
Design, Synthesis and Preliminary Biological Evaluation of Benzylsulfone Coumarin Derivatives as Anti-Cancer Agents

Abstract
:1. Introduction
2. Results and Discussion
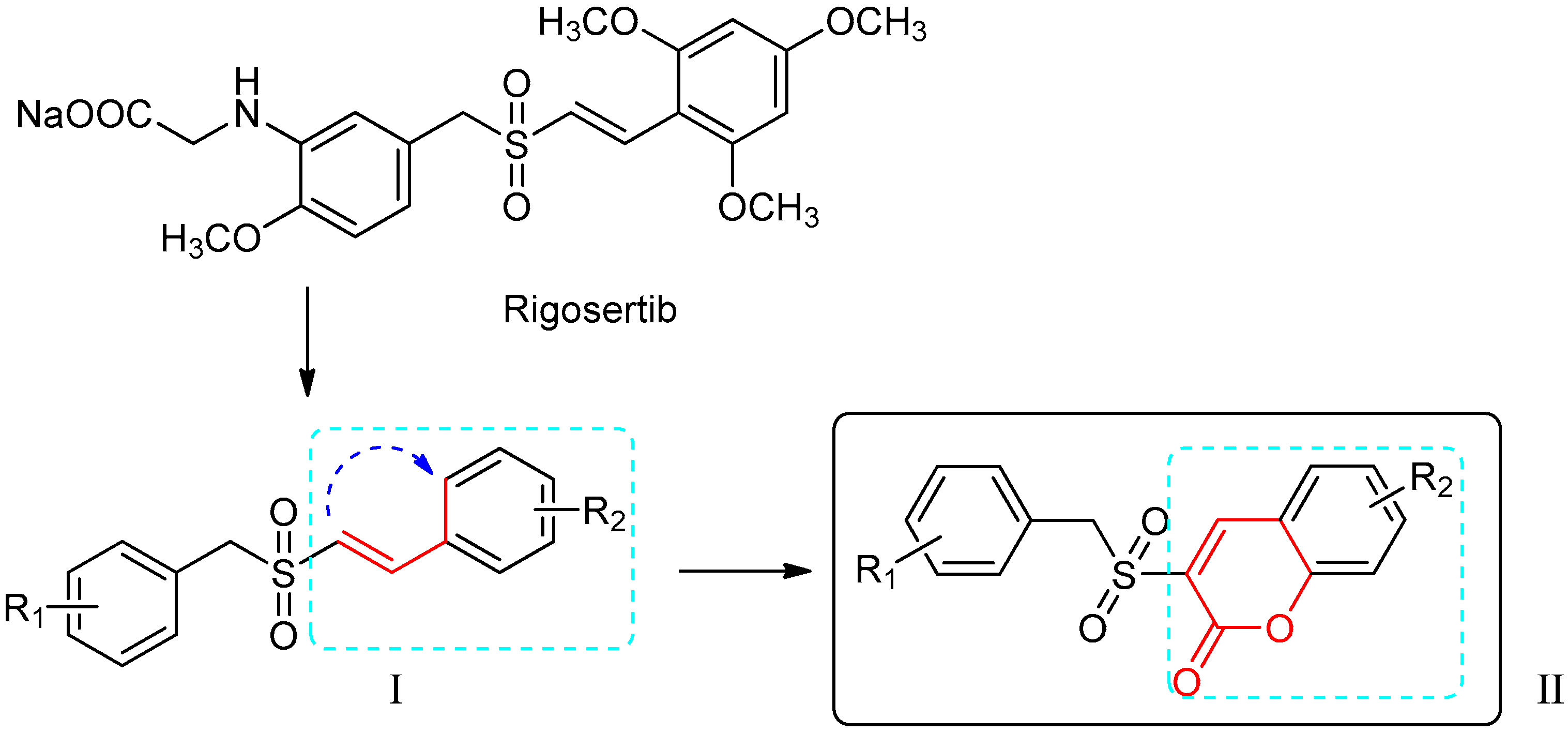
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. In vitro PI3K Inhibitory Assay
2.2.2. Cytotoxicity against Tumor Cells
2.2.3. Wound-Healing Assays of 5h and 5m
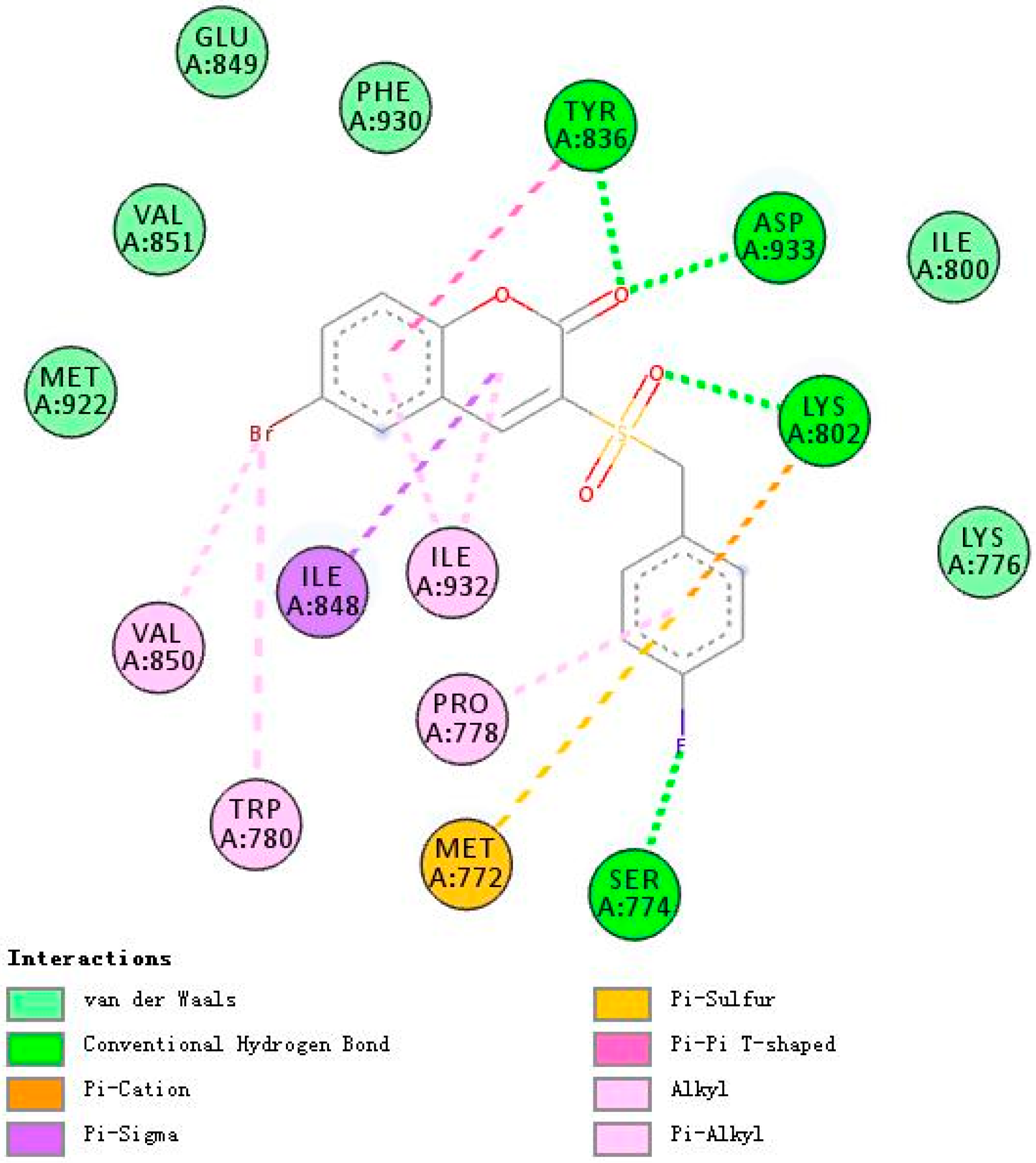
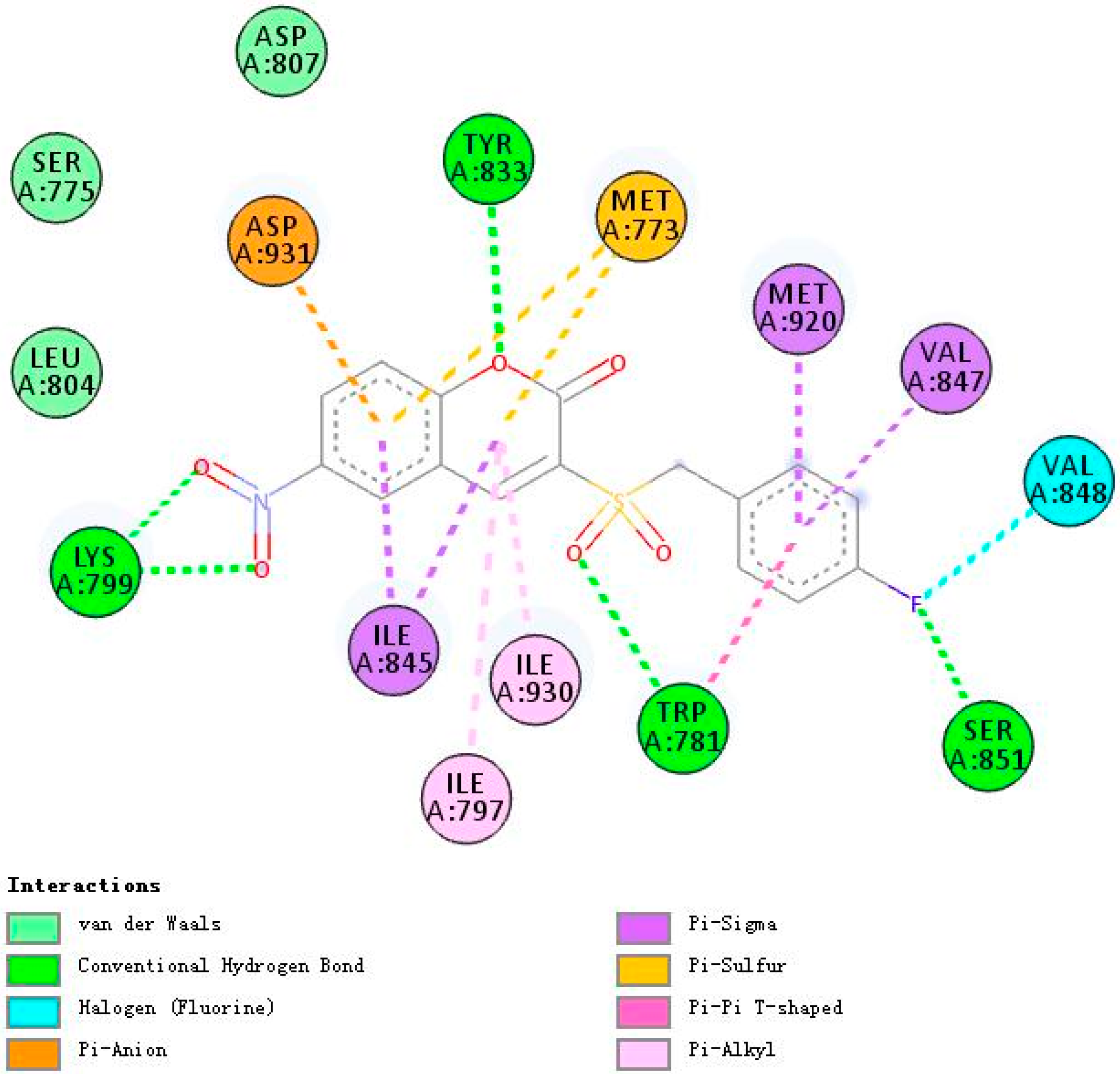
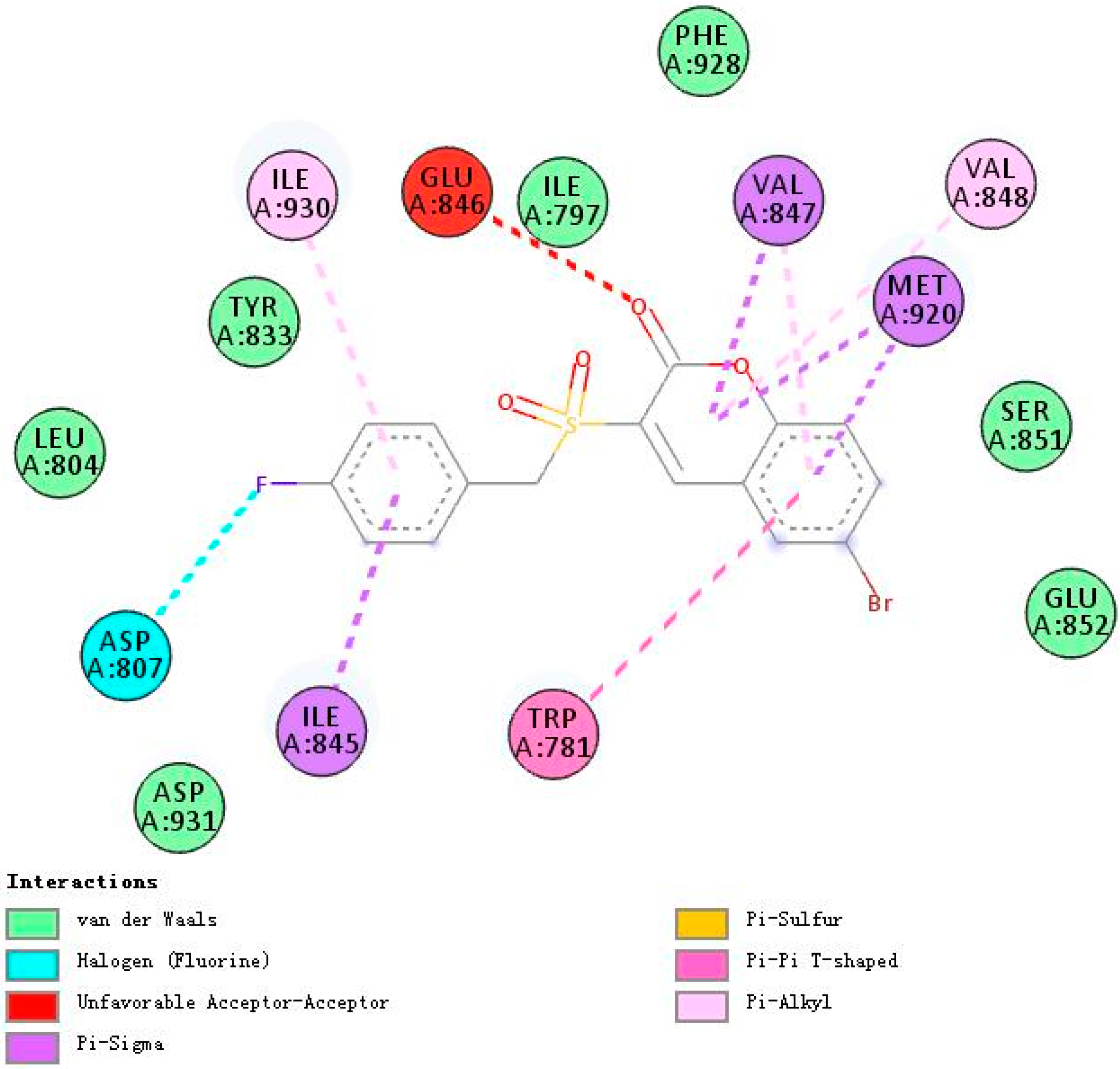
2.3. Molecular Docking Studies of 5h and 5m
2.3.1. Binding Modes of Different Compounds with PI3Kα (3HHM)
2.3.2. Binding Modes of Different Compounds with PI3Kβ (2Y3A)
3. Materials and Methods
3.1. Chemistry
Synthesis of The Target Compounds (5a–5o)
3.2. Biological Evaluation
3.2.1. PI3K Inhibitory Activity Assay
3.2.2. Cytotoxic Activity Assays
3.2.3. Wound Healing Assays
3.2.4. Molecular Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dienstmann, R.; Rodon, J.; Serra, V.; Tabernero, J. Picking the point of inhibition: A comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol. Cancer Ther. 2014, 13, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT signaling pathway and cancer: An updated review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M. Inhibition of the PI3K/AKT/mTOR Pathway in Solid Tumors. J. Clin. Oncol. 2016, 34, 3803–3815. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I.A.; Arteaga, C.L. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu. Rev. Med. 2016, 67, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Courtney, K.D.; Corcoran, R.B.; Engelman, J.A. The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 2010, 28, 1075–1083. [Google Scholar] [CrossRef]
- Bartholomeusz, C.; Gonzalez-Angulo, A.M. Targeting the PI3K signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 121–130. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Massi, D.; Teng, M.; Mandala, M. PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin. Cancer Biol. 2018, 48, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Rodon, J.; Dienstmann, R.; Serra, V.; Tabernero, J. Development of PI3K inhibitors: Lessons learned from early clinical trials. Nat. Rev. Clin. Oncol. 2013, 10, 143–153. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Y.; Zhou, Q.; Chen, M.; Zhang, Y.; Liang, H.; Zhao, J.; Zhong, W.; Wang, M. PI3K in cancer: Its structure, activation modes and role in shaping tumor microenvironment. Future Oncol. 2018, 14, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Katso, R.; Okkenhaug, K.; Ahmadi, K.; White, S.; Timms, J.; Waterfield, M.D. Cellular function of phosphoinositide 3-kinases: Implications for development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2001, 17, 615–675. [Google Scholar] [CrossRef] [PubMed]
- Janku, F. Phosphoinositide 3-kinase (PI3K) pathway inhibitors in solid tumors: From laboratory to patients. Cancer Treat. Rev. 2017, 59, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef]
- Wang, X.; Ding, J.; Meng, L.H. PI3K isoform-selective inhibitors: Next-generation targeted cancer therapies. Acta Pharmacol. Sin. 2015, 36, 1170–1176. [Google Scholar] [CrossRef]
- Mabuchi, S.; Kuroda, H.; Takahashi, R.; Sasano, T. The PI3K/AKT/mTOR pathway as a therapeutic target in ovarian cancer. Gynecol. Oncol. 2015, 137, 173–179. [Google Scholar] [CrossRef]
- Rodon, J.; Brana, I.; Siu, L.L.; De Jonge, M.J.; Homji, N.; Mills, D.; Di Tomaso, E.; Sarr, C.; Trandafir, L.; Massacesi, C.; et al. Phase I dose-escalation and-expansion study of buparlisib (BKM120), an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2014, 32, 670–681. [Google Scholar] [CrossRef]
- Sarker, D.; Ang, J.E.; Baird, R.; Kristeleit, R.; Shah, K.; Moreno, V.; Clarke, P.A.; Raynaud, F.I.; Levy, G.; Ware, J.A.; et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 77–86. [Google Scholar] [CrossRef]
- Tolaney, S.; Burris, H.; Gartner, E.; Mayer, I.A.; Saura, C.; Maurer, M.; Ciruelos, E.; Garcia, A.A.; Campana, F.; Wu, B.; et al. Phase I/II study of pilaralisib (SAR245408) in combination with trastuzumab or trastuzumab plus paclitaxel in trastuzumab-refractory HER2-positive metastatic breast cancer. Breast Cancer Res. Treat. 2015, 149, 151–161. [Google Scholar] [CrossRef]
- Juric, D.; Krop, I.; Ramanathan, R.K.; Wilson, T.R.; Ware, J.A.; Sanabria, B.S.; Savage, H.M.; Sampath, D.; Salphati, L.; Lin, R.S.; et al. Phase I Dose-Escalation Study of Taselisib, an Oral PI3K Inhibitor, in Patients with Advanced Solid Tumors. Cancer Discov. 2017, 7, 704–715. [Google Scholar] [CrossRef]
- Greenwell, I.B.; Flowers, C.R.; Blum, K.A.; Cohen, J.B. Clinical use of PI3K inhibitors in B-cell lymphoid malignancies: Today and tomorrow. Expert Rev. Anticancer Ther. 2017, 17, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Westin, J.R. Status of PI3K/Akt/mTOR pathway inhibitors in lymphoma. Clin. Lymphoma Myeloma Leuk. 2014, 14, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Gumireddy, K.; Reddy, M.V.; Cosenza, S.C.; Boominathan, R.; Baker, S.J.; Papathi, N.; Jiang, J.; Holland, J.; Reddy, E.P. ON01910, a non-ATP-competitive small molecule inhibitor of Plk1, is a potent anticancer agent. Cancer Cell 2005, 7, 275–286. [Google Scholar] [CrossRef] [Green Version]
- Prasad, A.; Park, I.W.; Allen, H.; Zhang, X.; Reddy, M.V.; Boominathan, R.; Reddy, E.P.; Groopman, J.E. Styryl sulfonyl compounds inhibit translation of cyclin D1 in mantle cell lymphoma cells. Oncogene 2009, 28, 1518–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, C.M.; Sun, X.; Roschewski, M.; Aue, G.; Farooqui, M.; Stennett, L.; Gibellini, F.; Arthur, D.; Perez-Galan, P.; Wiestner, A. ON 01910.Na is selectively cytotoxic for chronic lymphocytic leukemia cells through a dual mechanism of action involving PI3K/AKT inhibition and induction of oxidative stress. Clin. Cancer Res. 2012, 18, 1979–1991. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.V.; Venkatapuram, P.; Mallireddigari, M.R.; Pallela, V.R.; Cosenza, S.C.; Robell, K.A.; Akula, B.; Hoffman, B.S.; Reddy, E.P. Discovery of a clinical stage multi-kinase inhibitor sodium (E)-2-{2-methoxy-5-[(2′,4′,6′-trimethoxystyrylsulfonyl)methyl]phenylamino}acetate (ON 01910.Na): Synthesis, structure-activity relationship, and biological activity. J. Med. Chem. 2011, 54, 6254–6276. [Google Scholar] [CrossRef]
- Antonio, J.; Jing, L.; Messersmith, W.A.; Daniel, L.; Rudek, M.A.; Manoj, M.; Manuel, H.; Baker, S.D.; Donehower, R.C. Phase I study of ON 01910.Na, a novel modulator of the Polo-like kinase 1 pathway, in adult patients with solid tumors. J. Clin. Oncol. 2008, 26, 5504–5510. [Google Scholar]
- Jimeno, A.; Chan, A.; Cusatis, G.; Zhang, X.; Wheelhouse, J.; Solomon, A.; Chan, F.; Zhao, M.; Cosenza, S.C.; Ramana, R.M.; et al. Evaluation of the novel mitotic modulator ON 01910.Na in pancreatic cancer and preclinical development of an ex vivo predictive assay. Oncogene 2009, 28, 610–618. [Google Scholar] [CrossRef]
- Tian, Y.; Liang, Z.; Xu, H.; Mou, Y.; Guo, C. Design, Synthesis and Cytotoxicity of Novel Dihydroartemisinin-Coumarin Hybrids via Click Chemistry. Molecules 2016, 21, 758. [Google Scholar] [CrossRef]
- Morsy, S.A.; Farahat, A.A.; Nasr, M.; Tantawy, A.S. Synthesis, molecular modeling and anticancer activity of new coumarin containing compounds. Saudi Pharm. J. 2017, 25, 873–883. [Google Scholar] [CrossRef]
- Zhang, Z.; Gu, L.; Wang, B.; Huang, W.; Zhang, Y.; Ma, Z.; Zeng, S.; Shen, Z. Discovery of novel coumarin derivatives as potent and orally bioavailable BRD4 inhibitors based on scaffold hopping. J. Enzyme Inhib. Med. Chem. 2019, 34, 808–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimic, D.S.; Markovic, Z.S.; Saso, L.; Avdovic, E.H.; Dorovic, J.R.; Petrovic, I.P.; Stanisavljevic, D.D.; Stevanovic, M.J.; Potocnak, I.; Samolova, E.; et al. Synthesis and Characterization of 3-(1-((3,4-Dihydroxyphenethyl)amino)ethylidene)-chroman-2,4-dione as a Potential Antitumor Agent. Oxid. Med. Cell. Longev. 2019, 2019, 2069250. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.M.; Barreiro, E.J. Bioisosterism: A useful strategy for molecular modification and drug design. Curr. Med. Chem. 2005, 12, 23–49. [Google Scholar] [CrossRef] [PubMed]
- Ning, X.; Guo, Y.; Wang, X.; Ma, X.; Tian, C.; Shi, X.; Zhu, R.; Cheng, C.; Du, Y.; Ma, Z. Design, synthesis, and biological evaluation of (e)-3,4-dihydroxystyryl aralkyl sulfones and sulfoxides as novel multifunctional neuroprotective agents. J. Med. Chem. 2014, 57, 4302–4312. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Feng, T.; Shen, X.; Cui, J.; Wu, R.; Wang, L.; Wang, S.; Zhang, S.; Chen, H. Synthesis, characterization, and radioprotective activity of alpha, beta-unsaturated aryl sulfone analogs and their Tempol conjugates. Medchemcomm 2017, 8, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Freeman, F. Properties and reactions of ylidenemalononitriles. Chem. Rev. 1979, 80, 329–350. [Google Scholar] [CrossRef]
- Pedro, V.; Francisco, S.; Emilia, T. Knoevenagel reaction in [MMIm] [MSO4]: Synthesis of coumarins. Molecules 2011, 16, 4379–4388. [Google Scholar]
- Prasad, A.; Khudaynazar, N.; Tantravahi, R.V.; Gillum, A.M.; Hoffman, B.S. ON 01910.Na (rigosertib) inhibits PI3K/Akt pathway and activates oxidative stress signals in head and neck cancer cell lines. Oncotarget 2016, 7, 79388–79400. [Google Scholar] [CrossRef] [Green Version]
- Munikrishnappa, C.S.; Puranik, S.B.; Kumar, G.V.; Prasad, Y.R. Part-1: Design, synthesis and biological evaluation of novel bromo-pyrimidine analogs as tyrosine kinase inhibitors. Eur. J. Med. Chem. 2016, 119, 70–82. [Google Scholar] [CrossRef]
- Riahi, R.; Yang, Y.; Zhang, D.D.; Wong, P.K. Advances in wound-healing assays for probing collective cell migration. J. Lab. Autom. 2012, 17, 59–65. [Google Scholar] [CrossRef]
- Trepat, X.; Chen, Z.; Jacobson, K. Cell Migration. Compr. Physiol. 2012, 2, 2369–2392. [Google Scholar] [PubMed] [Green Version]
- Zhang, M.; Li, H.; Ma, H.; Qin, J. A simple microfluidic strategy for cell migration assay in an in vitro wound-healing model. Wound Repair Regen. 2013, 21, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Yarrow, J.C.; Perlman, Z.E.; Westwood, N.J.; Mitchison, T.J. A high-throughput cell migration assay using scratch wound healing, a comparison of image-based readout methods. BMC Biotechnol. 2004, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Mandelker, D.; Gabelli, S.B.; Schmidt-Kittler, O.; Zhu, J.; Cheong, I.; Huang, C.H.; Kinzler, K.W.; Vogelstein, B.; Amzel, L.M. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc. Natl. Acad. Sci. USA 2009, 106, 16996–17001. [Google Scholar] [CrossRef]
- Zhang, X.; Vadas, O.; Perisic, O.; Anderson, K.; Clark, J.; Hawkins, P.; Stephens, L.; Williams, R. Structure of Lipid Kinase p110β/p85β Elucidates an Unusual SH2-Domain-Mediated Inhibitory Mechanism. Mol. Cell 2011, 41, 567–578. [Google Scholar] [CrossRef]
- Tang, L.; Peng, T.; Wang, G.; Wen, X.; Sun, Y.; Zhang, S.; Liu, S.; Wang, L. Design, Synthesis and Preliminary Biological Evaluation of Novel Benzyl Sulfoxide 2-Indolinone Derivatives as Anticancer Agents. Molecules 2017, 22, 1979. [Google Scholar] [CrossRef]
- Yue, P.Y.; Leung, E.P.; Mak, N.K.; Wong, R.N. A simplified method for quantifying cell migration/wound healing in 96-well plates. J. Biomol. Screen. 2010, 15, 427–433. [Google Scholar] [CrossRef]
- Mu, C.; Wu, M.; Li, Z. Anti-Inflammatory Effect of Novel 7-Substituted Coumarin Derivatives through Inhibition of NF-kappaB Signaling Pathway. Chem. Biodivers. 2019, 16, e1800559. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 5a–5o are available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Inhibition Rates (%) | |
|---|---|---|
| 20 μM | 10 μM | |
| Rigosertib | 53.2 ± 3.7 | 40.9 ± 4.4 |
| 5a | 39.0 ± 5.1 | 28.8 ± 3.4 |
| 5b | 45.9 ± 6.7 | 26.4 ± 4.1 |
| 5c | 37.9 ± 7.5 | 25.1 ± 3.1 |
| 5d | 36.4 ± 0.4 | 18.7 ± 3.8 |
| 5e | 36.8 ± 4.1 | 23.4 ± 4.5 |
| 5f | 47.0 ± 1.4 | 31.7 ± 5.7 |
| 5g | 43.9 ± 3.4 | 23.4 ± 4.7 |
| 5h | 50.3 ± 4.8 | 38.0 ± 6.0 |
| 5i | 46.8 ± 4.4 | 20.0 ± 4.0 |
| 5j | 37.7 ± 4.5 | 25.5 ± 1.9 |
| 5k | 42.8 ± 2.4 | 22.8 ± 6.1 |
| 5l | 47.0 ± 5.7 | 24.1 ± 5.0 |
| 5m | 50.8 ± 7.0 | 40.3 ± 4.4 |
| 5n | 46.3 ± 6.3 | 23.7 ± 4.5 |
| 5o | 38.8 ± 7.7 | 27.2 ± 3.5 |
| Compound | IC50 (μM) | ||||
|---|---|---|---|---|---|
| Hela | HepG2 | H1299 | HCT-116 | MCF-7 | |
| 5a | 41.0 ± 4.5 | 43.0 ± 3.3 | 71.8 ± 5.7 | 83.9 ± 1.3 | 56.1 ± 0.7 |
| 5b | 39.5 ± 2.2 | 36.8 ± 1.1 | 54.3 ± 1.0 | 65.8 ± 2.0 | 32.9 ± 1.9 |
| 5c | 39.9 ± 0.9 | 36.8 ± 3.1 | 81.7 ± 5.5 | 33.5 ± 0.3 | >100 |
| 5d | 48.3 ± 4.3 | 33.2 ± 4.3 | 56.9 ± 1.0 | 56.6 ± 1.4 | 41.0 ± 0.4 |
| 5e | 51.8 ± 1.9 | 23.6 ± 2.4 | 38.0 ± 1.8 | 44.7 ± 0.9 | 39.9 ± 1.7 |
| 5f | 32.2 ± 0.7 | 29.6 ± 1.9 | 40.5 ± 2.1 | 30.6 ± 1.4 | 35.5 ± 0.9 |
| 5g | 41.4 ± 2.9 | 37.6 ± 3.7 | 46.3 ± 2.2 | 38.8 ± 1.8 | 41.1 ± 1.2 |
| 5h | 24.3 ± 2.8 | 18.1 ± 0.6 | 29.6 ± 0.7 | 32.6 ± 0.9 | 20.5 ± 0.8 |
| 5i | 37.5 ± 1.5 | 32.9 ± 4.1 | 32.9 ± 0.8 | 53.7 ± 1.4 | 36.6 ± 0.8 |
| 5j | 24.6 ± 1.0 | 31.1 ± 3.3 | 50.2 ± 2.8 | 41.8 ± 0.2 | 24.6 ± 1.7 |
| 5k | 35.9 ± 2.1 | 54.4 ± 1.4 | 63.9 ± 2.2 | >100 | 54.5 ± 4.3 |
| 5l | 22.6 ± 1.8 | 49.9 ± 1.5 | 62.6 ± 2.2 | 46.3 ± 1.0 | 50.7 ± 4.1 |
| 5m | 29.7 ± 2.1 | 30.6 ± 4.3 | 31.5 ± 1.1 | 42.1 ± 1.3 | 29.3 ± 1.8 |
| 5n | 33.8 ± 5.6 | 58.9 ± 2.1 | 81.9 ± 4.6 | 50.6 ± 1.4 | 69.5 ± 2.5 |
| 5o | 44.3 ± 4.3 | 41.9 ± 2.7 | 70.8 ± 7.2 | 54.7 ± 0.6 | 82.3 ± 5.4 |
| Regosertib | 0.01 ± 0 | 0.05 ± 0.01 | 0.14 ± 0.02 | 2.36 ± 0.21 | 0.83 ± 0.08 |
| Compound | Wound Closure (%) a | |
|---|---|---|
| 12 h | 24 h | |
| Control | 10.3 ± 2.8 | 19.5 ± 4.5 |
| 5h (10 μM) | 4.2 ± 1.1 ** | 9.9 ± 1.5 ** |
| 5h (5 μM) | 5.1 ± 1.1 ** | 14.4 ± 3.9 ** |
| 5m (10 μM) | 3.1 ± 1.0 ** | 8.2 ± 1.6 ** |
| 5m (5 μM) | 3.9 ± 6.6 ** | 9.1 ± 1.1 ** |
| Compound | Chemical Structure | △G (PI3Kα) Kcal/mole | △G(PI3Kβ) Kcal/mole |
|---|---|---|---|
| Rigosertib |  | −7.19 | −7.26 |
| 5h |  | −8.47 | −7.74 |
| 5m |  | −7.01 | −7.28 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, T.; Peng, T.; Wen, X.; Wang, G.; Sun, Y.; Liu, S.; Zhang, S.; Wang, L. Design, Synthesis and Preliminary Biological Evaluation of Benzylsulfone Coumarin Derivatives as Anti-Cancer Agents. Molecules 2019, 24, 4034. https://doi.org/10.3390/molecules24224034
Wang T, Peng T, Wen X, Wang G, Sun Y, Liu S, Zhang S, Wang L. Design, Synthesis and Preliminary Biological Evaluation of Benzylsulfone Coumarin Derivatives as Anti-Cancer Agents. Molecules. 2019; 24(22):4034. https://doi.org/10.3390/molecules24224034
Chicago/Turabian StyleWang, Tao, Tao Peng, Xiaoxue Wen, Gang Wang, Yunbo Sun, Shuchen Liu, Shouguo Zhang, and Lin Wang. 2019. "Design, Synthesis and Preliminary Biological Evaluation of Benzylsulfone Coumarin Derivatives as Anti-Cancer Agents" Molecules 24, no. 22: 4034. https://doi.org/10.3390/molecules24224034