI values was used for calibration of RP-HPLC [40].
4.6. Kinetic Solubility
Turbidimetric solubility allows for a rapid determination of kinetic solubility by using small amounts of the test compound. Assay controls were α-naphtoflavone (low solubility) and sulfaphenazole (expected solubility >100µM). DMSO stock solutions (10 mM) of each compound were first serially diluted 1 in 3 in DMSO and then spiked into an aqueous buffer (100 mM PBS), resulting in 5 following final concentration: 100, 30, 10, 3 and 1 µM (each concentration was tested in duplicate). The prepared solutions were then incubated at 37 °C for 2 h with gentle shaking, followed by absorbance measurements on microplate reader (Infinite F500, Tecan, CH) at 620 nm (at t = 0 and t = 2h). Absorbance is proportionally increased with the concentration of insoluble particles as compound precipitates. Compound samples were compared to a solvent control in an aqueous buffer (DMSO 1% final concentration), and a significant increase of sample absorbance was considered to have occurred when the absorbance reached a 3-fold standard deviation of the average DMSO absorbance. The calculation of solubility range data was made by Excel tool software. Results are expressed as an estimated solubility range.
4.7. Plasma Stability
The in vitro plasma stability of the test compounds was studied in human and rat plasma with propranolol, benflorex, and eucatropine as control substances. All compounds at final concentrations of 5 μM (0.5% DMSO) were incubated in a phosphate buffer (50 mM, pH 7.4) for 4 h at 37 °C together with plasma. Aliquots were taken at different time points (0, 30, 120 min and 4 h), followed by reaction termination by the addition of CH3CN:CH3OH 2:1 v/v., which contained diclofenac and warfarin as internal standards (IS). Samples were then centrifuged (at 1600 rpm at 4 °C for 30 min), and the resulting supernatants were subjected to LC/MS/MS analysis. Results are expressed as % remaining.
4.8. In Vitro Metabolic Stability in Rat and Liver Human Microsomes
Metabolic stability was assessed in rat and human liver microsomes, with testosterone and propranolol as positive controls. All compounds, at a final concentration of 1 μM (0.03% DMSO), were incubated in a phosphate buffer (50 mM, pH 7.4) for 60 min at 37 °C, together with liver microsomes in the absence and presence of NADPH generating system (nicotinamide adenine dinucleotide phosphate (NADP, 2 mM), glucose-6-phosphate (20 mM), glucose-6-phosphate dehydrogenase (6U) and magnesium chloride (2 mM)). Aliquots were taken at different time points (0, 10, 20, 30, 45 and 60 min), followed by reaction termination by addition of CH3CN:CH3OH 2:1 v/v, containing diclofenac as internal standard (IS). Samples were then centrifuged (at 2000 rpm at 4 °C for 30 min), and resulting supernatants were subjected to LC/MS/MS analysis. The in vitro half-life was calculated in GraphPadPrism software from % remaining vs. time regression using non-linear regression fit. In vitro intrinsic clearance, expressed as mL/min/g liver, was determined from in vitro half-life and normalized for the protein concentration in the incubation mixture, assuming a 52.5 mg/g liver. Finally, predicted in vivo hepatic clearance, expressed as %LBF (liver blood flow), was determined from in vitro intrinsic clearance normalized for the weight over whole body weight (87.5 g/kg for mouse and 25.7 g/kg for human) and liver blood flow (131 mL/min/kg for mouse and 21 mL/min/kg for human).
4.9. Plasma Protein Binding Assay
Plasma protein binding was measured using the equilibrium dialysis technique. Positive controls included in this assay were: Acebutolol (low binding) and nicardipine (very high binding). The assay was performed in a 96-well Teflon dialysis unit, where each well consisted of two chambers separated by a dialysis membrane. Plasma (human and rat, commercially obtained from Seralab) spiked with each compound (final concentration: 5 µM, 0.5% DMSO) were added to one chamber, and a blank buffer solution (60 mM Na2HPO4/14 mM KH2PO4/70 mM NaCl; pH 7.4) was added to the other side of the well. Each compound was analyzed in duplicate, for 4 h at 37 °C. After 4 h, both the plasma (free and bound fraction) and buffer (only free fraction) side of the well were sampled, matrix matched, then precipitated with a STOP solution (CH3CN:CH3OH 2:1 v/v, containing diclofenac as internal standard), and centrifuged at 2000 rpm for 30 min (+4 °C). The resulting supernatants were analyzed by LC-MS/MS. The fraction bound was calculated as the concentration difference (analyte vs. internal standard peak area ratio) in the plasma and buffer sides divided by the total concentration in the plasma side. Sampling was also performed from spiked plasma at t = 0 min in order to determine recovery, i.e., % of compound detected at t = 4 h in both the plasma and buffer chambers divided by the compound detected at t = 0 min.
5. Conclusions
Our strategy was to target the Thr209 and zinc atoms in the binding pocket of ATX, with the boronic acid isostere: Borinic acid. As well as similar enzyme potencies, we expected better ADME properties of such modified compounds. The group occupying the hydrophobic lipid-binding pocket was optimally that of the Pfizer molecule PF-8380. As the core spacer, we used saturated bicyclic systems (piperazino–piperidine, piperazino–pyrolidine and piperazino azetidine). This overall design was ratified by docking experiments, and the molecules were then synthesized (compounds 30–50). ATX inhibition in vitro and ADME properties were measured aiming for better overall profile than that possessed by HA130 and HA155. The compounds HA130 and HA155 were reported to be active in the in vivo study, lowering LPA significantly but with a very short half-life (less than five min) [10]. The four new compounds designed, synthesized and tested in this work (30, 32, 34, and 37) showed ATX inhibition in the nanomolar range with the IC50 value for the most active compound 37 being 130 nM, which is only two times less potent than HA155. Compound 37 has a core spacer more rigid than in HA155, designed for better stability. Most of ADME parameters for tested compounds were better than for HA155. The plasma stability for compound 37 showed 71.2% and 79.1% remaining in rat and human plasma, respectively, after four hours. HA155 showed 65.7% and 42.9% in rat and human plasma, respectively, also measured at four hours. Furthermore, the PPB of 37 was significantly lower than for HA155 (fraction unbound for 37 was 1.6% in rat and 2.1% in human plasma, while for HA155 was 0.4% and less than 0.1%, respectively). Based on our findings, we have identified potent ATX inhibitors with in vitro enzyme inhibition potencies similar to that of HA155 but with an improved ADME profile.
Based on these findings, additional experiments in vivo are ongoing to further evaluate the PK properties of the compound 37 head to head with HA155. If the PK data of compound 37 show better stability and half-life then HA155, the compound 37 will be tested in relevant animal oncology models.
Author Contributions
M.M. conceived and designed the study. K.K. performed the synthesis of compounds S.D. performed HRMS analysis. D.Ž. performed molecular modelling and docking experiments. D.J. and V.S. performed biological assays; A.C. performed ADME in vitro biological and kinetic solubility assays. M.M. and K.K. wrote the paper.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Barbayianni, E.; Magrioti, V.; Moutevelis-Minakakis, P.; Kokotos, G. Autotaxin inhibitors: A patent review. Expert Opin. Ther. Pat. 2013, 23, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Fotopoulou, S.; Oikonomou, N.; Grigorieva, E.; Nikitopoulou, I.; Paparountas, T.; Thanassopoulou, A.; Zhao, Z.; Xu, Y.; Kontoyiannis, D.L.; Remboutsika, E.; et al. ATX expression and LPA signalling are vital for the development of the nervous system. Dev. Biol. 2010, 339, 451–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, G.B.; Moolenaar, W.H. The emerging role of lysophosphatidic acid in cancer. Nat. Rev. Cancer 2003, 3, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, N.C.; Chun, J. Promising pharmacological directions in the world of lysophosphatidic acid signaling. Biomol. Ther. 2015, 23, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.J.; Park, S.Y.; Cho, K.H.; Sohn, J.S.; Lee, J.; Kim, Y.K.; Kang, J.; Park, C.G.; Han, J.W.; Lee, H.Y. The Rho/ROCK pathway for lysophosphatidic acid-induced proteolytic enzyme expression and ovarian cancer cell invasion. Oncogene 2012, 31, 4279–4289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leblanc, R.; Peyruchaud, O. New insights into the autotaxin/LPA axis in cancer development and metastasis. Exp. Cell Res. 2015, 333, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Okabe, T.; Okudaira, S.; Nishimasu, H.; Ishitani, R.; Kojima, H.; Nureki, O.; Aoki, J.; Nagano, T. Screening and X-ray crystal structure-based optimization of autotaxin (ENPP2) inhibitors, using a newly developed fluorescence probe. ACS Chem. Biol. 2013, 8, 1713–1721. [Google Scholar] [CrossRef]
- Matralis, A.N.; Afantitis, A.; Aidinis, V. Development and therapeutic potential of autotaxin small molecule inhibitors: From bench to advanced clinical trials. Med. Res. Rev. 2019, 39, 976–1013. [Google Scholar] [CrossRef]
- Hausmann, J.; Kamtekar, S.; Christodoulou, E.; Day, J.E.; Wu, T.; Fulkerson, Z.; Albers, H.M.H.G.; van Meeteren, L.A.; Houben, A.J.S.; van Zeijl, L.; et al. Structural basis of substrate discrimination and integrin binding by autotaxin. Nat. Struct. Mol. Biol. 2011, 18, 198–204. [Google Scholar] [CrossRef] [Green Version]
- Albers, H.M.H.G.; Dong, A.; van Meeteren, L.A.; Egan, D.A.; Sunkara, M.; van Tilburg, E.W.; Schuurman, K.; van Tellingen, O.; Morris, A.J.; Smyth, S.S.; et al. Boronic acid-based inhibitor of autotaxin reveals rapid turnover of LPA in the circulation. Proc. Natl. Acad. Sci. USA 2010, 107, 7257–7262. [Google Scholar] [CrossRef] [Green Version]
- Albers, H.M.H.G.; Hendrickx, L.J.D.; van Tol, R.J.P.; Hausmann, J.; Perrakis, A.; Ovaa, H. Structure-based design of novel boronic acid-based inhibitors of autotaxin. J. Med. Chem. 2011, 54, 4619–4626. [Google Scholar] [CrossRef] [PubMed]
- Lanier, M.; Cole, D.C.; Istratiy, Y.; Klein, M.G.; Schwartz, P.A.; Tjhen, R.; Jennings, A.; Hixon, M.S. Repurposing Suzuki coupling reagents as a directed fragment library targeting serine hydrolases and related enzymes. J. Med. Chem. 2017, 60, 5209–5215. [Google Scholar] [CrossRef] [PubMed]
- Tomsho, J.W.; Pal, A.; Hall, D.G.; Benkovic, S.J. Ring structure and aromatic substituent effects on the pKa of the benzoxaborole pharmacophore. ACS Med. Chem. Lett. 2012, 3, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk-Woźniak, A.; Borys, K.M.; Sporzyński, A. Recent developments in the chemistry and biological applications of benzoxaboroles. Chem. Rev. 2015, 115, 5224–5247. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Borrelli, K.W.; Greenwood, J.R.; Day, T.; Abel, R.; Farid, R.S.; Harder, E. Docking covalent inhibitors: A parameter free approach to pose prediction and scoring. J. Chem. Inf. Model. 2014, 54, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Castagna, D.; Budd, D.C.; Macdonald, S.J.F.; Jamieson, C.; Watson, A.J.B. Development of autotaxin inhibitors: An overview of the patent and primary literature: Miniperspective. J. Med. Chem. 2016, 59, 5604–5621. [Google Scholar] [CrossRef] [PubMed]
- Dokmanić, I.; Šikić, M.; Tomić, S. Metals in proteins: Correlation between the metal-ion type, coordination number and the amino-acid residues involved in the coordination. Acta Crystallogr. Sect. D Biol. Crystallogr. 2008, 64, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Perrakis, A.; Moolenaar, W.H. Autotaxin: Structure-function and signaling. J. Lipid Res. 2014, 55, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger LLC. Schrödinger Release 2018-1: Maestro; Schrödinger LLC: New York, NY, USA, 2018. [Google Scholar]
- Katsamakas, S.; Papadopoulos, A.; Hadjipavlou-Litina, D. Boronic acid group: A cumbersome false negative case in the process of drug design. Molecules 2016, 21, 1185. [Google Scholar] [CrossRef]
- Jones, S.B.; Pfeifer, L.A.; Bleisch, T.J.; Beauchamp, T.J.; Durbin, J.D.; Klimkowski, V.J.; Hughes, N.E.; Rito, C.J.; Dao, Y.; Gruber, J.M.; et al. Novel autotaxin inhibitors for the treatment of osteoarthritis pain: Lead optimization via structure-based drug design. ACS Med. Chem. Lett. 2016, 7, 857–861. [Google Scholar] [CrossRef]
- Dowdell, S.E.; Eidam, H.S.; Elban, M.; Fox, R.M.; Hammond, M.; Hilfiker, M.A.; Hoang, T.H.; Kallander, L.S.; Lawhorn, B.G.; Manns, S.; et al. Hydroxy Formamide Derivatives and Their Use. W.O. Patent 2015104684, 15 July 2015. [Google Scholar]
- Lee, M.H.; Lee, D.-Y.; Balupuri, A.; Jeong, J.-W.; Kang, N.S. Pharmacophoric site identification and inhibitor design for autotaxin. Molecules 2019, 24, 2808. [Google Scholar] [CrossRef]
- Imamura, S.; Horiuti, Y. Enzymatic determination of phospholipase D activity with choline oxidase. J. Biochem. 1978, 83, 677–680. [Google Scholar] [CrossRef] [PubMed]
- ACD/Percepta, ADC/Labs Release, Version 17.2; Advanced Chemistry Development, Inc.: Toronto, ON, Canada, 2017.
- Albers, H.M.H.G.; Ovaa, H. Chemical evolution of autotaxin inhibitors. Chem. Rev. 2012, 112, 2593–2603. [Google Scholar] [CrossRef]
- Akgun, B.; Hall, D.G. Fast and tight boronate formation for click bioorthogonal conjugation. Angew. Chem. Int. Ed. 2016, 55, 3909–3913. [Google Scholar] [CrossRef] [PubMed]
- Bian, H.; Chevalier, K.M.; Connolly, P.J.; Flores, C.M.; Lin, S.C.; Liu, L.; Mabus, J.; Macielag, M.J.; Mcdonnell, M.E.; Pitis, P.M.; et al. Azetidinyl Diamides as Monoacylglycerol Lipase Inhibitors. W.O. Patent WO2010124086, 28 October 2010. [Google Scholar]
- Lumeras, W.; Caturla, F.; Vidal, L.; Esteve, C.; Balagué, C.; Orellana, A.; Domínguez, M.; Roca, R.; Huerta, J.M.; Godessart, N.; et al. Design, synthesis, and structure−activity relationships of aminopyridine N -oxides, a novel scaffold for the potent and selective inhibition of p38 mitogen activated protein kinase. J. Med. Chem. 2009, 52, 5531–5545. [Google Scholar] [CrossRef] [PubMed]
- Wuttke, A.; Geyer, A. Self-assembly of peptide boroxoles on cis-dihydroxylated oligoamide templates in water: Self-assembly of peptide boroxoles in water. J. Pept. Sci. 2017, 23, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Kazmierski, W.M.; De La Rosa, M.; Samano, V. Inhibitors od Indoleamine 2,3-Dioxygenase. W.O. Patent 2017002078, 5 January 2017. [Google Scholar]
- Kuttruff, C.A.; Ferrara, M.; Bretschneider, T.; Hoerer, S.; Handschuh, S.; Nosse, B.; Romig, H.; Nicklin, P.; Roth, G.J. Discovery of BI-2545: A novel autotaxin inhibitor that significantly reduces LPA levels in vivo. ACS Med. Chem. Lett. 2017, 8, 1252–1257. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ji, M.; Zhu, Z.; Cao, R.; Chen, X.; Xu, B. Discovery of 2-substituted 1 H -benzo[d]immidazole-4-carboxamide derivatives as novel poly(ADP-ribose)polymerase-1 inhibitors with in vivo anti-tumor activity. Eur. J. Med. Chem. 2017, 132, 26–41. [Google Scholar] [CrossRef]
- Keune, W.-J.; Potjewyd, F.; Heidebrecht, T.; Salgado-Polo, F.; Macdonald, S.J.F.; Chelvarajan, L.; Abdel Latif, A.; Soman, S.; Morris, A.J.; Watson, A.J.B.; et al. Rational design of autotaxin inhibitors by structural evolution of endogenous modulators. J. Med. Chem. 2017, 60, 2006–2017. [Google Scholar] [CrossRef]
- Mori, M.; Dasso Lang, M.C.; Saladini, F.; Palombi, N.; Kovalenko, L.; De Forni, D.; Poddesu, B.; Friggeri, L.; Giannini, A.; Malancona, S.; et al. Synthesis and evaluation of bifunctional aminothiazoles as antiretrovirals targeting the HIV-1 nucleocapsid protein. ACS Med. Chem. Lett. 2019, 10, 463–468. [Google Scholar] [CrossRef]
- Crew, A.P.; Hornberger, K.R.; Snyder, L.B.; Zimmermann, K.; Wang, J.; Berlin, M.; Crews, C.M.; Dong, H. Compounds and Methods for the Targeted Degradation of Androgen Receptor. U.S. Patent 2018099940, 12 April 2018. [Google Scholar]
- Fairhurst, R.A.; Taylor, R.J. Purine Derivatives Acting as A2A Receptors Agonists. W.O. Patent 2006074925A1, 20 July 2006. [Google Scholar]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, R.J.; Green, D.V.S.; Luscombe, C.N.; Hill, A.P. Getting physical in drug discovery II: The impact of chromatographic hydrophobicity measurements and aromaticity. Drug Discov. Today 2011, 16, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Valkó, K.L. Lipophilicity and biomimetic properties measured by HPLC to support drug discovery. J. Pharm. Biomed. Anal. 2016, 130, 35–54. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |
Figure 1.
Lysophosphatidylcholine (LPC) is hydrolyzed by autotaxin (ATX) to lysophosphatidic acid (LPA); role of LPA on downstream signaling in cells.
Figure 1.
Lysophosphatidylcholine (LPC) is hydrolyzed by autotaxin (ATX) to lysophosphatidic acid (LPA); role of LPA on downstream signaling in cells.

Figure 2.
Design and development of small molecule ATX inhibitors.

Figure 3.
Formation of an adduct of benzoxaborole in the ATX active site according to literature [12]. (a) Nucleophilic attack on benzoxaboroles by catalytic threonine oxygen side chain; (b) reversible covalent tetrahedral adduct.
Figure 3.
Formation of an adduct of benzoxaborole in the ATX active site according to literature [12]. (a) Nucleophilic attack on benzoxaboroles by catalytic threonine oxygen side chain; (b) reversible covalent tetrahedral adduct.

Figure 4.
General structure of novel ATX inhibitors.

Figure 5.
Structures of the warheads (W) and the core spacers (CS).

Figure 6.
Overlay of HA155 boronic acid (green) inhibitor in the binding site of ATX (Protein Data Bank (PDB) ID: 2XRG [9] and re-docked pose of HA155 (magenta) using Covalent Docking v1.3. [15].

Figure 7.
Compound 37 docked in the binding site of ATX using covalent docking. The covalent bond between the oxygen atom of Thr209 and the boron atom of boronate is labelled in green (d(O–B) = 1.48 Å).
Figure 7.
Compound 37 docked in the binding site of ATX using covalent docking. The covalent bond between the oxygen atom of Thr209 and the boron atom of boronate is labelled in green (d(O–B) = 1.48 Å).

Figure 8.
Compound 37 docked in the binding site of ATX using covalent docking and overlapped with ligands from x-ray structures: (a) HA155 boronic acid inhibitor (green, PDB ID: 2XRG) [9]; (b) PF-8380 (magenta, PDB ID: 5L0K [21]).

Scheme 1.
Reagents and conditions: (i) Pinacol diborane, PdCl2(dppf), KOAc, dioxane, reflux; (ii) N-bromosuccinimide (NBS), azobisisobutyronitrile (AIBN), CCl4, reflux; (iii) Et2O, 20% KOH, rt; (ix) 0 °C, 6N HCl.
Scheme 1.
Reagents and conditions: (i) Pinacol diborane, PdCl2(dppf), KOAc, dioxane, reflux; (ii) N-bromosuccinimide (NBS), azobisisobutyronitrile (AIBN), CCl4, reflux; (iii) Et2O, 20% KOH, rt; (ix) 0 °C, 6N HCl.

Scheme 2.
Reagents and conditions: (i) LiAlH4, THF, 0 °C; (ii) tert-butyl piperazine-1-carboxylate, CDI, DMF, rt; (iii) TFA, dichloromethane (DCM), 0 °C to rt; (iv) Boc protected 4-, 5-, or 6-membered ketone, NaBH(OAc)3, DCM, 0 °C to rt; (v) TFA, DCM, 0 °C to rt.
Scheme 2.
Reagents and conditions: (i) LiAlH4, THF, 0 °C; (ii) tert-butyl piperazine-1-carboxylate, CDI, DMF, rt; (iii) TFA, dichloromethane (DCM), 0 °C to rt; (iv) Boc protected 4-, 5-, or 6-membered ketone, NaBH(OAc)3, DCM, 0 °C to rt; (v) TFA, DCM, 0 °C to rt.

Scheme 3.
Reagents and conditions: (i) N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide (EDAC), 4-dimethylaminopyridine (DMAP), DCM, rt.
Scheme 3.
Reagents and conditions: (i) N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide (EDAC), 4-dimethylaminopyridine (DMAP), DCM, rt.

Figure 9.
ATX inhibition results of biochemical testing of new benzoxaborole inhibitors. 11—concentrations dose–response curves, as average of duplicates, are presented.
Figure 9.
ATX inhibition results of biochemical testing of new benzoxaborole inhibitors. 11—concentrations dose–response curves, as average of duplicates, are presented.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Structures, inhibitory activities, calculated logP and determined Chrom logD values.
| Compound | Structure | a IC50 Value (µM) | b clog P | c Chrom logD | ||
|---|---|---|---|---|---|---|
| Warhead | Core | Lipophilic Tail | ||||
| W | CS | R | ||||
| 30 | W 1 | CS 1 | Cl | 0.18 ± 0.04 | 3.82 | 4.52 |
| 31 | W 4 | CS 1 | Cl | 7.5 ± 5.60 | 3.61 | 4.63 |
| 32 | W 2 | CS 1 | Cl | 0.37± 0.35 | 3.82 | 4.38 |
| 33 | W 3 | CS 1 | Cl | 8.0 ± 2.58 | 3.61 | 5.40 |
| 34 | W 1 | CS 2 | Cl | 0.92 ± 0.82 | 4.02 | 5.49 |
| 35 | W 2 | CS 2 | Cl | 1.8 ± 1.75 | 4.02 | 4.58 |
| 36 | W 4 | CS 2 | Cl | 1.8 ± 1.52 | 3.99 | 4.58 |
| 37 | W 1 | CS 3 | Cl | 0.13 ± 0.12 | 3.60 | 4.58 |
| 38 | W 2 | CS 3 | Cl | 2.0 ± 1.73 | 3.60 | 4.46 |
| 39 | W 1 | CS 1 | H | >50 ± 4.20 | 2.75 | 3.05 |
| 40 | W 2 | CS 1 | H | >50 ± 3.08 | 2.75 | 2.90 |
| 41 | W 1 | CS 3 | H | 23 ± 1.18 | 2.40 | 3.07 |
| 42 | W 2 | CS 3 | H | >50 ± 32.84 | 2.40 | 2.90 |
| 43 | W 1 | CS 1 | CH3 | 2.7 ± 2.11 | 3.17 | 4.13 |
| 44 | W 2 | CS 1 | CH3 | 4.7 ± 1.14 | 3.17 | 3.99 |
| 45 | W 1 | CS 3 | CH3 | 1.0 ± 1.15 | 2.90 | 4.17 |
| 46 | W 2 | CS 3 | CH3 | 5.1 ± 2.75 | 2.90 | 4.01 |
| 47 | W 1 | CS 1 | OCH3 | 14 ± 5.50 | 2.51 | 3.19 |
| 48 | W 2 | CS 1 | OCH3 | 15 ±6.07 | 2.51 | 3.03 |
| 49 | W 1 | CS 3 | OCH3 | 11 ± 4.59 | 2.48 | 3.21 |
| 50 | W 2 | CS 3 | OCH3 | 13 ± 3.26 | 2.48 | 3.07 |
| HA155 |  | 0.088 ± 0.04 0.0057 d | 3.76 | NA | ||
a IC50 values determined in choline release assay using LPC C16:0 as a substrate. The results are shown as the mean standard deviation from duplicate experiments. b calculated log P was the octanol/water partition coefficient calculated using Percepta. c Chrom logD was determined at pH 7.4 using Luna C18 (50 × 3 mm i.d., 5 µm) column. d IC50 values taken from literature [26].
Table 2.
In vitro ADME profile of selected compounds.
| Compound | a Solubility | b PS (rat/human) | c LMS (rat/human) | d PPB (rat/human) |
|---|---|---|---|---|
| 30 | >100 | 64.5/66.5 | 86/82 | 98.8/98.4 |
| 31 | >100 | 67.9/71.6 | 86/85 | NA e/97.8 |
| 34 | >100 | 61.6/70.6 | 77/89 | NA e/99.2 |
| 35 | >100 | 63.0/59.2 | 86/89 | NA e/98.3 |
| 37 | 30–100 | 72.2/79.1 | 62/85 | 98.4/97.9 |
| 38 | 30–100 | 60.1/79.1 | 82/79 | NA e |
| 43 | >100 | 59.5/79.2 | 80/85 | 96.1/97.8 |
| 45 | >100 | 68.6/72.8 | 61/84 | 94.4/95.9 |
| HA155 | 3–10 f 6.8 | 65.7/42.9 | <30/54 | 99.9/>99.9 |
a Kinetic solubility range after 1h [µM]. b Plasma stability after 4 h [%remaining]. c LMS: Metabolic stability in human/rat liver microsomes -> predicted in vivo hepatic clearance expressed as %LBF. d PPB: Plasma protein binding in human/rat -> fraction bound [%]. e recovery issue. f solubility value taken from literature [7].
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kraljić, K.; Jelić, D.; Žiher, D.; Cvrtila, A.; Dragojević, S.; Sinković, V.; Mesić, M. Benzoxaboroles—Novel Autotaxin Inhibitors. Molecules 2019, 24, 3419. https://doi.org/10.3390/molecules24193419
AMA Style
Kraljić K, Jelić D, Žiher D, Cvrtila A, Dragojević S, Sinković V, Mesić M. Benzoxaboroles—Novel Autotaxin Inhibitors. Molecules. 2019; 24(19):3419. https://doi.org/10.3390/molecules24193419
Chicago/Turabian StyleKraljić, Kristina, Dubravko Jelić, Dinko Žiher, Adam Cvrtila, Snježana Dragojević, Verona Sinković, and Milan Mesić. 2019. "Benzoxaboroles—Novel Autotaxin Inhibitors" Molecules 24, no. 19: 3419. https://doi.org/10.3390/molecules24193419