Palladium-Catalysed Synthesis and Transformation of Quinolones



Department of Chemistry & QOPNA and LAQV-REQUIMTE, Campus Universitário de Santiago, University of Aveiro, 3810-193 Aveiro, Portugal
*
Author to whom correspondence should be addressed.
Molecules 2019, 24(2), 228; https://doi.org/10.3390/molecules24020228
Submission received: 9 December 2018
/
Revised: 31 December 2018
/
Accepted: 4 January 2019
/
Published: 9 January 2019
(This article belongs to the Special Issue Frontiers in Metal-Catalysed Cross-Coupling Reactions for the Synthesis and Functionalisation of Heterocycles)
Abstract
:Palladium-catalysed reactions have had a large impact on synthetic organic chemistry and have found many applications in target-oriented synthesis. Their widespread use in organic synthesis is due to the mild conditions associated with the reactions together with their tolerance of a wide range of functional groups. Moreover, these types of reactions allow the rapid construction of complex molecules through multiple bond-forming reactions in a single step, the so-called tandem processes. Pd-catalysed reactions have been applied to the synthesis of a large number of natural products and bioactive compounds, some of them of complex molecular structures. This review article aims to present an overview of the most important Pd-catalysed reactions employed in the synthesis and transformations of quinolin-2(1H)-ones and quinolin-4(1H)-ones. These compounds are widely recognized by their diverse bioactivity, being privileged structures in medicinal chemistry and useful structural moieties for the development of new drug candidates. Furthermore, they hold significant interest due to their host–guest chemistry; applications in chemical, biochemical and environmental analyses and use in the development of new synthetic methods. In some cases, the quinolone formation step cannot be ascribed to a claimed Pd-catalysed reaction but this reaction is crucial to get the appropriate substrate for cyclization into the quinolone. Herein we present and discuss different economical, efficient and selective synthetic strategies to access quinolone-type compounds.
1. Introduction
Quinolin-2(1H)-ones and the isomeric quinolin-4(1H)-ones are benzo-α- and benzo-γ-pyridones, respectively, since they are constituted by a α- or γ-pyridone o-fused with a benzene ring (Figure 1). The quinolone motif is widely distributed in nature, being particularly found in alkaloids of Rutaceae family but can also be produced by different animal and bacterial species [1,2]. These compounds are privileged scaffolds in medicinal chemistry and are ubiquitous substructures associated with relevant biologically active natural products. For instance, quinolin-2(1H)-ones were reported as antiulcer (e.g., rebamipide), antihistaminic (e.g., repirinast) [3] and anticancer (e.g., tipifarnib) [4] agents and have also demonstrated activity as antivirals [5], for instance as inhibitors of HIV-1 reverse transcriptase [6,7]. In addition, they are useful intermediates in organic synthesis. Quinolin-4(1H)-ones are well-known as antibiotics (e.g., fluoroquinolones) and exhibit excellent antimicrobial activity (e.g., [8]). Moreover quinolin-4(1H)-ones possess other interesting biological properties such as antimalarial [9] and antitumoral [10,11] activities, among others. Up to now several methods for the synthesis of quinolones have been reported in the literature but the most commonly used involve the condensation of anilines with β-ketoesters followed by cyclization to give quinolin-2(1H)-ones (Conrad–Limpach–Knorr synthesis) or quinolin-4(1H)-ones (Conrad–Limpach synthesis). Other classical methods leading to the formation of the C3–C4 bond include the Friedlander synthesis, Camps modification and Niementowski reaction [12,13,14]. Most of these conventional methods often require harsh conditions, tedious workup and purification procedures, are regioselectivity compromised and the variety of substrates is limited. The diversity of the quinolone-containing structures encountered, as well as their biological and pharmaceutical relevance, have motivated research aimed at the development of new economical, efficient and selective synthetic strategies to access these compounds. Recently transition metal-catalysed (TM-catalysed) procedures for the synthesis of such compounds and further transformations have been developed providing increased tolerance toward functional groups and leading generally to higher reaction yields [15]. Many of these methods have proven to be the most powerful and are currently applied in target- or diversity-oriented syntheses. Among TM-catalysed reactions, Pd-catalysed reactions, especially cross-coupling reactions, have found wide application in organic synthesis. The use of Pd-catalysts to allow the rapid construction of complex molecules through multiple bond-forming reactions in a single step, the so-called tandem process, became a powerful tool for synthetic organic chemists. Such processes reduce the steps in the synthesis of certain molecules; thus, being attractive from the viewpoint of developing environmentally benign and economical synthetic methods. This review article is an attempt to compile the most important Pd-catalysed reactions that have been applied in the synthesis and transformation of quinolin-2(1H)-ones and quinolin-4(1H)-ones in the period ranging from 1990-2017. When describing the developments in this research field, emphasis will be given to the reaction conditions, including the catalysts, ligands and bases, the reaction’s scope, selectivity and in some cases to the mechanism.
2. Synthesis of Quinolin-2(1H)-ones
2.1. Heck Reaction
In 1978, Heck and co-workers described for the first time the synthesis of quinolin-2(1H)-ones from vinylic substitution of 1,2-disubstituted olefins with 2-iodoanilines [16]. Reaction of 2-iodoanilines 1 with dimethyl maleate 2 using Pd(OAc)2 as catalyst and Et3N as base, in acetonitrile at 100 °C, afforded as expected from previous reaction with 4-iodoaniline, the Z intermediate amino esters which in situ cyclize to quinolin-2(1H)-ones 3 in moderate to good yields (72, 55 and 30% for R = H, Br and OH, respectively) (Scheme 1). However, with diethyl fumarate the E intermediate amino ester was expected and cyclization should not occur without first isomerizing, since the β-carboxymethyl group was far from the amino group, although quinolin-2(1H)-one 3 was isolated in 47% yield along with 20% of aniline. In this case, isomerization of the intermediate should occur fairly easily or the σ-bonded Pd intermediate cyclizes readily in this reaction.
4-Phenylquinolin-2(1H)-one was obtained in 66% yield by reacting 2-iodoaniline with (Z)-N-phenylcinnamamide [16]. As previously referred, the use of (E)-N-phenylcinnamamide instead of the (Z)-isomer also gives the quinolone but only in 15% yield. An alternative route to prepare 4-phenylquinolin-2(1H)-one was the reaction of (E)-2-aminocinnamic acid (or esters or amides) with iodobenzene, which gives the expected quinolin-2(1H)-one in good yield (71%).
Cacchi and co-workers reported a straightforward domino Heck/cyclization reaction of methyl β-(2-acetamidophenyl)acrylates 4 with aryl iodides 5 in the presence of Pd(OAc)2 and KOAc in N,N-dimethylformamide (DMF) at 120 °C that afforded free NH 4-arylquinolin-2(1H)-ones 7 in 11–80% yield (Scheme 2) [17]. The vinylic substitution intermediate 6 was only observed when the reaction was performed at lower temperatures (25% at 80 °C and 30% at 60 °C).
The same authors demonstrated that β-(2-bromophenyl)acrylamide 8 can also be a good precursor for the synthesis of 4-arylquinolin-2(1H)-ones 10 by a sequential Heck-reaction-Cu-catalysed-cyclization, using neutral, electron-rich and electron-poor aryl iodides [Scheme 3, (i) and (ii)]. The optimized cyclization conditions involved CuI (20 mol%), NaI (2.0 equiv), K3PO4 (2.0 equiv), dimethylethylenediamine (DMEDA) (0.4 equiv) in 1,4-dioxane at 120 °C; the reaction did not work in the absence of CuI [18]. Later, they have shown that 4-arylquinolin-2(1H)-ones 10 can be prepared using a pseudo-domino process that involves a two mechanistically independent catalytic cycles, a Heck reaction of 2-bromocinnamamide 8 with aryl iodides 5, with formation of 9, followed by an intramolecular Buchwald-Hartwig C-N bond forming reaction [Scheme 3, (iii)] [19]. In the Heck reaction, phosphine-free Pd(OAc)2 (0.05 equiv) was used as the pre-catalyst and molten tetrabutylammonium acetate/tetrabutylammonium bromide (3.0 equiv of each one) as the reaction medium, at 120 °C.
Methoxyquinolin-2(1H)-ones were synthesized from iodo derivatives of methoxylated pivaloylaminobenzenes 11, which were obtained via metalation of the corresponding polymethoxypivaloylaminobenzenes with BuLi. The synthesis involves two steps; first the Heck coupling reaction of 11 with methyl acrylate using Pd(OAc)2 as catalyst and Et3N as base in acetonitrile at 100 °C afforded the corresponding methyl methoxypivaloylaminocinnamates 12 which then undergo cyclization to quinolin-2(1H)-ones 13, in acidic medium, in fairly good overall yields (19–62%) (Scheme 4) [20].
The coupling-cyclization between 2-iodoaniline 1 and α,β-unsaturated carbonyl compounds 14 with a catalytic amount of a Pd-catalyst along with a base in DMF at 100 °C afforded 3-substituted quinolin-2(1H)-ones 15 in moderate to good yields (67–76%) (Scheme 5) [21]. The best conditions found were 5 mol% Pd(OAc)2 as catalyst, 10 mol% PPh3 as ligand, NaOAc (6.0 equiv) as base. PdCl2(PPh3)2 was also effective in these conditions. If, a ketone group is linked to the vinyl carbon of the starting alkenes instead of a carboalkoxyl group, 2,3- or 2,4-disubstituted quinolines were obtained, depending if the α- or the β-position of the alkene is substituted.
A ligand free Pd-catalysed Heck-Matsuda reaction of ethyl cinnamate 16 with arene diazoniumtetrafluoroborate 17 gave β,β-diaryl acrylate 18 which undergo a Cu-catalysed asymmetric 1,4-reduction–cyclization affording 4-methoxyphenylquinolin-2(1H)-one 19 [Scheme 6, (i)] [22]. The presence of the base (in this case 2,6-di-t-butyl-4-methylpyridine) is important for the excellent stereoselectivity (>95:5 Z/E) due to the scavenging of PdH, preventing its reinsertion which is responsible for isomerization of Heck adducts. Based on the enantioselective synthesis of β,β-diaryl propanoates by Cu-catalysed 1,4-reduction of β,β-diaryl acrylate using Cu(OAc)2 and (R)-1-[(S)-2-(diphenylphosphino)ferrocenyl]ethyldicyclohexylphosphine (R-JOSIPHOS) as ligand, in the presence of an excess of polymethylhydrosiloxane (PMHS), the cyclization of intermediates 18 gave almost quantitatively quinolin-2(1H)-one 19 using PHMS/t-BuOH (4 equiv) (an intramolecular amidation reaction took place in preference to the 1,4-reduction) [Scheme 6, (iii)]. By using PHMS/t-BuOH (10 equiv), the expected dihydroquinolin-2(1H)-one 19 is obtained in good yield [Scheme 6, (ii)]. Dihydroquinolin-2(1H)-one 20 can also be prepared by subjecting quinolin-2(1H)-one 19 to a second Cu-catalysed 1,4-reduction [Scheme 6, (iv)].
The selective synthesis of 3,4-dihydroquinolin-2(1H)-ones 23 and 26 was achieved following a Pd-catalysed one-pot sequential Heck-reduction-cyclization (HRC) methodology, using either heterogeneous catalysts (Scheme 7) or mixed homogeneous/heterogeneous catalysts (Scheme 8) with Pd0/C generated in situ [23,24]. The overall reaction sequence proceeds under mild conditions with good to high isolated yields starting either from 2-(2-nitrophenyl)acrylates 21 and aryldiazonium salts 22 or 2-nitrobenzenediazonium salts 24 and acrylates 25. Recycling experiments showed that the reused heterogeneous Pd0/C catalyst was not able to promote another HRC sequence but was still highly active for hydrogenation reactions. 3,4-Dihydroquinolin-2(1H)-ones 26 could be easily dehydrogenated affording quinolin-2(1H)-one 27 under mild oxidative conditions.
The arylation of ethyl 3-furoate 28 with 1-bromo-2-nitrobenzene 29 in the presence of Pd(PPh3)4 afforded ethyl 2-(2-nitrophenyl)furan-3-carboxylate (80%) (Scheme 9) [25]. Hydrogenation of the nitro group with Pd/C as catalyst and subsequent cyclization occurred uneventfully to provide furo[3,2-c]quinolin-4(5H)-one 30 (75%). An overall yield of 60% was achieved for these two sequential Pd-catalysed reactions.
The successive Heck reaction on substituted 2-iodoaniline 1 with acrylic acid catalysed by Pd supported on nickel ferrite, as a heterogeneous and recyclable catalyst, followed by in situ cyclization afforded 4-arylquinolin-2(1H)-ones 32 (Scheme 10) [26]. Regarding the aryl halide 31, aryl iodides gave better yields of the desired product compared with aryl bromides. The scope of this methodology was extended to the synthesis of bioactive 3-alkenyl derivatives of 4-arylquinolin-2(1H)-ones 33 (Scheme 10).
Pd-catalysed intramolecular Heck cyclization of N-phenyl-1H-imidazole-4-carboxamide 34 afforded 5-butyl-1-methyl-1H-imidazo[4,5-c]quinolin-4(5H)-one 35 with 83% yield (Scheme 11) [27].
The intramolecular Heck cyclization of N-(hetero)arylcarboxamides afforded tricyclic fused quinolone derivatives (Scheme 12) [28]. These compounds were prepared by treatment of the acyl chlorides, generated in situ from the commercially available pyrrole-, thiophene- and furan-2/3-carboxylic acids 36, with the appropriate 2-iodoanilines 1, followed by N-methylation of the N-(hetero)arylcarboxamides 37 to avoid N-Pd complexation. Intramolecular Heck cyclization of 38 at the positions 2 or 3 of the heterocyclic nucleus with Pd(PPh3)4 as catalyst and KOAc as base afforded the corresponding tricyclic fused quinolones 39 in moderate to high yields (Scheme 12).
Pd-catalysed intramolecular Heck cyclization of N-acetyl-N-[2-(halomethyl)aryl]acrylamides 40 afforded 3-alkylquinolin-2(1H)-ones 41d and another three cyclization products 41a–c (Scheme 13) [29]. Compounds 41a and 41c are the acetylated products of 41b and 41d, respectively. Treatment of the reaction mixture with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) successfully converted 41a–c to 3-alkylquinolin-2(1H)-one 41d. Combination of Pd2(dba)3 and 1,1′-bis(diphenylphosphino)ferrocene (dppf) in the presence of Et3N in refluxing MeCN followed by treatment of the reaction mixture with DBU led to 3-alkylquinolin-2(1H)-one 41d in good to excellent yields (Scheme 13).
2.2. Sonogashira Reaction
Pd-catalysed coupling of ethyl N-(2-ethynyl)malonanilide 42 with aryl, heteroaryl, vinyl halides or vinyl triflates 43 gave derivatives 44, which upon intramolecular cyclization, under basic conditions, afforded 3,4-disubstituted quinolin-2(1H)-ones 45 (Scheme 14) [30]. The mechanism of this carbocyclization involves an intramolecular nucleophilic attack of the carbanion, generated from 44, on the carbon-carbon triple bond giving, after protonation, a six-membered ring methylidene intermediate that isomerizes to the quinolin-2(1H)-one 45. The success of this cyclization step is determined by the nature of the substituent in the acetylenic moiety. Good yields were obtained with aromatic rings bearing electron-withdrawing substituents (60–75%) while no quinolin-2(1H)-ones were obtained with electron-donating p-methoxyphenyl or butyl groups.
2.3. Carbonylative Annulation
Both 3- and 4-substituted quinolin-2(1H)-ones 48 and 49 were obtained by Pd-catalysed carbonylative annulation of terminal alkynes 47 with 2-iodoaniline derivatives 46, in the presence of carbon monoxide (CO), pyridine (Py) and Pd(OAc)2 (Scheme 15) [31]. Terminal alkynes bearing alkyl, phenyl, silyl, hydroxy, ester and cyano substituents gave quinolin-2(1H)-ones in moderate yields. Only the 3-substituted quinolin-2(1H)-ones 48 were obtained in the reaction with phenylacetylene and triethylsilylacetylene. Removal of the carbamate-protecting group by treating the crude reaction with 1 M of ethanolic NaOH is necessary to avoid the formation of a mixture of deprotected and protected quinolin-2(1H)-ones. Both 3- and 4-substituted quinolin-2(1H)-ones 48 and 49 were obtained in the reactions with terminal alkynes bearing long alkyl chains. An increase in the size of the substituent on the triple bond improves the regioselectivity. Furthermore, the ratio of isomers is around 4 to 1 when functionalized acetylenes are used compared to the 2.2 to 1 ratio obtained in most of the reactions with 1-hexyne. The formation of quinolin-2(1H)-ones and not quinolin-4(1H)-ones shows that the key step in this process is the insertion of the terminal alkyne into the carbon-Pd bond instead of undergoing a Sonogashira-type coupling [32,33]. The authors have conducted an isotope labelling experiment to unambiguously prove this reactivity pattern.
Later, the same authors reported the synthesis of 3,4-disubstituted quinolin-2(1H)-ones 51 and 52 by a ligand free Pd-catalysed annulation of internal alkynes 50 with N-substituted 2-iodoanilines 46 in the presence of CO (Scheme 15) [34]. The type of the substituent on the nitrogen was fundamental to obtain quinolin-2(1H)-ones in high yields. Better yields were obtained using mild electron-withdrawing substituents as p-toluenesulfonyl, trifluoroacetyl and alkoxycarbonyl. The nitrogen substituent is lost during the progress of the reaction affording N-unsubstituted quinolin-2(1H)-ones, except for aminocarbonyl and ethoxycarbonyl groups that originated the corresponding protected quinolin-2(1H)-one in 7% and 11% isolated yields, respectively. A wide variety of internal alkynes 50, bearing alkyl, aryl, heteroaryl, hydroxy and alkoxy substituents, were effective in this reaction; however, unsymmetrical alkynes originated mixtures of regioisomers 51 and 52, with low regioselectivity, owing to the two possible modes of alkyne insertion into the aryl-Pd bond. Electron-deficient alkynes are very poor substrates for the carbonylative annulation. Electron-rich 2-iodoanilines can react as annulating agents but when the substitution is para to the iodine the yield is lower. Carbonylative annulation of electron-poor 2-iodoanilines gives the corresponding quinolin-2(1H)-ones in lower yields than the parent system.
Pd-catalysed carbonylative annulation of unprotected 2-iodoanilines 1 and terminal alkynes 47, using the commercially available molybdenum hexacarbonyl [Mo(CO)6] as a convenient and solid CO source, also afforded 3- and 4-substituted quinolin-2(1H)-ones 48 and 49, the latter ones as minor products [Scheme 15, (iv)] [35]. The reactions were conducted at 160 °C for 30 min under microwave irradiation. Et3N was the best base in terms of yield and regioselectivity. Pd(OAc)2 exhibited good catalytic activity whereas 1,2-bis(diphenylphosphino)ethane (dppe) and tetrahydrofuran (THF) were the best ligand and solvent choice for regioselectivity and yields. Different substituted 2-iodoanilines and alkyl alkynes were used without significant loss in reaction yield or efficiency. Aryl-substituted alkynes gave only the 3-substituted quinolin-2(1H)-one 48 although the yield was moderate. Neither internal alkynes nor N-protected 2-iodoanilines can be used in this carbonylative annulation.
Protected iodoanilines 53 reacted with internal alkenes 54 or alkynes 56 and Mo(CO)6, affording 3,4-disubstituted (dihydro)quinolin-2(1H)-ones 55 and 57 (Scheme 16) [36]. The reaction is ligand-free and avoids the problematic use of gaseous CO. In addition, the annulation reactions proceed with insertion of unsaturated compounds into the arylpalladium bond in preference to insertion of CO where no isomeric quinolones were observed. For instance, norbornene adds to the arylpalladium intermediate in a cis-exo manner followed by CO insertion and intramolecular amination (Scheme 16). The slow CO liberation during the reaction also provides an efficient pressure of the gas to promote the carbonylative reaction. This protocol merges three Heck, carbonylation and amination reactions to construct two C-C and one C-N bonds in one pot. It is compatible with various amino protecting groups, such as ethoxycarbonyl, tosyl, sulfonyl and acyl groups; however, deprotection occurred during the reaction and workup procedure, affording the free quinolin-2(1H)-one. Nonetheless, it is important to note that the yields are better when using N-protected anilines and depend on the protecting group. Reactions of N-substituted anilines with dipropyl acetylene, 4-octyne and 5-decyne afforded the annulation product in moderate to very good yields. Unfortunately, diphenylacetylene did not participate in the annulation reaction with o-iodoaniline; only traces of the desired product were obtained.
The oxidative addition of 1-(2-iodophenyl)-3-p-tolylurea 58 to Pd(dba)2 in the presence of 1 equiv of N,N,N′,N′-tetramethyl-1,2-ethylenediamine (TMEDA) followed by the reaction of 59 with internal alkynes, originated vinyl-Pd complexes 60. These complexes reacted with CO to give Pd and two 3-substituted 4-phenylquinolin-2(1H)-ones 61 (R = Ph, 11% and R = CO2Me, 57%) (Scheme 17) [37]. The reaction mechanism should follow CO insertion into the Pd-C-vinyl bond of 60 to give an acyl Pd derivative, which undergoes a C,N coupling with loss of Pd and p-tolyl isocyanate.
Following the work of Larock, Jia and co-workers developed a protocol for the efficient synthesis of 4,5-fused tricyclic quinolin-2(1H)-ones 63 by intramolecular carbonylative annulation of alkyne-tethered o-iodoanilines 62 (Scheme 18) [38]. The use of PPh3 as ligand was found to be crucial for this reaction although its role is not clear. For most of the substrates tested, the reaction afforded a mixture of the N-protected and free (NH) fused quinolin-2(1H)-ones in different ratios. This problem was overwhelmed by treatment of the crude products with 1 M ethanolic NaOH to completely hydrolyse the N-substituted product thus obtaining only the expected fused quinolin-2(1H)-one 63. The electronic effect of the aryl groups on the internal alkyne on the reactivity was not significant. In addition, substrates leading to 7- and 8-membered ring fused quinolin-2(1H)-ones gave the desired products in good yields and the o-methyl substituent on 2-iodoaniline was well tolerated.
In 1979, Ban and co-workers described the Pd-catalysed carbonylation of vinyl bromides bearing an internal amide group 64 [39]. In the presence of the catalytic system Pd(OAc)2 and PPh3, the (Z)-isomer 64 of the vinyl bromide smoothly gave quinolin-2(1H)-one 65 with loss of the N-acetyl group (Scheme 19). In the same reaction conditions, the corresponding (E)-isomer gave only 7.7% yield of the same quinolin-2(1H)-one 65 because palladation occurs at the position of the halogen atom of the vinyl halide.
Asymmetric cyclocarbonylation of 2-(1-methylvinyl)anilines 66 using a catalyst system of Pd(OAc)2-2(−)-DIOP [2,3-O-isopropylidene-2,3-dihydroxy-1,4-bis(diphenylphosphino)butane] [Pd(OAc)2 (1 mol%), (−)-DIOP (2 mol%)], in CH2Cl2 at 100 °C for 48 h, under CO (500 psi) and hydrogen pressure (100 psi), originated chiral 4-methyl-3,4-dihydroquinolin-2(1H)-ones 67 in up to 54% ee (Scheme 20) [40]. Other chiral ligands [(2S,4S)-1-t-butoxycarbonyl-4-diphenylphosphino-2-(diphenylphosphinomethyl)pyrrolidine, 2,2′-bis(diphenylphosphino)-1,1′- binaphthyl, (2S,3S)-(−)-bis(diphenylphosphino)butane] [(−)-bppm, (+)-BINAP and R,R-BDPP] were inferior to (−)-DIOP in terms of enantioselectivity, which was not influenced by the amount of (−)-DIOP (1, 2, 6 equiv) or Pd-precursors [Pd2(dba)3.CHCl3, Pd(acac)2, Pd(CF3COO)2, п-allyl-Pd chloride dimer] employed. However, it was slightly sensitive to the ratio of CO and hydrogen pressure; an increase of this ratio reduces the enantioselectivity. In the absence of hydrogen, the yield is very low (37%) although the optical purity was still maintained (33% ee). Substituents at the 4- and 5- position of 2-(1-methylvinyl)anilines 66 showed some effect on the enantioselectivity (67: R2 = R3 = OMe, 20% ee; R2 = Br, 31% ee). Stronger effects were found in the case of 66: R1 = OMe, the reaction was not enantioselective affording the corresponding racemic product while the carbonylation of 66: R4 = OMe proceeded in a more enantioselective manner affording the corresponding quinolone 67 in nearly quantitative yield (99%, 54% ee). For 6-substituted anilines 66, excellent yields of 67: R4 = Me (98%) and 67: R4 = i-Pr (99%) were obtained whereas the carbonylation of aniline 66: R2 = R4 = Br afforded the corresponding 4-methyl-3,4-dihydroquinolin-2(1H)-one 67 in moderate yield (48%).
2.4. Ullmann Reaction
2.5. Buchwald-Hartwig Reaction
The coupling of the protected N-hydroxyamides 72 with benzaldehydes or benzoates by Pd-catalysed Buchwald-type C-N bond formation followed by a tandem cyclodehydration afforded the quinolin-2(1H)-ones 73 in good to excellent yields (63–96%) (Scheme 22) [42]. Pd2(dba)3 was the best catalyst in the presence of Cs2CO3 and 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (Xantphos) as the ligand. Various aryl and naphthyl substituents were tolerated under the reaction conditions. Treatment of compounds 73 with trifluoroacetic acid (TFA) in dichloromethane afforded the corresponding N-hydroxyquinolin-2(1H)-ones 74 in 57–78% yield.
Starting from symmetrical α-(2-bromobenzyl)malonamides 75, desymmetrized 3,4-dihydroquinolin-2(1H)-ones 76 were synthesized in nearly quantitative yields and enantiomeric ratios up to 88:12 by an unusual enantioselective Buchwald-Hartwig reaction (Scheme 23) [43]. Using enantiopure (R)-77 as ligand, 76: R = Bn was isolated in 85% yield but in a 57:43 er. When (R)-78 was employed in the same reaction, the yield was comparable to that obtained with (R)-77 but enantioselectivity was improved significantly to a 79:21 ratio. Different ratios of Pd to (R)-78 were investigated from 1:1 to 1:3 because this monophosphine may complex Pd differently from other bisphosphines. The yield was slightly lower for 1:3 ratio but the enantioselectivity was nearly the same for all ratios studied. In 1,4-dioxane or THF the product was afforded in excellent yields and good enantiomeric ratios compared to more polar solvents such as MeCN and DMSO. A series of bases were screened using refluxing THF but the best results were obtained with K3PO4 and Cs2CO3 affording the product in nearly quantitative yield with good enantioselectivity. While most of quinolin-2(1H)-ones 76 were obtained in 99%, more sterically demanding amide nitrogen substituents led to lower reaction yields (i-Pr, 90%; 2,6-Me2Ph, 54%; 1-naphthyl, 79%), although the enantioselectivity increased with the steric bulk of the substituent (ranging from 69:31 er for Me up to 85:15 er and 88:12 er for i-Pr and 4-OMeBn, respectively). In the case of N-aryl substrates the same enantiomeric ratios of N-phenyl and N-benzylquinolin-2(1H)-ones (79:21 er) were observed.
Enantioselective intramolecular double N-arylation of malonamides 79 bearing 2-bromoarylmethyl groups catalysed by a Pd-BINAP complex afforded C2-symmetric spirobis[3,4-dihydroquinolin-2(1H)-ones] 80 in 99% yield and up to 70% ee (Scheme 24) [44]. The scope of this reaction was investigated using a variety of malonamides 79 and it was demonstrated that substrates having 2-chlorobenzyl groups were less reactive (8% yield, 6% ee). Although the reaction with the iodide analogues took place smoothly to give quinolin-2(1H)-ones 80 in high yield (94%), its enantioselectivity was as low as 38% ee. The reaction enantioselectivity is also affected by the substituent on the aromatic ring, moderate optical purity being obtained for 5-Cl substituents (85% yield, 48% ee) whereas nearly racemic products were produced for 5-OMe- (90% yield, 6% ee) and 3-NO2-substituted malonamides (42% yield, 5% ee). It was also possible to introduce a 1,3-benzodioxole ring leading to quantitative formation of the corresponding spirobis[3,4-dihydroquinolin-2(1H)-one] (99% yield, 57% ee) and no selectivity was observed for sterically more demanding substrates.
The mechanism proposed for the formation of compounds 80 seems to be initiated by the oxidative addition of one of the bromoarenes in 79 to a catalytically active Pd(0) complex. Then desymmetrisation of the amide groups of the resulting Pd(II) complex 81 occurs affording palladacycle 82. The subsequent C-N bond-forming reductive elimination yields monocyclized compound 80′ with regeneration of the Pd catalyst. Then intramolecular N-arylation of 80′ affords the expected spirobis[3,4-dihydroquinolin-2(1H)-one] 80. According to the authors, the enantioselectivity is determined in the first cyclization. They also confirmed that a kinetic resolution process is involved in the second cyclization (Scheme 25) [44].
2.6. Intramolecular C-H Alkenylation
Pd(II)-catalysed selective 6-endo intramolecular C-H alkenylation of N-phenylacrylamides 83, in the conditions shown in Scheme 26, allows the construction of the quinolin-2(1H)-one core 84 [45]. In all cases, the quinolin-2(1H)-one 84 was the unique isolated product and the formation of the indolin-2-one, due to a 5-exo cyclization, was not detected. The reaction was efficient with an unsubstituted alkene and substitution on the 3-position of the acrylamide is well tolerated, leading to 4-substituted quinolin-2(1H)-ones. However, the presence of an ester or ketone moiety led to the expected product in low isolated yields, 10% and 22%, respectively. A better yield of 30% was achieved for the ketone but in the absence of Cu(OAc)2. 2,3-Disubstituted acrylamides can also be used leading to 3,4-disubstituted quinolin2(1H)-ones. N-demethylated acrylamides also gave the corresponding quinolin-2(1H)-ones but in lower yields. Although acetic acid is a good solvent, the reaction can also be performed in aqueous media at room temperature, using a 2% aqueous solution of polyoxyethanyl-α-tocopheryl sebacate (PTS) or even in water, with good yields but for a prolonged reaction time (24 h).
The mechanism of this intramolecular cyclization seems to involve an electrophilic palladation of the aromatic ring to form an aryl-Pd(II) intermediate (Scheme 27). This arylpalladium would undergo a syn insertion into the activated alkene moiety in a 6-endo mode, followed by β-hydride elimination to afford the quinolone framework. Given the (E)-configuration of the starting alkenes, the subsequent syn β-hydride elimination would require the prior epimerization of the α-carbon through the formation of an O-Pd intermediate.
2.7. Intramolecular Amidation of C(sp2)-H Bonds
Pd-catalysed amidation of halo aromatics substituted in the o-position by a carbonyl functional group or its equivalent 85 with primary or secondary amides 86 gave substituted quinolin-2(1H)-ones 87 (Scheme 28) [46]. Both aryl bromides and chlorides are effective in this reaction but aryl chlorides required longer reaction times than bromides (6 h versus 2 h). On the other hand, electron-rich aryl bromides afforded the product in moderate yield (47%). The reaction was not limited to o-halo aldehydes; enolizable methyl ketones, such as 2-bromoacetophenone, 2-bromobenzophenone, methyl 2-bromobenzoate, methyl 2-bromo-5-phenylbenzoate, 2-bromobenzonitrile also coupled with 2-phenylacetamide to give 4-substituted quinolin-2(1H)-one derivatives 88 in moderate to good yields (33–74%). Primary aryl and heterocyclic acetamides worked well with the exception of 2-pyridylacetamide that did not react in the reaction conditions presented in Scheme 28. With the secondary amide, N-methyl-2-phenylacetamide, the product was obtained in good yield (60%) however, neither the more sterically demanding N-isopropyl-2-phenylacetamide nor the N-arylamide N-phenyl-2-phenylacetamide was coupled with 2-bromobenzaldehyde using these conditions. Primary and secondary alkyl amides, such as propionamide and 2-cyclopropylacetamide, undergo coupling but failed the in situ cyclization; in addition, other attempts to cyclize the coupling products in several acidic or basic conditions failed. 2-Methoxyacetamide and N-methylpropionamide were coupled with 2-bromobenzaldehydes but also failed the cyclization in the coupling reaction conditions. However, they cyclized into the expected quinolin-2(1H)-ones, in low yields (51% and 32%, respectively), by the addition of NaO-t-Bu in t-BuOH.
Pd-catalysed intramolecular amidation of N-substituted-3,3-diarylacrylamides 89 was efficiently performed in the presence of a catalytic amount of PdCl2 and Cu(OAc)2 under O2 atmosphere, affording a range of diversely substituted 4-arylquinolin-2(1H)-ones 90 and 91 (Scheme 29) [47,48]. The cyclization of the 3,3-diarylacrylamides having tosyl, acetyl or a t-butyl group on the nitrogen atom of the amide moiety proceeded with concomitant loss of the protecting group, whereas the use of free amide or alkyl amides led to the product in poor yields. The benzene ring is also a suitable substituent on the nitrogen atom and, in this case, the reaction yield increases as the electron density of the benzene ring decreases, suggesting that the nucleophilicity of the nitrogen atom plays an important role in the process. This method is applicable to a wide range of symmetrical 3,3-diarylacrylamides (R2 = R3) possessing various functional groups on the benzene ring (Scheme 29). Unsymmetrical substrates (R2 ≠ R3) having a cyano group at the p-position of the benzene ring yielded only one regioisomer. In the case of the m-cyano substituted compounds the (E)-isomer was much more reactive than the (Z)-isomer (Scheme 29).
A tandem process consisting of two mechanistically independent sequential Pd(II)-catalysed reactions, the oxidative Heck reaction of cinnamamides 92 with arylboron compounds (Ar′–“B”) followed by the intramolecular C–H amidation reaction, represented a facile and novel route to various substituted 4-arylquinolin-2(1H)-one derivatives (Scheme 30) [48,49]. The coupling of N-methoxycinnamamide 92 with phenylboronic acid using a catalytic combination of Pd(OAc)2/1,10-phenanthroline, Cu(TFA)2·nH2O (200 mol%) and Ag2O (100 mol%) in AcOH successfully delivered the expected quinolin-2(1H)-one 93. In this case, demethoxylation unexpectedly occurred during the reaction, resulting in the formation of N-free quinolin-2(1H)-one 93 in fairly good yield (60%). The use of increased amounts of Ag2O (e.g., 300 mol% or more) completely inhibited demethoxylation and resulted in the formation of quinolin-2(1H)-one 94 in 52–81% yield (Scheme 30). The best result was obtained when 800 mol% of Ag2O was employed, affording 94 in high yields (81%). During the process, the use of a large excess of re-oxidants may inhibit the undesired N–O bond cleavage in which palladium and/or copper salt(s) are involved. This tandem process is also applicable to the synthesis of quinolin-2(1H)-ones via the formation of symmetrical 3,3-diarylacrylamides [48,49]. Reactions of cinnamamides with a methyl group or a halogen atom at the p-position of the benzene ring proceeded efficiently (59–76%), whereas that of cinnamamide with a methoxy group at the same position, resulted in the formation of the corresponding quinolin-2(1H)-one 95 (R1 = 4-OMe) in only 12% yield, along with the recovery of the starting material in 85% yield. For cinnamamides with a methyl group at the m-position of the benzene ring, C–H cyclization occurred at the less-hindered site; in contrast, the introduction of a methyl group at the o-position of the benzene ring did not result in the formation of the coupling or cyclized product (Scheme 30).
Reactions via the formation of unsymmetrical 3,3-diarylacrylamides were also investigated (Scheme 30) [48,49]. Although 4-methoxyphenylboronic acid was not suitable for this process, other arylboronic acids possessing a trifluoromethyl group, a methoxycarbonyl group or a halogen atom on the benzene ring (R2) successfully underwent reaction, affording different substituted 4-arylquinolin-2(1H)-ones 96 and/or 96′. For example, cinnamamide 92 (R1 = H) reacted with the 3-trifluoromethylphenylboronic acid (R2 = 3-CF3) under optimized conditions to successfully afford quinolin-2(1H)-one 96 (R1 = H, R2 = 3-CF3) as the sole product in 67% yield. No regioisomer was identified, which suggests that the alkene isomerization of the Heck product formed in situ did not occur during the process. Likewise, the reaction of cinnamamide 92 (R1 = 3-OMe) with 4-chlorophenylboronic acid (R2 = 4-Cl) afforded only quinolin-2(1H)-one 96 (R1 = 3-OMe, R2 = 4-Cl) in 72% yield. However, with cinnamamide 92 (R1 = 4-OMe) sometimes a lower selectivity was observed, giving rise to a mixture of the corresponding quinolin-2(1H)-ones 96 and 96′, indicating that the (E→Z)-isomerization could be occurring during the reaction course [49].
2.8. Intramolecular Hydroarylation of C-C Triple Bonds
Various aryl alkynanilides 99, prepared from the corresponding alkynoic acids 97 and anilines 98, undergo fast intramolecular reaction, at room temperature, in the presence of a catalytic amount of Pd(OAc)2 in a mixed solvent containing TFA, affording quinolin-2(1H)-ones 100 in very good yields (82–91%), with more than 1000 turnover numbers (TON) to Pd (Scheme 31) [50]. The methodology tolerated a number of functional groups such as Br and CHO. On the basis of isotope experiments, a possible mechanism involving ethynyl chelation-assisted electrophilic metalation of aromatic C-H bonds by in situ generated cationic Pd(II) species was proposed. The involvement of vinyl-cationic species was also suggested [50]. On the other hand, it was demonstrated that the intermolecular reaction of alkynoic acids 97 and anilines 98 did not afford quinolin-2(1H)-ones 100, presumably because the amino groups in anilines are converted in TFA to ammonium ions which act as electron-withdrawing groups and consequently deactivate the aromatic rings (Scheme 31) [50].
2.9. [3 + 3] Annulation
The Pd-catalysed [3 + 3] annulation between diarylamines 101 and α,β-unsaturated acids 102 through C-H activation allowed direct access to 4-substituted-quinolin-2(1H)-ones 103 in high yield (Scheme 32) [51]. The use of TFA was crucial for suppressing facile decarboxylation of α,β-unsaturated acids. This method proved general and versatile for the synthesis of a variety of 4-substituted-quinolin-2(1H)-ones with aromatic, aliphatic and heterocyclic substituents at the 4-position. Although all of the substrates reacted successfully, electron-rich cinnamic acids gave better yields. Monoarylamines (ArNHR, R=H, Me, Et, i-Pr, Ts and Ac) either formed mixtures of inseparable compounds or gave low yields of the desired quinolones. Interestingly, all o-substituted unsymmetrical diarylamines gave single regioisomeric products. Based on preliminary mechanistic studies, involving competition experiments, intermediate studies and deuterium labelling, a reaction sequence was proposed, involving o-palladation, п-coordination, β-migratory insertion and β-hydride elimination [51].
2.10. Other Reactions
Benzothieno[2,3-c]quinolin-6(5H)-ones 106 were prepared by one-pot three steps Pd-catalysed borylation, Suzuki coupling and amidation (lactamization), starting from o-haloanilines 104 and alkyl 3-bromobenzo[b]tiophene-2-carboxylates 105 (Scheme 33) [52]. The amidation reaction probably occurs in the Suzuki coupling intermediate, through a nucleophilic attack of the nitrogen atom of the amine on the carbonyl of the ester group, with loss of methanol or ethanol, giving the corresponding tetracyclic quinolone. The use of an electron-rich sterically hindered ligand, such as 2-(dicyclohexylphosphanyl)biphenyl and Ba(OH)2·8H2O as base, is convenient for sterically hindered substrates. In addition, the borylation should be performed in the component bearing an o-electron-donating group with the other Suzuki coupling component having an o-electron-withdrawing group. This method allows the use of two bromo components conveniently substituted as starting materials. The in situ Pd-catalysed borylation avoids the preparation and isolation of boronic acids or esters and occurs with atom economy. The borylation occurred either using o-bromo or o-chloroanilines enhancing the scope of the reaction.
N-(1′-Alkoxy)cyclopropyl-2-haloanilines 107 were converted to 3,4-dihydroquinolin-2(1H)-ones 109 via Pd-catalysed cyclopropane ring expansion (Scheme 34) [53]. Good yields of 108 were obtained using electron-rich biphenyl-based phosphine ligands; however, the more sterically demanding 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl (XPhos) provides the better yield. The use of K2CO3 as base led to the best result while KOt-Bu did not afford the desired compound. A lower reaction temperature resulted in a lower yield and DMF provided better yields than toluene or 1,4-dioxane. Bromo- and iodoaniline derivatives gave better yields in a shorter reaction time than chloroaniline. Hydrolysis of 2-alkoxy-3,4-dihydroquinoline 108 resulted in the formation of 3,4-dihydroquinolin-2(1H)-ones 109 in good yields. The reaction tolerates a variety of functional groups such as ester, nitrile, ether and ketone groups. The substrate bearing a nitro group did not give the desired compound.
A plausible mechanism for the formation of 108 starts with the oxidative addition of aryl halide 107 to Pd(0) followed by intramolecular ligand exchange to give a four-membered azapalladacycle. Further Pd rearrangement accompanied with cyclopropane ring opening gave the energetically favourable seven-membered azapalladacycle [53]. Finally, reductive elimination produces 2-methoxy-3,4-dihydroquinoline 108, with concomitant regeneration of the reactive Pd(0) species. The hydrolysis of 2-methoxy-3,4-dihydroquinolines 108 furnishes the 3,4-dihydroquinolin-2(1H)-ones 109 [53].
Liu and co-workers reported a concise and general strategy for the synthesis of quinolin-2(1H)-ones 112 by a one-pot catalysed cascade that includes successive ammonolysis, C-H bond activation and cyclization reactions, using simple anilines 110 as substrates (Scheme 35) [54]. Under the optimized reaction conditions, a wide range of quinolin-2(1H)-ones 112 was prepared in good to excellent yields. Anilines containing electron-donating, electron-withdrawing groups or halogens as substituents are effective in the reaction. For m-toluidine and 3-trifluoromethylaniline, C−H bond activation occurred solely at p-position to the methyl or trifluoromethyl group to provide the corresponding products in 96% and 53% yields, respectively. However, the reactions with anilines bearing trifluoromethyl, nitro or carboxyl groups at the p-position, did not give the desired quinolin-2(1H)-ones. When using cinnamates, the electronic character of the substituents on the aryl ring significantly influenced the efficiency of the cyclization. Cinnamates with electron-donating groups on the aryl moiety displayed higher reactivity than those with electron-withdrawing groups. No product was obtained when using ethyl (2E)-3-(2-trifluoromethylphenyl)propenoate but heterocyclic substituents containing propenoate and alkyl-substituted propenoates, such as ethyl crotonate and ethyl methacrylate, afforded the desired quinolin-2(1H)-ones 112 in low to moderate yields (37–68%). The utility of this method was demonstrated by a formal synthesis of Tipifarnib, an orally active inhibitor of farnesylprotein transferase with potent activity against neoplastic diseases, antineoplastic activity in solid tumours, such as breast cancer and in haematological malignancies found in leukaemia (Scheme 35).
A plausible mechanism for this cascade process involves the formation of an aryl-Pd complex II via C−H activation at the C2-position of the substrate assisted by an in situ N-acetylation. Next, Heck-type coupling of II with acrylate through Pd complex III forms intermediate IV and releases the Pd(0) species. Then oxidation of Pd(0) by Na2S2O8 to Pd(II) completes the metal catalytic cycle. Under acidic conditions, the intermediate IV could be transferred to intermediate V and followed the ammonolysis of the ester with the amide to give the intermediate VI, which after hydrolysis gave the expected quinolin-2(1H)-one (Scheme 36) [54].
3,3-Disubstituted-3,4-dihydroquinolin-2(1H)-ones 115 were synthesized from easily available o-halogenated acetylide derivatives 114 by the Pd-catalysed oxidative-addition-initiated activation and arylation of inert C(sp3)-H bonds (Scheme 37) [55]. Pd(OAc)2 and P(o-tol)3 were used as the catalyst and ligand, respectively, to improve the efficiency of the reaction. Polar solvents, such as N-methyl-2-pyrrolidone (NMP) gave the best results. The use of PivOH (Piv = pivaloyl) as additive is not essential; however the product was obtained in a slightly better yield in the presence of this additive. Regarding the reactivity of the C-X bonds, chloro-substituted substrates showed a low reactivity and iodo-substituted compounds also gave a lower yield than bromo-substituted ones. When there are substituents at 3- or 6-position, the cyclized products were obtained in low yields, which indicate that steric hindrance plays a key role in influencing the reaction efficiency. Furthermore, some sensitive functional groups, such as nitro, ester and acyl groups were not tolerated. Nonetheless, by changing the acyl group from the pivaloyl group to a 1-methylcyclopentanecarbonyl or a 1-methylcyclohexanecarbonyl group, it is possible to synthesize polycyclic compounds with spiro-centres. However, in the presence of a C(sp2)-H bond located at a suitable position for intramolecular C-H activation, the desired product of the inert C(sp3)-H activation could not be observed. A further advantage of this reaction is that it could be performed in air. Mechanistic studies showed that a relatively rare seven-membered palladacycle is a key intermediate of the catalytic cycle [55].
A wide range of α-carbamoyl ketene dithioacetals 116 readily reacted with arynes 117 to selectively afford functionalized quinolin-2(1H)-ones 118 in high yields under neutral reaction conditions by a C-S activation/aryne insertion/intramolecular coupling sequence catalysed by Pd(OAc)2 (Scheme 38) [56]. The use of dppf, as a ligand, dramatically improved the aryne annulation and both palladium and the ligand play a key role in the insertion of benzyne into a C-S bond. Other phosphine-containing ligands, such as PCy3 (Cy = cyclohexyl), PPh3 and Xantphos, were found to be less efficient than dppf for this annulation. Additionally, either decreasing the amount of the catalyst or lowering the reaction temperature resulted in unsatisfactory yields of 118a (R1 = Me, R2 = 4-NO2C6H4, R3 = Bn, R4 = H). The reaction scope is quite general, however for dithioacetals which do not have a substituent at the α-position (R2 = H) the reaction was more complex and the quinolin-2(1H)-one 118 (R2 = H) was obtained in a low yield (27%), along with quinolin-2(1H)-one 118 (R2 = Ph) that results from the first α-phenylation of the dithioacetal (R2 = H) with benzyne by a Pd-catalysed α-C-H activation and subsequent annulation with the benzyne. Finally, the reaction was found to be sensitive to substrates 116 bearing a free NH2; in this case, the obtained product was only detected by NMR and HRMS-ESI studies of the reaction mixture after 18 h of reaction time. When the unsymmetrical aryne 4-methoxy-2-(trimethylsilyl)-phenyl trifluoromethanesulfonate was employed, a mixture of two isomeric quinolin-2(1H)-ones was obtained [56].
The Pd-catalysed oxidative annulation of electron-deficient acrylamides 119 with benzyne precursors 120, in the presence of Pd(OAc)2, Cu(OAc)2 and CsF, in a mixture of dioxane and DMSO as solvent at 80 °C, allowed the formation of quinolin-2(1H)-one 121 in good yields (Scheme 39) [57]. Pd(PPh3)4 can also be employed but the product is generated in lower yield. Tetrabutylammonium bromide (TBAB) was found to be an important additive leading to highly improved yields. Acrylamides 119 bearing alkyl or aromatic substituents at α-position are good substrates in the reaction with benzyne. Cyclic substrates also react efficiently to give the expected fused-quinolin-2(1H)-ones 121 in moderate to good yields (55–74%) but cinnamide and crotonyl amide did not react. Substituted arynes bearing both electron-donating and electron-withdrawing substituents reacted with acrylamide 119: R1 = Ph, R2 = H, generating the corresponding quinolin-2(1H)-ones 121 in low to moderate yield (35–61%); with the asymmetric aryne, a 1:1 mixture of regioisomers was obtained. The methoxy group on the nitrogen atom is a very important protecting group. It was easily removed by NaH to give free quinolin-2(1H)-one 121 in 93% yield. The oxidative annulation of an N-methylacrylamide with benzyne gave the corresponding quinolin-2(1H)-one in only 25% yield under the same reaction conditions.
Although two possible reaction pathways for the oxidative annulation are possible [57], the most probable one involves the N-H activation of acrylamide 119 followed by aminopalladation of intermediate 122 to benzyne originating the palladium intermediate 123, which went through a Heck-type reaction, insertion to the electron-deficient double bond and subsequent β-hydrogen elimination, producing the expected quinolin-2(1H)-one 121 (Scheme 40).
Pd-catalysed intermolecular aminocarbonylation/intramolecular amidation cascade sequences were employed to efficiently and selectively convert a range of 2-(2-haloalkenyl)aryl halides 124 to the corresponding quinolin-2(1H)-ones 126 (Scheme 41) [58]. In the presence of Pd2(dba)3 and Cs2CO3 a variety of ligands, such as P(i-Pr)2 127, diphosphine dppp 128 and P(t-Bu)3 129, efficiently delivered quinolin-2(1H)-ones 126 in reasonable yields. Alkyl amines and p-methoxybenzylamine were suitable N-nucleophiles for this reaction. With allylamine, the reaction was performed at 50 °C for 2 h before heating at 100 °C. With O-phenylethanolamine CO atmosphere had to be removed after 3 h of reaction in the standard conditions. The use of p-anisidine required Xantphos as ligand and afforded the N-arylquinolone in only 33% yield.
A better yield was obtained following a discrete two-step process involving isolation of the intermediate N-arylamide and then re-subjection of the amide to ring closure conditions. Xantphos was employed as the ligand for both steps and allowed the isolation of the corresponding quinolin-2(1H)-one in 65% yield for the two steps. A range of electron-donating and electron-withdrawing substituents are tolerated in the aryl ring of the dihalide substrate. This method also allows the synthesis of quinolin-2(1H)-ones having aryl or styryl substituents at C-3 position.
Isoquinoline derivatives can be obtained using the same conditions but delaying the introduction of the CO atmosphere [58]. A limitation of this approach is the requirement to employ a sterically demanding N-nucleophile, as the use of less hindered coupling partners results in competitive indole formation.
3. Synthesis of Quinolin-4(1H)-ones
3.1. Sonogashira Reaction
2-Iodoanilides 130 reacted with terminal propargyl alcohols 131 under Pd-catalysed conditions to yield the corresponding 2-substituted anilides 132 (Scheme 42) [59,60]. PdCl2(PPh3)2 was found to be the best catalyst. Propargyl alcohols 131 in the presence of Et3N appear to be capable to reduce Pd(II) to Pd(0) before the oxidative addition of Pd(0) to the aryl halide could occur. Only in one case (R1 = CF3; R2 = R3 = H) the corresponding indole, obtained by spontaneously cyclization of 2-substituted anilides 132, was isolated. By acid-catalysed Meyer-Schuster rearrangement, 2-substituted anilides 132 were converted to N-substituted 2′-aminochalcones 133 which upon alkaline hydrolysis, deprotection and cyclization, under acidic conditions, afforded 2-aryl-2,3-dihydroquinolin-4(1H)-ones 134 (Scheme 42).
The Sonogashira coupling of 2-(pseudo)halogenated anilines 135 with propargyl alcohols 131 followed by a Brønsted acid-catalysed cyclization of the resulting 2-(3-hydroxypropynyl)anilines 136 afforded 2,3-dihydroquinolin-4(1H)-ones 137 (Scheme 43) [61]. Anilines 136 were converted into quinolin-4(1H)-ones by acid-catalysed tandem Rupe rearrangement-Donnelly-Farrell ring-closure reaction upon treatment with concentrated HCl-H2O (1:1, v/v), at 120 °C, followed by basic workup. The Rupe rearrangement of 136, in the case of free aniline, involves a regioselective hydration-dehydration rearrangement of the alkyne moiety, probably via aldol 138, to give the corresponding α,β-unsaturated ketone, which by acid catalysed 6-endo-trig Michael-type ring closure is converted into dihydroquinolin-4(1H)-ones 137 (Scheme 43). The cyclization of the N-acetyl derivatives occurs via Rupe rearrangement followed by acetamide hydrolysis, in situ, immediately prior to cyclization [61]. This reaction allows the introduction of different groups at C-2 of the quinolin-4(1H)-one skeleton (e.g., R3 = H, Me; R4 = Ph) thus providing access to a wide variety of 2-substituted 2,3-dihydroquinolin-4(1H)-ones 137.
The Pd-catalysed carbonylative coupling of 2-haloanilines 139 with terminal arylacetylenes 47, initially reported by Torii and Kalinin [32,33,62], appears to be the most versatile method for the synthesis of 2-substituted quinolin-4(1H)-ones 141. The desired compounds were obtained in good yields using either PdCl2(PPh3)2 or PdCl2(dppf) and an excess of Et2NH which acts as solvent and base and plays a key role in the cyclization step (Scheme 44). 2-Iodoanilines are preferred to 2-bromoanilines and react both as a free base and in the hydrochloride form although the latter in lower yield [62]. The reaction of arylacetylenes generally gave better yields than aliphatic acetylenes, which may be due to the acidity of the acetylene proton. Functional groups, such as thiophenyl, acetal, tetrahydropyran (THP), ester, keto and ether, tolerate the reaction conditions. When the nitrogen of the anilines has a substituent, decrease of the nucleophilicity retards the cyclization. In fact, reaction with alkylated aniline, under the same reaction conditions, led to the corresponding enamine 142 as main product (52%) and only 20% of quinolin-4(1H)-one 141 was obtained, although subsequent treatment of the enamine with sodium hydride in THF led to quinolin-4(1H)-one 141 quantitatively [32,33]. Decrease of CO pressure or temperature drops the reaction yield.
Pd-catalysed carbonylative Sonogashira coupling of 2-iodo-5-methoxyaniline 144 with thiazolylacetylene 143 afforded quinolin-4(1H)-one 146, a key substructure of the hepatitis C virus NS3 protease inhibitor BILN2061 (Scheme 45) [63]. This approach may also provide a practical and general access to polysubstituted quinolones related to structure 146.
Pd-catalysed carbonylative Sonogashira coupling between 2-iodoanilines 1, alkynes 47 and CO (5 bar), using Et3N as the base in the presence of PdCl2(dppp) [dppp = 1,3-bis(diphenylphosphino)propane] as catalyst selectively afforded intermediate 147. In a second step, an organocatalyzed cyclization occurs after the addition of Et2NH to give the expected quinolin-4(1H)-ones 148 in high selectivity and yields [Scheme 46, (i)] [64]. Although successful, this one-pot two-step multi-catalysis method suffers from the need of homogenous catalysts, which are tedious to remove and could result in high Pd and ligand contamination of the final products that is not acceptable when dealing with animal and human health. The use of heterogeneous catalysts associating the [Pd(PNP)]@SBA-15 catalyst to a grafted amine catalyst as [NH2]@SBA-3 in a one-pot tandem [Pd/amine] mode allowed, for example, the selective synthesis of 2-phenylquinolin-4(1H)-one in a suitable 61% isolated yield [Scheme 46, (ii)] [65]. This approach resulted in a strong decrease of Pd-contamination in the final products as only 3 to 5 ppm of Pd was found in the crude quinolin-4(1H)-ones, while 40 ppm was measured when using homogeneous catalytic system. The overall reaction time was also reduced from 7 to 3 days (in the same reaction conditions). Recycling of the {[Pd(PNP)]@SBA-15/[NH2]@SBA-3} catalyst mixture was successful for 3 runs [66].
Two different protocols were developed for the Sonogashira coupling of 2-iodoanilines 1 with alkynes 47 and cyclization toward functionalized quinolin-4(1H)-ones 149 (Scheme 47) [67]. In the first protocol (Method A), quinolin-4(1H)-ones 149 were obtained after 20 min of microwave (MW) heating at 120 °C using Mo(CO)6, Pd2(dba)3 with an excess dppf, as ligand and Cs2CO3 in diethylamine. The reaction scope is quite general, however, when using 2-iodo-4-nitroaniline, the corresponding product was obtained in low yield, due to the thermally induced nitro group reduction by Mo(CO)6. To overcome this limitation, a second protocol (Method B) was developed using milder conditions, extending the scope of the reaction to reduction-prone or other sensitive groups, such as nitro and bromo substituents. The coupling reaction was performed at room temperature using a different catalytic system, Mo(CO)6, Pd(OAc)2, [HP(t-Bu)3]BF4 followed by the addition of diethylamine to promote the cyclization of the arylalkynone intermediate 150. The formation of quinolin-2(1H)-ones was never detected using these two methods, on contrary to the previous observations of Chen and co-workers [35], who reported a Mo(CO)6 protocol for the formation quinolin-2(1H)-ones starting from 1 and 47.
3.2. Buchwald-Hartwig Reaction
An efficient Pd-catalysed tandem amination approach, starting from easily accessible 2-haloaryl acetylenic ketones 151 and primary amines 125, involving a sequential double C-N bond formation, furnished functionalized quinolin-4(1H)-ones 152 in good to excellent yields in one step (Scheme 48) [68]. The reaction of 151 (X = Br, R1 =H) with aniline in the presence of Pd(PPh3)4 in 1,4-dioxane, using K2CO3 as base, gave the corresponding quinolin-4(1H)-one 152 in 71% yield. A similar result was obtained using the PdCl2(dppf)-CH2Cl2 complex as catalyst. Improvements were made using Pd2(dba)3-CHCl3 as catalyst combined with Xantphos or dppp, however PPh3 proved to be the best ligand affording quinolin-4(1H)-one 152 in 84% yield. A range of commercially available aryl amines can be employed to give the corresponding products in moderate to good yields (61–93%). Even those with active amino or hydroxy groups remain intact under the reaction conditions. However, with aliphatic amines such as butylamine the product was obtained in moderate yield (42%). This reaction is also compatible with different types of ynones substituted with aryl and pyridyl groups.
Two pathways were proposed for this reaction (Scheme 49), which may involve either oxidative addition of Pd(0) to the C-Br bond in 151 (intermediate I in Path A) or conjugate addition of aniline to 151 (intermediate II in Path B) in the first step. The formed intermediate I presumably leads to III through Buchwald-Hartwig amination or activation of a C≡C bond in I through coordination to the Pd and attack by aniline to form intermediate IV. Both pathways will go through intermediates IV and V, followed by reductive elimination of Pd(0) to give the desired quinolin-4(1H)-one 152. Based on some experiments conducted by Xu and Zhao [68], path A could be the major pathway to afford the target quinolone.
An efficient Pd-catalysed tandem amination protocol for the synthesis of 1,2-disubstituted quinolin-4(1H)-ones 154 was developed starting from easily accessible chalcones 153 and primary amines 125, in which the Pd-catalyst [Pd(OAc)2] plays a dual role, namely, in the Buchwald–Hartwig coupling and catalytic dehydrogenation (Scheme 50) [69]. Pd2(dba)3 that was a good catalyst in the Pd-catalysed tandem amination of 2-haloaryl acetylenic ketones 151 and primary amines 125 [68] was less effective than Pd(OAc)2 in this transformation (55% and 74%, respectively). The procedure using PPh3 as a ligand in refluxing anhydrous 1,4-dioxane and K2CO3 as base was efficient with aromatic, heteroaromatic and aliphatic amines. Arylamines containing electron-donating groups gave higher yields (79–87%) than those with electron-withdrawing groups (54–78%) and aliphatic amines gave the expected quinolin-4(1H)-ones 154 in slightly lower yields (45–52%). Different functional groups (R1 and R2) of chalcones 153 were all compatible with the reaction conditions. Due to the low oxidative addition reactivity of the C–Cl bond, 2-chloro-substituted chalcones gave quinolin-4(1H)-ones 154 in lower yield than the corresponding bromo-derivatives, even upon raised temperature.
3.3. Stille Reaction
Pd-catalysed cross-coupling reaction of 5-(tributylstannyl)-3-methylisoxazole 156 with 2-bromonitrobenzene 155 followed by the catalytic hydrogenation of 157 over Raney nickel afforded 2-methylquinolin-4(1H)-one 158 in 57% overall yield (Scheme 51) [70].
The Pd-catalysed carbonylative coupling of ethyl 2-iodophenylcarbanylate 159 with (Z)-tributyl(2-ethoxyvinyl)stannane 160, under CO atmosphere, afforded ethyl (E)-2-(3-ethoxy-1-oxoprop-2-en-1-yl)phenyl carbanylate 161. Then compound 161 underwent cyclization under acidic conditions to give quinolin-4(1H)-one 162 (Scheme 52) [71].
3.4. Cyclocarbonylation Reaction
Carbonylation of 3-substituted 3-(2-haloarylamino)prop-2-enoates 163 in the presence of Pd-catalyst under CO atmosphere, at 120 °C, resulted in heterocyclization to form a variety of 2-substituted quinolin-4(1H)-one-3-carboxylates 164 (Scheme 53) [72]. Cyclocarbonylation of iodoenamine 163 (X = I) was effective at 20 kg·cm−2, whereas 30 kg·cm−2 were required for the bromoenamine 163 (X = Br). After treatment with diazomethane, to avoid the presence of quinolin-4(1H)-one-3-carboxylic acid, quinolin-4(1H)-ones 164 were obtained in moderate to good yields (55-82%) although derivatives involving 2-methoxycarbonyl group or 6,7-difluoride gave lower yields (24% and 37%, respectively) [Scheme 53, (i)]. Carbonylative heterocyclization of other functionalized enamines 165 resulted in the successful formation of the desired products 166 in 82% and 87% yield; furthermore, the hydrolysis of lactone 166 may provide other intriguing quinolin-4(1H)-one carboxylic acids [72]. This method is of practical importance because of the wide choice of the group R3 in addition to the fact that the benzene ring may carry a diverse number of substituents. Under similar conditions but using CO at atmospheric pressure, Stanforth and co-workers reported the synthesis of three 2-trifluoromethylated quinolin-4(1H)-ones in 54–77% yield [Scheme 53, (ii)] [73,74].
Pd-catalysed cyclization-alkoxycarbonylation of 1-[(2-trimethylsilyl-ethynyl)phenyl]urea 167 using Pd/C-Bu4NI as catalyst in the presence of KF, for the in situ deprotection, gave quinolin-4(1H)-one 168 (Scheme 54) [75,76]. Formation of the compound 168 is quite intriguing and it was described as arising from some sort of rearrangement, probably via intermediate formation of a benzoxazine derivative. A reasonable mechanistic hypothesis was proposed by the authors but requires further investigation [45,75,76]. The nature of the substituent was found to be crucial for the product formation. In the case of an unsubstituted urea 167 quinolin-4(1H)-one 168 was the only isolated reaction product; but when it is substituted, no quinolin-4(1H)-one was obtained [75].
Pd-catalysed carbonylative cyclization of readily available N-(2-iodoaryl)enaminones 170 afforded 2-substituted-3-aroylquinolin-4(1H)-ones 171 in very satisfactory yields (37–91%) (Scheme 55) [77]. Pd2(dba)3 was found to be the best catalyst but the use of a bulky monodentate ligand such as 2-dicyclohexylphosphino-2’,6’-dimethoxy-1,1′-biphenyl (SPhos) or XPhos is required as well as 20 atm of CO. Good to high yields are usually obtained with enaminones bearing both electron-donating and electron-withdrawing as well as neutral aromatic rings. A moderate yield of the desired 3-aroylquinolin-4(1H)-one derivative 171 is obtained with an N-(2-iodoaryl)-β-enaminone containing a bromine substituent in the aniline moiety. The synthesis of 2-substituted-3-aroylquinolin-4(1H)-ones 171 is also possible without isolation of the enaminone intermediate by adding Pd2(dba)3, XPhos, Cs2CO3, MeCN and CO (20 atm) to the crude mixture derived from the reaction of 2-iodoanilines 1 with α,β-ynones 169 after evaporation of the volatile materials. Under these conditions 171a (R1 = H, Ar1 = Ar2 = Ph) was isolated in 61% overall yield.
Highly functionalized 2,3-dihydroquinolin-4(1H)-ones 173 were obtained in one step, with moderate to good yields, by Pd-catalysed intermolecular cyclocarbonylation of 2-iodoanilines 1 with diethyl ethoxycarbonylbutendienoate 172 (Scheme 56) [78]. The method involves a Michael addition and subsequent carbonylation reactions using the catalytic system of Pd2(dba)3/2-(di-t-butylphosphino)biphenyl in MeCN at 80 °C under 500 psi of CO. Solvents like CH2Cl2 or THF promote the formation of the Michael addition product 174, in more than 80% yield, being the unique reaction product since no carbonylation occurs. In general, better yields were obtained with electron-donating phosphines, such as trialkylphosphines and dialkylarylphosphines; however, 2-(di-t-butylphosphino)biphenyl was the better ligand. Xantphos behaved differently promoting the formation of 175 much more selectively than when using any other ligands. The reaction is sensitive to the electronic nature of the substituents at the p-position relatively to the iodide group. Highly electron-donating or electron-withdrawing groups, such as methoxy and chlorine, afforded 173 in lower yields (39% and 26%, respectively). Additionally, substituents have influence on the rate of the carbonylation and Michael addition steps and both can take place independently. Thus, a favourable balance of the rate between these two reactions is important to achieve successful results. In the reaction of 1 with a tetrasubstituted olefin the initial Michael addition cannot take place, probably due to steric effects and therefore no product was isolated. Other types of Michael acceptors 172c, 172d and 172e were tested leading to unsatisfactory yields, even after a brief screening (of phosphines and bases) to optimize the reaction conditions (14–35%).
A plausible reaction mechanism suggests the occurrence of a Michael addition of 2-iodoaniline 1 to diethyl ethoxycarbonylbutendienoate 172, as the first step, to produce the Michael adduct 174. Then the phosphine-ligated Pd(0) species undergoes oxidative addition to the C-I bond of 174, followed by CO insertion giving the aroyl-Pd intermediate I. Nucleophilic attack of the internal malonate anion on this intermediate I ends the catalytic cycle affording quinolin-4(1H)-ones 173 regenerating the Pd(0) species. Formation of 175 was explained by intermolecular double carbonylation of 1 with 174 formed in situ. First intermolecular carbonylation takes place between 1 and 174 affording an acyclic amide II and/or III, which can then undergo a second intramolecular carbonylation to give the final product 175 (Scheme 57) [78].
Quinolin-4(1H)-ones 177 were obtained by a Pd(0)-catalysed termolecular queuing process involving oxidative addition to aryl iodide 176, followed by carbonylation, allene insertion and capture of the resulting п-allyl-Pd(II) species by an internal N-nucleophile [79]. CO at atmospheric pressure was used in contrast to Alper′s similar reported cascades [80], which were performed at high pressures of CO (20 atm). Under optimal conditions, using allene (1 atm) and CO (1 atm), a (3 + 1 + 2)-cycloaddition reaction of 2-iodo-1-tosylaniline occurred giving quinolin-4(1H)-ones 177 in good yields (55–99%) (Scheme 58). Substituents are tolerated on both the allene and the aryl iodide allowing access to a variety of heterocycles with s-cis-enone moieties.
In the first step of the catalytic cycle, the Pd(0) catalyst undergoes oxidative addition to the aryl iodide 176 bond followed by coordination and insertion of CO. Then addition of acyl-Pd(II) intermediate I to the allene at the central carbon atom originated a п-allyl-Pd(II) species II which undergoes nucleophilic attack by the internal nucleophile to give the enone product 177 (Scheme 59) [79]. These authors also described the one-pot quinolin-4(1H)-one synthesis-Michael addition starting from 2-iodo-1-tosylaniline 176, CO (1 atm) and allene (1atm) in toluene at 45 °C during 48 h. CO was released before the addition of the nucleophile and subsequent Michael addition completed after further 24 h (Scheme 60) [79]. Both aliphatic and heteroaromatic N-nucleophiles were effective and in the case of 1,2,4-triazole only the 1-substituted triazole was observed. As the obtained Michael adducts were sensitive to retro-Michael addition, their reduction with LiAlH4 was performed without prior isolation, obtaining the corresponding γ-amino alcohols 178 as single stereoisomers.
3.5. Oxidative Carbonylation
Ley and co-workers synthesized quinolin-4(1H)-ones 182 via Pd-catalysed oxidative carbonylation starting from simple and commercially available amines 179 and ketones 180 using CO at atmospheric pressure (Scheme 61) [81]. First, an imine 181 is formed from ketone and amine by dehydration condensation. Then, enamine derived from imine/enamine isomerization, undergoes electrophilic attack by Pd(II) to form an intermediate which undergoes intramolecular C−H activation and CO insertion. Subsequent reductive elimination affords the final product and releases Pd(0), which is oxidized by copper and O2 to regenerate Pd(II) and complete the catalytic cycle [81]. From different palladium salts, Pd(dba)2 was found to be the best option and the use of Xantphos as a ligand improves the product yield. CuBr(Me2S), PhCO2Na and KI are necessary for the reaction which could also proceed smoothly to give the desired product under nonexplosive conditions (CO/O2 = 15/1). For m-substituted anilines 179 the reaction with acetophenones gave the desired products in moderate to good yields but with poor selectivity. However, this protocol provides a straightforward route to access useful quinolin-4(1H)-ones from inexpensive chemicals. As a wide range of functional groups is well tolerated, various ketones, heterocyclic ketones and amines are workable substrates that can be employed to generate quinolin-4(1H)-ones in good yields.
3.6. Other Reactions
A mild, one pot sequential Pd-catalysed amidation of 2′-bromoacetophenones 183 followed by base-promoted intramolecular cyclization, afforded 2-substituted quinolin-4(1H)-ones 185 in 29–96% yields over two steps (Scheme 62) [82]. The best solvent-base combination was 1,4-dioxane/Cs2CO3. The addition of a strong base, either NaOH or NaOt-Bu, was found to be necessary to avoid the hydrolysis of the amide before cyclization. The reaction scope was quite general for both coupling partners although acyclic secondary amides were not successful. Indeed, this method allows the synthesis of a wide range of 2-substituted quinolin-4(1H)-ones 185.
The regioselective Pd-catalysed intramolecular N-arylation of (Z)-enamine 186 afforded quinolin-4(1H)-one 187 in 88% yield, which was further reduced to the corresponding 2,3-dihydroquinolin-4(1H)-one 188 (Scheme 63) [83].
Pd-catalysed allylic amination-thiazolium salt-catalysed Stetter reaction between 2-aminobenzaldehydes 189 and allylic acetates 190 is an interesting method to prepare 3-substituted 2,3-dihydroquinolin-4(1H)-ones 192 (Scheme 64) [84,85]. The key to the success of this method is the compatibility of the second catalysis with the conditions of the first Pd-catalysis. Under optimized conditions, the one-pot sequential two-step multi-catalytic cascade process afforded 2,3-dihydroquinolin-4(1H)-ones 192 in excellent yields (94–99%). This procedure tolerates 2-aminobenzaldehydes 189 bearing electron-donating and electron-withdrawing substituents on the aromatic ring, although, in contrast to that of γ-acetoxy-α,β-unsaturated esters 190 (R2 = CO2Et, CO2t-Bu) the reaction did not proceed for γ-acetoxy-α,β-unsaturated nitrile 190 (R2 = CN) [Scheme 64, (i)]. When performing the one-pot sequential multi-catalytic cascade process in the presence of both catalysts (one-step procedure), the reaction proceeded well with the three different allylic acetates 190 giving 2,3-dihydroquinolin-4(1H)-ones 192 in excellent yields (98–99%), but, on the other hand, the allylic amination occurred only for the unsubstituted 2-aminobenzaldehyde 189: (R1 = H, 98%) [Scheme 64, (ii)].
Pd(0)-catalysed intramolecular coupling of β-(2-iodoanilino)esters 193 afforded several 2,3-dihydroquinolin-4(1H)-ones 194 in moderate to good yields (Scheme 65) [86]. The use of Pd(PPh3)4 as the catalyst and Cs2CO3 as base in THF resulted exclusively in the reduction product 195. In contrast, using K3PO4 as base in combination with Et3N and toluene led to an increase in the yield of dihydroquinolin-4(1H)-ones 194. These compounds 194 resulted from the nucleophilic substitution at the alkoxycarbonyl group. Substituents on the aromatic ring have little effect on the carbopalladation reaction evidencing that the nucleophilicity of the aryl-Pd species does not appear to be affected by the electronic properties of the substituent. The low yield for the product derived from 193 (R1 = CO2Me) is mainly a consequence of an increase in the rate of the competitive retro-Michael fragmentation of the β-aminoester. Benzyl ester counterpart of 193 (R4 = Bn) can also be used as substrate; however, the desired product is formed in better yields when using methyl ester (R4 = Me) (50% and 65%, respectively). The reaction of amino esters 193 without α-hydrogen atoms to the carbonyl group proceeded smoothly to give exclusively the corresponding dihydroquinolin-4(1H)-ones 194 in high yields (79–88%). On the other hand, no competition between nucleophilic attack at the carbonyl group and α-arylation was observed in the reactions of amino esters 193 which contain α-hydrogen atoms to the carbonyl group; however, dihydroquinolin-4(1H)-ones 194 (35–67%) were obtained together with the reduction products 195 (25–45%).
This Pd(0)-catalysed cyclization involves the oxidative addition of the aryl iodide to a Pd(0) species affording a four-membered azapalladacycle I. A carbopalladation between the α-aryl Pd moiety and the alkoxycarbonyl group would give the alkoxide-Pd(II) chelate III. The coordination of the N atom to the Pd centre in I brings the carbonyl group nearer to the metal to facilitate the formation of a transient chelated intermediate II in which the carbonyl group is coordinated to the Pd centre and increases the electron density on the Pd centre to enable the otherwise unfavourable carbopalladation reaction. β-Alcoxide elimination from III would afford the dihydroquinolin-4(1H)-one 194 and a Pd(II) alkoxide, which would finally undergo β-hydride elimination regenerating the Pd(0) catalyst. The reduction sequence is supported by the isolation of significant amounts of benzaldehyde in the reaction of benzyl ester 193 (R4 = Bn) (Scheme 66).
4. Transformation of Quinolin-2(1H)-ones
4.1. Heck Reaction
Pd-catalysed Heck reaction of 3-iodoquinolin-2(1H)-one 196 with 2-methyl-3-buten-2-ol 197 afforded the quinolone dimers paraensidimerins′ precursor 198 (Scheme 67) [87]. The reaction of 4-acetoxyquinolin-2(1H)-one 196 (R = Ac), obtained by acetylation of 4-hydroxy-3-iodo-1-methyl quinolin-2(1H)-one 196 (R = H), gave three products: (i) the expected quinolone allylic alcohol 198 (30%); (ii) the diene 200 (11%), probably obtained by dehydration of allylic alcohol 198 by the Et3NHI salt by-product formed in the reaction mixture; and (iii) the known alkaloid N-methylflindersine 199, probably formed by deacetylation and subsequent intramolecular cyclization during the course of the reaction.
Successful Heck vinylations of 4-aryl-3-bromo-1-methylquinolin-4(1H)-ones 201 with acrylates 202 were achieved by Kappe and co-workers using Pd(PPh3)4 and Et3N in DMF, affording 3-substituted-quinolin-4(1H)-ones 203 (Scheme 68) [88]. Similar transformations were performed by Wu and co-workers using a different catalyst system, Pd(OAc)2 in the presence of tris(2,6-dimethoxyphenyl)phosphine and TBAB, using also Et3N and DMF (Scheme 68) [89].
The highly α-regioselective Heck coupling of 4-tosylquinolin-2(1H)-one 204 with butyl vinyl ether afforded 4-acetylquinolin-2(1H)-one 206 in very good yield (93%) (Scheme 69) [90]. The α-product 205 was obtained in very high α/β regioselectivity (>99/1) and after acidic treatment led to 4-acetylquinolin-2(1H)-one 206. Although α-regioselectivity in Heck coupling can be obtained with triflates as substrates, tosylates are less toxic, less expensive and, importantly, more stable.
The intramolecular Heck reaction of the benzylallylquinolin-2(1H)-one precursor 207 originated a benzoxocine-fused quinolin-2(1H)-one 208 (Scheme 70) [91]. The reaction led to the regioselective formation of the 8-exo cyclization product 208 and no 9-endo product was observed in this case.
Majumdar and co-workers reported a Pd-catalysed intramolecular Heck reaction of inactivated allylic quinolin-2(1H)-ones 209 that led to the formation of quinolin-2(1H)-one annulated benzoazocines 210 (Scheme 71) [92]. The intramolecular Heck reaction afforded exclusively the endo-cyclic product 210 in good yields. Once exclusively cyclization via the 8-exo mode is unusual, in this case, it was suggested that the formation of the endo-cyclic products may occur via two possible pathways; the 8-exo mode of cyclization followed by double-bond isomerization, or, alternatively, a double-bond isomerization prior to the Heck reaction leading to the subsequent 8-endo-Heck cyclization (Scheme 71) [92].
Later, the same authors described the synthesis of quinolin-2(1H)-one annulated benzazocinones 212 by applying the same methodology to inactivated allylic quinolin-2(1H)-ones 211 (Scheme 72) [93]. In this case, the optimal reaction conditions required Pd(PPh3)4 as the catalyst. This highly regioselective ligand-free Heck coupling reaction leads exclusively to the expected 8-exo-Heck product 212, in good yields, without any contamination of the 9-endo-Heck or 8-exo-isomerized product. The formation of the 8-exo-Heck product is favoured because of the lower steric and transannular interactions. These authors have also prepared two quinolin-2(1H)-one annulated benzazoninones 214 starting from inactivated allylic quinolin-2(1H)-ones 213 (Scheme 72) [94]. The Heck reaction proceeded in reasonably good yields with shorter reaction times and only the product corresponding to the 9-exo mode of cyclization was obtained.
Pd-catalysed intramolecular Heck cyclization of previously synthesized 3- and 4-(2-bromobenzyloxy)quinolin-2(1H)-ones 215 and 217 [95,96], under Jeffery’s two-phase protocol afforded four tetracyclic quinolones 216 and 218 in very good yields (Scheme 73) [97].
Intramolecular regioselective arylation of N-methylquinolin-2(1H)-one 219 (X = Br) was achieved using a simplified catalyst system, Pd(OAc)2, TBAB and KOAc in toluene and Jeffrey’s conditions for the Mizoroki-Heck-type reaction, affording 12-methyl-6H-isochromeno[4,3-c] quinolin-11(12H)-one 220 in 65% yield (Scheme 74) [98]. The same compound 220 was obtained, in good yield (60%), by intramolecular direct arylation of quinolin-2(1H)-one 219 (X = I) which was performed using a Pd(0) source and pivalic acid as a crucial additive (Scheme 74) [99]. Mechanistic studies suggested that the reaction proceeds through an initial oxidative addition followed by a pivalate assisted Concerted-Metalation-Deprotonation (CMD)-type mechanism, although a SEAr route was not ruled out [99].
A ligand free intramolecular Heck reaction of 6-amino-5-bromoquinolin-2(1H)-ones 221 catalysed by Pd(OAc)2 gave new pyrrole-fused quinolin-2(1H)-ones 222 (Scheme 75) [100]. This methodology is applicable to secondary and tertiary amines. Under similar reaction conditions the intramolecular Heck reaction of quinolin-2(1H)-one 223 afforded the corresponding fused quinolin-2(1H)-one 224.
Other pyrrolo[3,2-f]quinolin-7(6H)-ones 227 were synthesized, in very good yields, by ligand free Pd(OAc)2-catalyzed intramolecular Heck reaction of enamines, prepared in situ from the condensation of 6-amino-5-bromo-1-methylquinolin-2(1H)-ones 225 with cyclic and acyclic ketones 226 (Scheme 76) [101]. The reaction was only effective when using DMF as solvent and other Pd sources [PdCl2, PdCl2(PPh3)2, Pd(PPh3)4] were less efficient than Pd(OAc)2. From the screened bases (DABCO, KOAc, NaOAc, Et3N), DABCO was found the most suitable and it was not necessary to protect the amino group. First, substrates 225 condense with the ketone to generate I and produce the enamines II in the presence of base. The formed enamines subsequently underwent intramolecular Pd-catalysed Heck reaction to afford the products 227 (Scheme 76).
4.2. Suzuki-Miyaura Reaction
An efficient convergent synthesis of the potent and selective KDR inhibitor 3-{5-[[4-(methylsulfonyl)-1-piperazinyl]methyl]-1H-indol-2-yl}quinolin-2(1H)-one 231 involved a Suzuki coupling of two substrates 228 and 229, as the main step to construct the indol-2-ylquinolin-2(1H)-one moiety 230, the key pharmacophore of this type of KDR inhibitors. The cross-coupling was run in THF with Pd(OAc)2 (0.5 mol%) and PPh3; however, the usual basic aqueous system led to substantial deboronation. Dicyclohexylamine was found to be an excellent activator and slow addition of boronic acid to the catalyst/bromoquinolinone mixture minimized deboronation (Scheme 77) [102]. Formerly, Fraley and co-workers have also adopted the Suzuki-Miyaura cross-coupling of diversely substituted indolylboronic acids with 3-iodoquinolin-2(1H)-ones, using Pd(PPh3)4 as catalyst, to synthesize other related KDR inhibitors [103].
Pd-catalysed cross-coupling reactions of 3-bromo-4-trifloxyquinolin-2(1H)-ones 232 with arylboronic acids afforded 4- and 3,4-substituted quinolin-2(1H)-ones 233 and 234 (Scheme 78) [104]. Regiocontrolled cross-coupling reaction of 3-bromo-4-trifloxyquinolin-2(1H)-one 232 was achieved by tuning the temperature and the amount of arylboronic acid. When using PdCl2(PPh3)2 as catalyst in the reaction with 4-methoxyphenylboronic acid (1.5 equiv) at 50 °C, the corresponding product 233 was obtained in 20% yield together with disubstituted compound 234 (60%). At room temperature, 233 was obtained as the major product (78%) and only traces of 234 were detected. Reducing the amount of 4-methoxyphenylboronic acid to 1.1 equiv, 233 was the only product (81%). At 60 °C with a higher excess of 4-methoxyphenylboronic acid (2.5–3.0 equiv), only 234 was generated (87%). Both electron-withdrawing and electron-donating-substituted arylboronic acids are suitable coupling partners, giving similar reaction yields. Compound 233 (R1 = 3-CF3Ph) was further elaborated by Suzuki-Miyaura reaction with different arylboronic acids affording the corresponding products 235 in good yields (Scheme 78) [105]. The reaction of 3-bromo-4-trifloxyquinolin-2(1H)-one 232 with arylboronic acids, catalysed by PdCl2(PPh3)2, afforded 4-aryl-3-bromoquinolin-2(1H)-ones 233 (Scheme 78) [105]. Various aryl groups could be easily introduced at the 4-position of quinolin-2(1H)-one scaffold in good to excellent yields.
Derivatization of 4-chloro- and 4-trifloxy-1-methylquinolin-4(1H)-ones 236, by introducing 4-aryl substituents, was performed by Pd-catalysed Suzuki-Miyaura reaction. The best conversions and isolated product yields (83–91%), for the reaction of 236 (R = Cl) with arylboronic acids, were achieved by using a combination of Pd(OAc)2 (5 mol%), PPh3, a 3:1 mixture of 1,2-dimethoxyethane (DME):water as solvent, together with either Na2CO3 or Et3N as base, at 150 °C (Scheme 79) [88]. When employing 236 (R = OTf) as electrophile, the coupling product was isolated in 88% yield, using PdCl2(PPh3)2, Na2CO3 and a mixture of THF and water as solvent (Scheme 79) [89]. The presence of water in the solvent mixture was found to be crucial for these reactions to proceed properly.
Pd-catalysed one-pot borylation-Suzuki cross-coupling reaction of 4-chloroquinolin-2(1H)-ones 238, under controlled MW irradiation, gave functionalized 4,4′-bisquinolones 239 in good to excellent yields (Scheme 80) [106]. Excellent conversions were achieved using a strong base such as KOH in combination with PdCl2(dppf). Among the tested solvents, DMSO, DMF or toluene promote the formation of the dehalogenated product while 1,4-dioxane, dichloromethane and 1,2-dichloroethane (DCE) minimized dehalogenation; however, 1-chlorobutane proved to be the best solvent in this particular case.
4.3. Sonogashira Reaction
Sequential coupling and cyclization reactions of aryl halides with (trimethylsilyl)acetylene with concurrent elimination of the trimethylsilyl (TMS) substituent furnished pyrrolo[3,2-f]quinolin-7(6H)-ones 246 in excellent yields (Scheme 82) [108]. Acetylenic amines possessing an electron-donating group on the nitrogen atom also underwent Cu(I)-catalysed cyclization. Quinolones 243 were prepared from commercially available quinolines [109,110], then brominated and the resulting bromo-derivatives 244 were transformed into the required precursors for heteroannulation 245 by a Sonogashira coupling with (trimethylsilyl)acetylene using PdCl2(PPh3)2 as catalyst and CuI as the co-catalyst in THF/DMF mixed solvent containing Et3N. The reactions were optimized by smoothly heating the reaction in a sealed tube. Heteroannulation of the acetylenic amines to give 246 was achieved by refluxing the precursors 245 in DMF in the presence of 50 mol% of CuI. Another work reported the synthesis of acetylenic amines 245 and 247 by Sonogashira coupling of the corresponding 6-amino-5-bromoquinolin-2(1H)-ones 244 with (trimethylsilyl)acetylene or phenylacetylene, respectively. Both reactions were performed using PdCl2(PPh3)2 as catalyst and CuI as co-catalyst, although in slightly different experimental conditions (Scheme 82) [111].
Acetylenic amines 248, obtained from 244 by a Sonogashira coupling, underwent an intramolecular hydroamination reaction, catalysed by PdCl2/FeCl3, to give pyrrolo[3,2-f]quinolin-7(6H)-ones 249 (Scheme 82) [112]. PdCl2(PPh3)2 and Pd(OAc)2 were also tested as Pd(II) sources but were ineffective, unlike PdCl2 that was efficient in only 1 mol%. FeCl3 is necessary for this hydroamination reaction to occur and may facilitate the reoxidation of Pd(0) to Pd(II) in the catalytic cycle. This cyclization proceeded well in 1,2-dichloroethane at reflux, in the presence of aromatic- and aliphatic-substituted alkynes and with both protected and unprotected amines.
Pd-catalysed Sonogashira reaction of 1-methyl-3-nitro-4-trifloxyquinolin-2(1H)-one 250 with various alkynes, in the presence of Pd(PPh3)2Cl2/CuI in THF at room temperature, using K2CO3 as the base, afforded the coupling products 251 in moderate to good yields (Scheme 83) [113]. Aliphatic alkynes are excellent substrates, whereas no product was detected when using aromatic alkynes. A strategy was developed to install aromatic substituents at the alkynes moiety at the C-4 of the quinolin-2(1H)-one core by subjecting compound 253 (R = H) to Sonogashira coupling with aryl iodides. This cross-coupling reaction afforded the corresponding 3-amino-4-arylethynyl quinolin-2(1H)-ones 252 in high yields with various aryl iodides. Reduction of the nitro group of 251, mediated by iron powder in AcOH/H2O at room temperature, generated the 4-alkynyl-3-aminoquinolin-2(1H)-ones 252 in excellent yields. Finally the intramolecular hydroamination of 3-amino-4-aryl-ethynylquinolin-2(1H)-ones 252 in DMF, in the presence of PdCl2, gave the desired 3H-pyrrolo[2,3-c]quinolin-4(5H)-ones 254 in moderate to good yields; however, arylacetylenic substrates with electron-withdrawing groups on the R position were less reactive than those with electron-donating groups (Scheme 83) [113]. The one-pot reaction between 3-amino-4-ethynylquinolin-2(1H)-one 252 (R = H) and p-iodoanisole was also successfully achieved affording 3H-pyrrolo[2,3-c]quinolin-4-(5H)-one 254 in 71% yield (Scheme 83) [113].
According to Wu and co-workers, the Sonogashira reaction of quinolin-2(1H)-ones 255 with 2-ethynylaniline 256 proceeded smoothly in the presence of PdCl2 (5 mol%), CuI (5 mol%) and PPh3 (10 mol%) in Et3N at 50 °C to afford the desired products 257 in 89% yield. CuI-mediated cyclization of 257 led to the desired 1H-indol-2-yl-(4-aryl)quinolin-2(1H)-ones 258 in very good yield (95%) (Scheme 84) [105]. These authors have also attempted the one-pot reaction of 3-bromo-4-(4-methoxyphenyl)quinolin-2(1H)-one 255 (Ar = 4-MeOC6H4) with 2-ethynylaniline 256. After completion of the reaction of 255 with 256, CuI (1.0 equiv) was added directly in the reaction mixture and it was stirred at 160 °C for another 1 h affording the expected product 257 (Ar = 4-MeOC6H4) in 79% yield. They have demonstrated the generality of this one-pot procedure by preparing a series of other derivatives which were obtained in moderate to good yields (65–80%) (Scheme 84) [105].
4-Alkynyl-3-bromoquinolin-2(1H)-ones 260, prepared by Sonogashira reaction of 3-bromo-4-trifloxyquinolin-2(1H)-ones 259 with terminal alkynes 47, underwent Pd-catalysed domino reaction with amines affording 3H-pyrrolo[2,3-c]quinolin-4(5H)-ones 261 (Scheme 85) [114]. Both aryl and alkyl substituents in the quinolin-2(1H)-one 260 and in the terminal alkyne 47 were well tolerated with exception of trimethylsilyl acetylene. The electronic nature of the arylamine does not influence the outcome of the domino reaction. In addition, o-substituted anilines are well tolerated. Substrates 260 with an alkyl group as R1 were more reactive than with an aryl group. An attempt to perform the synthesis of 3H-pyrrolo[2,3-c]quinolin-4(5H)-one 261 in a one-pot and sequential manner, from of 3-bromo-4-trifloxyquinolin-2(1H)-one 259, phenylacetylene and aniline, afforded the expected product in 32% yield. Diversity of 3H-pyrrolo[2,3-c]quinolin-4(5H)-ones 261 was increased after a bromination of pyrrole ring with NBS and Suzuki coupling with p-tolylboronic acid that led to 5-methyl-2,3-diphenyl-1-(p-tolyl)-3H-pyrrolo[2,3-c]quinolin-4(5H)-one 262 in 99% yield (Scheme 85) [114].
4.4. Buchwald-Hartwig Reaction
Pd-catalysed C–N coupling reaction, starting from 3-bromoquinolin-2(1H)-ones 263, originated 3-(N-substituted)aminoquinolin-2(1H)-ones 264–267 in good to excellent yields (Scheme 86) [115]. Several nucleophiles, including amines, amides and benzylcarbamate, as well as the less nucleophilic alkyl- or arylsulfonamides, were effective in this transformation. In all the cases, the reactions occurred rapidly in 1,4-dioxane, using Pd(OAc)2 as a catalyst, Xantphos as a ligand and Cs2CO3 as a base (Scheme 86). In the presence of two carbon–bromine bonds, as in the quinolin-2-(1H)-ones 263 (R2 = Br), the coupling proceeded at the more activated C-3 position and yielded the mono-substituted product (44–48%) together with the disubstituted one (15–27%). The C-N bond forming reaction was also performed with urea, in the same experimental conditions, leading to the double heteroarylated coupling product 267 in good yield (61%) (Scheme 86) [115]. Also, Pd-catalysed coupling reaction of 3-bromo-1-methylquinolin-2(1H)-one 263, as the electrophilic partner, with indole and 5-methoxyindoles 268 afforded the coupling products 269 in excellent yields (Scheme 86) [116].
4.5. Aminocarbonylation Reaction
Pd-catalysed aminocarbonylation of 4-hydroxyquinolin-2(1H)-one 270 afforded quinolin-2(1H)-one-6-carboxamides 275 which are potent antagonists of gonadotropin releasing hormone receptors [117]. O-Alkylation of 270 with either (±)-271 or (S)-271 using the Mitsunobu protocol afforded the O-alkyl ethers (±)-272 and (S)-272. Then Pd-catalysed aminocarbonylation of the iodo compound 272 with a variety of primary and secondary amines 273 led to the protected quinolin-2(1H)-one-6-carboxamides 274. Removal of Boc protecting group of piperidines 274 using trifluoroacetic acid afforded the targeted amides 275 in quantitative yields (Scheme 87) [117]. Quinolin-2(1H)-one-6-carboxamide 278 was also prepared by alkylation of 4-hydroxyquinolin-2(1H)-one 270 with (S)-2-(2-chloroethyl)-1-methylpiperidine 276 followed by aminocarbonylation of intermediate 277 with 4-aminopyrimidine (Scheme 87) [117].
Kappe and co-workers carried out the microwave-assisted aminocarbonylation of 6-bromo-4-phenylquinolin-2(1H)-one 279 with benzylamine. The reaction was performed using molybdenum hexacarbonyl as a solid source of CO and Herrmann’s palladacycle in combination with Fu′s salt [(t-Bu)3PH.BF4] as a catalyst system, in acetonitrile at 170 °C for 25 min. affording the expected amide 280 in 61% yield (Scheme 88) [88,89].
Aminocarbonylation of 4-aryl-3-bromoquinolin-2(1H)-one 281 under the conditions reported by Wu and co-workers, [Pd2(dba)3 (3 mol%), (R)-BINAP (4.5 mol%), Cs2CO3, toluene, 80 °C], afforded the expected 3-amino derivatives 282 in good to excellent yields (Scheme 89) [89]. Similar results were obtained for electron-donating and electron-withdrawing groups attached on the aromatic ring of substrates. Aliphatic amines were found to be good partners whereas no reaction occurred with bulky amines.
4.6. Other Reactions
Quinolin-2(1H)-ones 283 were functionalized at the 3-position through an intermolecular C-H alkenylation reaction with acrylates or acrylamide, in the presence of Pd(OAc)2 and AgOAc as oxidant, leading to the corresponding 3-alkenyl-4-substituted-quinolin-2(1H)-ones 284 in good yields and with complete regio- and stereoselectivity, even with 4-unsubstituted quinolin-2(1H)-ones (Scheme 90) [45].
The alkylthio functional group of quinolin-2(1H)-one 285 was functionalized by Pd-catalysed cross-coupling with phenylboronic acid in the presence of copper-(I)-thiophene-2-carboxylate (CuTC) affording 4-phenylquinolin-2(1H)-one 286 in 79% yield (Scheme 91) [56].
Quinolin-2(1H)-one-based diaryl methane derivatives 292 and 293 were prepared via basic alumina supported Pd-catalysed cross-coupling of halogenated quinolones 287–290 (X = Cl, Br, I) with freshly prepared benzyl indium organometallic reagent 291, using microwave heating (Scheme 92) [118]. Only 0.5 mol% of Pd(PPh3)4 with basic alumina could afford high yield of product just in 10 min. Increase of the reaction time did not alter the product yield. No addition of base from outside is required to promote the catalytic reaction in basic alumina, which itself acts as the base during the course of the reaction, whereas neutral alumina or silica gels are very ineffective in absence of base. When the reaction was performed using an oil bath at 120 °C low yield of product was achieved even after overnight heating. Basic alumina can be reused after calcinations at 150 °C for 5 h (after washing with water and acetone) and could be recycled at least 3–4 times with minimum loss on its activity. The procedure offers a broad synthetic route for the construction of new polynuclear heteroaromatic systems having potential biological activity.
5. Transformation of Quinolin-4(1H)-ones
5.1. Heck Reaction
The Heck reaction of 3-iodoquinolin-4(1H)-ones 294 with styrene derivatives 295 afforded (E)-3-styrylquinolin-4(1H)-ones 296 (Scheme 93) [119]. The reaction of 3-iodoquinolin-4(1H)-one 294 (R1 = H) with styrene afforded the (E)-3-styrylquinolin-4(1H)-one 296 (R1 = R2 = R3 = H) in moderated yield (46%) using Pd(PPh3)4 as catalyst, PPh3 as ligand and Et3N as base in NMP at 100 °C. Change of the Pd source [Pd(OAc)2 or Pd/C], the ligand [P(o-tol)3] [tol = 4-methylphenyl(tolyl)] or the base (NaOAc or K2CO3) did not improve the yields; instead when P(o-tol)3 was used, the branched regioisomer 3-(1-phenylethenyl)quinolin-4(1H)-one 297 was obtained as the main product (296: 10%; 297: 16%). This Heck procedure revealed to be efficient only when 3-iodoquinolin-4(1H)-one was N-methylated. The Heck reaction of 3-iodo-1-methylquinolin-4(1H)-one 294 led to the (E)-1-methyl-3-styrylquinolin-4(1H)-one 296 in better yield (55%) although the branched regioisomer 297 was also isolated (14%). When the reaction was performed under MW, shortening of the reaction time occurred but lower yields were obtained (40% in 1.5 h). The best catalyst found for the unsubstituted styrene [Pd(PPh3)4] did not work well with substituted styrenes, except for styrene 295 [R2 = NO2; R3 = H (CH: 65%; MW: 45%]. For other styrenes 295, PdCl2 proved to be more efficient (R2 = H; R3 = OMe, CH: 59%, MW: 36%; R2 = R3 = OMe, CH: 55%, MW: 30%; R2 = H; R3 = F, CH: 56%; MW: 48%). Later, Silva and co-workers have performed the Heck reaction of 3-iodoquinolin-4(1H)-ones 294 with styrene derivatives 295 in an ohmic heating (ΩH) reactor [120,121], in aqueous media, using Pd(OAc)2 as catalyst and TBAB as phase transfer catalyst in the presence of an inorganic base [122]. Butyl acrylate was also used as a coupling partner to give a different 3-substituted quinolin-4(1H)-one 298 (Scheme 93) [122]. This methodology is environmentally friendly, due to the use of water as solvent and there is no need for costly and toxic phosphine ligands. Yields were dependent on the amount of styrene or acrylate and on the electronic and steric effects of the substituents on the styrene moiety. Moreover, neutral and electron-donating substituents favoured the formation of the branched isomer. In general, yields were moderate to good and in most cases better than those obtained by the conventional procedures that used organic solvents. In addition, ohmic heating proved to be more efficient than conventional and microwave heating.
The same authors described the Heck reaction of (E)-3-iodo-2-styrylquinolin-4(1H)-ones 299 with styrenes 295, leading to (E,E)-2,3-distyrylquinolin-4(1H)-ones 300 in good yields (58–65%) (Scheme 94) [123,124]. Pd(PPh3)4 was the best catalyst [Pd(OAc)2 and PdCl2 proved to be unsuccessful], MeCN was the most appropriated solvent and Et3N the most suitable base. When heated at high temperatures, the obtained (E,E)-2,3-distyrylquinolin-4(1H)-ones 300 cyclized in two different ways (Scheme 94). Electrocyclization and further in situ oxidation leads to 2,3-diarylacridin-9(10H)-ones 301, whereas tautomerisation, cyclization by nucleophilic attack of the oxygen of the hydroxy group to the β-position of the 3-styryl group and further in situ oxidation produced (E)-2-phenyl-4-styrylfuro[3,2-c]quinolines 303. When the reaction was performed in refluxing 1,2,4-trichlorobenzene, acridin-9(10H)-ones 301 were obtained in low yields (16–37%) and (E)-2-phenyl-4-styrylfuro[3,2-c]quinolines 303 as the main products (35–66%). The use of acidic medium, by addition of p-toluenesulfonic acid (PTSA), in order to displace the tautomerism of 4-hydroxyquinoline 302 to the quinolone 300 and the addition of iodine to facilitate the isomerization of the double bonds improved the yield of acridin-9(10H)-ones 301 (2–40%), although (E)-2-phenyl-4-styrylfuro[3,2-c]quinolines 303 were still obtained as main products (36–60%) [123,124]. Another method, which gave only 2,3-diaryl-10-methylacridin-9(10H)-ones 301 (R1 = Me), involves the Heck reaction of N-protected (E)-3-iodo-1-methyl-2-styrylquinolin-4(1H)-ones 299 (R1 = Me) and styrenes 295, leading to (E,E)-1-methyl-2,3-distyrylquinolin-4(1H)-ones 300 (R1 = Me), which when heated at high temperatures cyclized through tandem-electrocyclization and oxidation processes affording the expected acridin-9(10H)-ones 301 (R1 = Me) (Scheme 94)[124].
The Heck reaction of 6-iodoquinolin-4(1H)-ones 305 with macrolides (from azithromycin) 304 allowed the synthesis of compounds with high antibacterial activity [125]. Macrolones (macrolide + quinolone) 306 were isolated in good to excellent yields when using the catalytic system Pd(OAc)2/P(o-tol)3. The optimized protocol required 2.5 mol excess of quinolone in DMF as the solvent and two different temperatures (65 °C and 75 °C). The desirable macrolones 307 were isolated with > 92% purity after complete hydrogenation of 306 catalysed by Pd/C (Scheme 95) [125].
5.2. Suzuki-Miyaura Reaction
3-(4-Methoxyphenyl)-5-trifluoromethanesulfonatequinolin-4(1H)-ones 308 were converted into derivatives 309, in moderate to good yields, by a modified Suzuki methodology (Scheme 96) [126]. This protocol was successfully applied to the synthesis of 2-(3-methoxyphenyl)quinolin-4(1H)-one analogues 310 [127].
Mono- and/or sequential Suzuki-Miyaura cross-coupling reactions of quinolin-4(1H)-ones 311 with boronic acids afforded novel medicinally important 3-substituted-quinolin-4(1H)-ones 312 (Scheme 97) [128]. When using a Pd/SPHOS catalytic system, the 3-substituted-quinolin-4(1H)-ones 312 were obtained in high yields; however, better yields were obtained with 3-iodo- than with 3-bromoquinolin-4(1H)-ones. For the less soluble quinolin-4(1H)-ones, DMF was required as solvent, while the use of toluene for N-methylated quinolone substrates was found to be superior. This reaction was effective for coupling quinolin-4(1H)-ones with all major subclasses of substrates including aryl, heteroaryl, both electron-donating and electron-withdrawing, alkenyl, fluorinated and pyridyl, as well as sterically hindered substrates, with moderate to excellent yields.
The divergent nature of this synthetic route was demonstrated using 6-chloro-3-iodoquinolin-4(1H)-ones 311, taking advantage of the reactivity differences between the iodo- and chloro- substituents. Previously, the 3-position was subjected to Suzuki-Miyaura coupling (Scheme 97) and then a subsequent coupling with different boronic acids at 6-position was performed, generating complex quinolin-4(1H)-ones 313 (Scheme 97). Very good yields were obtained for 1-methylquinolin-4(1H)-ones (70–94%) while the N-H containing 6-chloro-3-(2-furanyl)quinolin-4(1H)-ones only produced modest yields of the coupling product (41–47%) [128].
Sequential Suzuki-Miyaura arylation at 3- and 6-position of 1-substituted 6-bromo-3-iodoquinolin-4(1H)-ones 314 was carried out regioselectively under standard conditions and controlled reaction temperature [129]. The chemoselective 3-functionalization of the 6-bromo-3-iodoquinolin-4(1H)-ones 314 with arylboronic acids afforded 3-aryl-6-bromoquinolin-4(1H)-ones 315 in good yields (80–84%), except when 2-furanboronic acid (49%) and 4-methoxyphenylvinylboronic acid (47%) were employed, without concurrent formation of regioisomeric and/or bis-coupling products [Scheme 98, (i)]. Further elaboration of compounds 315 into trisubstituted quinolin-4(1H)-ones 316 was achieved through sequential Suzuki-Miyaura cross-coupling reaction by exploiting the reactivity of bromine at C-6 position [Scheme 98, (ii)]. The reaction was performed under the same conditions used to generate compounds 315 by increasing the reaction temperature to 80 °C. The trisubstituted quinolin-4(1H)-ones 316 were obtained in good yields (67–99%) with complete conversion of the substrates 315 except when using 4-(trifluoromethoxy)phenylboronic acid (50%) [129]. The one-pot sequential Suzuki-Miyaura reaction was also performed to afford the trisubstituted derivatives 316 in overall yields comparable or slightly lower than those obtained in the stepwise synthesis [Scheme 98, (i, ii); 70% for 316 (R1 = Me, R2 = Ph, R3 = 3-ClPh and 72% for 316: R1 = Me, R2 = 4-Tol, R3 = 3-CNPh)] in a total reaction time of 10 min, without the isolation of the monoarylated intermediate [Scheme 98, (i, iii)] [129].
The 6-bromoquinolin-4(1H)-one 317 was converted to the corresponding vinyl derivative 318 via a Suzuki coupling with potassium vinyltrifluoroborate (Scheme 99) [130].
Potential bioactive 3-arylquinolin-4(1H)-ones 320 were synthesized, using ohmic heating, following an efficient and ligand-free protocol for the Suzuki−Miyaura coupling of 1-substituted-3-iodoquinolin-4(1H)-ones 294 with several arylboronic acids 319, in water, using Pd(OAc)2 as a catalyst and TBAB as the phase transfer catalyst (Scheme 100) [131]. The reaction is sensitive to the electronic and steric effects of the substituents in the arylboronic acid; in general, arylboronic acids having electron-donating substituents gave better yields. However, the extensive substrate generality, ease of execution and short reaction time make this method exploitable for the generation of libraries of substituted 3-arylquinolin-4(1H)-ones 320. The reaction of 294 (R1 = Me) with phenylboronic acid was also performed in classical heating conditions and using microwave irradiation but, for a reaction time of 15 min, ohmic heating was the most efficient heating method [131]. After a simple workup, the Pd/catalyst-H2O-TBAB system could be reused for at least seven cycles without significant loss of activity [131].
Porphyrin-quinolin-4(1H)-one conjugates 323 were synthesized by Suzuki-Miyaura coupling reaction of a β-borylated porphyrin 321, prepared by borylation of the corresponding 3-bromotetraphenylporphyrinatozinc(II) with pinacol borane in the presence of PdCl2(PPh3)2 in dicloroethane at 65 °C for 18 h [132], with 6- or 7-bromoquinolin-4(1H)-ones 322 containing N-ethyl and N-D-ribofuranosyl substituents (Scheme 101) [132]. Quinolones 322 bearing N-ethyl substituent were more reactive than those bearing N-ribonucleosides providing the corresponding porphyrin-quinolin-4(1H)-one conjugates 323 in very good yields (82–89%). The other conjugates 323 were isolated in lower yields (50–51%) but nearly 50% of the starting porphyrin 321 was recovered in both cases. Attempts to improve these yields (e.g., by increasing the reaction time and by increasing the number of equivalents of the bromoquinolin-4(1H)-ones) were not successful, probably due to steric effects caused by the ribofuranosyl group. Basic hydrolysis and deprotection of the ribose moieties ester and benzoyl groups in conjugates 323 followed by the acid demetallation afforded the corresponding conjugates that were evaluated as singlet oxygen generators.
The enantioselective conjugate addition of arylboronic acids 319 to N-carboxybenzyl(Cbz)- quinolin-4(1H)-ones 324 utilizing the Pd/PyOX catalyst system, under an atmosphere of air, afforded novel 2-aryl-2,3-dihydroquinolin-4(1H)-ones 325 with high enantioselectivity in moderate to excellent yields (Scheme 102) [133]. Nitrogen-containing heteroaromatic and simpler boronic acid derivatives were successfully employed as nucleophiles in the 1,4-addition to quinolin-4(1H)-ones. Alkyl- and halogen-substituted boronic acids gave reasonable yields (45–65%) and enantioselectivities (67–89% ee) and disubstituted boronic acids were also well tolerated and gave similar results. More sterically demanding boronic acids led to lower product yield.
5.3. Sonogashira Reaction
The tandem coupling-cyclization process, involving Sonogashira reaction followed by the electrophilic or transition-metal-mediated cyclization of the resulting alkynes, having a suitable nucleophilic group in the proximity of the triple bond, generated diverse 2-substituted furo[3,2-c]quinolines 327 (Scheme 103) [134,135]. Better yields were obtained when using Pd(PPh3)4 (85%) or PdCl2(PPh3)2 (80%) instead of Pd/C-PPh3 (70%), although the latter is cheaper. DMF was found to be a better solvent when compared to THF or MeCN and CuI is crucial in this reaction; otherwise only de-iodinated product is formed. The absence of PPh3 when using Pd/C resulted in a poor yield (22%). Good yields of the furo[3,2-c]quinolines 327 were obtained regardless the nature of terminal alkynes used. The key features of the present tandem-coupling-cyclization process are the transition-metal-mediated activation of the triple bond of the 3-alkynyl quinoline generated in situ followed by an intramolecular attack of the oxygen on the activated triple bond with subsequent proton transfer and release of the metal ion to give the desired furoquinolines 327. The NH of the quinolin-4(1H)-one ring has a critical role facilitating the participation of C-4 quinoline oxygen in the cyclization step. Indeed, when 3-iodo-1-methyl-4-oxo-1,4-dihydroquinoline-2-carboxylic acid methyl esters 326 reacted with terminal alkynes, under the same conditions, only 3-alkynylquinolin-4(1H)-ones 328 were isolated as a result of a normal Sonogashira coupling reaction and formation of furoquinolines 327 was not observed, even in trace amounts (Scheme 103).
Diverse macrolone derivatives, which possess high antibacterial activity, were prepared starting from 6-iodoquinolin-4(1H)-ones 305 via Sonogashira reaction [125,136,137]. For example, macrolones 330 were prepared by Sonogashira reaction of 6-iodoquinolin-4(1H)-ones 305 with macrolides 329. Then, Pd/C catalysed hydrogenation was performed to obtain the desirable macrolones 331 (Scheme 104) [125].
Another group of macrolones (with tricyclic quinolone moiety) 336 (R3 = H) and 337 were also synthesized by Sonogashira approach although, only when ethyl esters 332 of the parent acids were used, the reaction proceeded in shorter reaction times and better yields [Scheme 105, (i): R3 = H, (i)*: R3 = Et]. After hydrogenation [Scheme 105, (ii), esters 336, R3 = Et, were hydrolysed to the corresponding carboxylic acids 337, Scheme 105, (iii)*] [125].
The Sonogashira reaction of 6-bromo-3-iodoquinolin-4(1H)-ones 314 with TMSA afforded the expected coupling products in very satisfactory yields, with no further conversion to furo[3,2-c]quinoline derivatives (Scheme 106) [129]. Suzuki and Sonogashira cross-coupling reactions at 6-position of substrates 338 yielded the trisubstituted quinolin-4(1H)-ones 339. In general, harder conditions were necessary to functionalize the 6-position due to the lower reactivity of bromine compared to iodine but, on the other hand, the increased structural complexity made substrates 338 more prone to decomposition and side products’ formation. Therefore, cross-coupling reactions at 6-position proceeded with slightly lower efficiency, leading to products 339 in moderate to low yields.
Another 6-bromoquinolin-4(1H)-one 314 (R1 = 4-FPh, X = CO2Et) was converted to the corresponding alkyne derivatives via Sonogashira couplings; the coupling of 314 with a terminal alkyne (R2CCH) gave 340 directly, whereas, the coupling with trimethylsilylacetylene followed by fluoride mediated silyl cleavage gave a 6-alkyne intermediate that was coupled to an aryl bromide (R2Br) to afford 340 (Scheme 106) [130]. The Pd-catalysed hydrogenation of the alkyne moiety of 340 provided derivative 341 [130].
A practical synthesis of 2-substituted 6-oxopyrrolo[3,2,1-ij]quinolines 343 was achieved following a single-step Pd/C-mediated coupling-cyclization strategy. The methodology involves the reaction of 8-iodo-4-oxo-1,4-dihydroquinoline-3-carboxylic acid ethyl ester 342 with a variety of terminal alkynes 47 in the presence of 10% Pd/C–PPh3–CuI as a catalyst system in EtOH (Scheme 107) [138]. This process was found to be general since both aliphatic and aromatic alkynes are tolerated, as well as alkyl side chains containing primary, secondary or tertiary hydroxyl group or a cyano group.
5.4. Stille Reaction
A series of 1-cyclopropylquinolin-4(1H)-ones 346 (R5 = c-Pr), bearing a vinyl, a 1-cyclopentenyl or a 1,2,3,6-tetrahydropyridin-4-yl group at C-7, were synthesized by Pd-catalysed cross-coupling of 7-quinolyltriflate 344 (X = OTf) with an appropriately functionalized vinylstannane 345 (1.2 equiv) [Scheme 108, (i)] [139,140]. The reaction with a range of vinylstannanes 345 bearing different functional groups afforded the corresponding quinolin-4(1H)-ones 346 in moderate to good yields (22–88%) with complete chemo- and regioselectivity; the coupling takes place exclusively at the C-7 position of the quinolone nucleus even in the presence of the C-6 fluorine or the C-2 α,β-unsaturated keto-ester moiety. In some cases, additional amounts of the tin reagent and/or the catalyst [either PdCl2(PPh3)2 or Pd(PPh3)4] were necessary to improve the yield of coupled product. The low yield obtained with the cyclopentenylstannane possessing a bulky t-butoxycarbonyl group at the 3-position (31%) was attributed to unfavourable steric hindrance. Later, novel 7-pyridinylquinolin-4(1H)-ones (346, R1 = pyridinyl group) were synthesized, following the same methodology but starting from 7-bromo- or 7-chloroquinolin-4(1H)-ones 344 (X=Br or Cl), [Scheme 108, (ii)] [141]. Likewise, the quinolin-4(1H)-one esters 346 were hydrolysed to the corresponding acids (50–73%), with sodium hydroxide or hydrochloric acid, with concomitant removal of the N-protecting groups.
5-Substituted-3-(4-methoxyphenyl)quinolin-4(1H)-ones 309 have been synthesized in good yields via Stille cross-coupling reaction of the corresponding 3-(4-methoxyphenyl)-5-trifluoromethanesulfonatequinolin-4(1H)-ones 308 with tributyl(vinyl)stannane (67–85%) or with tributyl(thien-2-yl)stannane (64%) (Scheme 109) [126]. This protocol was successfully applied to the synthesis of 2-(3-methoxyphenyl)quinolin-4(1H)-one analogue 310 [127].
5.5. Buchwald-Hartwig Reaction
Four 6-substituted-quinolin-4(1H)-one nucleosides linked to aniline or phenol via N- or heteroatom-bridges were synthesized by Pd-catalysed Buchwald-Hartwig cross-coupling reactions starting from 6-bromoquinolin-4(1H)-one nucleosides 347 and 348. Different standard deprotection steps resulted in free ribose for all compounds 353–357 and either the free acid 356, the amides 353 and 357 or the esters 354 and 355 in the 3-position of quinolin-4(1H)-one (Scheme 110) [142].
Pd-catalysed amination reactions of 6-bromoquinolin-4(1H)-ones 322 containing N-ethyl, N-pentyl and N-ribofuranosyl substituents with (2-amino-5,10,15,20-tetraphenylporphyrinato) nickel(II) 358 followed by demetallation, allowed the access to porphyrin-quinolin-4(1H)-one conjugates 361 and 362 (Scheme 111) [143]. The use of Pd(OAc)2 in combination with rac-BINAP, as the ligand, led to complete consumption of starting material 322 and to the formation of the conjugates 359 in moderate to very good yields (63–89%). The yields were lower and longer reaction time was required in the case of quinolones having bulky groups, such as pentyl and ribofuranosyl groups, probably due to steric effects. The formation of the by-products 360 is promoted by the transfer of a rac-BINAP phenyl group to the metal ion centre followed by a reductive-elimination step during the catalytic cycle. The conjugates 361a demonstrated good to high capability to generate singlet oxygen evidencing potential application in the inactivation of the Gram-positive bacteria Staphylococcus aureus [143].
Buchwald-Hartwig amination of 6-bromoquinolin-4(1H)-one 314 with a commercially available amine afforded quinolin-2(1H)-one 363 in 42% yield (Scheme 112) [130].
5.6. Aminocarbonylation Reaction
A Pd-catalysed carbonylation of 6-bromo-3-iodoquinolin-4(1H)-ones 314 with amines, using Pd(OAc)2 as catalyst and Mo(CO)6 as the CO source, provided compounds 364 in low yields (25–38%) [Scheme 113, (i)] [129]. Slight modifications of the experimental conditions did not improve the reaction yield; even when gaseous CO and Pd/C were used as the catalyst [Scheme 113, (ii)] the aminocarbonylation product was obtained in similar yield (35%).
5.7. Other Reactions
Pd-catalysed 3-alkenylation of quinolin-4(1H)-ones 365 with compounds 366 was efficiently achieved with 1% catalyst loading affording 3-substituted-quinolin-4(1H)-ones 367 (Scheme 114) [144]. From a series of tested catalysts [Pd(OAc)2, PdCl2, Pd(TFA)2], PdCl2 was the most efficient at low loadings. The addition of Cu(OAc) (10 mol%) was necessary to achieve high yield of the product. The presence of O2 in the reaction medium, which acts as a co-oxidant, is favourable. Regarding the reaction scope, a substituent is required on the nitrogen; coupling of free (NH) quinolone with acrylate gave no product. The presence of electron-donating groups at the C-6 position provided high yields while electron-withdrawing groups gave lower yields; however, high yield was obtained when CF3 group was at that position and with halogens (6-F, 6-Cl and 6-Br). 8-Substituted quinolones have a similar reactivity as 6-substituted ones but generally with lower yields, probably due to steric effects. Even the 6,7-(methylenedioxy)-substituted quinolone gave the product in modest yield. Terminal acrylates, N,N-dimethylacrylamide, styrene and 2-vinylnaphthalene gave the corresponding products in high yields while modest to reasonable yields were obtained with acrylic acid, sterically hindered acrylates, diethyl fumarate, methacrylonitrile, vinyl phosphonate and methyl vinyl sulfone.
An efficient and practical metal-catalysed decarboxylative cross-coupling reaction of quinolin-4(1H)-one 3-carboxylic acids 368 with various (hetero)-aryl halides was described (Scheme 115) [145]. An extensive screening of various reaction parameters (Pd, ligand, solvent, base and temperature) showed that PdI2, Pd(OAc)2 and PdCl2 were less effective than PdBr2. The nature of the phosphine ligand has an important influence on the reaction selectivity and optimal reaction conditions of 368 (R1 = Ph; R2 = H) with 4-iodoanisole involved the combination of the bidentate phosphine DPEphos with PdBr2 in toluene/dimethylacetamide (DMA) at 150 °C. The use of microwave irradiation provided shortening of reaction time and increase of quinolin-4(1H)-ones 369 yield (MW: 1 h, 81%; CH: 8 h, 77% and 1 h, 60%). The bimetallic system PdBr2/Ag2CO3 is necessary for the coupling to occur; no product could be formed in the absence of PdBr2 or when Ag2CO3 was replaced by other bases. Using optimal conditions under microwave irradiation, electron-rich and electron-deficient, o-, m- and p-substituted aryl iodides and bromides, all efficiently underwent decarboxylative coupling in good yields (40–99%) and the coupling with heterocyclic halides was also successful (40% for 3-bromocoumarin and 57% for 3-bromoquinolin-2(1H)-one). Both N-alkyl- and N-arylquinolin-4(1H)-one 3-carboxylic acids 368 having electron-donating or electron-withdrawing groups on the aromatic nucleus led to the formation of the corresponding coupled products 369 in good yields (60–90%). Excellent chemical selectivity was observed for 368 (R2 = Cl) preserving the C-Cl bond, which could undergo further metal-catalysed functionalization reactions. This protocol is an attractive alternative to the existing methods for the synthesis of 3-(hetero)-arylquinolin-4(1H)-ones 369 and was also applied to the synthesis of 1-methyl-3-phenylquinolin-2(1H)-one (44%).
The ligand-free palladium-catalysed decarboxylative functionalization of quinolinone-3-carboxylic acids 370 and 372 with aryl halides afforded biologically important 3-arylquinolin-4(1H)-ones 371 and 373 in moderate to high yields (Scheme 116) [146]. The reaction proceeded smoothly under an argon atmosphere at relatively low temperature by using Pd(OAc)2 as the catalyst and Ag2CO3 as the oxidant. Aryl iodides gave better results than aryl bromides and various functionalities were compatible under the reaction conditions. Electronic effects strongly influenced the reaction. The coupling of aryl iodides bearing electron-donating substituents provided the corresponding products in high yields but iodobenzene derivatives with electron-deficient groups were significantly less reactive.
Pd-catalysed direct thioetherification of quinolone derivatives 370 and 376 with diaryl disulfides 374 via decarboxylative C-S cross-couplings proceeded smoothly under air in the presence of Pd(OAc)2 and Ag2CO3 in DMSO (Scheme 117) [147]. The solvent has an important role in the reaction, DMSO being superior to other solvents and no product was isolated when the reaction was performed under argon or nitrogen atmosphere. Disulfides 374 substituted with electron-withdrawing groups gave the product in lower yields. In addition, when some radical scavengers such as TEMPO or HQ were used in this C-S coupling, the reaction was inhibited and no product was detected. The reaction mechanism initiates with the formation of an organometallic species via decarboxylation by reaction with the silver salt. Subsequently, the Pd(II) reacts with the diaryl disulfides to generate a Pd(II) species. Then, an aryl-Pd(II) species was produced by way of a transmetalation reaction between the organometallic and Pd(II) species. Finally, the reductive elimination of the aryl Pd(II) afforded the target product and Pd(0) species, which can be oxidized to Pd(II) by oxygen and continue the catalytic cycle [147]. This protocol is an alternative to existing approaches to construct aryl sulphides of quinolone derivatives which may be used as important intermediates in the synthesis of new drug candidates.
Pd-catalysed hydroxylation of 6-bromoquinolin-4(1H)-one 314 allowed the synthesis of quinolin-4(1H)-one 378 with a 6-hydroxy group (Scheme 118) [130].
6-Chloroquinolin-4(1H)-one 312 was efficiently converted into 3-furanyl-6-cyanoquinolin-4(1H)-one 379, in high yields, using a Pd/dppf catalyst with Zn(0) as the in situ reductant (Scheme 119) [128]. The same conditions failed to catalyse the cyanation of 6-chloro-3-iodoquinolin-4(1H)-one 311. However, using a copper iodide/DMEDA system with sodium cyanide resulted in 3-cyanation with excellent yield (Scheme 119) [128].
Pd-catalysed addition of quinolin-4(1H)-ones 380 to terminal allenes 381, using an achiral Pd-catalyst, furnished linear N-allylated quinolin-4(1H)-ones 382 in high yields and high (E)-selectivity (Scheme 120) [148]. Compatible functional groups on the quinolin-4(1H)-ones were methyl, methoxy, a thiophene and a boronic ester, enabling further functionalization through cross-coupling reactions.
Quinolin-4(1H)-ones 383 undergo Pd-catalysed cross-coupling reaction with ethyl bromodifluoroacetate, in the presence of copper mediators, providing difluoro-containing quinolin-4(1H)-ones 384 (Scheme 121) [149]. Among the tested Pd-catalysts, Pd(dppf)Cl2 provided compounds 384 in less time and higher yields. Solvent has a significant impact on the yields with DMSO giving the best result. This method was employed in the modification of graveolinine, a natural quinolin-4(1H)-one alkaloid with interesting antibacterial, spasmolysis and antitumor activities. Treatment of 3-iodograveolinine (383, R1 = benzo[d][1,3]dioxole) with ethyl bromodifluoroacetate gave the target compound in 78% yield. Since a Pd(II) catalyst is employed in this reaction, first it may be reduced by copper to an active Pd(0) species, which then reacts via an oxidative addition into the C−I bond of compound 383 to form intermediate I. Meanwhile, the unstable copper ethyl difluoroacetate complex II is formed and intermediate III is generated rapidly via a reaction between intermediates I and II. Finally, reduction elimination affords the expected quinolin-4(1H)-one 384, regenerating the Pd(0) catalyst (Scheme 122) [149].
6. Conclusions
Pd-catalysed reactions are of paramount importance in the synthesis and transformation of quinolin-2(1H)-ones and quinolin-4(1H)-ones, as it was evidenced by the several examples presented along these review article. In some of these examples, especially in the synthesis of quinolones, the claimed Pd-catalysed reaction is crucial for the formation of the appropriate substrate for cyclization into the desired quinolone, in spite of not being used in the cyclization step.
Among the Pd-catalysed reactions developed for the synthesis and transformation of quinolones, the cross-coupling reactions, which require the use of an already activated counterpart, for instance a halogenated derivative, which is coupled to another appropriate substrate, are the most common. More recently, several works have been focused on the development of protocols for direct C-H functionalization, thus allowing the construction of more complex and highly substituted quinolone derivatives in a more straightforward way. In addition, efforts have been made to develop new protocols for already known Pd-catalysed reactions, aiming to meet the green chemistry requirements and to facilitate the reactions′ scale-up. Some important advances have been achieved in this area by replacing homogeneous Pd-catalysts by heterogeneous Pd-catalysts, by using ligand-free conditions or room temperature. Another interesting example, in the carbonylation reactions, is the replacement of gaseous CO by more convenient solid sources of CO, such as molybdenum hexacarbonyl [Mo(CO)6], facilitating the reactions’ scale-up. All the Pd-catalysed reactions herein presented have profoundly changed the protocols for the construction of various quinolone-type compounds, some of them of recognized importance due to their relevant biological activities. In spite of the great advances of Pd-catalysed reactions achieved in the last 20 years, this field of research is still wide-open for innovation and will continue to advance as even more versatile transformations are developed.
Author Contributions
Both authors contributed to this overview.
Funding
Thanks are due to the University of Aveiro and FCT/MEC for the financial support to the QOPNA research project (FCT UID/QUI/00062/2013), financed by national funds and when appropriate co-financed by FEDER under the PT2020 Partnership Agreement. Vera L. M. Silva also thanks to the Integrated Programme of SR&TD “pAGE—Protein Aggregation Across the Lifespan” (reference CENTRO-01-0145FEDER-000003), co-funded by Centro 2020 program, Portugal 2020 and European Union, through the European Regional Development Fund.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Michael, J.P. Quinoline, quinazoline and acridone alkaloids. Nat. Prod. Rep. 1991, 8, 53–68. [Google Scholar] [CrossRef]
- Michael, J.P. Quinoline, quinazoline and acridone alkaloids. Nat. Prod. Rep. 2008, 25, 166–187. [Google Scholar] [CrossRef]
- Uchida, M.; Tabusa, F.; Komatsu, M.; Morita, S.; Kanbe, T.; Nakagawa, K. Studies on 2(1H)-quinolinone derivatives as gastric antiulcer active agents. Synthesis and antiulcer activities of optically active alpha-amino acid derivatives of 2(1H)-quinolinone and oxindole. Chem. Pharm. Bull. 1987, 35, 853–856. [Google Scholar] [CrossRef] [PubMed]
- Norman, P. Tipifarnib (Janssen Pharmaceutica). Curr. Opin. Investig. Drugs 2002, 3, 313–319. [Google Scholar] [PubMed]
- Richter, S.; Parolin, C.; Palumbo, M.; Palù, G. Antiviral properties of quinolone-based drugs. Curr. Drug Targets Infect. Disord. 2004, 4, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Ren, J.; Milton, J.; Hazen, R.J.; Chan, J.H.; Stuart, D.I.; Stammers, D.K. Design of non-nucleoside inhibitors of HIV-1 reverse transcriptase with improved drug resistance properties. 1. J. Med. Chem. 2004, 47, 5912–5922. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.A.; Andrews, C.W.; Hopkins, A.L.; Lowell, G.S.; Schaller, L.T.; Cowan, J.R.; Gonzales, S.S.; Koszalka, G.W.; Hazen, R.J.; Boone, L.R.; et al. Design of non-nucleoside inhibitors of HIV-1 reverse transcriptase with improved drug resistance properties. 2. J. Med. Chem. 2004, 47, 5923–5936. [Google Scholar] [CrossRef]
- Huse, H.; Whiteley, M. 4-Quinolones: Smart phones of the microbial world. Chem. Rev. 2011, 111, 152–159. [Google Scholar] [CrossRef]
- Winter, R.; Kelly, J.X.; Smilkstein, M.J.; Hinrichs, D.; Koop, D.R.; Riscoe, M.K. Optimization of endochin-like quinolones for antimalarial activity. Exp. Parasitol. 2011, 127, 545–551. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Yang, Z.Y.; Xia, P.; Bastow, K.F.; Nakanishi, Y.; Nampoothiri, P.; Hamel, E.; Brossi, A.; Lee, K.H. Antitumor agents. Part 226: Synthesis and cytotoxicity of 2-phenyl-4-quinolone acetic acids and their esters. Bioorg. Med. Chem. Lett. 2003, 13, 2891–2893. [Google Scholar] [CrossRef]
- Wang, S.W.; Pan, S.L.; Huang, Y.C.; Guh, J.H.; Chiang, P.C.; Huang, D.Y.; Kuo, S.C.; Lee, K.H.; Teng, C.M. CHM-1, a novel synthetic quinolone with potent and selective antimitotic antitumor activity against human hepatocellular carcinoma in vitro and in vivo. Mol. Cancer Ther. 2008, 7, 350–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joule, J.A.; Mills, K. Quinolines and isoquinolines: Reaction and synthesis. In Heterocyclic chemistry, 4th ed.; Blackwell Science: Oxford, MA, USA, 2000; Volume 6, pp. 1–589. [Google Scholar]
- Fuson, R.C.; Burness, D.N. A new synthesis of 2-aryl-4-hydroxyquinolines. J. Am. Chem. Soc. 1946, 68, 1270–1272. [Google Scholar] [CrossRef] [PubMed]
- Boteva, A.A.; Krasnykh, O.P. The methods of synthesis, modification and biological activity of 4-quinolones (review). Chem. Heterocycl. Comp. 2009, 45, 757–785. [Google Scholar] [CrossRef]
- Seixas, R.S.G.R.; Silva, V.L.M.; Silva, A.M.S. Metal-catalysed cross-coupling reactions in the synthesis and transformations of quinolones and acridones, in synthesis and modification of heterocycles by metal-catalyzed cross-coupling reactions. Top. Heterocycl. Chem. 2016, 45, 159–230. [Google Scholar] [CrossRef]
- Cortese, N.A.; Ziegler, C.B.J.; Hrnjez, B.J.; Heck, R.F. Palladium-catalyzed synthesis of 2-quinolones derivatives from 2-iodoanilines. J. Org. Chem. 1978, 43, 2952–2958. [Google Scholar] [CrossRef]
- Bernini, R.; Cacchi, S.; Fabrizi, G.; Sferrazza, A. 4-Aryl-2-quinolones via a domino Heck reaction/cyclization process. Heterocycles 2006, 69, 99–105. [Google Scholar] [CrossRef]
- Bernini, R.; Cacchi, S.; De Salve, I.; Fabrizi, G. The Heck reaction of β-arylacrylamides: An approach to 4-aryl-2-quinolones. Synlett 2006, 2947–2952. [Google Scholar] [CrossRef]
- Battistuzzi, G.; Bernini, R.; Cacchi, S.; De Salve, I.; Fabrizi, G. 4-Aryl-2-quinolones through a pseudo-domino Heck/Buchwald–Hartwig reaction in a molten tetrabutylammonium acetate/tetrabutylammonium bromide mixture. Adv. Synth. Catal. 2007, 349, 297–302. [Google Scholar] [CrossRef]
- Fourquez, J.M.; Godard, A.; Marsais, F.; Quéquiner, G. Regioselectivity of the metalation of polymethoxylated pivaloylaminobenzenes. Synthesis of methoxy-2(1H)-quinolones. J. Heterocycl. Chem. 1995, 32, 1165–1170. [Google Scholar] [CrossRef]
- Cho, C.S.; Kim, J.U. An approach for quinolines via palladium-catalyzed Heck coupling followed by cyclization. Tetrahedron Lett. 2007, 48, 3775–3778. [Google Scholar] [CrossRef]
- Taylor, J.G.; Correia, C.R.D. Stereoselective synthesis of unsymmetrical β,β-diarylacrylates by a Heck-Matsuda reaction: Versatile building blocks for asymmetric synthesis of β,β-diphenylpropanoates, 3-aryl-indole and 4-aryl-3,4-dihydro-quinolin-2-one and formal synthesis of (−)-indatraline. J. Org. Chem. 2011, 76, 857–869. [Google Scholar] [CrossRef]
- Felpin, F.X.; Coste, J.; Zakri, C.; Fouquet, E. Preparation of 2-quinolones by Heck reduction-cyclization (HRC) reactions by using a multitask palladium catalyst. Chem. Eur. J. 2009, 15, 7238–7245. [Google Scholar] [CrossRef]
- Djakovitch, L.; Batail, N.; Genelot, M. Recent advances in the synthesis of N-containing heteroaromatics via heterogeneously transition metal catalysed cross-coupling reactions. Molecules 2011, 16, 5241–5267. [Google Scholar] [CrossRef]
- Glover, B.; Harvey, K.A.; Liu, B.; Sharp, M.J.; Tymoschenko, M.F. Regioseletive palladium-catalyzed arylation of 3-carboalkoxy furan and thiophene. Org. Lett. 2003, 5, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Borhade, S.R.; Waghmode, S.B. An efficient synthesis of 4-arylquinolin-2(1H)-ones and 3-alkenyl-4-arylquinolin-2(1H)-one using a Pd/NiFe2O4-catalyzed consecutive Heck reaction. Can. J. Chem. 2011, 89, 1355–1363. [Google Scholar] [CrossRef]
- Kuroda, T.; Suzuki, F. Synthesis of 1H-imidazo[4,5-c]quinolin-4(5H)-one via palladium-catalyzed cyclization of N-(2-bromophenyl)-1H-imidazole-4-carboxamide. Tetrahedron Lett. 1991, 32, 6915–6918. [Google Scholar] [CrossRef]
- Beccalli, E.M.; Broggini, G.; Martinelli, M.; Paladino, G.; Zoni, C. Synthesis of tricyclic quinolones and naphthyridones by intramolecular Heck cyclization of functionalized electron-rich heterocycles. Eur. J. Org. Chem. 2005, 2005, 2091–2096. [Google Scholar] [CrossRef]
- Liu, Z.; Shi, C.; Chen, Y. Synthesis of 3-alkyl-1H-quinolin-2-ones via palladium-catalyzed intramolecular cyclization of benzyl halides and α,β-unsaturated amides. Synlett 2008, 1734–1736. [Google Scholar] [CrossRef]
- Arcadi, A.; Cacchi, S.; Fabrizi, G.; Manna, F.; Pace, P. Ethyl N-(o-ethynyl)malonanilide as a useful building block for the preparation of 3,4-disubstituted-2(1H)-quinolones, 3,4-disubstituted- and 2,3,4-trisubstituted quinolones. Synlett 1998, 446–448. [Google Scholar] [CrossRef]
- Kadnikov, D.V.; Larock, R.C. Palladium-catalyzed carbonylative annulation of terminal alkynes: Synthesis of coumarins and 2-quinolones. J. Organomet. Chem. 2003, 687, 425–435. [Google Scholar] [CrossRef]
- Torii, S.; Okumoto, H.; Xu, L.H. Palladium-catalyzed carbonylation to form 2-substituted 1,4-dihydro-4-oxo-quinoline. Tetrahedron Lett. 1991, 32, 237–240. [Google Scholar] [CrossRef]
- Torii, S.; Okumoto, H.; Xu, L.H.; Sadakane, M.; Shostakovsky, M.V.; Ponomaryov, A.B.; Kalinin, V.N. Synthesis of chromones and quinolones via Pd-catalyzed carbonylation of o-iodophenols and anilines in the presence of acetylenes. Tetrahedron 1993, 49, 6773–6784. [Google Scholar] [CrossRef]
- Kadnikov, D.V.; Larock, R.C. Synthesis of 2-quinolones via palladium-catalyzed carbonylative annulation of internal alkynes by N-substituted o-iodoanilines. J. Org. Chem. 2004, 69, 6772–6780. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-R.; Liao, J.; Xiao, W.-J. Microwave-assisted, palladium-catalyzed carbonylative cyclization—Rapid synthesis of 2-quinolones from unprotected 2-iodoanilines and terminal alkynes. Can. J. Chem. 2010, 88, 331–337. [Google Scholar] [CrossRef]
- Jafarpour, F.; Otaredi-Kashani, A. Carbonylative annulation of unsaturated compounds using molybdenum hexacarbonyl: An efficient synthesis of 2(1H)-quinolones. Arkivoc 2014, 193–203. [Google Scholar] [CrossRef]
- Vicente, J.; Abad, J.-A.; López-Serrano, J.; Jones, P.G.; Nájera, C.; Botella-Segura, L. Synthesis and reactivity of ortho-palladated arylureas. Synthesis and catalytic activity of a C,N,C pincer complex. Stoichiometric syntheses of some N-heterocycles. Organometallics 2005, 24, 5044–5057. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, H.; Jia, Y. A palladium-catalyzed intramolecular carbonylative annulation reaction for the synthesis of 4,5-fused tricyclic 2-quinolones. Chem. Commun. 2016, 52, 7665–7667. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Chiba, K.; Ohta, N.; Ban, Y. A novel synthesis of cyclic imides and quinolone by use of palladium catalyzed carbonylation. Heterocycles 1979, 13, 329–332. [Google Scholar] [CrossRef]
- Okuro, K.; Kai, H.; Alper, H. Palladium-catalyzed asymmetric cyclocarbonylation of 2-(1-methylvinyl) anilines. Tetrahedron Asymm. 1997, 8, 2307–2309. [Google Scholar] [CrossRef]
- Banwell, M.G.; Lupton, D.W.; Ma, X.; Renner, J.; Sydnes, M.O. Synthesis of quinolines, 2-quinolones, phenanthridines and 6(5H)-phenanthridinones via palladium[0]-mediated Ullmann cross-coupling of 1-bromo-2-nitroarenes with β-halo-enals, -enones or –esters. Org. Lett. 2004, 6, 2741–2744. [Google Scholar] [CrossRef]
- Teng, Y.; Suwanarusk, R.; Ngai, M.H.; Srinivasan, R.; Ong, A.S.M.; Ho, B.; Laurent, R.; Chai, C.L.L. An amidation/cyclization approach to the synthesis of N-hydroxyquinolinones and their biological evaluation as potential anti-plasmodial, anti-bacterial and iron(II)-chelating agents. Bioorg. Med. Chem. Lett. 2015, 25, 607–610. [Google Scholar] [CrossRef]
- Porosa, L.; Viirre, R.D. Desymmetrization of malonamides via an enantioselective intramolecular Buchwald-Hartwig reaction. Tetrahedron Lett. 2009, 50, 4170–4173. [Google Scholar] [CrossRef]
- Takenaka, K.; Itoh, N.; Sasai, H. Enantioselective synthesis of C2-symmetric spirobilactams via Pd-catalyzed intramolecular double N-arylation. Org. Lett. 2009, 11, 1483–1486. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-de-Elguea, V.; Sotomayor, N.; Lete, E. Two consecutive palladium(II)-promoted C-H alkenylation reactions for the synthesis of 3-alkenylquinolones. Adv. Synth. Catal. 2015, 357, 463–473. [Google Scholar] [CrossRef]
- Manley, P.J.; Bilodeau, M.T. A new synthesis of naphthyridinones and quinolinones: Palladium-catalyzed amidation of o-carbonyl-substituted aryl halides. Org. Lett. 2004, 14, 2433–2435. [Google Scholar] [CrossRef]
- Inamoto, K.; Saito, T.; Hiroya, K.; Doi, T. Palladium-catalysed intramolecular amidation of C(sp2)-H bonds: Synthesis of 4-aryl-2-quinolinones. J. Org. Chem. 2010, 75, 3900–3903. [Google Scholar] [CrossRef] [PubMed]
- Inamoto, K. Synthesis of heterocyclic compounds through palladium-catalyzed C–H cyclization processes. Chem. Pharm. Bull. 2013, 61, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Inamoto, K.; Kawasaki, J.; Hiroya, K.; Kondo, Y.; Doi, T. Tandem-type Pd(II)-catalyzed oxidative Heck reaction/intramolecular C–H amidation sequence: A novel route to 4-aryl-2-quinolinones. Chem. Commun. 2012, 48, 4332–4334. [Google Scholar] [CrossRef]
- Jia, C.; Piao, D.; Kitamura, T.; Fujiwara, Y. New method for preparation of coumarins and quinolinones via Pd-catalyzed intramolecular hydroarylation of C-C triple bonds. J. Org. Chem. 2000, 65, 7516–7522. [Google Scholar] [CrossRef]
- Kancherla, R.; Naveen, T.; Maiti, D. Palladium-catalyzed [3 + 3] annulation between diarylamines and α,β-unsaturated acids through C-H activation: Direct access to 4-substituted 2-quinolinones. Chem. Eur. J. 2015, 21, 8360–8364. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, M.J.R.P.; Castanheira, E.M.S.; Lopes, T.C.T.; Cruz, Y.K.; Kirsch, G. Synthesis of fluorescent tetracyclic lactams by a “one pot” three steps palladium-catalyzed borylation, Suzuki coupling (BSC) and lactamization DNA and polynucleotides binding studies. J. Photochem. Photobiol. A Chem. 2007, 190, 45–52. [Google Scholar] [CrossRef]
- Tsuritani, T.; Yamamoto, Y.; Kawasaki, M.; Mase, T. Novel Approach to 3,4-dihydro-2(1H)-quinolinone derivatives via cyclopropane ring expansion. Org. Lett. 2009, 11, 1043–1045. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xiang, S.; Zeng, J.; Leow, M.; Liu, X.W. Practical route to 2-quinolinones via a Pd-catalysed C-H bond activation/C-C bond formation/ cyclisation cascade reaction. Org. Lett. 2015, 17, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.X.; Li, H.; Liu, X.W.; Shi, J.L.; Wang, X.; Shi, Z.J. Palladium-catalyzed C(sp3)-H activation: A facile method for the synthesis of 3,4-dihydroquinolinone derivatives. Angew. Chem. Int. Ed. 2014, 53, 4945–4949. [Google Scholar] [CrossRef]
- Dong, Y.; Liu, B.; Chen, P.; Liu, Q.; Wang, M. Palladium-catalyzed C-S activation/aryne insertion/coupling sequence: Synthesis of functionalized 2-quinolinones. Angew. Chem. Int. Ed. 2014, 53, 3442–3446. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Peng, X.; Qin, X.; Zhao, X.; Ma, C.; Tung, C.H.; Xu, Z. Synthesis of quinolinones with palladium-oxidative annulation between acrylamides and arynes. J. Org. Chem. 2015, 80, 2835–2841. [Google Scholar] [CrossRef] [PubMed]
- Tadd, A.C.; Matsuno, A.; Fielding, M.R.; Willis, M.C. Cascade palladium-catalyzed alkenyl aminocarbonylation/intramolecular aryl amidation: An annulative synthesis of 2-quinolones. Org. Lett. 2009, 11, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Kundu, N.G.; Mahanty, J.S.; Das, P.; Das, B. Synthesis of quinolines and 2,3-dihydro-4(1H)-quinolones. Palladium catalysed reaction of o-iodoanilides with acetylenic carbinols. Tetrahedron Lett. 1993, 34, 1625–1628. [Google Scholar] [CrossRef]
- Mahanty, J.S.; De, M.; Das, P.; Kundu, N.G. Palladium catalysed heteroannulation with acetylenic carbinols as synthons—Synthesis of quinolines and 2,3-dihydro-4(1H)-quinolones. Tetrahedron 1997, 53, 13397–13418. [Google Scholar] [CrossRef]
- Pisaneschi, F.; Sejberg, J.J.P.; Blain, C.; Ng, W.H.; Aboagye, E.O.; Spivey, A.C. 2-Substituted-2,3-dihydro- -1H-quinolin-4-ones via acid-catalyzed tandem Rupe rearrangement-Donnelly-Farrell ring closure of 2-(3’-hydroxy-propynyl)anilines. Synlett 2011, 2, 241–244. [Google Scholar] [CrossRef]
- Kalinin, V.N.; Shostakovsky, M.V.; Ponomaryov, A.B. A new route to 2-aryl-4-quinolones via palladium catalyzed carbonylative coupling of o-iodoanilines with terminal arylacetylenes. Tetrahedron Lett. 1992, 33, 373–376. [Google Scholar] [CrossRef]
- Haddad, N.; Tan, J.; Farina, V. Convergent synthesis of the quinolone substructure of BILN 2061 via carbonylative Sonogashira coupling/cyclization. J. Org. Chem. 2006, 71, 5031–5034. [Google Scholar] [CrossRef]
- Genelot, M.; Bendjeriou, A.; Dufaud, V.; Djakovitch, L. Optimised procedures for the one-pot selective syntheses of indoxyls and 4-quinolones by a carbonylative Sonogashira/cyclisation sequence. Appl. Catal. A 2009, 369, 125–132. [Google Scholar] [CrossRef]
- Genelot, M.; Dufaud, V.; Djakovitch, L. Heterogeneous metallo-organocatalysis for the selective one-pot synthesis of 2-benzylidene-indoxyl and 2-phenyl-4-quinolone. Tetrahedron 2011, 67, 976–981. [Google Scholar] [CrossRef]
- Batail, N.; Genelot, M.; Dufaud, V.; Joucla, L.; Djakovitch, L. Palladium-based innovative catalytic procedures: Designing new homogeneous and heterogeneous catalysts for the synthesis and functionalisation of N-containing heteroaromatic compounds. Catal. Today 2011, 173, 2–14. [Google Scholar] [CrossRef]
- Åkerbladh, L.; Nordeman, P.; Wejdemar, M.; Odell, L.R.; Larhed, M. Synthesis of 4-Quinolones via a Carbonylative Sonogashira Cross-Coupling Using Molybdenum Hexacarbonyl as a CO Source. J. Org. Chem. 2015, 80, 1464–1471. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Xu, B. Palladium-catalyzed tandem amination reaction for the synthesis of 4-quinolones. Org. Lett. 2010, 12, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Fei, X.D.; Zhou, Z.; Li, W.; Zhu, Y.M.; Shen, J.K. Buchwald–Hartwig coupling/Michael addition reactions: One-pot synthesis of 1,2-disubstituted 4-quinolones from chalcones and primary amines. Eur. J. Org. Chem. 2012, 2012, 3001–3008. [Google Scholar] [CrossRef]
- Sakamoto, T.; Kondo, Y.; Uchiyama, D.; Yamanaka, H. Condensed heteroaromatic ring systems. XIX. Synthesis and reactions of 5-(tributylstannyl)isoxazoles. Tetrahedron 1991, 47, 5111–5118. [Google Scholar] [CrossRef]
- Sakamoto, T.; Yasuhara, A.; Kondo, Y.; Yamanaka, H. Condensed heteroaromatic ring systems. XX. Palladium-catalyzed carbonylative coupling of iodobenzenes with (Z)-1-ethoxy-2-(tributylstannyl)ethane. Chem. Pharm. Bull. 1992, 40, 1137–1139. [Google Scholar] [CrossRef]
- Torii, S.; Okumoto, H.; Xu, L.H. A direct approach to 2-substituted 1,4-dihidro-4-oxo-quinoline-3- carboxylates by palladium-catalyzed carbonylative cyclization. Tetrahedron Lett. 1990, 31, 7175–7178. [Google Scholar] [CrossRef]
- Latham, E.J.; Stanforth, S.P. Synthesis of indoles and quinolones by sequential Wittig and Heck reactions. Chem. Commun. 1996, 2253–2254. [Google Scholar] [CrossRef]
- Latham, E.J.; Stanforth, S.P. Synthesis of indoles and quinolones by sequential Wittig and Heck reactions. J. Chem. Soc. Perkin Trans. 1997, 1, 2059–2063. [Google Scholar] [CrossRef]
- Costa, M.; Cà, N.D.; Gabriele, B.; Massera, C.; Salerno, G.; Soliani, M. Synthesis of 4H-3,1-benzoxazines, quinazolin-2-ones and quinolin-4-ones by palladium-catalyzed oxidative carbonylation of 2-ethynylaniline derivatives. J. Org. Chem. 2004, 69, 2469–2477. [Google Scholar] [CrossRef] [PubMed]
- Gabriele, B.; Salerno, G.; Costa, M. PdI2-catalyzed synthesis of heterocycles. Synlett 2004, 2468–2483. [Google Scholar] [CrossRef]
- Alfonsi, R.; Botta, B.; Cacchi, S.; Di Marcotullio, L.; Fabrizi, G.; Faedda, R.; Goggiamani, A.; Iazzetti, A.; Mori, M. Design, palladium-catalyzed synthesis and biological investigation of 2-substituted 3-aroylquinolin-4(1H)-ones as inhibitors of the Hedgehog signaling pathway. J. Med. Chem. 2017, 60, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Okuro, K.; Alper, H. Palladium-catalyzed intermolecular cyclocarbonylation of 2-iodoanilines with the Michael acceptor, diethyl ethoxycarbonylbutendienoate. J. Org. Chem. 2012, 77, 4420–4424. [Google Scholar] [CrossRef] [PubMed]
- Grigg, R.; Liu, A.; Shaw, D.; Suganthan, S.; Woodall, D.E.; Yoganathan, G. Synthesis of quinol-4-ones and chroman-4-ones via a palladium-catalysed cascade carbonylation-allene insertion. Tetrahedron Lett. 2000, 41, 7125–7128. [Google Scholar] [CrossRef]
- Okuro, K.; Alper, H. Palladium-catalyzed carbonylation of o-iodophenols with allenes. J. Org. Chem. 1997, 62, 1566–1567. [Google Scholar] [CrossRef]
- Wu, J.; Zhou, Y.; Wu, T.; Zhou, Y.; Chiang, C.W.; Lei, A. From ketones, amines and carbon monoxide to 4-quinolones: Palladium-catalyzed oxidative carbonylation. Org. Lett. 2017, 19, 6432–6435. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chen, Y.; King, A.O.; Dilmeghani, M.; Larsen, R.D.; Faul, M.M. A mild, one-pot synthesis of 4-quinolones via sequential Pd-catalyzed amidation and base-promoted cyclization. Org. Lett. 2008, 10, 2609–2612. [Google Scholar] [CrossRef] [PubMed]
- Park, K.Y.; Lee, J.; Park, S.J.; Heo, J.N.; Lim, H.J. Regioselective synthesis of 1,2- and 1,4-dihydroquinolines by palladium-catalyzed intramolecular N-arylation. Adv. Synth. Catal. 2015, 357, 3917–3926. [Google Scholar] [CrossRef]
- Nemoto, T.; Fukuda, T.; Hamada, Y. Efficient synthesis of 3-substituted 2,3-dihydroquinolin-4-ones using a one-pot sequential multi-catalytic process: Pd-catalyzed allylic amination-thiazolium salt-catalyzed Stetter reaction cascade. Tetrahedron Lett. 2006, 47, 4365–4368. [Google Scholar] [CrossRef]
- Barluenga, J.; Rodríguez, F.; Fañanás, F.J. Recent advances in the synthesis of indole and quinoline derivatives through cascade reactions. Chem. Asian J. 2009, 4, 1036–1048. [Google Scholar] [CrossRef] [PubMed]
- Solé, D.; Serrano, O. Palladium-catalyzed intramolecular nucleophilic substitution at the alkoxycarbonyl group. Angew. Chem. Int. Ed. 2007, 46, 7270–7272. [Google Scholar] [CrossRef] [PubMed]
- Neville, C.F.; Barr, S.A.; Grundon, M.F. Approaches to the syntheses of dimeric quinolinone alkaloids. Tetrahedron Lett. 1992, 33, 5995–5998. [Google Scholar] [CrossRef]
- Glasnov, T.N.; Stadlbauer, W.; Kappe, C.O. Microwave-assisted multistep synthesis of functionalized 4-arylquinolin-2(1H)-ones using palladium-catalyzed cross-coupling chemistry. J. Org. Chem. 2005, 70, 3864–3870. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, B.; Wu, J. Diversity-oriented synthesis of functionalized quinolin-2(1H)-ones via Pd-catalyzed site-selective cross-coupling reactions. J. Comb. Chem. 2007, 9, 811–817. [Google Scholar] [CrossRef]
- Valente, S.; Kirsch, G. Facile synthesis of 4-acetyl-coumarins, -thiocoumarin and -quinolin-2(1H)-one via very high α-regioselective Heck coupling on tosylates. Tetrahedron Lett. 2011, 52, 3429–3432. [Google Scholar] [CrossRef]
- Chattopadhyay, S.K.; Neogi, K.; Singha, S.K.; Dey, R. Sequential Claisen rearrangement and intramolecular Heck reaction as a route to medium-ring oxacycle-fused heterocycles. Synlett 2008, 1137–1140. [Google Scholar] [CrossRef]
- Majumdar, K.C.; Nandi, R.K.; Samanta, S.; Chattopadhyay, B. Novel synthesis of heterocycle-annulated azocine derivatives of biological relevance by aromatic aza-Claisen rearrangement and intramolecular Heck reaction. Synthesis 2010, 985–990. [Google Scholar] [CrossRef]
- Majumdar, K.C.; Samanta, S.; Ghosh, T. Synthesis of coumarin- and quinolone-annulated benzazocinone frameworks by a palladium-catalyzed intramolecular Heck reaction. Synthesis 2012, 1711–1717. [Google Scholar] [CrossRef]
- Majumdar, K.C.; Ghosh, T.; Samanta, S. Facile synthesis of coumarin- and quinolone-annulated benzazoninone derivatives by an intramolecular Heck reaction strategy via 9-exo-trig cyclization. Synthesis 2011, 1569–1574. [Google Scholar] [CrossRef]
- Majumdar, K.C.; Mukhopadhyay, P.P. Regioselective synthesis of 2H-benzopyrano[3,2-c]quinolin-7(8H)-ones by radical cyclization. Synthesis 2003, 97–100. [Google Scholar] [CrossRef]
- Majumdar, K.C.; Mukhopadhyay, P.P.; Basu, P.K. Regioselective synthesis of coumarin and quinolone-annulated spiro heterocycles via aryl radical cyclization. Synthetic Commun. 2005, 35, 1291–1299. [Google Scholar] [CrossRef]
- Majumdar, K.C.; Pal, A.K.; Taher, A.; Debnath, P. Highly effective regioselective method for the synthesis of substituted coumarin- and quinolone-annulated heterocycles using a palladium(0)-catalyzed reaction. Synthesis 2007, 1707–1711. [Google Scholar] [CrossRef]
- Nolan, M.T.; Bray, J.T.W.; Eccles, K.; Cheung, M.S.; Lin, Z.; Lawrence, S.E.; Whitwood, A.C.; Fairlamb, I.J.S.; McGlacken, G.P. Pd-catalysed intramolecular regioselective arylation of 2-pyrones, pyridones, coumarins and quinolones by C-H bond functionalization. Tetrahedron 2014, 70, 7120–7127. [Google Scholar] [CrossRef]
- Pardo, L.M.; Prendergast, A.M.; Nolan, M.T.; Ó Muimhneacháin, E.; McGlacken, G.P. Pd/pivalic acid mediated direct arylation of 2-pyrones and related heterocycles. Eur. J. Org. Chem. 2015, 2015, 3540–3550. [Google Scholar] [CrossRef]
- Majumdar, K.C.; Chakravorty, S.; Shyam, P.K.; Taher, A. Palladium-mediated Heck reaction to the synthesis of 3-substituted indoles and indolones. Synthesis 2009, 403–408. [Google Scholar] [CrossRef]
- Majumdar, K.C.; Ganai, S.; Chattopadhyay, B.; Ray, K. Palladium-catalyzed one-pot synthesis of pyrrole-annulated coumarin, quinolone and 7-aza-indole derivatives. Synlett 2011, 2369–2373. [Google Scholar] [CrossRef]
- Payack, J.F.; Vazquez, E.; Matty, L.; Kress, M.H.; McNamara, J. A concise synthesis of a novel antiangiogenic tyrosine kinase inhibitor. J. Org. Chem. 2005, 70, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Fraley, M.E.; Arrington, K.L.; Buser, C.A.; Ciecko, P.A.; Coll, K.E.; Fernandes, C.; Hartman, G.D.; Hoffman, W.F.; Lynch, J.J.; McFall, R.C.; et al. Optimization of the indolyl quinolinone class of KDR (VEGFR-2) kinase inhibitors: Effects of 5-amido- and 5-sulphonamido-indolyl groups on pharmacokinetics and hERG binding. Bioorg. Med. Chem. Lett. 2004, 14, 351–355. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, L.; Sun, X. Synthesis of 3,4-disubstituted quinolin-2(1H)-ones via palladium-catalyzed regioselective cross-coupling reactions of 3-bromo-4-trifloxyquinolin-2(1H)-one with arylboronic acids. Chem. Lett. 2005, 34, 550–551. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, J. Synthesis of 1H-indol-2-yl-(4-aryl)-quinolin-2(1H)-ones via Pd-catalyzed regioselective cross-coupling reaction and cyclization. Tetrahedron 2008, 64, 1736–1742. [Google Scholar] [CrossRef]
- Hashim, J.; Glasnov, T.N.; Kremsner, J.M.; Kappe, C.O. Symmetrical bisquinolones via metal-catalyzed cross-coupling and homocoupling reactions. J. Org. Chem. 2006, 71, 1707–1710. [Google Scholar] [CrossRef] [PubMed]
- Hao, F.; Asahara, H.; Nishiwaki, N. Direct and efficient functionalization of the 1-methyl-2-quinolone framework. Proc. Engin. 2017, 174, 1058–1066. [Google Scholar] [CrossRef]
- Majumdar, K.C.; Mondal, S. A new strategy for the synthesis of coumarin- and quinolone-annulated pyrroles via Pd(0) mediated cross-coupling followed by Cu(I) catalyzed heteroannulation. Tetrahedron Lett. 2008, 49, 2418–2420. [Google Scholar] [CrossRef]
- Majumdar, K.C.; Ghosh, S.K. Studies on amine oxide rearrangements: Regioselective synthesis of pyrano[3,2-e]indol-7-one. J. Chem. Soc. Perkin Trans. 1994, 1, 2889–2894. [Google Scholar] [CrossRef]
- Majumdar, K.C.; Biswas, P.; Jana, G.H. Studies on amine oxide rearrangements: Regioselective synthesis of pyrrolo[3,2-f]quinolin-7-ones. J. Chem. Res. 1997, 310–311. [Google Scholar] [CrossRef]
- Majumdar, K.C.; Chatoppadhyay, B.; Samanta, S. A short route to the synthesis of pyrrolocoumarin and pyrroloquinolone derivatives by Sonogashira cross-coupling and gold-catalyzed cycloisomerization of acetylenic amines. Synthesis 2009, 311–317. [Google Scholar] [CrossRef]
- Majumdar, K.C.; De, N.; Roy, B. Iron/palladium-catalyzed intramolecular hydroamination: An expedient synthesis of pyrrole-annulated coumarin and quinolone derivatives. Synthesis 2010, 24, 4207–4212. [Google Scholar] [CrossRef]
- Wang, Z.; Xing, X.; Xue, L.; Gao, F.; Fang, L. Synthesis of 3H-pyrrolo[2,3-c]quinolin-4(5H)-ones via Pd-catalyzed cross-coupling reaction and cyclization. Org. Biomol. Chem. 2013, 11, 7334–7341. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xue, L.; He, Y.; Weng, L.; Fang, L. Access to functionalized 3H-pyrrolo[2,3-c]quinolin-4(5H)-ones and thieno[2,3-c]quinolin-4(5H)-ones via domino reaction of 4-alkynyl-3-bromoquinolin-2(1H)-ones. J. Org. Chem. 2014, 79, 9628–9638. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, S.; Audisio, D.; Brion, J.D.; Alami, M. Rapid access to 3-(N-substituted)-aminoquinolin-2(1H)-ones using palladium-catalyzed C–N bond coupling reaction. Tetrahedron 2007, 63, 10202–10210. [Google Scholar] [CrossRef]
- Soussi, M.A.; Audisio, D.; Messaoudi, S.; Provot, O.; Brion, J.D.; Alami, M. Palladium-catalyzed coupling of 3-halo-substituted coumarins, chromenes and quinolones with various nitrogen-containing nucleophiles. Eur. J. Org. Chem. 2011, 2011, 5077–5088. [Google Scholar] [CrossRef]
- Walsh, T.F.; Toupence, R.B.; Young, J.R.; Huang, S.X.; Ujjainwalla, F.; DeVita, R.J.; Goulet, M.T.; Wyvratt, M.J., Jr.; Fisher, M.H.; Lo, J.L.; et al. Potent antagonists of gonadotropin releasing hormone receptors derived from quinolone-6-carboxamides. Bioorg. Med. Chem. Lett. 2000, 10, 443–447. [Google Scholar] [CrossRef]
- Sahu, K.B.; Maity, A.; Mondal, S.; Paira, R.; Saha, P.; Naskar, S.; Hazra, A.; Benerjee, S.; Mondal, N.B. Basic alumina-supported synthesis of aryl-heteroarylmethanes via palladium catalyzed cross-coupling under microwave irradiation. J. Heterocycl. Chem. 2013, 50, E148. [Google Scholar] [CrossRef]
- Almeida, A.I.S.; Silva, A.M.S.; Cavaleiro, J.A.S. Reactivity of 3-iodo-4-quinolones in Heck reactions: Synthesis of novel (E)-3-styryl-4-quinolones. Synlett 2010, 462–466. [Google Scholar] [CrossRef]
- Pinto, J.; Silva, V.L.M.; Silva, A.M.G.; Silva, A.M.S.; Costa, J.C.S.; Santos, L.M.N.B.F.; Enes, R.; Cavaleiro, J.A.S.; Vicente, A.A.M.O.S.; Teixeira, J.A.C. Ohmic heating as a new efficient process for organic synthesis in water. Green Chem. 2013, 15, 970–975. [Google Scholar] [CrossRef]
- Silva, V.L.M.; Santos, L.M.N.B.F.; Silva, A.M.S. Ohmic heating: An emerging concept in organic synthesis. Chem. Eur. J. 2017, 23, 7853–7865. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.; Silva, V.L.M.; Santos, L.M.N.B.F.; Silva, A.M.S. Synthesis of (E)-3-styrylquinolin-4(1H)-ones in water by ohmic heating: A comparison with other methodologies. Eur. J. Org. Chem. 2016, 2016, 2888–2896. [Google Scholar] [CrossRef]
- Silva, V.L.M.; Silva, A.M.S.; Cavaleiro, J.A.S. New synthesis of 2,3-diarylacridin-9(10H)-ones and (E)-2-phenyl-4-styrylfuro[3,2-c]quinolones. Synlett 2010, 2565–2570. [Google Scholar] [CrossRef]
- Silva, V.L.M.; Silva, A.M.S. New synthetic methods for 2,3-diarylacridin-9(10H)-one and (E)-2-aryl-4-styrylfuro[3,2-c]quinoline derivatives. Tetrahedron 2014, 70, 5310–5320. [Google Scholar] [CrossRef]
- Jakopović, I.P.; Kragol, G.; Forrest, A.K.; Frydrych, C.S.V.; Štimac, V.; Kapić, S.; Škugor, M.M.; Ilijaš, M.; Paljetak, H.C.; Jelić, D.; et al. Synthesis and properties of macrolones characterized by two ether bonds in the linker. Bioorg. Med. Chem. 2010, 18, 6578–6588. [Google Scholar] [CrossRef] [PubMed]
- Joseph, B.; Béhard, A.; Lesur, B.; Guillaumet, G. Convenient synthetic routes to 5-substituted 3-(4-methoxyphenyl)- 4(1H)-quinolones. Synlett 2003, 1542–1544. [Google Scholar] [CrossRef]
- Pain, C.; Célanire, S.; Guillaumet, G.; Joseph, B. Synthesis of 5-substituted 2-(4- or 3-methoxyphenyl)-4(1H)-quinolones. Tetrahedron 2003, 59, 9627–9633. [Google Scholar] [CrossRef]
- Cross, R.M.; Manetsch, R. Divergent route to access structurally diverse 4-quinolones via mono or sequential cross-couplings. J. Org. Chem. 2010, 75, 8654–8657. [Google Scholar] [CrossRef]
- Mugnaini, C.; Falciani, C.; De Rosa, M.; Brizzi, A.; Pasquini, S.; Corelli, F. Regioselective functionalization of quinolin-4(1H)-ones via sequential palladium-catalyzed reactions. Tetrahedron 2011, 67, 5776–5783. [Google Scholar] [CrossRef]
- Felts, A.S.; Rodriguez, A.L.; Smith, K.A.; Engers, J.L.; Morrison, R.D.; Byers, F.W.; Blobaum, A.L.; Locuson, C.W.; Chang, S.; Venable, D.F.; et al. Design of 4-oxo-1-aryl-1,4-dihydroquinoline-3-carboxamides as selective negative allosteric modulators of metabotropic glutamate receptor subtype 2. J. Med. Chem. 2015, 58, 9027–9040. [Google Scholar] [CrossRef]
- Pinto, J.; Silva, V.L.M.; Silva, A.M.G.; Santos, L.M.N.B.F.; Silva, A.M.S. Ohmic heating-assisted synthesis of 3-arylquinolin-4(1H)-ones by a reusable and ligand-free Suzuki−Miyaura reaction in water. J. Org. Chem. 2015, 80, 6649–6659. [Google Scholar] [CrossRef]
- Gomes, A.T.P.C.; Cunha, A.C.; Domingues, M.R.M.; Neves, M.G.P.M.S.; Tomé, A.C.; Silva, A.M.S.; Santos, F.C.; Souza, M.C.B.V.; Ferreira, V.F.; Cavaleiro, J.A.S. Synthesis and characterization of new porphyrin/4-quinolone conjugates. Tetrahedron 2011, 67, 7336–7342. [Google Scholar] [CrossRef]
- Holder, J.C.; Marziale, A.N.; Gatti, M.; Mao, B.; Stoltz, B.M. Palladium-catalyzed asymmetric conjugate addition of arylboronic acids to heterocyclic acceptors. Chem. Eur. J. 2013, 19, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, S.; Barange, D.K.; Pal, M. One-pot synthesis of 2-substituted furo[3,2-c]quinolines via tandem coupling-cyclization under Pd/C-copper catalysis. Tetrahedron Lett. 2006, 47, 7317–7322. [Google Scholar] [CrossRef]
- Pal, M. Palladium-catalyzed alkynylation of aryl and hetaryl halides: A journey from conventional palladium complexes or salts to palladium/carbon. Synlett 2009, 18, 2896–2912. [Google Scholar] [CrossRef]
- Škugor, M.M.; Štimac, V.; Palej, I.; Lugarić, D.; Paljetak, H.Č.; Filić, D.; Modrić, M.; Dilović, I.; Gembarovski, D.; Mutak, S.; et al. Synthesis and biological activity of 4′′-O-acyl derivatives of 14- and 15-membered macrolides linked to ω-quinolone-carboxylic unit. Bioorg. Med. Chem. 2010, 18, 6547–6558. [Google Scholar] [CrossRef] [PubMed]
- Kapić, S.; Paljetak, H.Č.; Alihodžić, S.; Antolović, R.; Haber, V.E.; Jarvest, R.L.; Holmes, D.J.; Broskey, J.P.; Hunt, E. 6-Alkylquinolone-3-carboxylic acid tethered to macrolides synthesis and antimicrobial profile. Bioorg. Med. Chem. 2010, 18, 6569–6577. [Google Scholar] [CrossRef]
- Layek, M.; Gajare, V.; Kalita, D.; Islam, A.; Mukkanti, K.; Pal, M. Pd/C–Cu in coupling-cyclization process: A general synthesis of 2-substituted 6-oxopyrrolo[3,2,1-ij]quinoline derivatives. Tetrahedron Lett. 2009, 50, 3867–3871. [Google Scholar] [CrossRef]
- Laborde, E.; Leshaski, L.E.; Kiely, J.S. Palladium-catalyzed intermolecular vinylic arylation of cycloalkenes. Applications to the synthesis of quinolone antibacterials. Tetrahedron Lett. 1990, 31, 1837–1840. [Google Scholar] [CrossRef]
- Kiely, J.S.; Laborde, E.; Lesheski, L.E.; Bucsh, R.A. Synthesis of 7-(alkenyl, cycloalkenyl and 1,2,3,6-tetrahydro-4-pyridinyl)quinolones. J. Heterocycl. Chem. 1991, 28, 1581–1585. [Google Scholar] [CrossRef]
- Reuman, M.; Daum, S.J.; Singh, B.; Wentland, M.P.; Perni, R.B.; Pennock, P.; Carabateas, P.M.; Gruett, M.D.; Saindane, M.T.; Dorff, P.H.; et al. Synthesis and antibacterial activity of some novel l-substituted 1,4-dihydro-4-oxo-7-pyridinyl-3-quinolinecarboxylic acids. Potent antistaphylococcal agents. J. Med. Chem. 1995, 38, 2531–2540. [Google Scholar] [CrossRef]
- Wicke, L.; Engels, J.W.; Gambari, R.; Saab, A.M. Synthesis and antiproliferative activity of quinolone nucleosides against the human myelogenous leukemia K-562 cell line. Arch. Pharm. Chem. Life Sci. 2013, 346, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Batalha, P.N.; Gomes, A.T.P.C.; Forezi, L.S.M.; Costa, L.; Souza, M.C.B.V.; Boechat, F.C.S.; Ferreira, V.F.; Almeida, A.; Faustino, M.A.F.; Neves, M.G.P.M.S.; et al. Synthesis of new porphyrin/4-quinolone conjugates and evaluation of their efficiency in the photoinactivation of Staphylococcus aureus. RSC Adv. 2015, 5, 71228–71239. [Google Scholar] [CrossRef]
- Li, M.; Li, L.; Ge, H. Direct C-3-alkenylation of quinolones via palladium-catalyzed C-H functionalization. Adv. Synth. Catal. 2010, 352, 2445–2449. [Google Scholar] [CrossRef]
- Messaoudi, S.; Brion, J.D.; Alami, M. Palladium-catalyzed decarboxylative coupling of quinolone-3-carboxylic acids and related heterocyclic carboxylic acids with (hetero)aryl halides. Org. Lett. 2012, 14, 1496–1499. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Yang, Y.; Wei, Z.; Yu, W.; Liao, H.; Shen, C.; Zhang, P. Palladium-catalyzed decarboxylative Csp2–Csp2 cross-coupling reactions: An efficient route for synthesis of azaisoflavone derivatives. Catal. Lett. 2015, 145, 1634–1642. [Google Scholar] [CrossRef]
- Xia, C.; Wei, Z.; Yang, Y.; Yu, W.; Liao, H.; Shen, C.; Zhang, P. Palladium-catalyzed thioetherification of quinolone derivatives via decarboxylative C-S cross-couplings. Chem. Asian J. 2016, 11, 360–366. [Google Scholar] [CrossRef]
- Schmidt, J.P.; Li, C.; Breit, B. Transition-metal-catalyzed regiodivergent and stereoselective access to branched and linear allylated 4-pyridones. Chem. Eur. J. 2017, 23, 6531–6534. [Google Scholar] [CrossRef]
- Han, X.; Yue, Z.; Zhang, X.; He, Q.; Yang, C. Copper-mediated, palladium-catalyzed cross-coupling of 3-iodochromones, thiochromones and quinolones with ethyl bromodifluoroacetate. J. Org. Chem. 2013, 78, 4850–4856. [Google Scholar] [CrossRef]
Figure 1.
Structures of quinolin-2(1H)-one and quinolin-4(1H)-one.

Scheme 1.
Synthesis of quinolin-2(1H)-ones 3 by Heck reaction of 2-iodoanilines 1 with dimethyl maleate 2 [16].
Scheme 1.
Synthesis of quinolin-2(1H)-ones 3 by Heck reaction of 2-iodoanilines 1 with dimethyl maleate 2 [16].

Scheme 2.
Synthesis of 4-arylquinolin-2(1H)-ones 7 through a domino Heck/cyclization of methyl β-(2-acetamidophenyl)acrylates 4 with aryl iodides 5 [17].
Scheme 2.
Synthesis of 4-arylquinolin-2(1H)-ones 7 through a domino Heck/cyclization of methyl β-(2-acetamidophenyl)acrylates 4 with aryl iodides 5 [17].

Scheme 3.
Synthesis of 4-arylquinolin-2(1H)-ones 10 through a sequential Heck/Cu-catalysed cyclization process (i, ii) or through a pseudo-domino Heck/Buchwald-Hartwig process (iii) [18,19].

Scheme 4.
Synthesis of quinolin-2(1H)-ones 13 from pivaloylaminobenzenes 11 via Heck coupling and cyclization [20].
Scheme 4.
Synthesis of quinolin-2(1H)-ones 13 from pivaloylaminobenzenes 11 via Heck coupling and cyclization [20].

Scheme 5.
Synthesis of 3-substituted quinolin-2(1H)-ones 15 via Heck coupling-cyclization of 2-iodoaniline 1 with α,β-unsaturated carbonyl compounds 14 [21].
Scheme 5.
Synthesis of 3-substituted quinolin-2(1H)-ones 15 via Heck coupling-cyclization of 2-iodoaniline 1 with α,β-unsaturated carbonyl compounds 14 [21].

Scheme 6.
Synthesis of 4-arylquinolin-2(1H)-one 19 via a Pd-catalysed Heck-Matsuda reaction of ester 16 with 17 followed by a Cu-catalysed asymmetric 1,4-reduction/cyclization [22].
Scheme 6.
Synthesis of 4-arylquinolin-2(1H)-one 19 via a Pd-catalysed Heck-Matsuda reaction of ester 16 with 17 followed by a Cu-catalysed asymmetric 1,4-reduction/cyclization [22].

Scheme 7.
Heterogeneous tandem HRC of 2-(2-nitrophenyl)acrylates 21 with aryldiazonium salts 22: synthesis of 3,4-dihydroquinolin-2(1H)-ones 23 [23].
Scheme 7.
Heterogeneous tandem HRC of 2-(2-nitrophenyl)acrylates 21 with aryldiazonium salts 22: synthesis of 3,4-dihydroquinolin-2(1H)-ones 23 [23].

Scheme 8.
Homogeneous/heterogeneous sequential HRC of 2-nitrobenzenediazonium salts 24 with acrylates 25: Synthesis of 3,4-dihydroquinolin-2(1H)-ones 26 [23].
Scheme 8.
Homogeneous/heterogeneous sequential HRC of 2-nitrobenzenediazonium salts 24 with acrylates 25: Synthesis of 3,4-dihydroquinolin-2(1H)-ones 26 [23].

Scheme 9.
Synthesis of furo[3,2-c]quinolin-4(5H)-one 30 through two sequential Pd-catalysed reactions of ethyl 3-furoate 28 and 1-bromo-2-nitrobenzene 29 [25].
Scheme 9.
Synthesis of furo[3,2-c]quinolin-4(5H)-one 30 through two sequential Pd-catalysed reactions of ethyl 3-furoate 28 and 1-bromo-2-nitrobenzene 29 [25].

Scheme 10.
Synthesis of 4-arylquinolin-2(1H)-ones 32 by Heck reaction of substituted 2-iodoanilines 1 with acrylic acid followed by in situ cyclization [26].
Scheme 10.
Synthesis of 4-arylquinolin-2(1H)-ones 32 by Heck reaction of substituted 2-iodoanilines 1 with acrylic acid followed by in situ cyclization [26].

Scheme 11.
Synthesis of quinolone 35 by an intramolecular Heck cyclization of carboxamide 34 [27].
Scheme 11.
Synthesis of quinolone 35 by an intramolecular Heck cyclization of carboxamide 34 [27].

Scheme 12.
Intramolecular Heck cyclization of N-(hetero)arylcarboxamides 38 into quinolones 39 [28].
Scheme 12.
Intramolecular Heck cyclization of N-(hetero)arylcarboxamides 38 into quinolones 39 [28].

Scheme 13.
Synthesis of quinolin-2(1H)-ones 41d by intramolecular Heck cyclization of acrylamides 40 [29].
Scheme 13.
Synthesis of quinolin-2(1H)-ones 41d by intramolecular Heck cyclization of acrylamides 40 [29].

Scheme 14.
Synthesis of 3,4-disubstituted quinolin-2(1H)-ones 48 by a Pd-catalysed reaction of ethyl N-(2-ethynyl)malonanilide 45 followed by an intramolecular carbocyclization [30].
Scheme 14.
Synthesis of 3,4-disubstituted quinolin-2(1H)-ones 48 by a Pd-catalysed reaction of ethyl N-(2-ethynyl)malonanilide 45 followed by an intramolecular carbocyclization [30].

Scheme 15.
Synthesis of 3- and 4-substituted quinolin-2(1H)-ones 48 and 49 and 3,4-disubstituted quinolin-2(1H)-ones 51 and 52 via Pd-catalysed carbonylative annulation of 2-iodoanilines 46 and 1 with terminal alkynes and with internal alkynes, respectively [31,34,35].

Scheme 16.
Synthesis of 3,4-disubstituted (dihydro)quinolin-2(1H)-ones 55 and 57 by carbonylative annulation of o-iodoanilines 53 [36].
Scheme 16.
Synthesis of 3,4-disubstituted (dihydro)quinolin-2(1H)-ones 55 and 57 by carbonylative annulation of o-iodoanilines 53 [36].

Scheme 17.
Synthesis of 3-substituted 4-phenylquinolin-2(1H)-ones 61 by reaction of vinyl-Pd complexes 60 with CO [37].
Scheme 17.
Synthesis of 3-substituted 4-phenylquinolin-2(1H)-ones 61 by reaction of vinyl-Pd complexes 60 with CO [37].

Scheme 18.
Synthesis of 4,5-fused tricyclic quinolin-2(1H)-ones 63 by intramolecular carbonylative annulation of alkyne-tethered o-iodoanilines 62 [38].
Scheme 18.
Synthesis of 4,5-fused tricyclic quinolin-2(1H)-ones 63 by intramolecular carbonylative annulation of alkyne-tethered o-iodoanilines 62 [38].

Scheme 19.
Synthesis of quinolin-2(1H)-one 65 by Pd-catalysed carbonylation of vinyl bromides 64 [39].
Scheme 19.
Synthesis of quinolin-2(1H)-one 65 by Pd-catalysed carbonylation of vinyl bromides 64 [39].

Scheme 20.
Synthesis of 4-methyl-3,4-dihydroquinolin-2(1H)-ones 67 by asymmetric cyclocarbonylation of 2-(1-methylvinyl)anilines 66 [40].
Scheme 20.
Synthesis of 4-methyl-3,4-dihydroquinolin-2(1H)-ones 67 by asymmetric cyclocarbonylation of 2-(1-methylvinyl)anilines 66 [40].

Scheme 21.
Synthesis of quinolin-2(1H)-one 71 via Pd0-mediated Ullmann cross-coupling/reductive cyclization sequence [41].
Scheme 21.
Synthesis of quinolin-2(1H)-one 71 via Pd0-mediated Ullmann cross-coupling/reductive cyclization sequence [41].

Scheme 22.
Synthesis of N-hydroxyquinolin-2(1H)-ones 74 by Pd-catalysed Buchwald-Hartwig reaction of protected N-hydroxyamides 72 with benzaldehydes or benzoates followed by cyclodehydration and deprotection [42].
Scheme 22.
Synthesis of N-hydroxyquinolin-2(1H)-ones 74 by Pd-catalysed Buchwald-Hartwig reaction of protected N-hydroxyamides 72 with benzaldehydes or benzoates followed by cyclodehydration and deprotection [42].

Scheme 23.
Synthesis of enantiopure 3,4-dihydroquinolin-2(1H)-ones 76 by an enantioselective Buchwald-Hartwig reaction [43].
Scheme 23.
Synthesis of enantiopure 3,4-dihydroquinolin-2(1H)-ones 76 by an enantioselective Buchwald-Hartwig reaction [43].

Scheme 24.
Synthesis of spirobis[3,4-dihydroquinolin-2(1H)-ones] 80 by intramolecular double N-arylation of malonamides 79 [44].
Scheme 24.
Synthesis of spirobis[3,4-dihydroquinolin-2(1H)-ones] 80 by intramolecular double N-arylation of malonamides 79 [44].

Scheme 25.
Plausible catalytic cycle [44].
Scheme 25.
Plausible catalytic cycle [44].

Scheme 26.
Synthesis of quinolin-2(1H)-ones 84 by selective 6-endo intramolecular C-H alkenylation of N-phenylacrylamides 83 [45].
Scheme 26.
Synthesis of quinolin-2(1H)-ones 84 by selective 6-endo intramolecular C-H alkenylation of N-phenylacrylamides 83 [45].

Scheme 27.
Mechanism proposed for the Pd(II)-catalysed 6-endo intramolecular C-H alkenylation [45].
Scheme 27.
Mechanism proposed for the Pd(II)-catalysed 6-endo intramolecular C-H alkenylation [45].

Scheme 28.
Synthesis of quinolin-2(1H)-ones 87 and 88 by tandem Pd(0)-catalysed amidation/aldol condensation of appropriate aryl halides 85 with acetamides 86 [46].
Scheme 28.
Synthesis of quinolin-2(1H)-ones 87 and 88 by tandem Pd(0)-catalysed amidation/aldol condensation of appropriate aryl halides 85 with acetamides 86 [46].

Scheme 29.
Synthesis of 4-arylquinolin-2(1H)-ones 90, 90′ and 91 by Pd-catalysed intramolecular amidation of 3,3-diarylacrylamides 89 [47,48].

Scheme 30.
Synthesis of 4-arylquinolin-2(1H)-ones 93, 94 and 95 via symmetrical 3,3-diarylacrylamides 92 and 96 and/or 96′ via unsymmetrical 3,3-diarylacrylamides 92 [48,49].

Scheme 31.
Synthesis of quinolin-2(1H)-ones 100 by Pd-catalysed intramolecular addition reactions of the corresponding alkynanilides 99 [50].
Scheme 31.
Synthesis of quinolin-2(1H)-ones 100 by Pd-catalysed intramolecular addition reactions of the corresponding alkynanilides 99 [50].

Scheme 32.
Synthesis of 4-substituted quinolin-2(1H)-ones 103 and 103′ by Pd-catalysed [3 + 3] annulation between diarylamines 101 and α,β-unsaturated acids 102 [51].
Scheme 32.
Synthesis of 4-substituted quinolin-2(1H)-ones 103 and 103′ by Pd-catalysed [3 + 3] annulation between diarylamines 101 and α,β-unsaturated acids 102 [51].

Scheme 33.
Synthesis of benzothieno[2,3-c]quinolin-6(5H)-ones 106 by one-pot three steps Pd-catalysed borylation, Suzuki coupling and amidation [52].
Scheme 33.
Synthesis of benzothieno[2,3-c]quinolin-6(5H)-ones 106 by one-pot three steps Pd-catalysed borylation, Suzuki coupling and amidation [52].

Scheme 34.
Synthesis of 3,4-dihydroquinolin-2(1H)-ones 109 via Pd-catalysed cyclopropane ring expansion [53].
Scheme 34.
Synthesis of 3,4-dihydroquinolin-2(1H)-ones 109 via Pd-catalysed cyclopropane ring expansion [53].

Scheme 35.
Synthesis of quinolin-2(1H)-ones 112 by a one-pot catalysed successive ammonolysis, C-H bond activation and cyclization reactions, using simple anilines 110 as substrates [54].
Scheme 35.
Synthesis of quinolin-2(1H)-ones 112 by a one-pot catalysed successive ammonolysis, C-H bond activation and cyclization reactions, using simple anilines 110 as substrates [54].

Scheme 36.
Plausible mechanism for the successive ammonolysis, C-H bond activation and cyclization of anilines 110 [54].
Scheme 36.
Plausible mechanism for the successive ammonolysis, C-H bond activation and cyclization of anilines 110 [54].

Scheme 37.
Synthesis of 3,3-disubstituted 3,4-dihydroquinolin-2(1H)-ones 115 by Pd-catalysed, oxidative-addition-initiated activation and arylation of inert C(sp3)-H bonds [55].
Scheme 37.
Synthesis of 3,3-disubstituted 3,4-dihydroquinolin-2(1H)-ones 115 by Pd-catalysed, oxidative-addition-initiated activation and arylation of inert C(sp3)-H bonds [55].

Scheme 38.
Synthesis of quinolin-2(1H)-ones 118 by Pd-catalysed C-S activation/aryne insertion/coupling sequence [56].
Scheme 38.
Synthesis of quinolin-2(1H)-ones 118 by Pd-catalysed C-S activation/aryne insertion/coupling sequence [56].

Scheme 39.
Synthesis of quinolones 121 by Pd-oxidative annulation between acrylamides 119 and arynes 120 [57].
Scheme 39.
Synthesis of quinolones 121 by Pd-oxidative annulation between acrylamides 119 and arynes 120 [57].

Scheme 40.
Plausible mechanism for the formation of quinolones 121 [57].
Scheme 40.
Plausible mechanism for the formation of quinolones 121 [57].

Scheme 41.
Synthesis of quinolin-2(1H)-ones 126 by Pd-catalysed aminocarbonylation/ intramolecular amidation cascade sequence starting from 2-(2-haloalkenyl)aryl halides 124 [58].
Scheme 41.
Synthesis of quinolin-2(1H)-ones 126 by Pd-catalysed aminocarbonylation/ intramolecular amidation cascade sequence starting from 2-(2-haloalkenyl)aryl halides 124 [58].

Scheme 42.
Synthesis of 2,3-dihydroquinolin-4(1H)-ones 134 from 2-substituted anilides 132 obtained through Pd-catalysed reaction of 2-iodoanilides 130 with acetylenic carbinols 131 [59,60].

Scheme 43.
Synthesis of 2,3-dihydroquinolin-4(1H)-ones 137 via Sonogashira coupling of 2-(pseudo)haloanilines 135 with propargyl alcohols 131 followed by acid-catalysed tandem Rupe rearrangement-Donnelly-Farrell ring closure of anilines 136 [61].
Scheme 43.
Synthesis of 2,3-dihydroquinolin-4(1H)-ones 137 via Sonogashira coupling of 2-(pseudo)haloanilines 135 with propargyl alcohols 131 followed by acid-catalysed tandem Rupe rearrangement-Donnelly-Farrell ring closure of anilines 136 [61].

Scheme 44.
Synthesis of 2-substituted quinolin-4(1H)-ones 141 by Pd-catalysed carbonylative coupling of 2-haloanilines 139 with terminal acetylenes 47 [32,33,62].

Scheme 45.
Synthesis of the quinolin-4(1H)-one substructure 146 by Pd-catalysed carbonylative Sonogashira coupling of 2-iodo-5-methoxyaniline 144 with thiazolylacetylene 143 [63].
Scheme 45.
Synthesis of the quinolin-4(1H)-one substructure 146 by Pd-catalysed carbonylative Sonogashira coupling of 2-iodo-5-methoxyaniline 144 with thiazolylacetylene 143 [63].

Scheme 46.
Synthesis of 2-substituted quinolin-4(1H)-ones 148 by a carbonylative Sonogashira coupling reaction under homogeneous and heterogeneous Pd-catalysis [24,64,65,66].

Scheme 47.
Synthesis of quinolin-4(1H)-ones 149 via carbonylative Sonogashira cross-coupling reaction using molybdenum hexacarbonyl as a CO source [67].
Scheme 47.
Synthesis of quinolin-4(1H)-ones 149 via carbonylative Sonogashira cross-coupling reaction using molybdenum hexacarbonyl as a CO source [67].

Scheme 48.
Synthesis of quinolin-4(1H)-ones 152 by a Pd-catalysed tandem amination reaction [68].
Scheme 48.
Synthesis of quinolin-4(1H)-ones 152 by a Pd-catalysed tandem amination reaction [68].

Scheme 49.
Plausible mechanism for the synthesis of 152 from 151 [68].
Scheme 49.
Plausible mechanism for the synthesis of 152 from 151 [68].

Scheme 50.
Synthesis of 1,2-disubstituted-quinolin-4(1H)-ones 154 through a Buchwald-Hartwig coupling and catalytic dehydrogenation sequence [69].
Scheme 50.
Synthesis of 1,2-disubstituted-quinolin-4(1H)-ones 154 through a Buchwald-Hartwig coupling and catalytic dehydrogenation sequence [69].

Scheme 51.
Synthesis of 2-methylquinolin-4(1H)-one 158 via Stille reaction followed by catalytic hydrogenation [70].
Scheme 51.
Synthesis of 2-methylquinolin-4(1H)-one 158 via Stille reaction followed by catalytic hydrogenation [70].

Scheme 52.
Synthesis of quinolin-4(1H)-one 162 via Stille reaction followed by cyclization [71].
Scheme 52.
Synthesis of quinolin-4(1H)-one 162 via Stille reaction followed by cyclization [71].

Scheme 53.
Synthesis of quinolin-4(1H)-ones 164 and 166 via Pd-catalysed carbonylative cyclization of 2-haloenamines 163 and 165 [72,73,74].

Scheme 54.
Synthesis of quinolin-4(1H)-one 168 via Pd-catalysed oxidative carbonylation of 2-ethynylaniline 167 [75,76].

Scheme 55.
Two-step and sequential syntheses of 3-aroylquinolin-4(1H)-ones 171 by Pd-catalysed carbonylative cyclization of N-(2-iodoaryl)enaminones 170 [77].
Scheme 55.
Two-step and sequential syntheses of 3-aroylquinolin-4(1H)-ones 171 by Pd-catalysed carbonylative cyclization of N-(2-iodoaryl)enaminones 170 [77].

Scheme 56.
Synthesis of 2,3-dihydroquinolin-4(1H)-ones 173 by Pd-catalysed intermolecular cyclocarbonylation of 2-iodoanilines 1 and diethyl ethoxycarbonyl butendienoate 172 [78].
Scheme 56.
Synthesis of 2,3-dihydroquinolin-4(1H)-ones 173 by Pd-catalysed intermolecular cyclocarbonylation of 2-iodoanilines 1 and diethyl ethoxycarbonyl butendienoate 172 [78].

Scheme 57.
Probable reaction mechanism for the formation of quinolin-4(1H)-ones 173 [78].
Scheme 57.
Probable reaction mechanism for the formation of quinolin-4(1H)-ones 173 [78].

Scheme 58.
Synthesis of quinolin-4(1H)-ones 177 via a Pd-catalysed cascade carbonylation-allene insertion [79].
Scheme 58.
Synthesis of quinolin-4(1H)-ones 177 via a Pd-catalysed cascade carbonylation-allene insertion [79].

Scheme 59.
Proposed reaction mechanism for the formation of quinolin-4(1H)-ones 177 [79].
Scheme 59.
Proposed reaction mechanism for the formation of quinolin-4(1H)-ones 177 [79].

Scheme 60.
Synthesis of γ-aminoalcohols 178 via Pd-catalysed cascade carbonylation-allene insertion-Michael addition followed by reduction of the corresponding quinolin-4(1H)-ones [79].
Scheme 60.
Synthesis of γ-aminoalcohols 178 via Pd-catalysed cascade carbonylation-allene insertion-Michael addition followed by reduction of the corresponding quinolin-4(1H)-ones [79].

Scheme 61.
Synthesis of quinolin-4(1H)-ones 182 by Pd-catalysed oxidative carbonylation of ketones with amines in the presence of carbon monoxide at atmospheric pressure [81].
Scheme 61.
Synthesis of quinolin-4(1H)-ones 182 by Pd-catalysed oxidative carbonylation of ketones with amines in the presence of carbon monoxide at atmospheric pressure [81].

Scheme 62.
Synthesis of 2-substituted quinolin-4(1H)-ones 185 via sequential Pd-catalysed amidation followed by base-promoted intramolecular cyclization [82].
Scheme 62.
Synthesis of 2-substituted quinolin-4(1H)-ones 185 via sequential Pd-catalysed amidation followed by base-promoted intramolecular cyclization [82].

Scheme 63.
Synthesis of 2,3-dihydroquinolin-4(1H)-one 188 by Pd-catalysed intramolecular N-arylation of Z-enamine 186 and further reduction [83].
Scheme 63.
Synthesis of 2,3-dihydroquinolin-4(1H)-one 188 by Pd-catalysed intramolecular N-arylation of Z-enamine 186 and further reduction [83].

Scheme 64.
Synthesis of 3-substituted 2,3-dihydroquinolin-4(1H)-ones 192 by Pd-catalysed allylic amination-thiazolium salt-catalysed Stetter cascade reaction [84].
Scheme 64.
Synthesis of 3-substituted 2,3-dihydroquinolin-4(1H)-ones 192 by Pd-catalysed allylic amination-thiazolium salt-catalysed Stetter cascade reaction [84].

Scheme 65.
Synthesis of 2,3-dihydroquinolin-4(1H)-ones 194 by Pd(0)-catalysed intramolecular coupling of β-(2-iodoanilino)esters 193 [86].
Scheme 65.
Synthesis of 2,3-dihydroquinolin-4(1H)-ones 194 by Pd(0)-catalysed intramolecular coupling of β-(2-iodoanilino)esters 193 [86].

Scheme 66.
Proposed mechanism for the formation of dihydroquinolin-4(1H)-ones 194 [86].
Scheme 66.
Proposed mechanism for the formation of dihydroquinolin-4(1H)-ones 194 [86].

Scheme 67.
Heck reaction of 3-iodoquinolin-2(1H)-one 196 with 2-methyl-3-buten-2-ol 197 [87].
Scheme 67.
Heck reaction of 3-iodoquinolin-2(1H)-one 196 with 2-methyl-3-buten-2-ol 197 [87].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
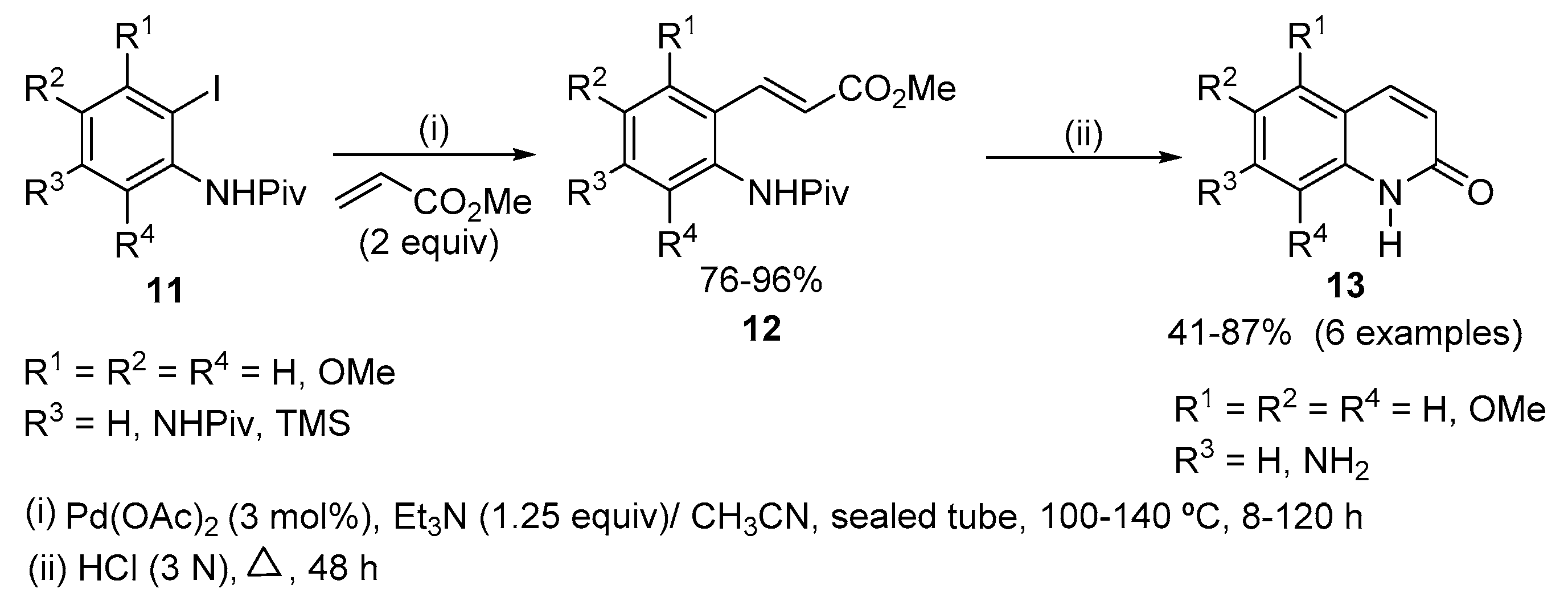{kind=link}
{kind=link}
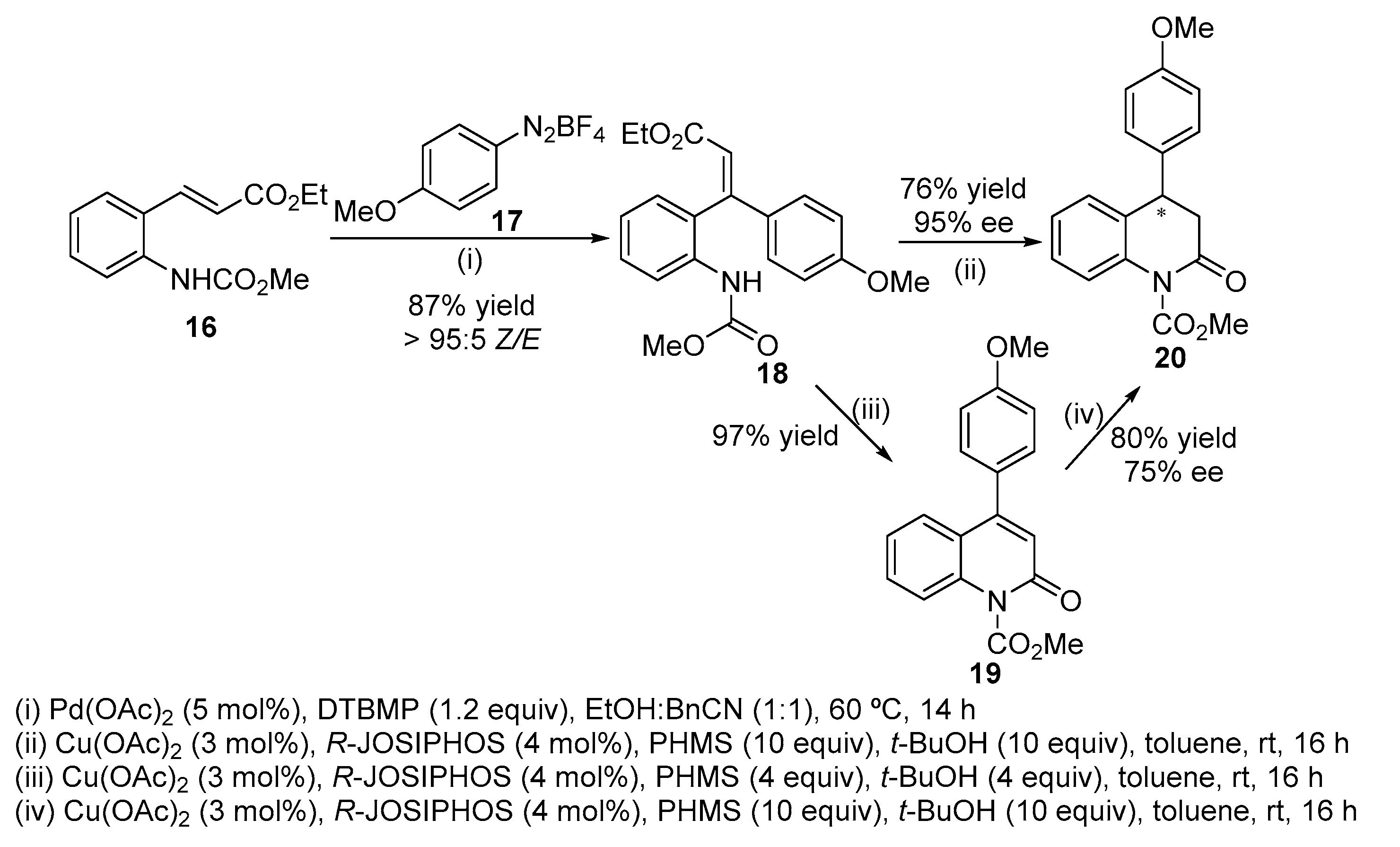{kind=link}
{kind=link}
{kind=link}
{kind=link}
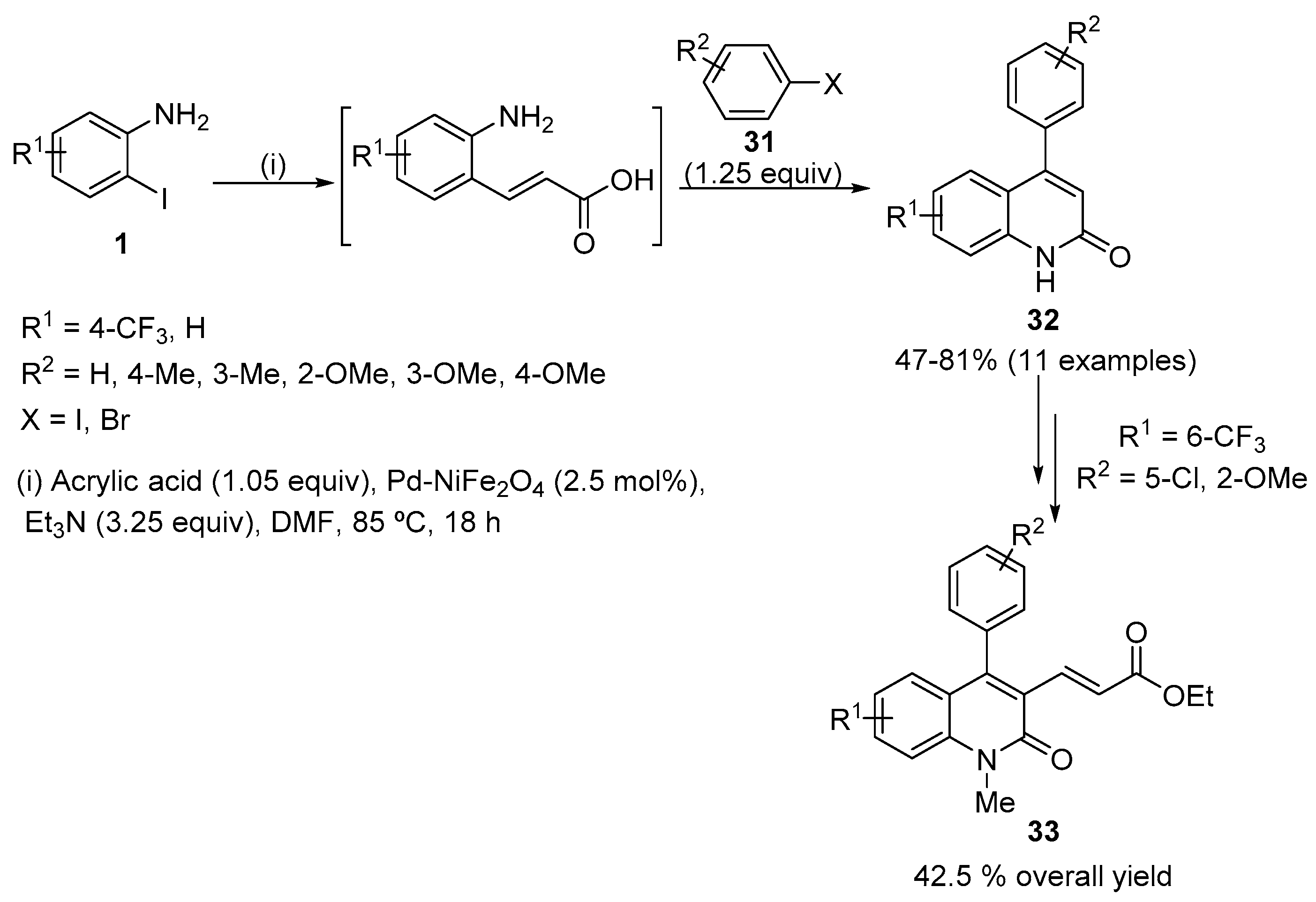{kind=link}
{kind=link}
{kind=link}
{kind=link}
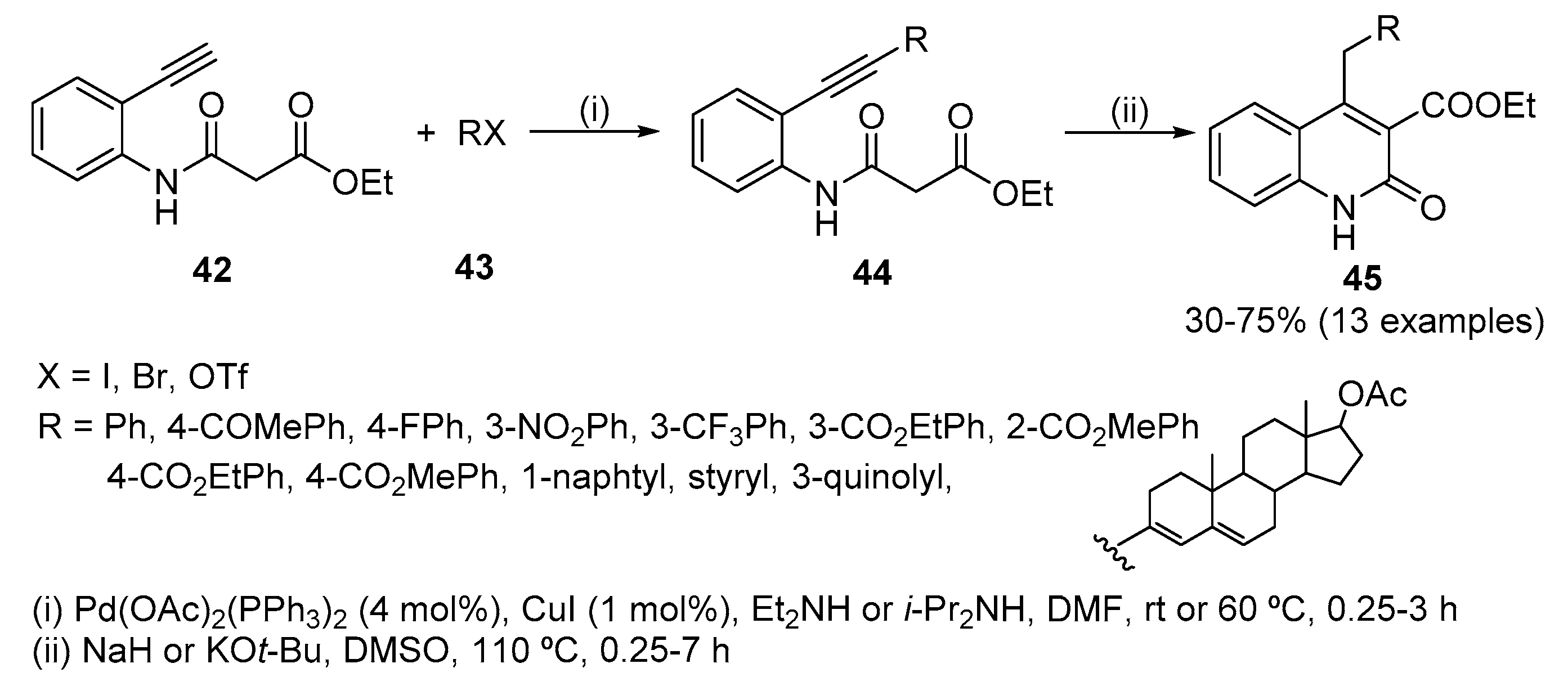{kind=link}
{kind=link}
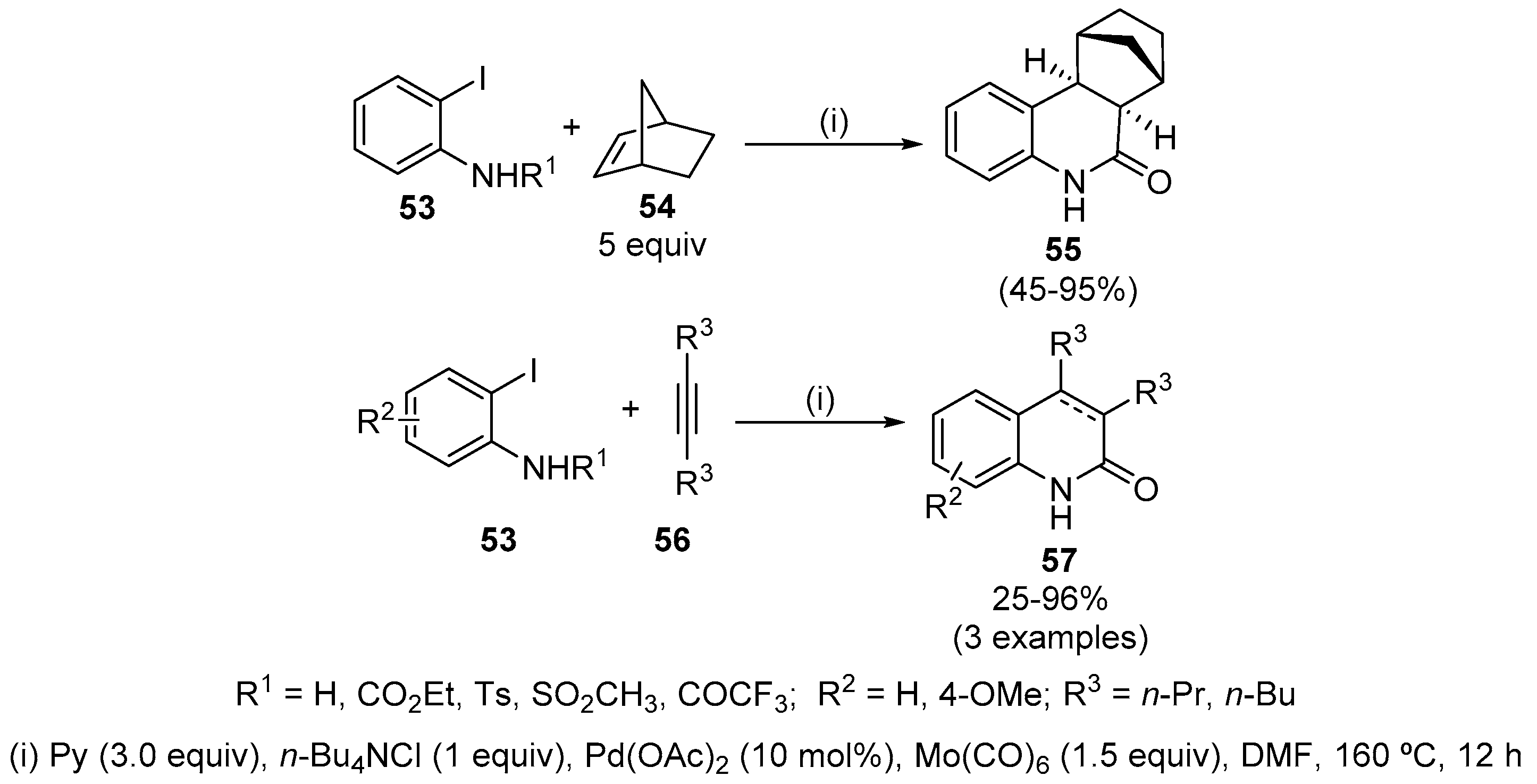{kind=link}
{kind=link}
{kind=link}
{kind=link}
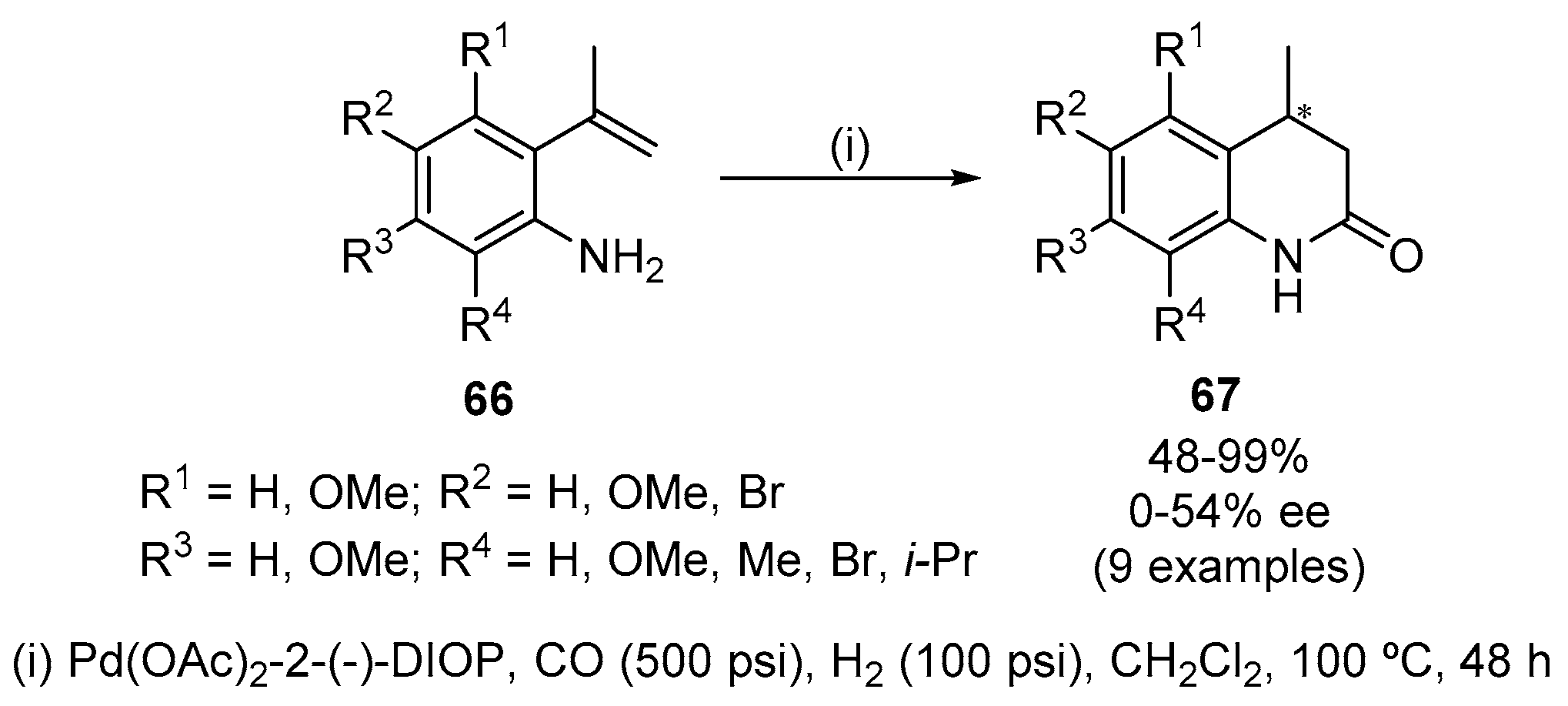{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
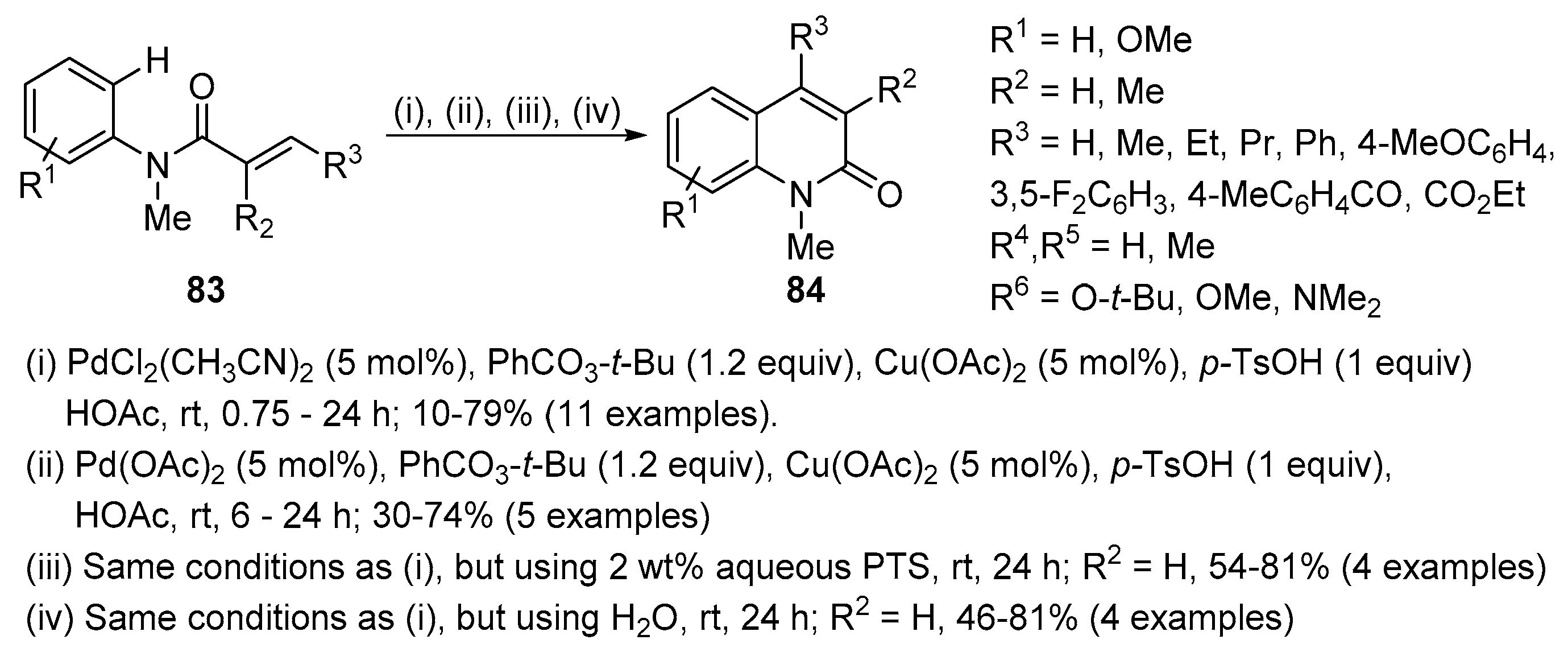{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
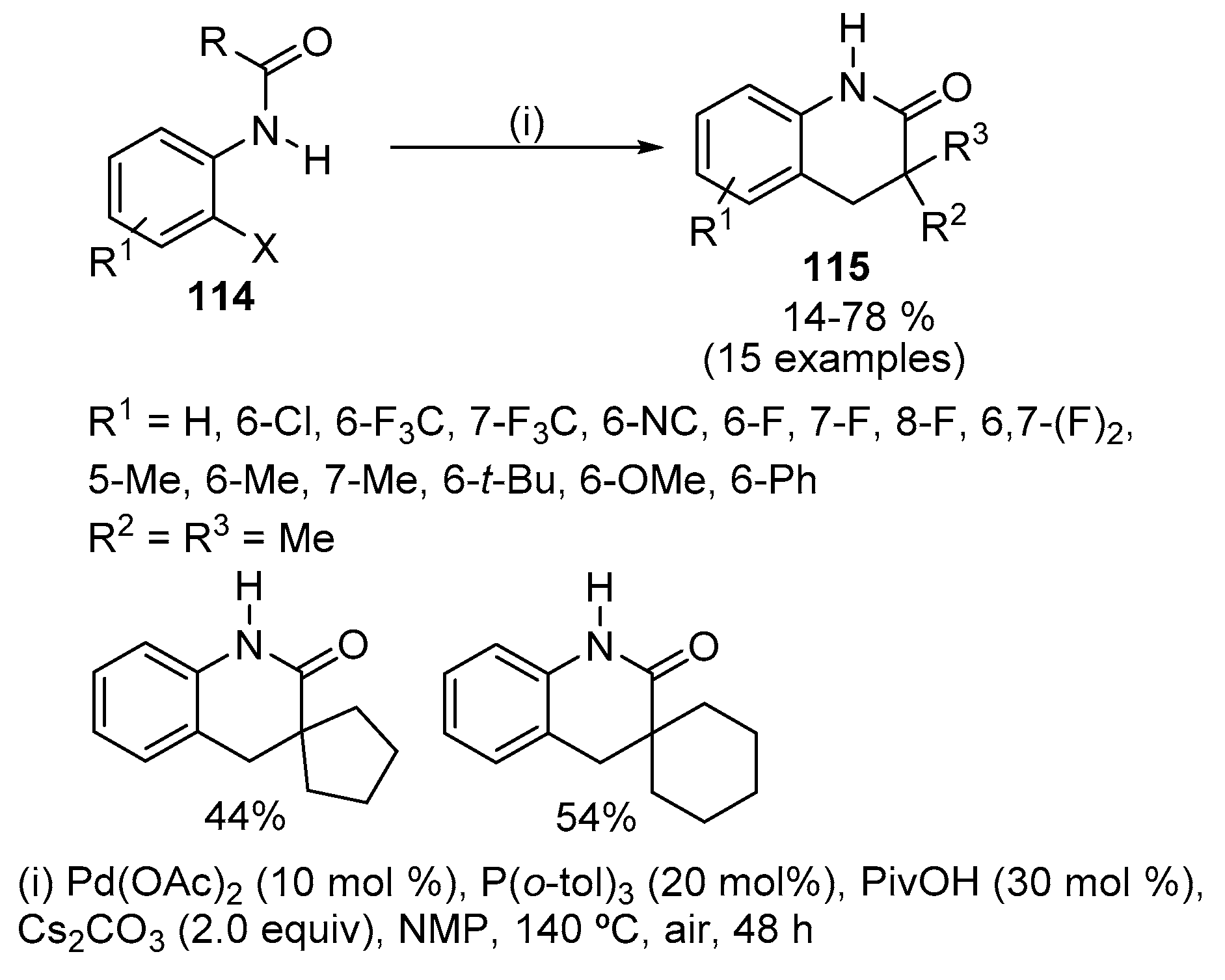{kind=link}
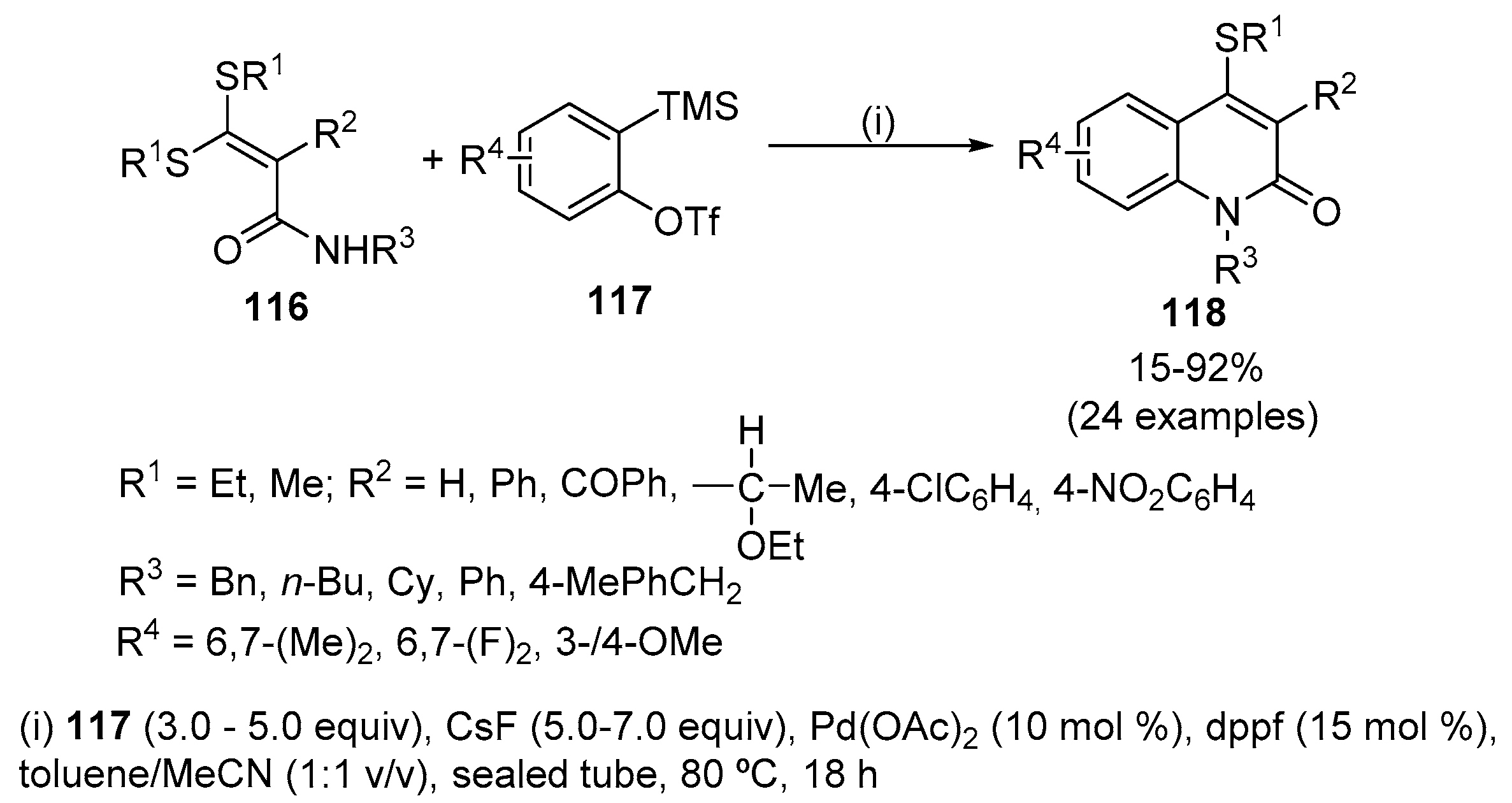{kind=link}
{kind=link}
{kind=link}
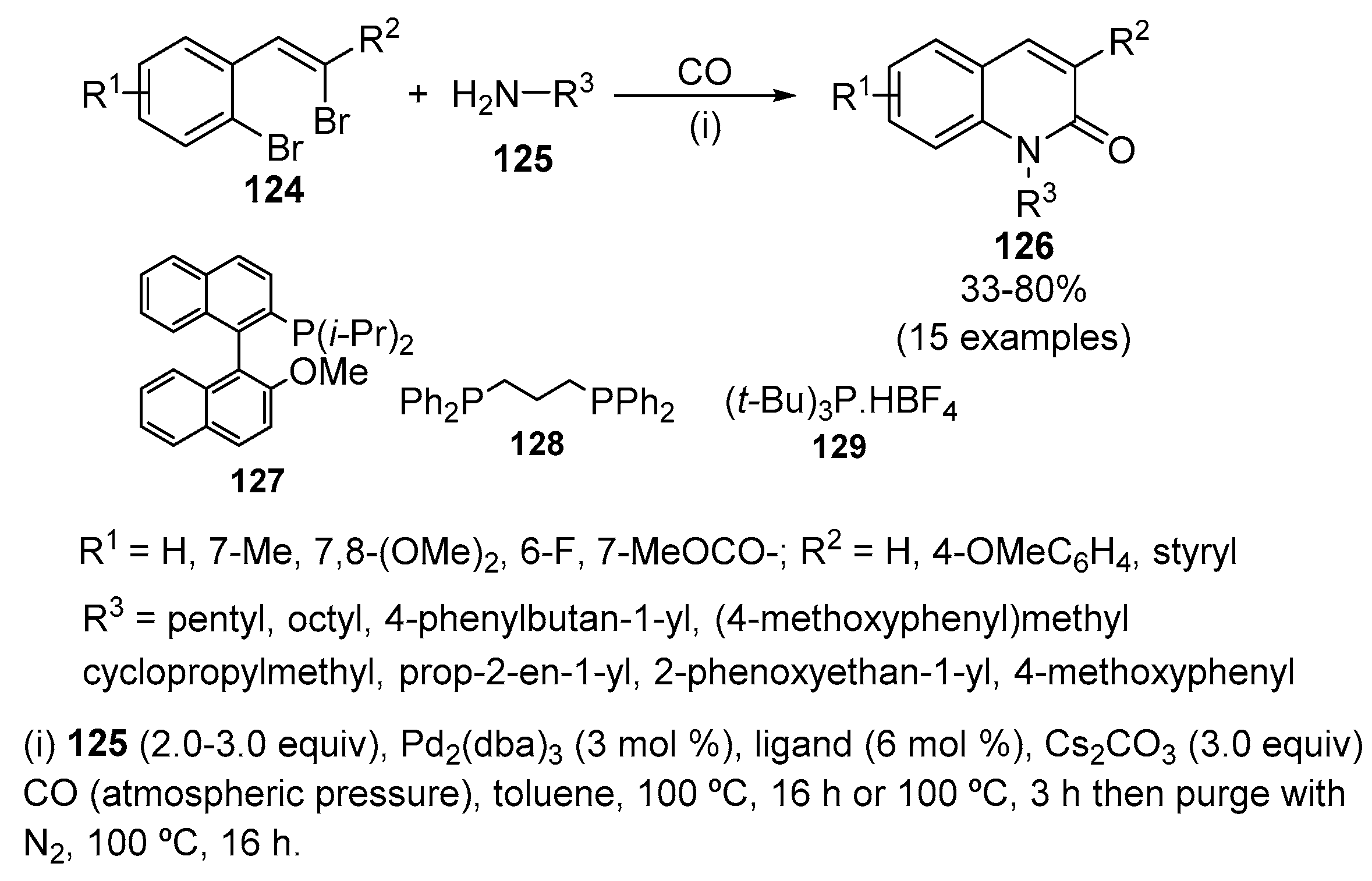{kind=link}
{kind=link}
{kind=link}
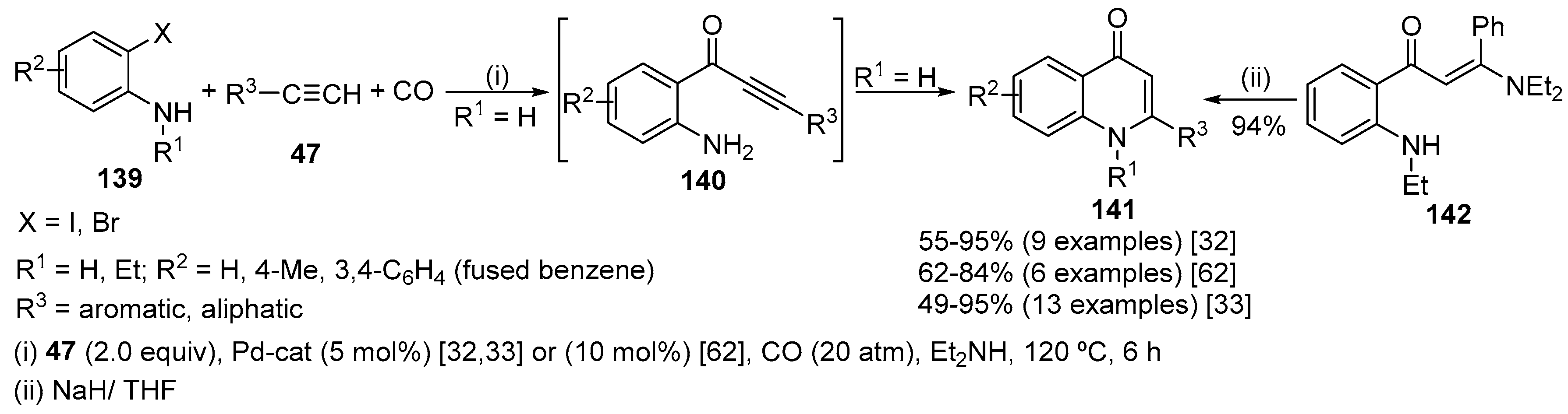{kind=link}
{kind=link}
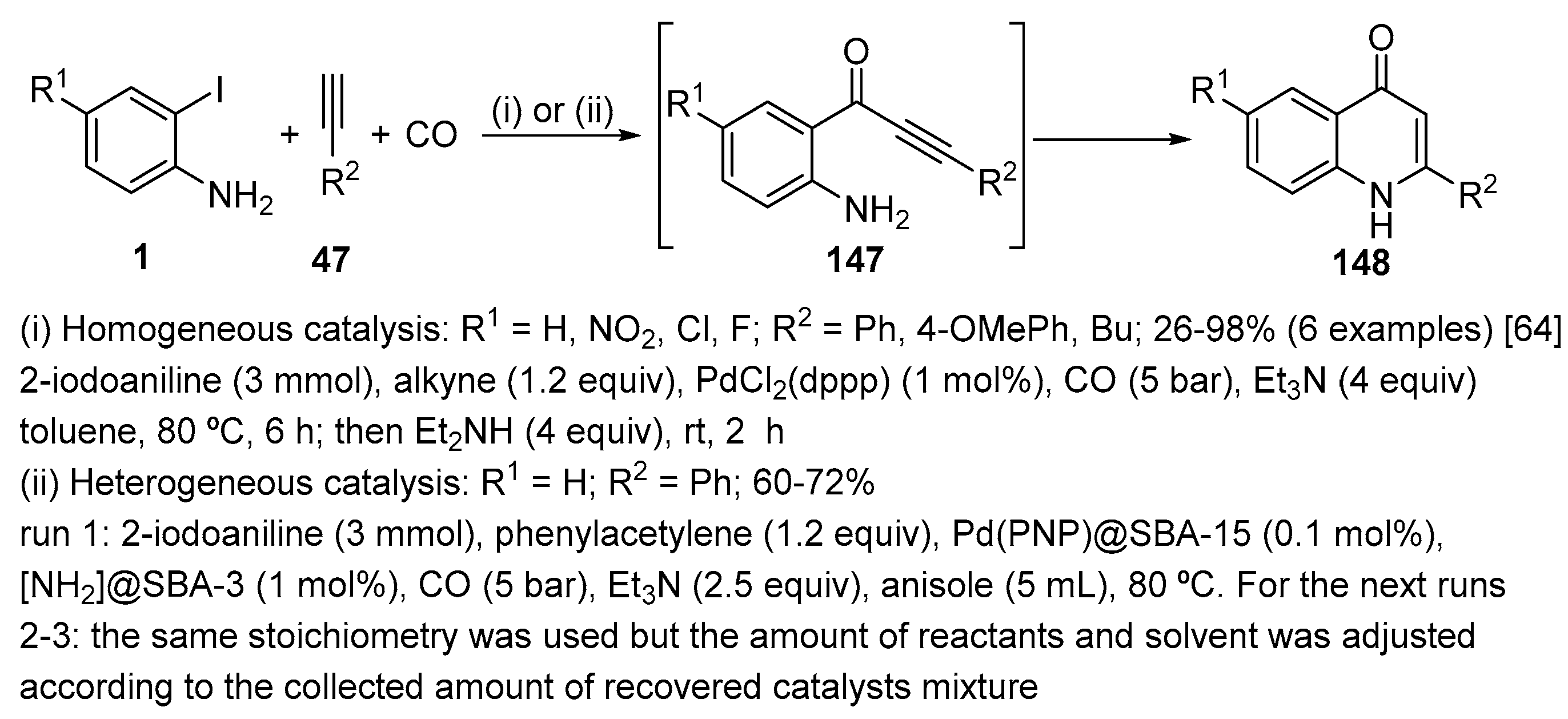{kind=link}
{kind=link}
{kind=link}
{kind=link}
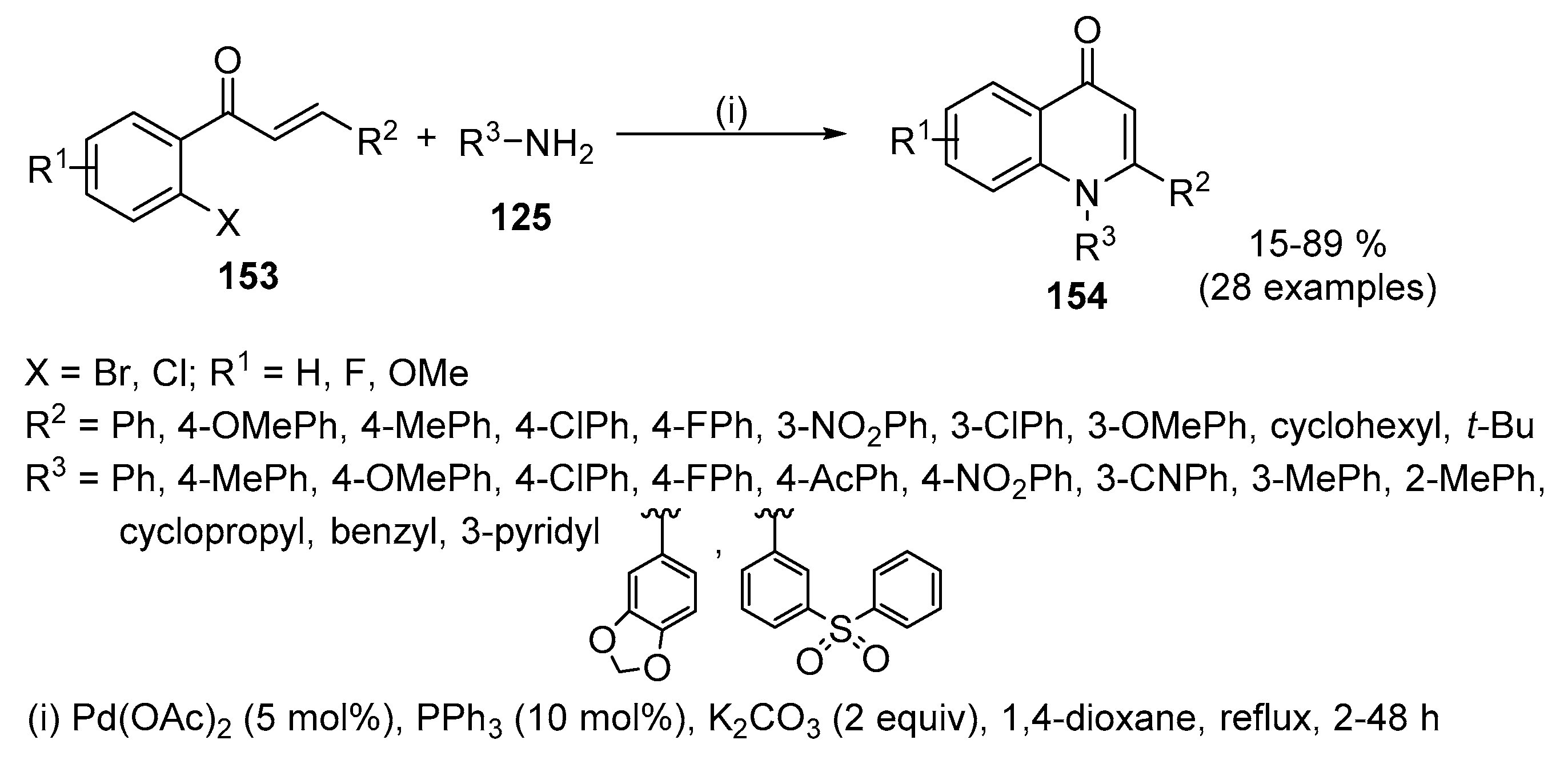{kind=link}
{kind=link}
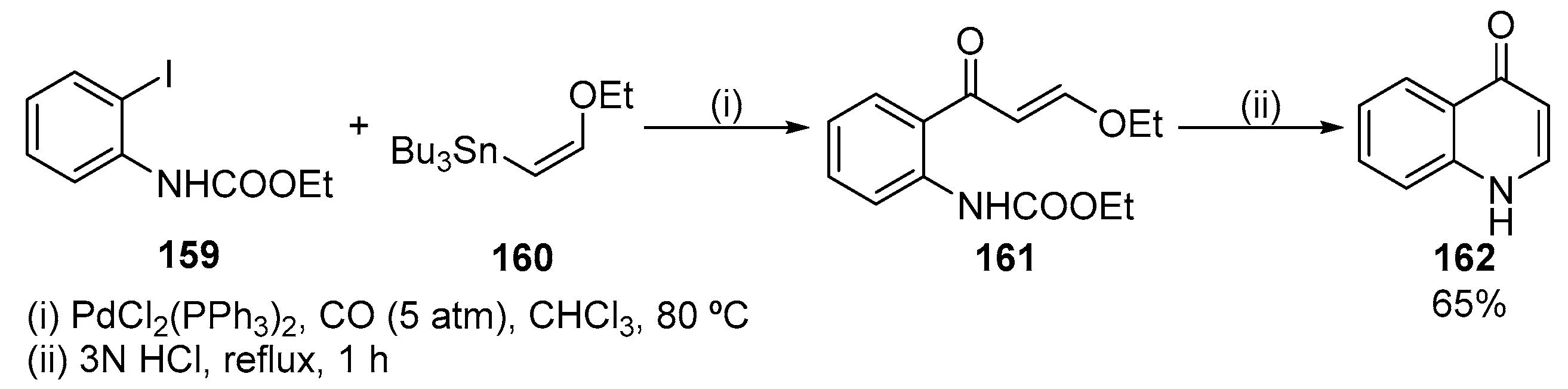{kind=link}
{kind=link}
{kind=link}
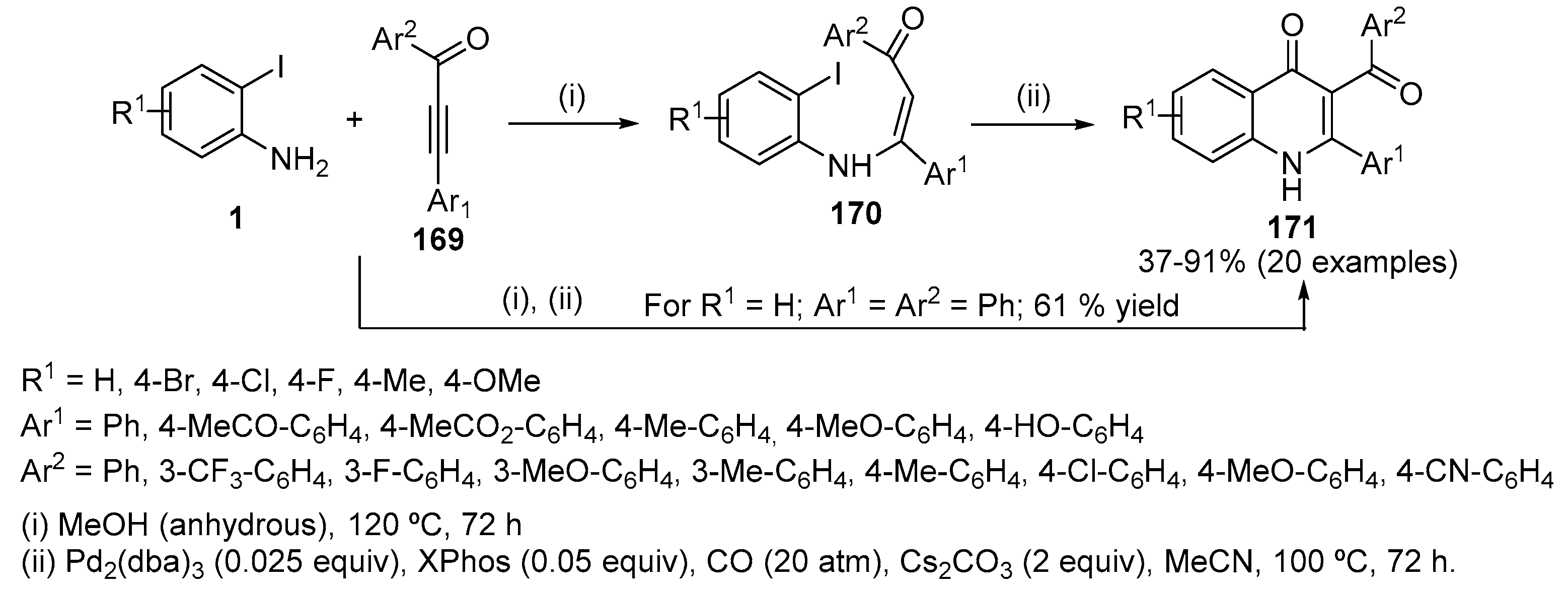{kind=link}
{kind=link}
{kind=link}
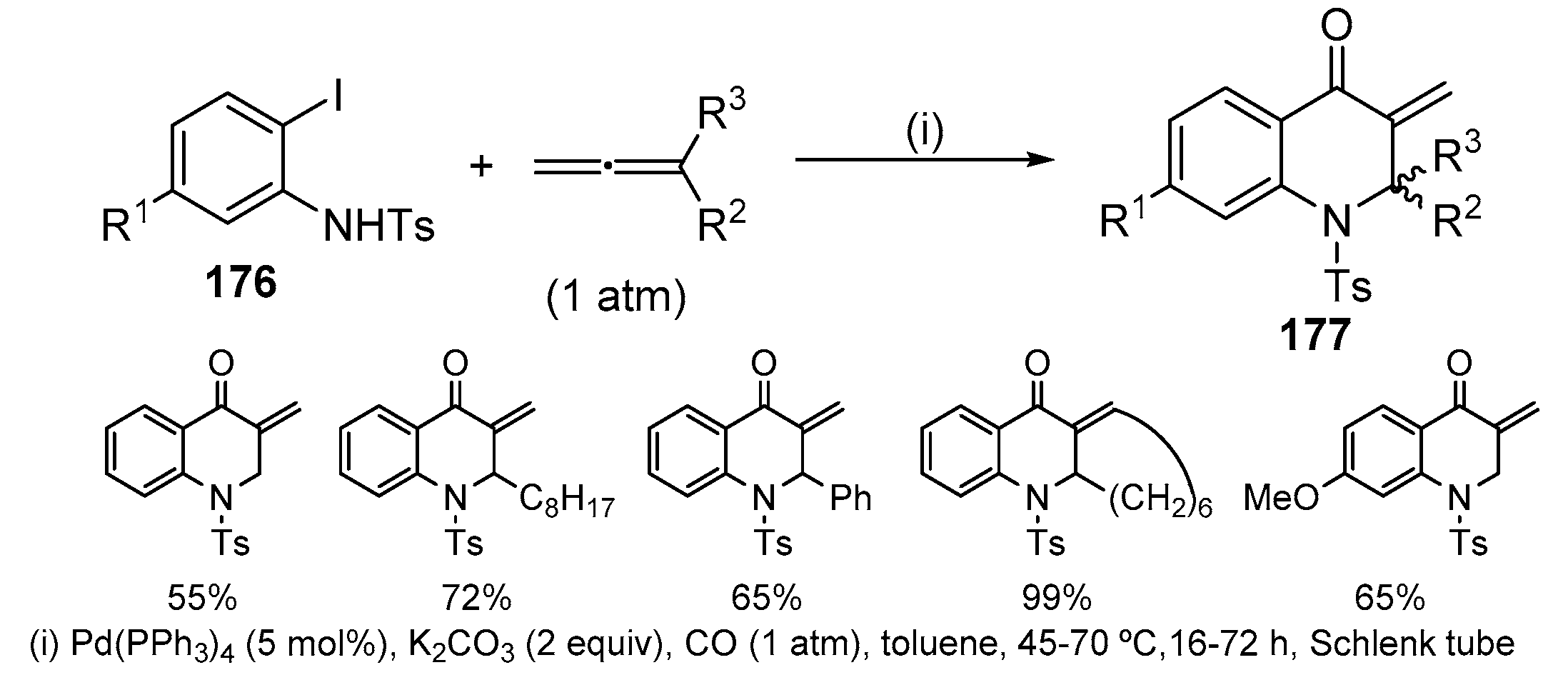{kind=link}
{kind=link}
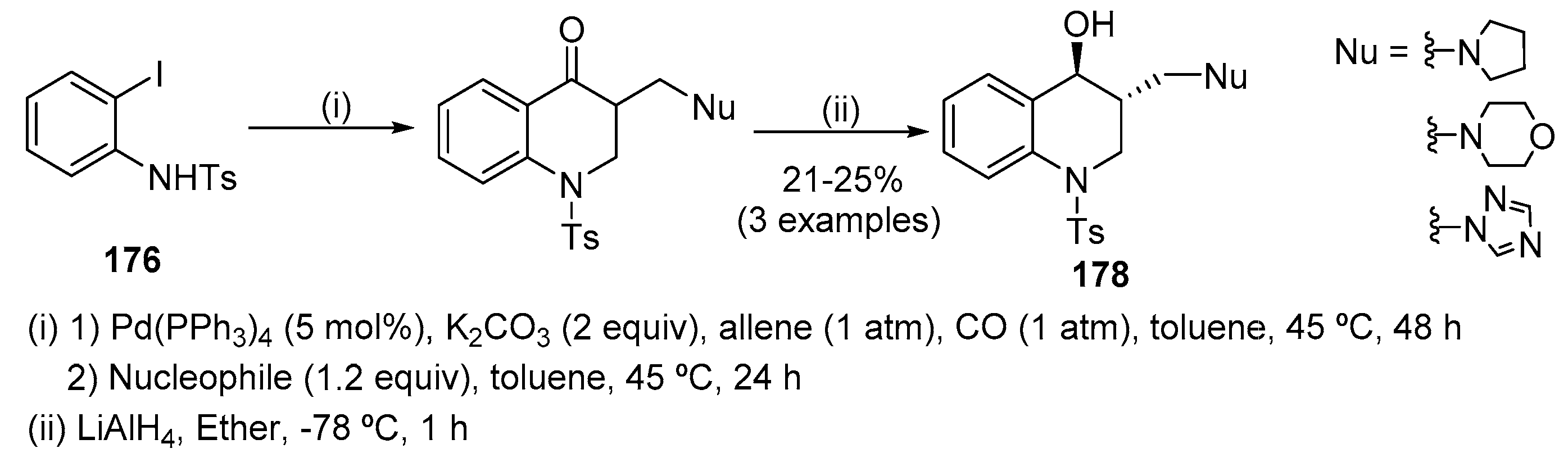{kind=link}
{kind=link}
{kind=link}
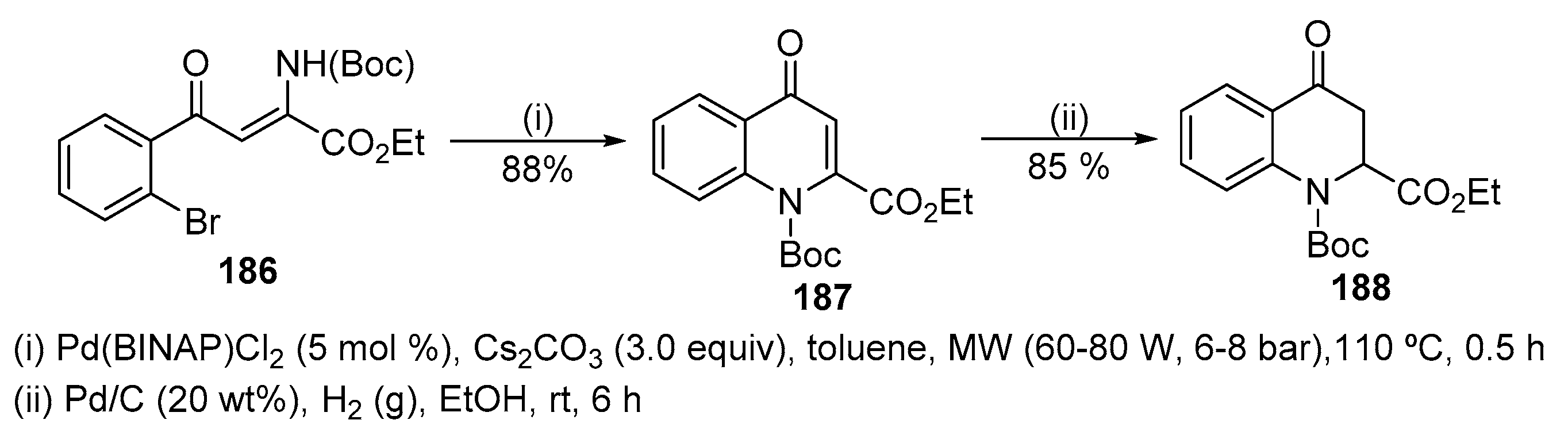{kind=link}
{kind=link}
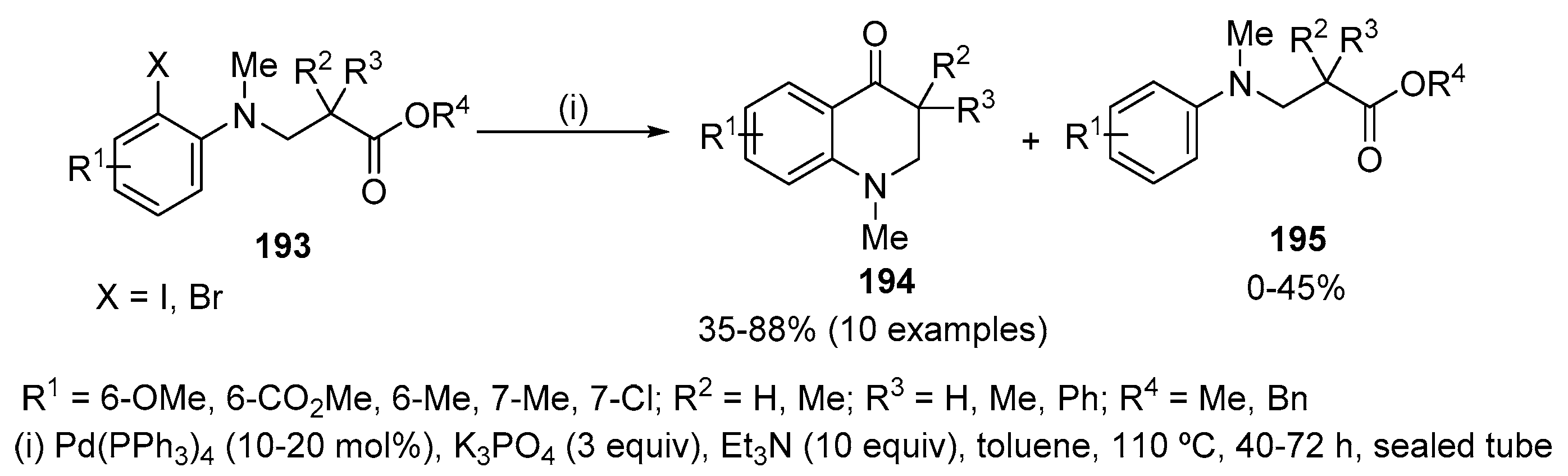{kind=link}
{kind=link}
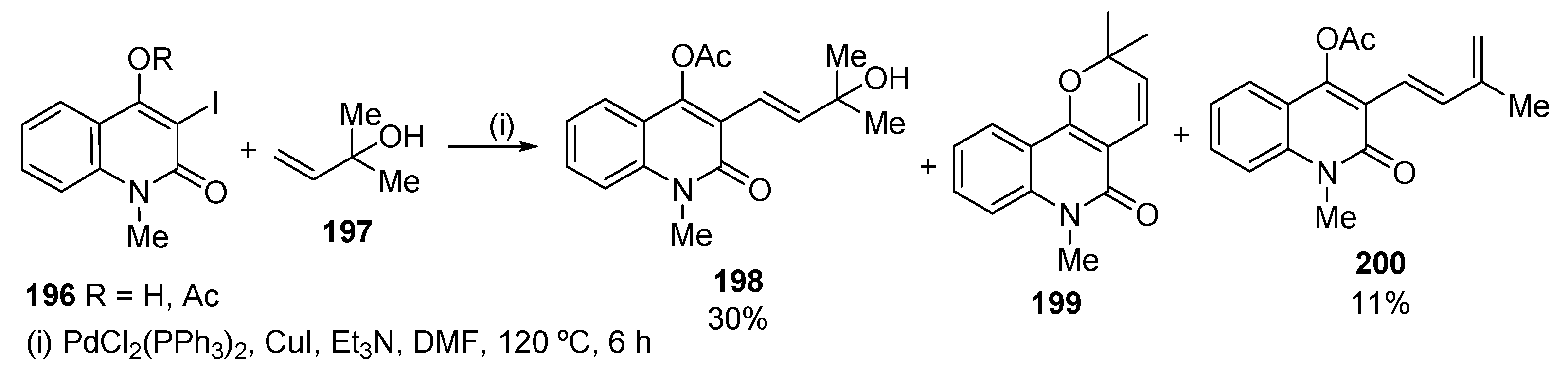{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
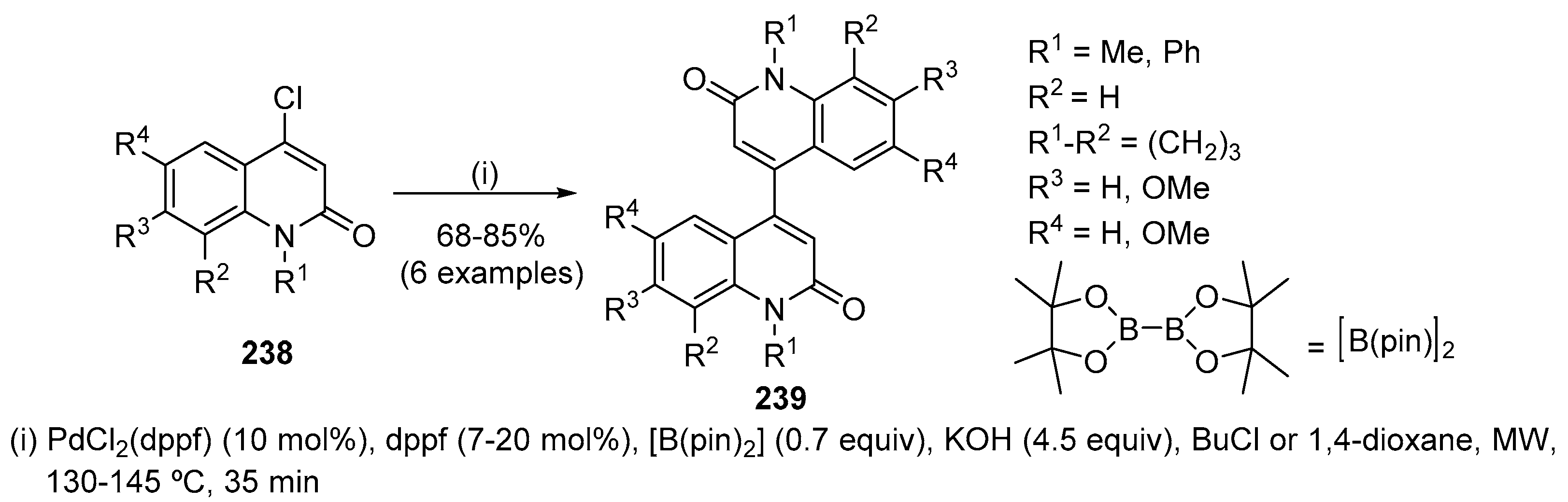{kind=link}
{kind=link}
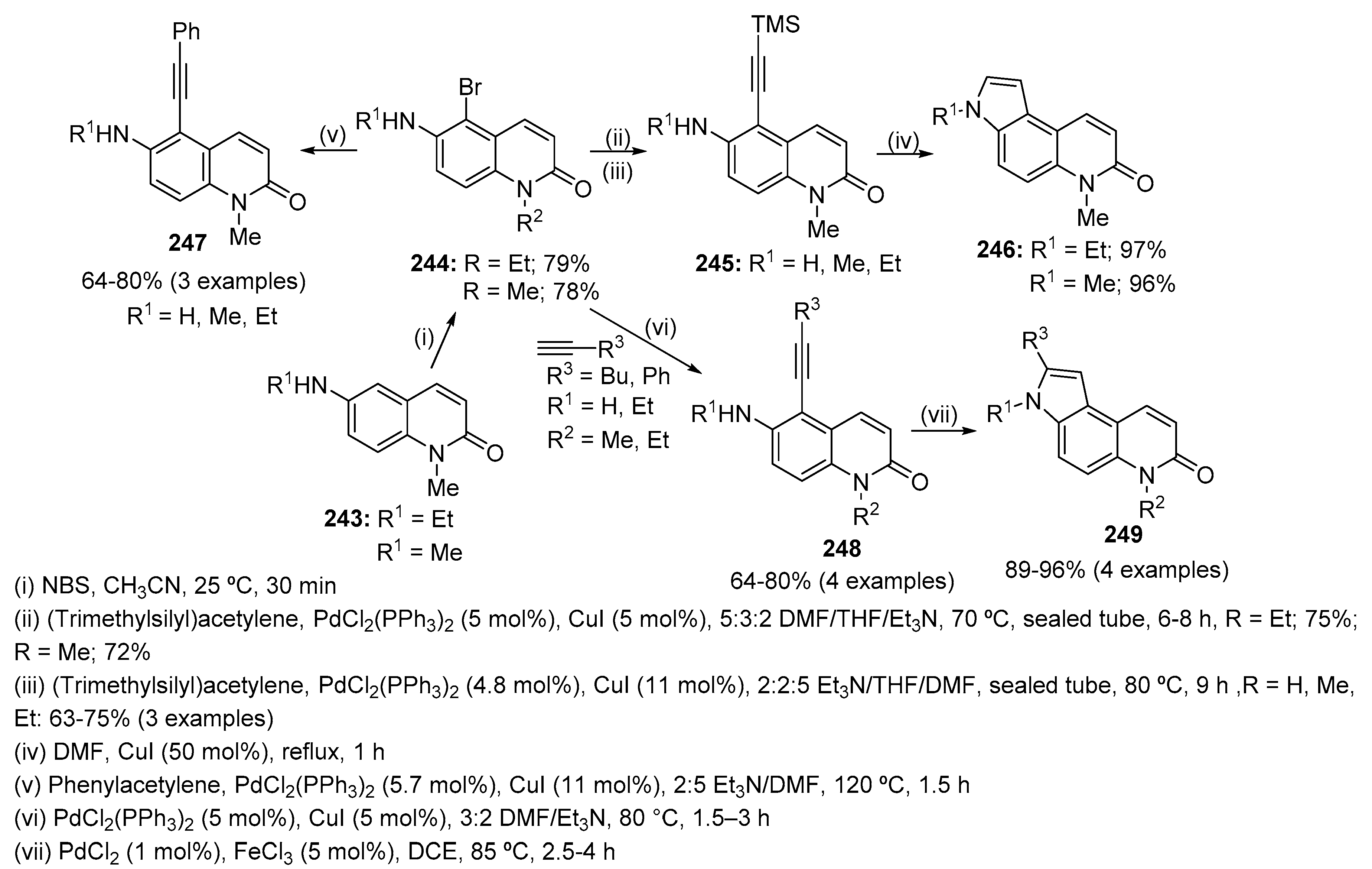{kind=link}
{kind=link}
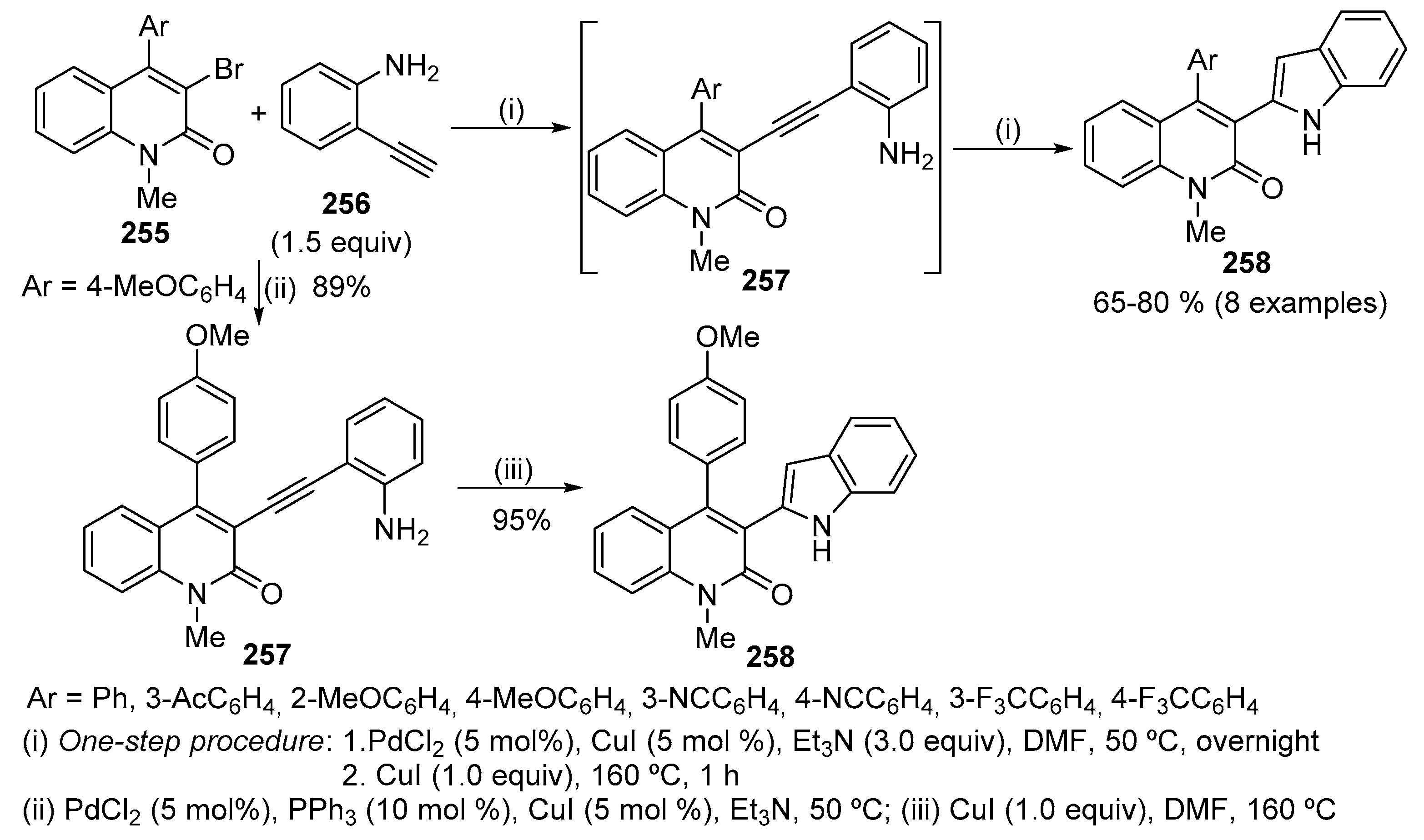{kind=link}
{kind=link}
{kind=link}
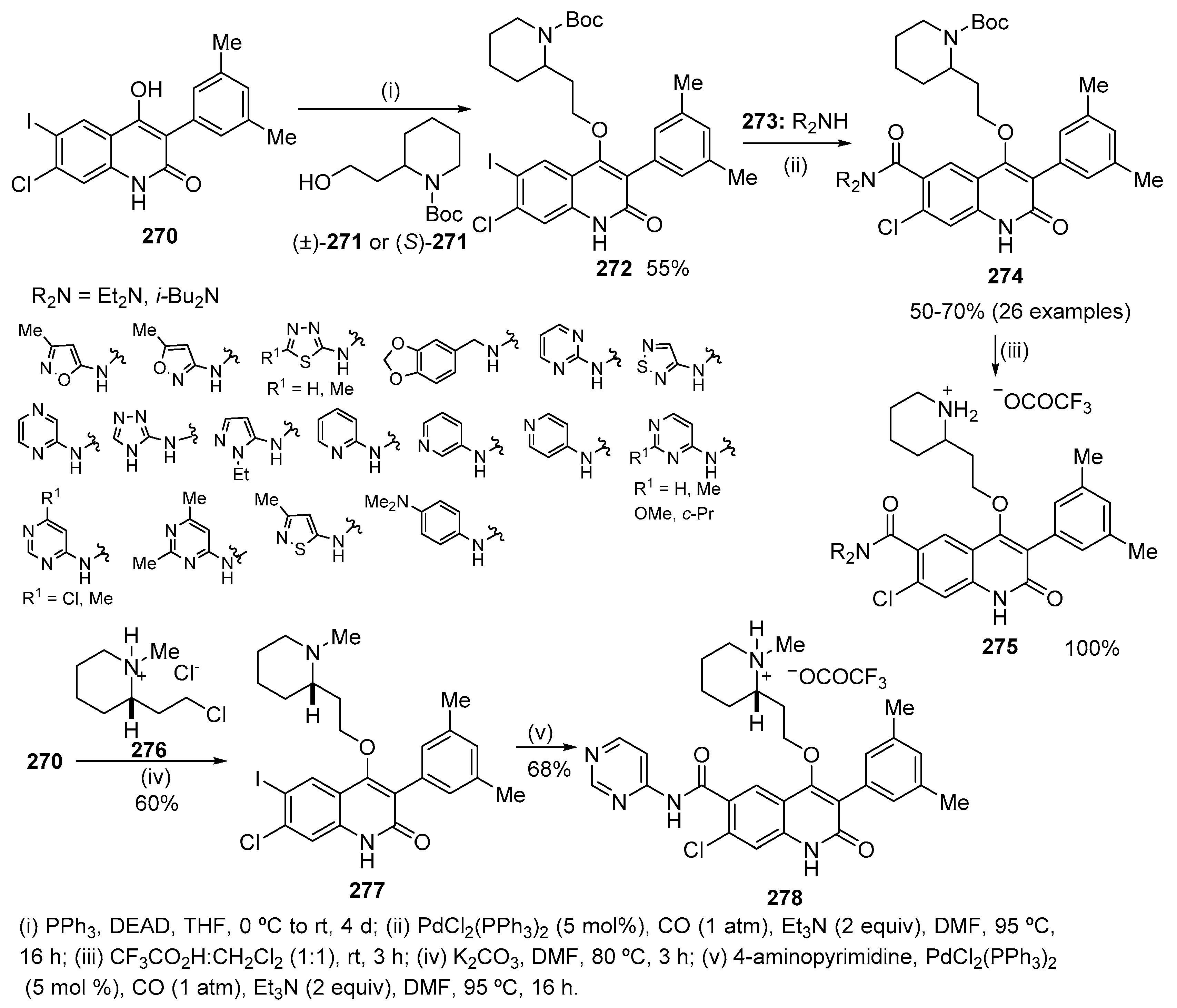{kind=link}
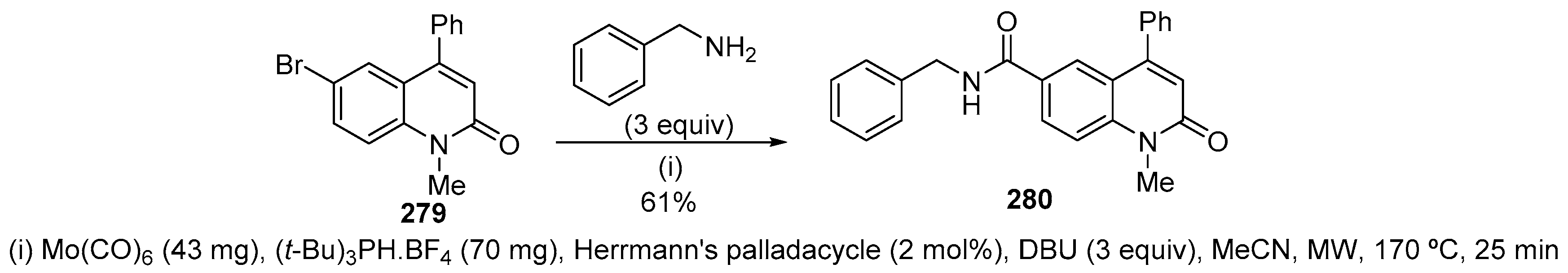{kind=link}
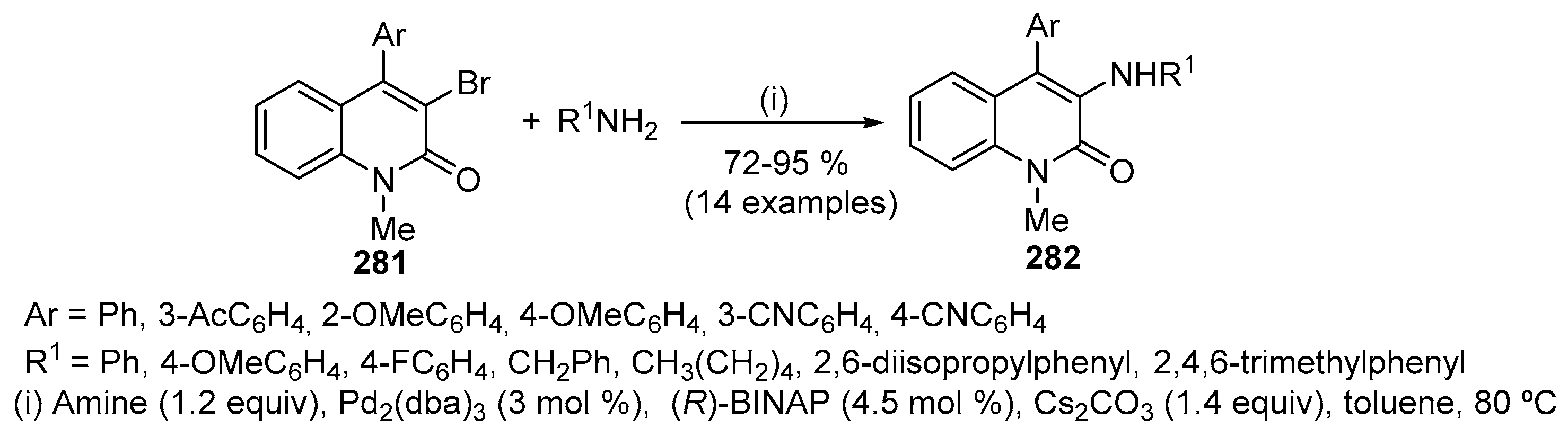{kind=link}
{kind=link}
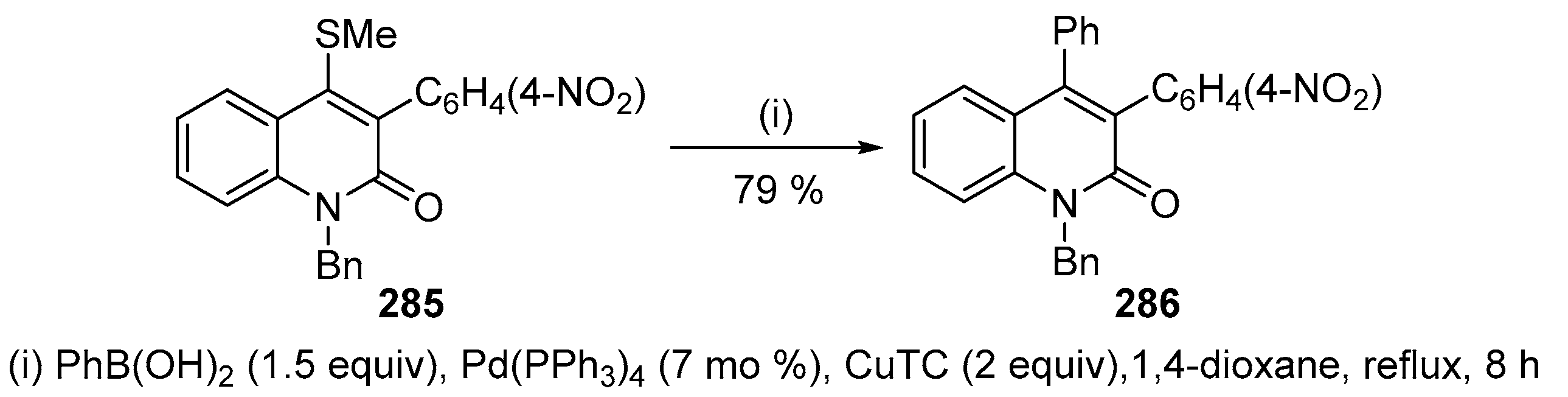{kind=link}
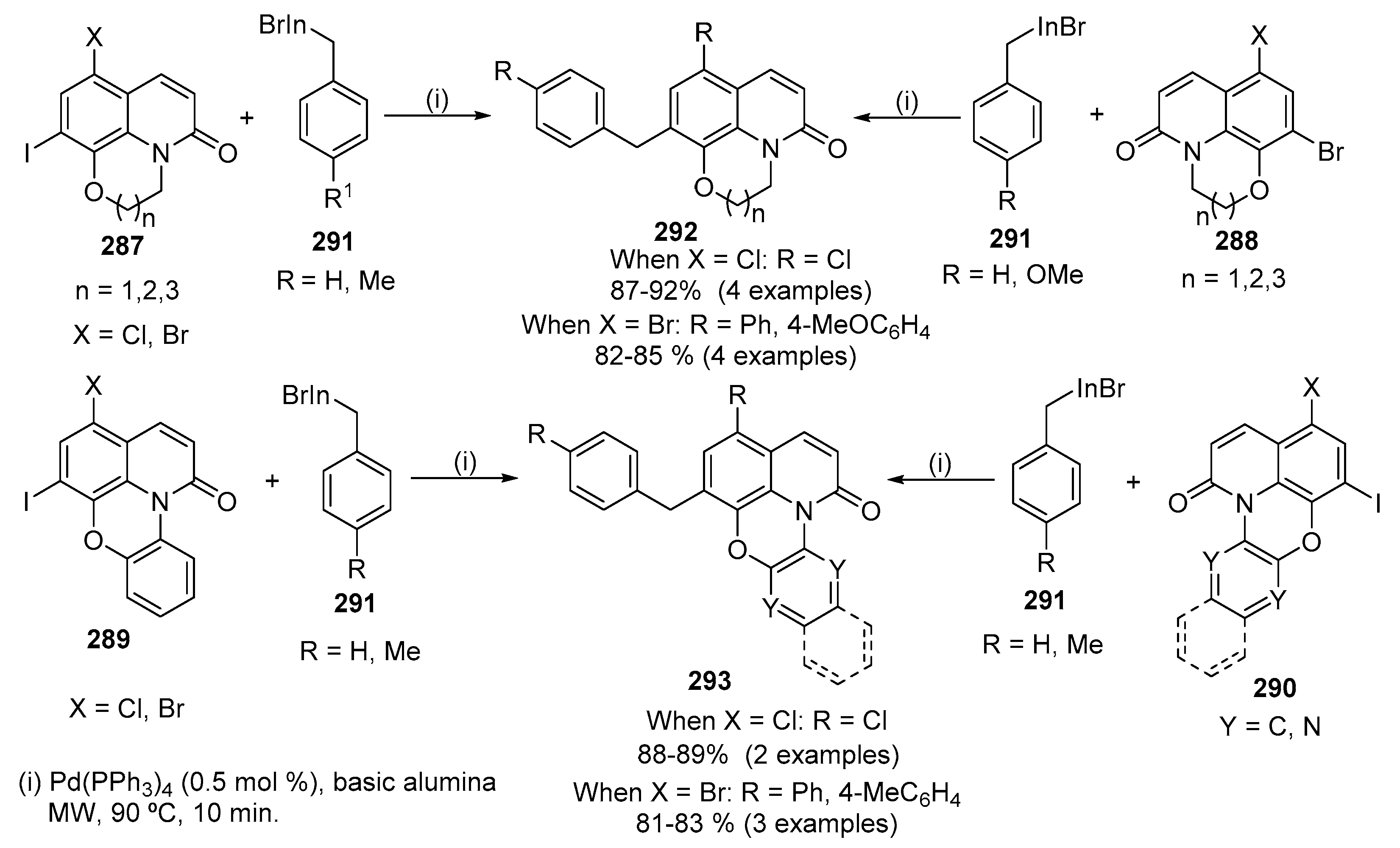{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
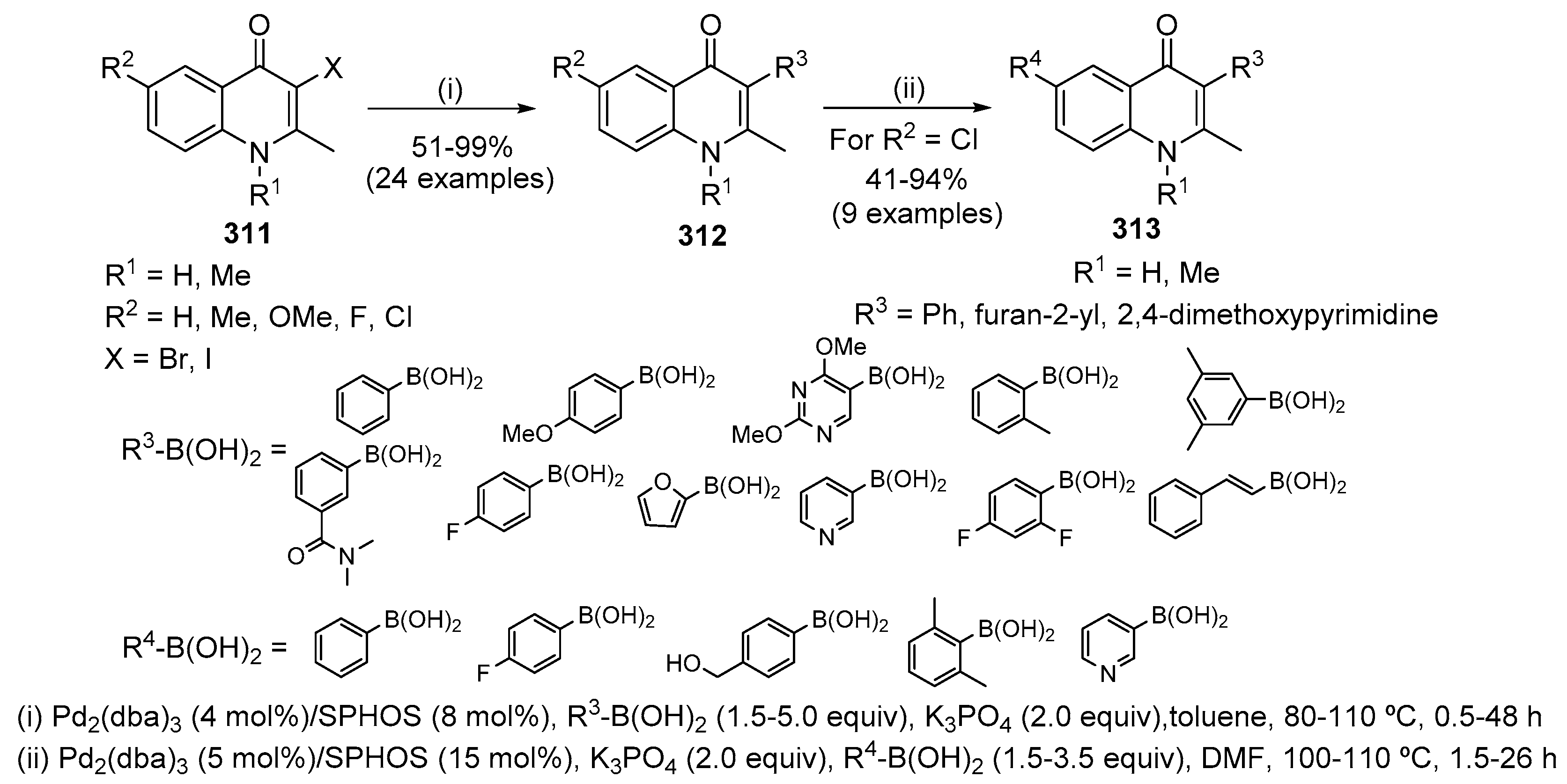{kind=link}
{kind=link}
{kind=link}
{kind=link}
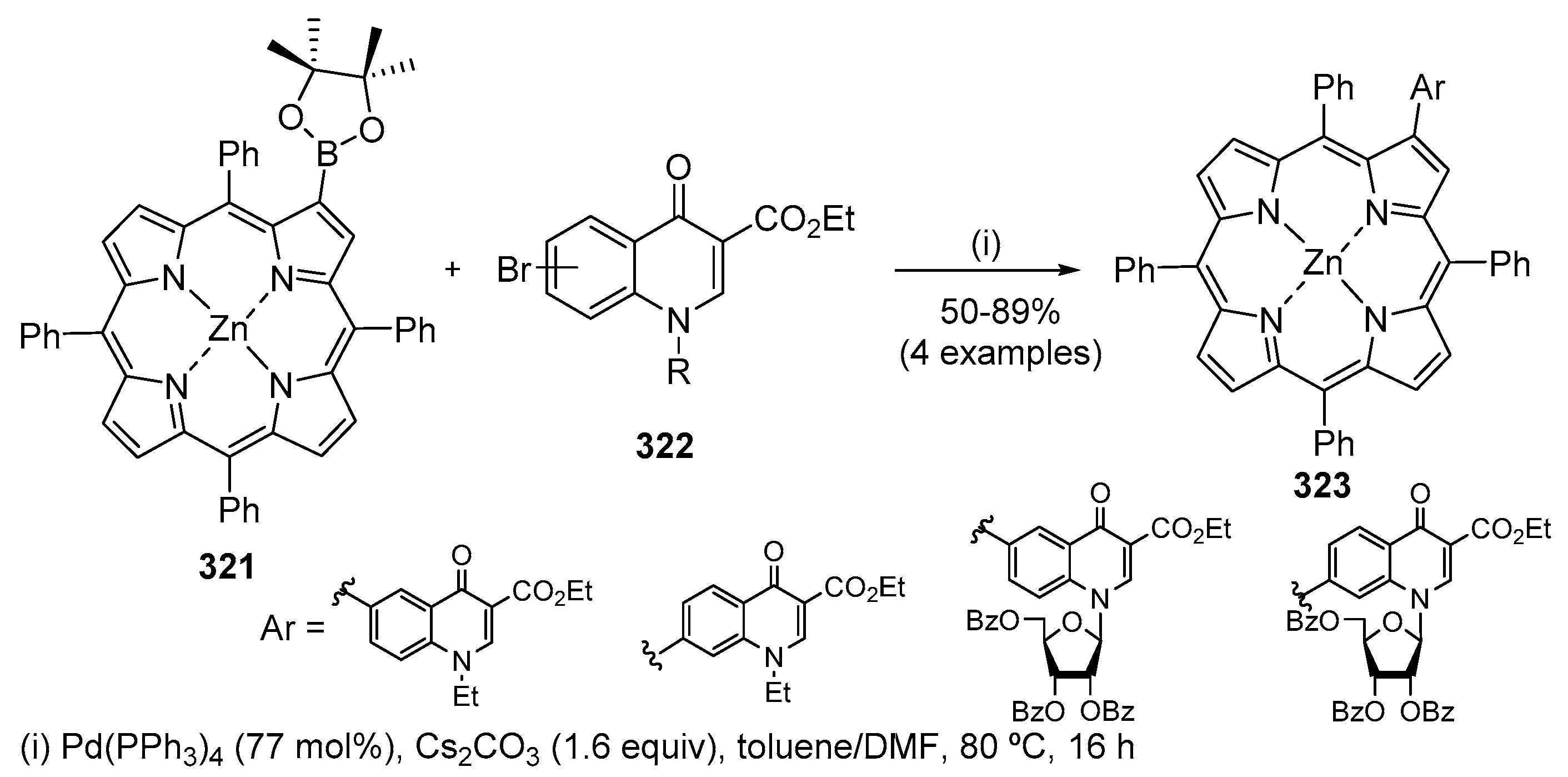{kind=link}
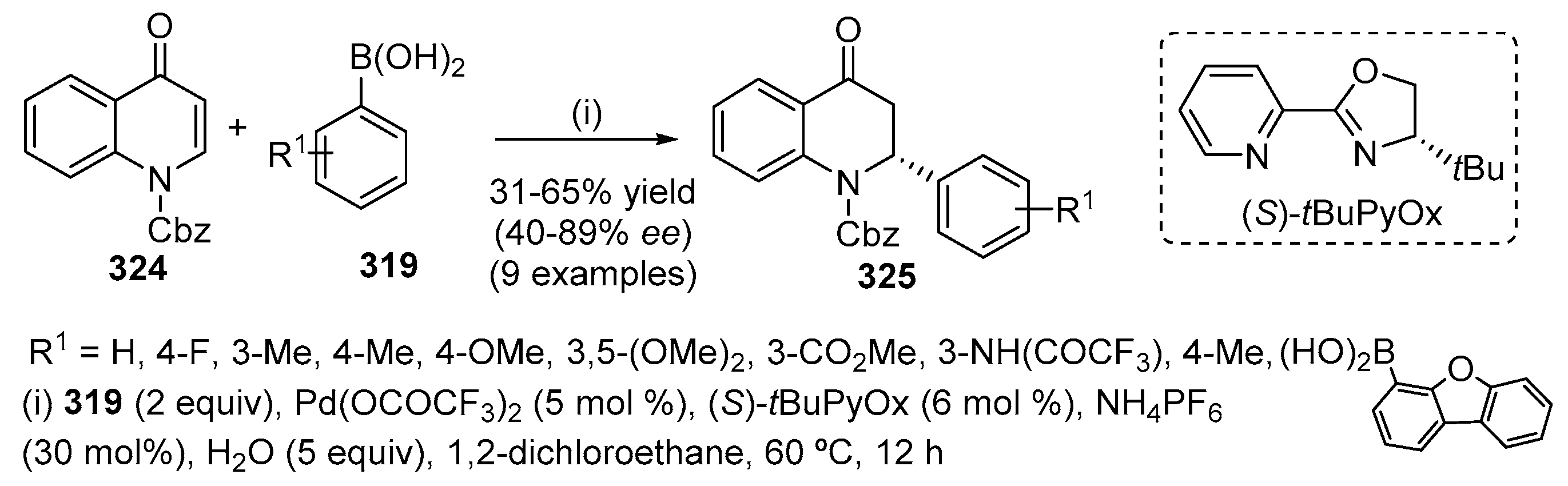{kind=link}
{kind=link}
{kind=link}
{kind=link}
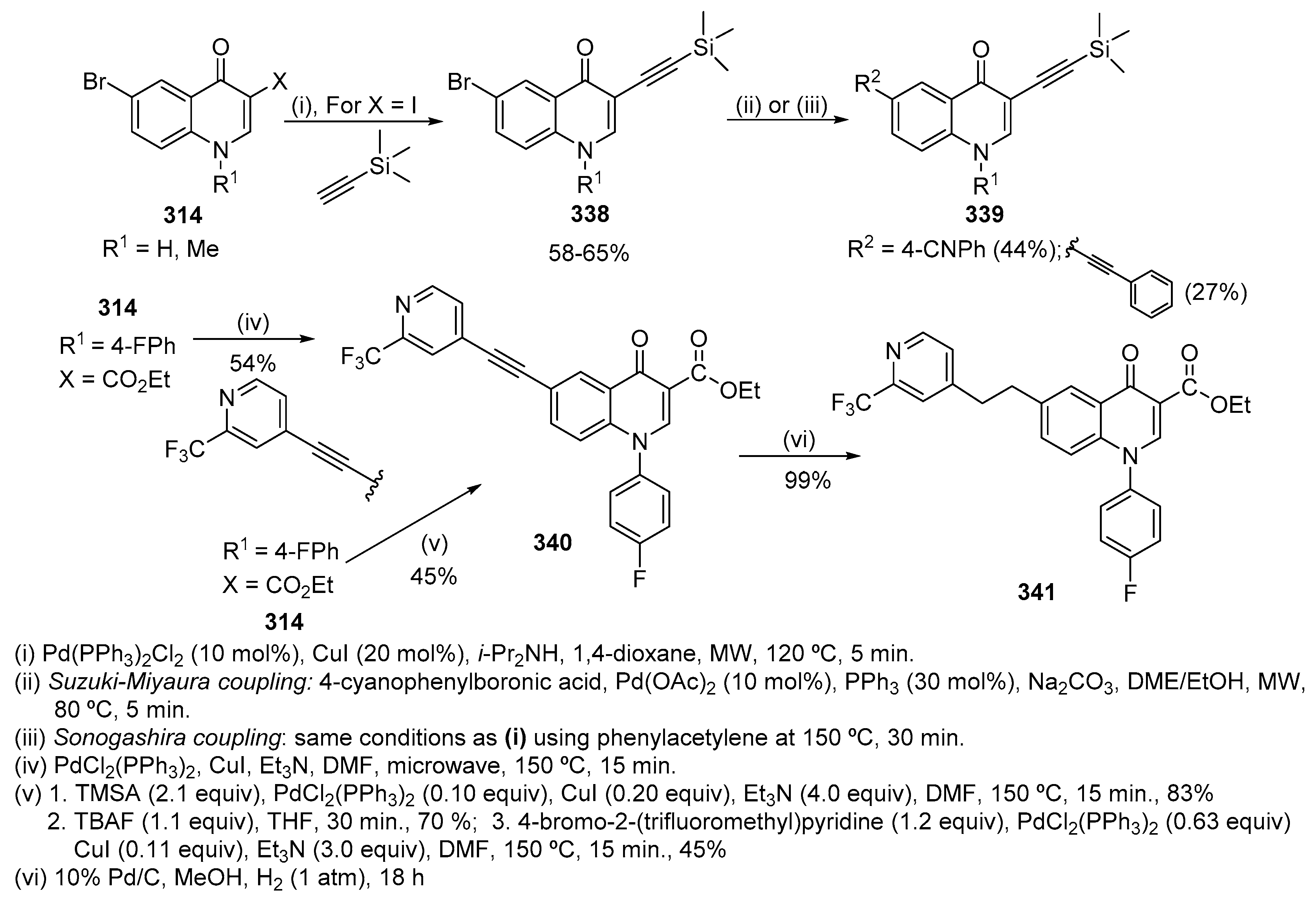{kind=link}
{kind=link}
{kind=link}
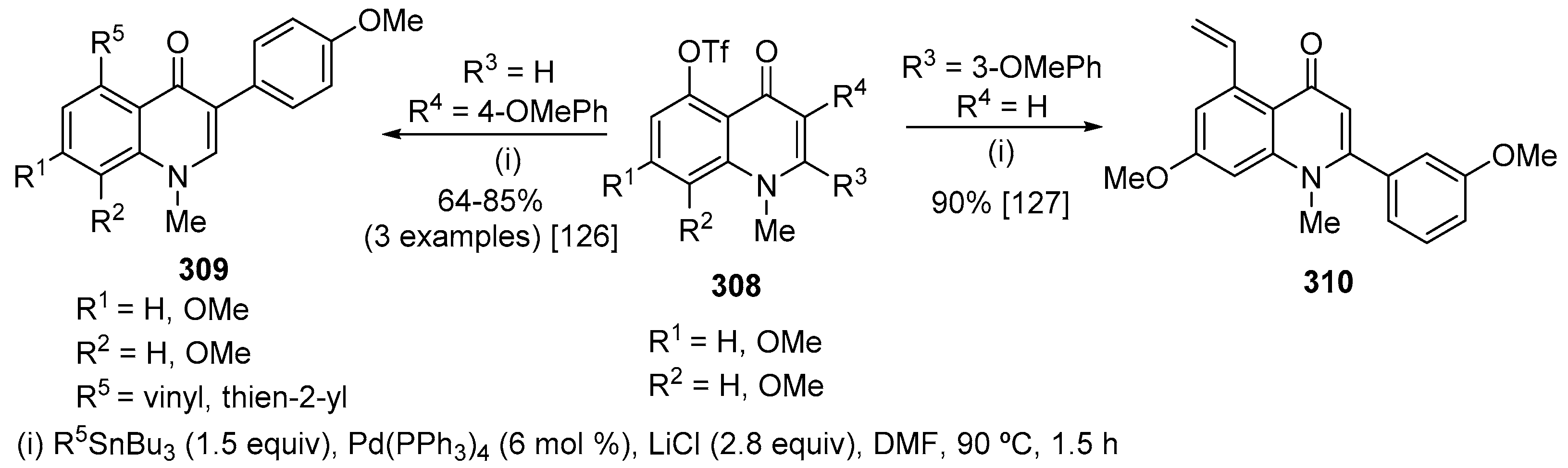{kind=link}
{kind=link}
{kind=link}
{kind=link}
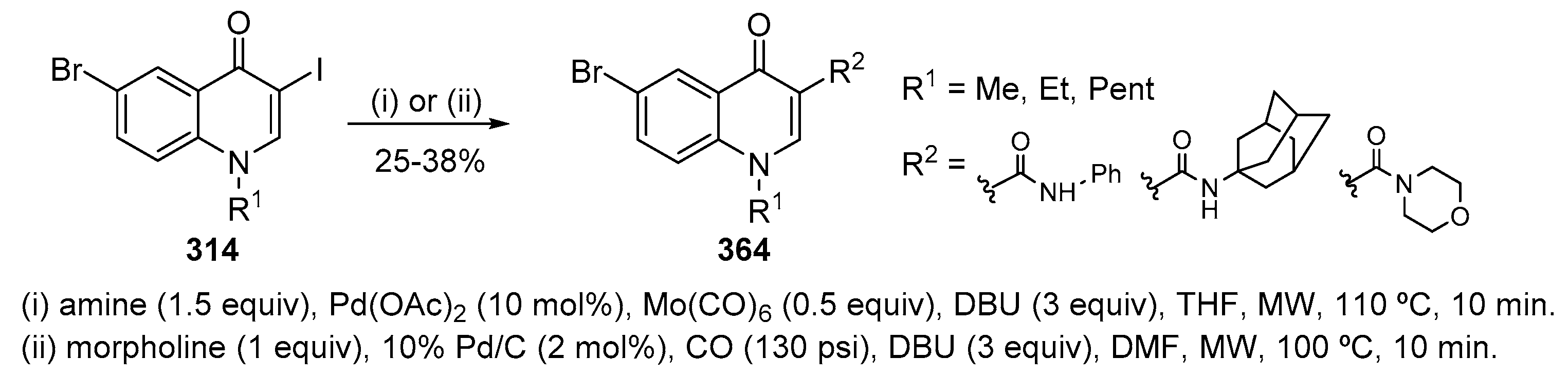{kind=link}
{kind=link}
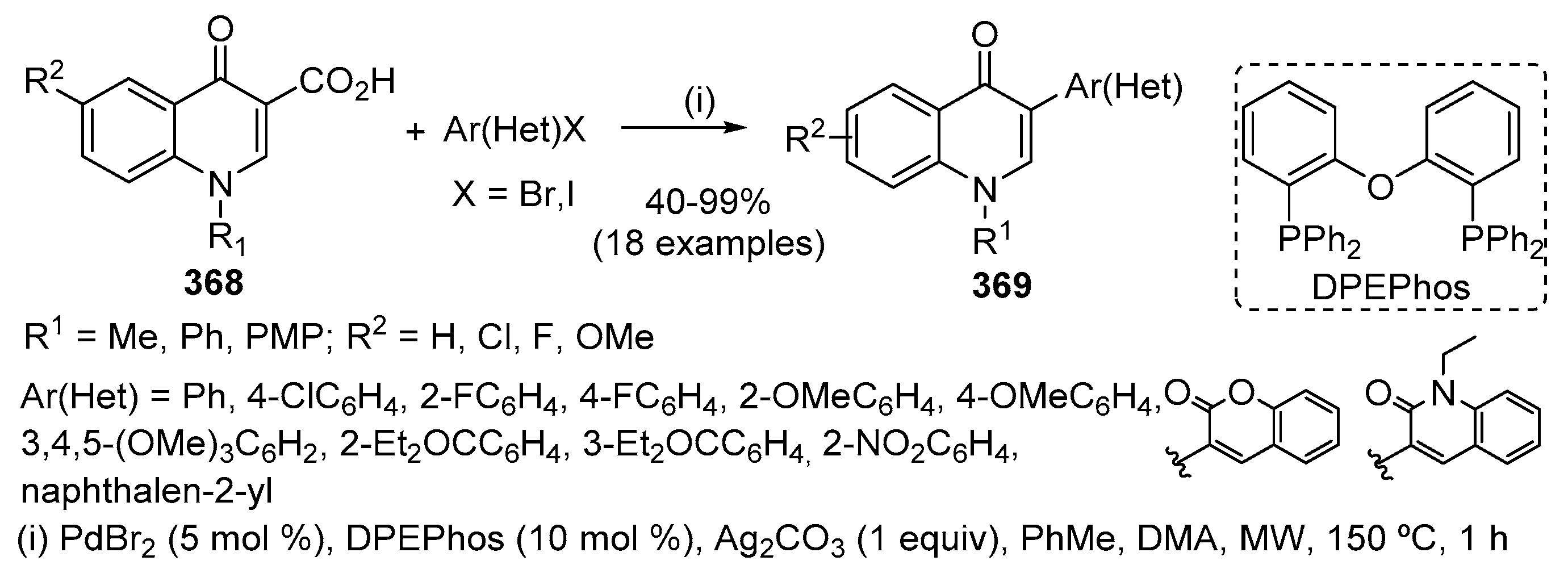{kind=link}
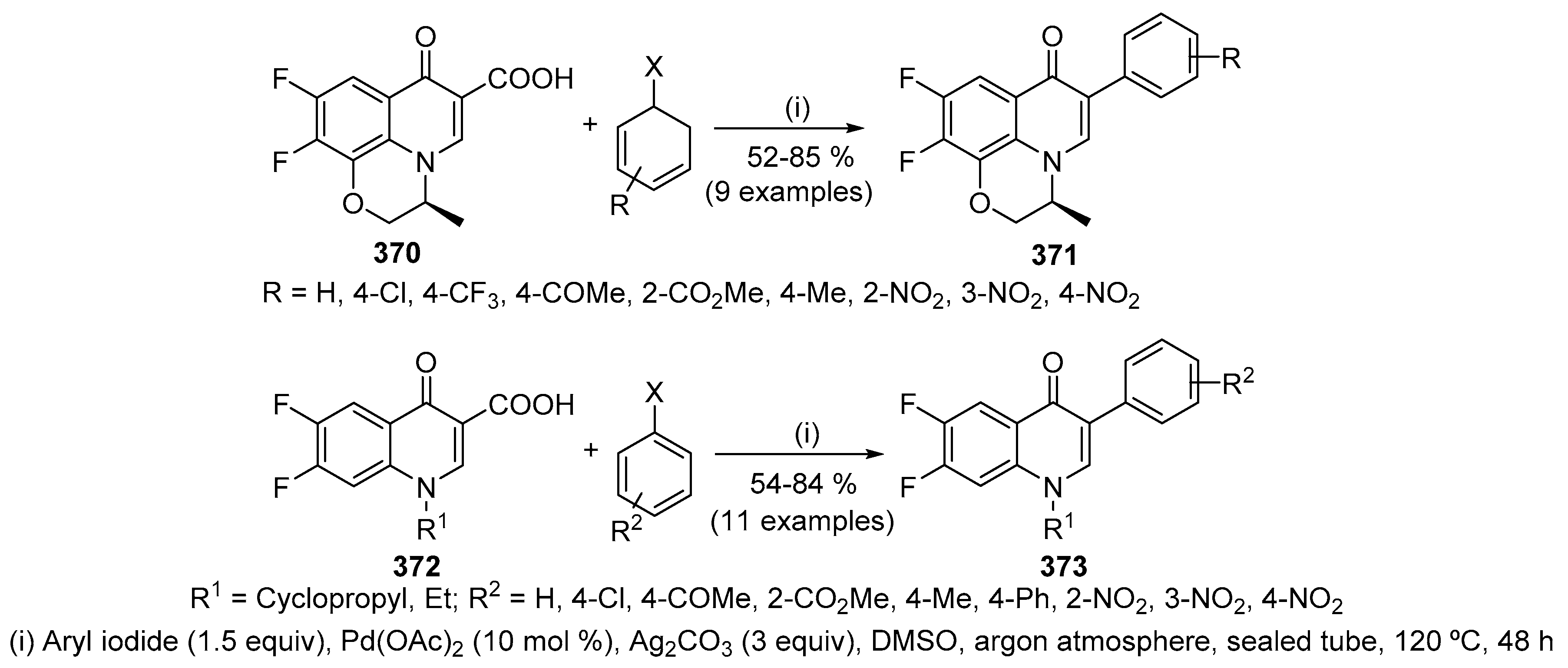{kind=link}
{kind=link}
{kind=link}
{kind=link}
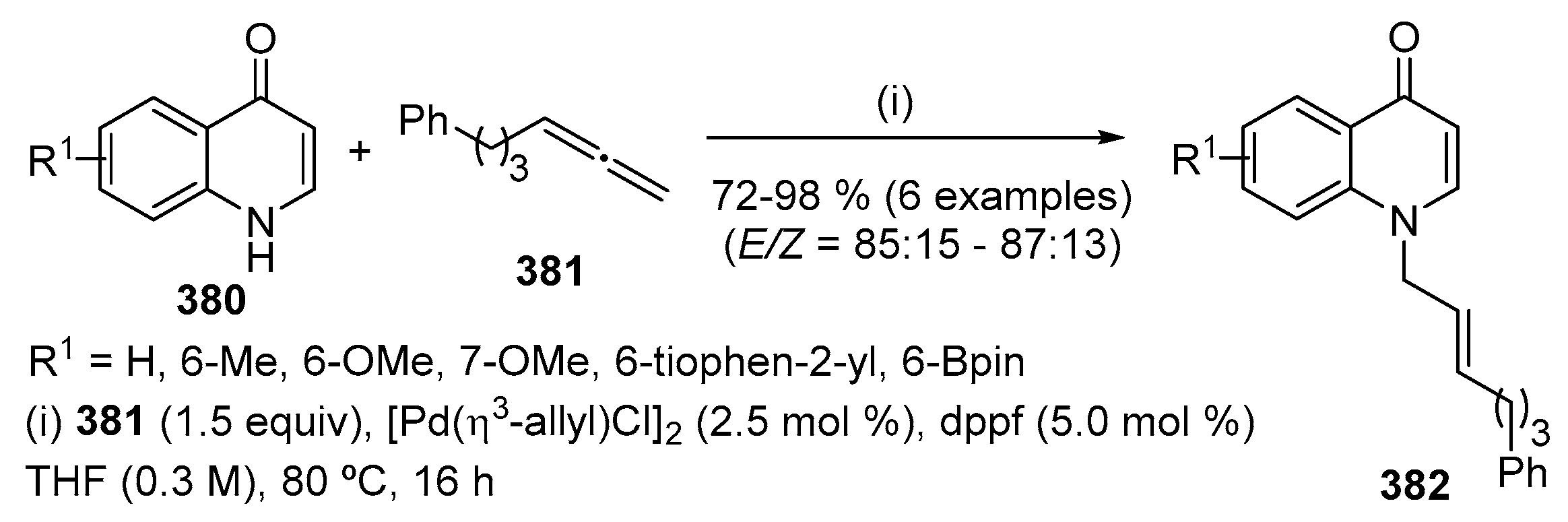{kind=link}
{kind=link}
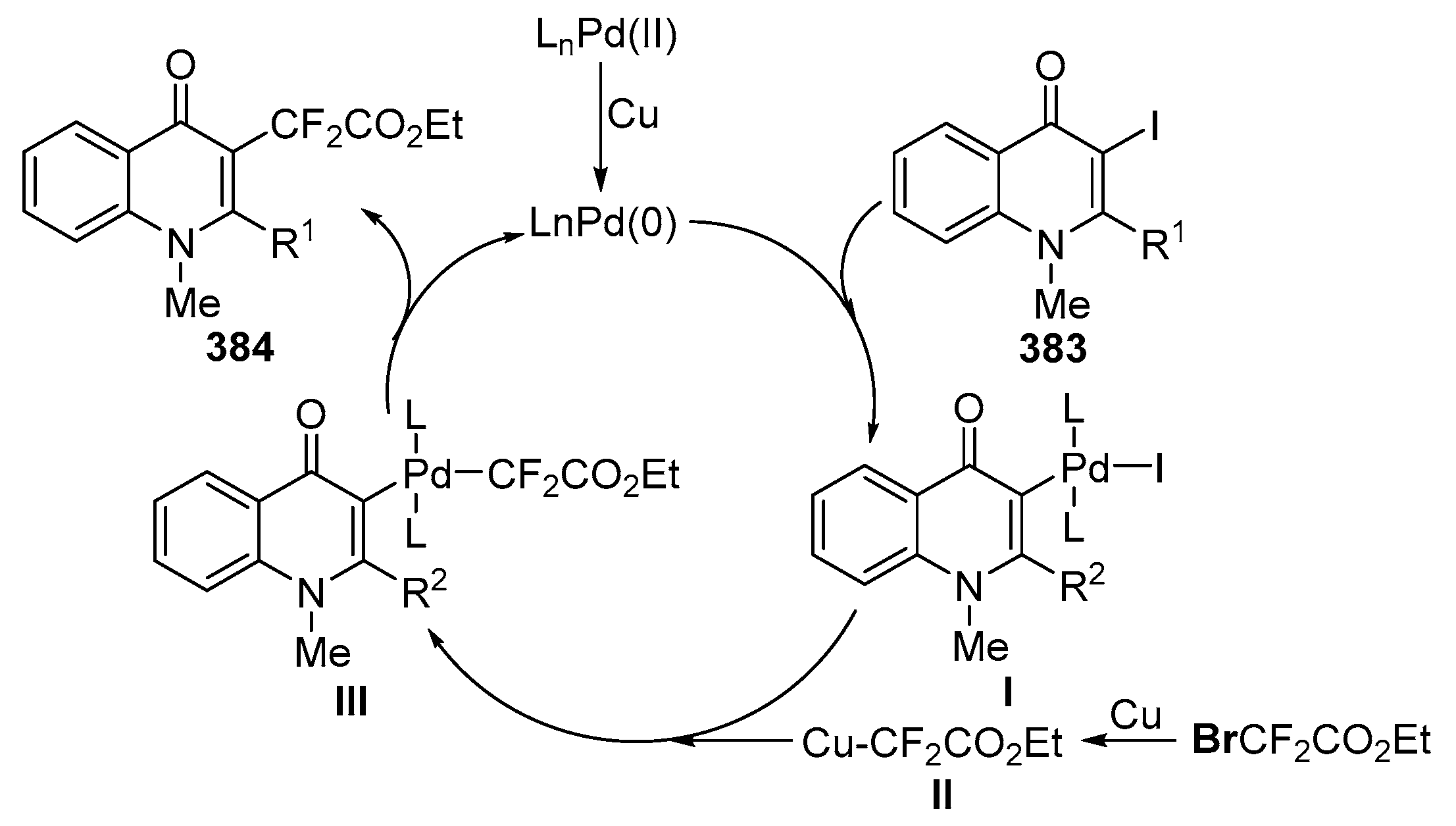{kind=link}
Scheme 69.
Synthesis of 4-acetylquinolin-2(1H)-one 206 via Heck reaction of 4-tosylquinolin-2(1H)-one 204 with butyl vinyl ether followed by acidic treatment [90].
Scheme 69.
Synthesis of 4-acetylquinolin-2(1H)-one 206 via Heck reaction of 4-tosylquinolin-2(1H)-one 204 with butyl vinyl ether followed by acidic treatment [90].

Scheme 70.
Synthesis of benzoxocine-fused quinolin-2(1H)-one 208 by intramolecular Heck reaction of benzylallylquinolin-2(1H)-one 207 [91].
Scheme 70.
Synthesis of benzoxocine-fused quinolin-2(1H)-one 208 by intramolecular Heck reaction of benzylallylquinolin-2(1H)-one 207 [91].

Scheme 71.
Probable mechanistic pathway for the synthesis of quinolin-2(1H)-one annulated benzoazocines 210 by intramolecular Heck reaction of allylic quinolin-2(1H)-ones 209 [92].
Scheme 71.
Probable mechanistic pathway for the synthesis of quinolin-2(1H)-one annulated benzoazocines 210 by intramolecular Heck reaction of allylic quinolin-2(1H)-ones 209 [92].

Scheme 72.
Synthesis of quinolin-2(1H)-one annulated benzazocinones 212 and quinolin-2(1H)-one annulated benzazoninones 214 by intramolecular Heck reaction of allylic quinolin-2(1H)-ones 211 and 213 [93,94].

Scheme 73.
Synthesis of tetracyclic quinolones 216 and 218 by intramolecular Heck reaction of 3- and 4-(2-bromobenzyloxy)quinolin-2(1H)-ones 215 and 217 [97].
Scheme 73.
Synthesis of tetracyclic quinolones 216 and 218 by intramolecular Heck reaction of 3- and 4-(2-bromobenzyloxy)quinolin-2(1H)-ones 215 and 217 [97].

Scheme 74.
Synthesis of 12-methyl-6H-isochromeno[4,3-c]quinolin-11(12H)-one 220 by intramolecular direct arylation of quinolin-2(1H)-ones 219 [98,99].

Scheme 75.
Synthesis of pyrrolo[3,2-f]quinolin-7(6H)-ones 222 and 224 by intramolecular Heck reaction of 5-bromoquinolin-2(1H)-ones 221 and 223 [100].
Scheme 75.
Synthesis of pyrrolo[3,2-f]quinolin-7(6H)-ones 222 and 224 by intramolecular Heck reaction of 5-bromoquinolin-2(1H)-ones 221 and 223 [100].

Scheme 76.
Proposed mechanism for the formation of pyrrolo[3,2-f]quinolin-7(6H)-ones 227 from intramolecular Heck reaction of enamines prepared in situ [101].
Scheme 76.
Proposed mechanism for the formation of pyrrolo[3,2-f]quinolin-7(6H)-ones 227 from intramolecular Heck reaction of enamines prepared in situ [101].

Scheme 77.
Synthesis of indol-2-ylquinolin-2(1H)-one 230 by Suzuki-Miyaura cross-coupling reaction of quinolin-2(1H)-one 228 with boronic acid derivative 229 [102].
Scheme 77.
Synthesis of indol-2-ylquinolin-2(1H)-one 230 by Suzuki-Miyaura cross-coupling reaction of quinolin-2(1H)-one 228 with boronic acid derivative 229 [102].

Scheme 78.
Synthesis of 4- and 3,4-substituted quinolin-2(1H)-ones 233 and 234 via regioselective Suzuki-Miyaura reactions of 3-bromo-4-trifloxyquinolin-2(1H)-ones 232 and further transformation of quinolin-2(1H)-one 233 [104,105].

Scheme 79.
Synthesis of 4-arylquinolin-2(1H)-ones 237 via Suzuki-Miyaura cross-coupling reaction of quinolin-2(1H)-ones 236 with arylboronic acids [88,89].

Scheme 80.
Synthesis of 4,4′-bisquinolones 239 by one-pot borylation-Suzuki cross-coupling of 4-chloroquinolin-2(1H)-ones 238 [106].
Scheme 80.
Synthesis of 4,4′-bisquinolones 239 by one-pot borylation-Suzuki cross-coupling of 4-chloroquinolin-2(1H)-ones 238 [106].

Scheme 81.
Synthesis of quinolin-2(1H)-one 242 by Suzuki-Miyaura cross-coupling of dinitroquinolin-2(1H)-one 240 with 4-methylphenylboronic acid 241 [107].
Scheme 81.
Synthesis of quinolin-2(1H)-one 242 by Suzuki-Miyaura cross-coupling of dinitroquinolin-2(1H)-one 240 with 4-methylphenylboronic acid 241 [107].

Scheme 82.
Sonogashira coupling of 6-amino-5-bromoquinolin-2(1H)-ones 244 with different acetylenes and cyclization of the resulting compounds into pyrrolo[3,2-f]quinolin-7(6H)-ones 246 and 249 [108,111,112].

Scheme 83.
Synthesis of 3H-pyrrolo[2,3-c]quinolin-4(5H)-ones 254 from 1-methyl-3-nitro-4-trifloxyquinolin-2(1H)-one 250 by Pd-catalysed Sonogashira cross-coupling reaction and cyclization [113].
Scheme 83.
Synthesis of 3H-pyrrolo[2,3-c]quinolin-4(5H)-ones 254 from 1-methyl-3-nitro-4-trifloxyquinolin-2(1H)-one 250 by Pd-catalysed Sonogashira cross-coupling reaction and cyclization [113].

Scheme 84.
Synthesis of 1H-indol-2-yl-(4-aryl)quinolin-2(1H)-ones 258 via Sonogashira coupling of quinolin-2(1H)-ones 255 with 2-ethynylaniline 256 and CuI-mediated cyclization of 257 [105].
Scheme 84.
Synthesis of 1H-indol-2-yl-(4-aryl)quinolin-2(1H)-ones 258 via Sonogashira coupling of quinolin-2(1H)-ones 255 with 2-ethynylaniline 256 and CuI-mediated cyclization of 257 [105].

Scheme 85.
Synthesis of 3H-pyrrolo[2,3-c]quinolin-4(5H)-ones 261 via Pd-catalysed domino reaction of 4-alkynyl-3-bromoquinolin-2(1H)-ones 260 [114].
Scheme 85.
Synthesis of 3H-pyrrolo[2,3-c]quinolin-4(5H)-ones 261 via Pd-catalysed domino reaction of 4-alkynyl-3-bromoquinolin-2(1H)-ones 260 [114].

Scheme 86.
Synthesis of 3-(N-substituted)-aminoquinolin-2(1H)-ones 264–267 and 269 by Pd-catalysed C–N coupling reaction of 3-bromoquinolin-2(1H)-ones 263 [115,116].

Scheme 87.
Synthesis of quinolin-2(1H)-one-6-carboxamides 275 and 278 by Pd-catalysed aminocarbonylation of 4-hydroxyquinolin-2(1H)-one 270 with diverse amines 273 and 4-aminopyrimidine [117].
Scheme 87.
Synthesis of quinolin-2(1H)-one-6-carboxamides 275 and 278 by Pd-catalysed aminocarbonylation of 4-hydroxyquinolin-2(1H)-one 270 with diverse amines 273 and 4-aminopyrimidine [117].

Scheme 88.
Synthesis of quinolin-2(1H)-one 280 via aminocarbonylation of 6-bromo-4-phenylquinolin-2(1H)-one 279 with benzylamine [88].
Scheme 88.
Synthesis of quinolin-2(1H)-one 280 via aminocarbonylation of 6-bromo-4-phenylquinolin-2(1H)-one 279 with benzylamine [88].

Scheme 89.
Pd-catalysed reaction of 4-aryl-3-bromoquinolin-2(1H)-one 281 with amines [89].
Scheme 89.
Pd-catalysed reaction of 4-aryl-3-bromoquinolin-2(1H)-one 281 with amines [89].

Scheme 90.
Synthesis of 3-alkenyl-4-substituted-quinolin-2(1H)-ones 284 by Pd-catalysed intermolecular C-H alkenylation with acrylates or acrylamide [45].
Scheme 90.
Synthesis of 3-alkenyl-4-substituted-quinolin-2(1H)-ones 284 by Pd-catalysed intermolecular C-H alkenylation with acrylates or acrylamide [45].

Scheme 91.
Functionalization of alkylthioquinolin-2(1H)-one 285 by Pd-catalysed cross-coupling reaction with phenylboronic acid [56].
Scheme 91.
Functionalization of alkylthioquinolin-2(1H)-one 285 by Pd-catalysed cross-coupling reaction with phenylboronic acid [56].

Scheme 92.
Basic alumina-supported synthesis of quinolin-2(1H)-one-based diarylmethanes 292 and 293 via Pd-catalysed cross-coupling reactions with benzyl indium reagents 291 [118].
Scheme 92.
Basic alumina-supported synthesis of quinolin-2(1H)-one-based diarylmethanes 292 and 293 via Pd-catalysed cross-coupling reactions with benzyl indium reagents 291 [118].

Scheme 93.
Synthesis of (E)-3-substituted-quinolin-4(1H)-ones 296, 297 and 298 by Heck reaction of 3-iodoquinolin-4(1H)-ones 294 with styrenes 295 and butyl acrylate, respectively [119,122].

Scheme 94.
Synthesis of 2,3-diarylacridin-9(10H)-ones 301 and (E)-2-aryl-4-styrylfuro[3,2-c]quinolines 303 via Heck reaction of (E)-3-iodo-2-styrylquinolin-4(1H)-ones 299 with styrenes 295 [123,124].

Scheme 95.
Synthesis of macrolones 307 through Heck reaction of macrolide 304 with 6-iodoquinolin-4(1H)-ones 305 followed by Pd/C hydrogenation [125].
Scheme 95.
Synthesis of macrolones 307 through Heck reaction of macrolide 304 with 6-iodoquinolin-4(1H)-ones 305 followed by Pd/C hydrogenation [125].

Scheme 96.
Synthesis of quinolin-4(1H)-ones 309 and 310 by Suzuki-Miyaura coupling reaction of 5-trifluoromethanesulfonatequinolin-4(1H)-ones 308 with boronic acids [126,127].

Scheme 97.
Synthesis of 3-substituted-quinolin-4(1H)-ones 312 and their transformation into quinolin-4(1H)-ones 313 via Suzuki-Miyaura cross-coupling reactions with boronic acids [128].
Scheme 97.
Synthesis of 3-substituted-quinolin-4(1H)-ones 312 and their transformation into quinolin-4(1H)-ones 313 via Suzuki-Miyaura cross-coupling reactions with boronic acids [128].

Scheme 98.
Synthesis of trisubstituted quinolin-4(1H)-ones 316 by sequential Suzuki-Miyaura (i, ii) or one-pot double Suzuki-Miyaura (i, iii) reactions of 1-substituted-6-bromo-3-iodoquinolin-4(1H)-ones 314 with arylboronic acids [129].
Scheme 98.
Synthesis of trisubstituted quinolin-4(1H)-ones 316 by sequential Suzuki-Miyaura (i, ii) or one-pot double Suzuki-Miyaura (i, iii) reactions of 1-substituted-6-bromo-3-iodoquinolin-4(1H)-ones 314 with arylboronic acids [129].

Scheme 99.
Synthesis of 6-vinylquinolin-4(1H)-one 318 by Suzuki-Miyaura coupling of 6-bromoquinolin-4(1H)-one 317 with potassium vinyltrifluoroborate [130].
Scheme 99.
Synthesis of 6-vinylquinolin-4(1H)-one 318 by Suzuki-Miyaura coupling of 6-bromoquinolin-4(1H)-one 317 with potassium vinyltrifluoroborate [130].

Scheme 100.
Ohmic heating-assisted synthesis of 3-arylquinolin-4(1H)-ones 320 by Suzuki-Miyaura reaction of 1-substituted 3-iodoquinolin-4(1H)-ones 294 with arylboronic acids 319 [131].
Scheme 100.
Ohmic heating-assisted synthesis of 3-arylquinolin-4(1H)-ones 320 by Suzuki-Miyaura reaction of 1-substituted 3-iodoquinolin-4(1H)-ones 294 with arylboronic acids 319 [131].

Scheme 101.
Synthesis of porphyrin-quinolin-4(1H)-one conjugates 323 by Suzuki-Miyaura reaction of β-borylated porphyrin 321 with 6- or 7-bromoquinolin-4(1H)-ones 322 [132].
Scheme 101.
Synthesis of porphyrin-quinolin-4(1H)-one conjugates 323 by Suzuki-Miyaura reaction of β-borylated porphyrin 321 with 6- or 7-bromoquinolin-4(1H)-ones 322 [132].

Scheme 102.
Synthesis of 2-aryl-2,3-dihydroquinolin-4(1H)-ones 325 by Pd-catalysed asymmetric conjugate addition of arylboronic acids 319 to quinolin-4(1H)-ones 324 [133].
Scheme 102.
Synthesis of 2-aryl-2,3-dihydroquinolin-4(1H)-ones 325 by Pd-catalysed asymmetric conjugate addition of arylboronic acids 319 to quinolin-4(1H)-ones 324 [133].

Scheme 103.
Pd/C-PPh3 mediated synthesis of 2-substituted furo[3,2-c]quinolines 327 and 3-alkynyl quinolin-4(1H)-ones 328 [134].
Scheme 103.
Pd/C-PPh3 mediated synthesis of 2-substituted furo[3,2-c]quinolines 327 and 3-alkynyl quinolin-4(1H)-ones 328 [134].

Scheme 104.
Synthesis of macrolones 331 via Sonogashira reaction of macrolide 329 with 6-iodoquinolin-4(1H)-ones 305 followed by Pd/C hydrogenation [125].
Scheme 104.
Synthesis of macrolones 331 via Sonogashira reaction of macrolide 329 with 6-iodoquinolin-4(1H)-ones 305 followed by Pd/C hydrogenation [125].

Scheme 105.
Synthesis of macrolones 336 and 337 via Sonogashira reaction followed by Pd/C hydrogenation and hydrolysis of the corresponding esters 336 [125].
Scheme 105.
Synthesis of macrolones 336 and 337 via Sonogashira reaction followed by Pd/C hydrogenation and hydrolysis of the corresponding esters 336 [125].

Scheme 106.
Sonogashira reaction of 6-bromo-3-iodoquinolin-4(1H)-ones 314 and subsequent Suzuki and Sonogashira reactions at 6-position of quinolin-4(1H)-ones 338 [129,130].

Scheme 107.
Synthesis of 2-substituted 6-oxopyrrolo[3,2,1-ij]quinolines 343 by Pd/C-mediated coupling-cyclization of 8-iodo-4-oxo-1,4-dihydroquinoline-3-carboxylic acid ethyl ester 342 with terminal alkynes 47 [138].
Scheme 107.
Synthesis of 2-substituted 6-oxopyrrolo[3,2,1-ij]quinolines 343 by Pd/C-mediated coupling-cyclization of 8-iodo-4-oxo-1,4-dihydroquinoline-3-carboxylic acid ethyl ester 342 with terminal alkynes 47 [138].

Scheme 108.
Pd-catalysed Stille cross-coupling reaction of 7-quinolyltriflates 344 with functionalized stannanes 345 [139, 140, 141].

Scheme 109.
Synthesis of quinolin-4(1H)-ones 309 and 310 by Stille coupling reaction between 5-trifluoromethanesulfonatequinolin-4(1H)-ones 308 and tributyl(vinyl)stannanes [126,127].

Scheme 110.
Synthesis of 6-substituted-quinolin-4(1H)-one nucleosides by Pd-catalysed Buchwald-Hartwig cross-coupling reactions of 6-bromoquinolin-4(1H)-one nucleosides 347 and 348 with aniline and phenol [142].
Scheme 110.
Synthesis of 6-substituted-quinolin-4(1H)-one nucleosides by Pd-catalysed Buchwald-Hartwig cross-coupling reactions of 6-bromoquinolin-4(1H)-one nucleosides 347 and 348 with aniline and phenol [142].

Scheme 111.
Synthesis of porphyrin-quinolin-4(1H)-one conjugates 361 and 362 by Pd-catalysed amination of 6-bromoquinolin-4(1H)-ones 322 with (2-amino-5,10,15,20-tetraphenylporphyrinato) nickel(II) 358 followed by demetallation [143].
Scheme 111.
Synthesis of porphyrin-quinolin-4(1H)-one conjugates 361 and 362 by Pd-catalysed amination of 6-bromoquinolin-4(1H)-ones 322 with (2-amino-5,10,15,20-tetraphenylporphyrinato) nickel(II) 358 followed by demetallation [143].

Scheme 112.
Synthesis of quinolin-4(1H)-one 363 by Pd-catalysed amination of 6-bromoquinolin-4(1H)-one 314 [130].
Scheme 112.
Synthesis of quinolin-4(1H)-one 363 by Pd-catalysed amination of 6-bromoquinolin-4(1H)-one 314 [130].

Scheme 113.
Pd-catalysed aminocarbonylation of 6-bromo-3-iodoquinolin-4(1H)-ones 314 [129].
Scheme 113.
Pd-catalysed aminocarbonylation of 6-bromo-3-iodoquinolin-4(1H)-ones 314 [129].

Scheme 114.
Direct 3-alkenylation of quinolin-4(1H)-ones 365 [144].
Scheme 114.
Direct 3-alkenylation of quinolin-4(1H)-ones 365 [144].

Scheme 115.
Metal-catalysed decarboxylative coupling of quinolin-4(1H)-one 3-carboxylic acids 368 with (hetero)-aryl halides [145].
Scheme 115.
Metal-catalysed decarboxylative coupling of quinolin-4(1H)-one 3-carboxylic acids 368 with (hetero)-aryl halides [145].

Scheme 116.
Synthesis of 3-arylquinolin-4(1H)-ones 371 and 373 by Pd-catalysed decarboxylative cross-coupling of quinolin-4(1H)-one 3-carboxylic acids 370 and 372 with aryl halides [146].
Scheme 116.
Synthesis of 3-arylquinolin-4(1H)-ones 371 and 373 by Pd-catalysed decarboxylative cross-coupling of quinolin-4(1H)-one 3-carboxylic acids 370 and 372 with aryl halides [146].

Scheme 117.
Pd-catalysed direct thioetherification of quinolin-4(1H)-one-3-carboxylic acids 370 and 376 [147].
Scheme 117.
Pd-catalysed direct thioetherification of quinolin-4(1H)-one-3-carboxylic acids 370 and 376 [147].

Scheme 118.
Synthesis of 6-hydroxyquinolin-4(1H)-one 378 by Pd-catalysed hydroxylation [130].
Scheme 118.
Synthesis of 6-hydroxyquinolin-4(1H)-one 378 by Pd-catalysed hydroxylation [130].

Scheme 119.
Cyanation of 6-chloro-1-methylquinolin-4(1H)-ones 311 and 312 [128].
Scheme 119.
Cyanation of 6-chloro-1-methylquinolin-4(1H)-ones 311 and 312 [128].

Scheme 120.
Synthesis of N-allylated quinolin-4(1H)-ones 382 by Pd-catalysed addition of quinolin-4(1H)-ones 380 to terminal allenes 381 [148].
Scheme 120.
Synthesis of N-allylated quinolin-4(1H)-ones 382 by Pd-catalysed addition of quinolin-4(1H)-ones 380 to terminal allenes 381 [148].

Scheme 121.
Synthesis of difluoro-containing quinolin-4(1H)-ones 384 by Pd-catalysed cross-coupling reaction of 3-iodo-quinolin-4(1H)-one 383 with ethyl bromodifluoroacetate [149].
Scheme 121.
Synthesis of difluoro-containing quinolin-4(1H)-ones 384 by Pd-catalysed cross-coupling reaction of 3-iodo-quinolin-4(1H)-one 383 with ethyl bromodifluoroacetate [149].

Scheme 122.
Mechanism of the copper-mediated Pd-catalysed cross-coupling reaction [149].
Scheme 122.
Mechanism of the copper-mediated Pd-catalysed cross-coupling reaction [149].

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Silva, V.L.M.; Silva, A.M.S. Palladium-Catalysed Synthesis and Transformation of Quinolones. Molecules 2019, 24, 228. https://doi.org/10.3390/molecules24020228
AMA Style
Silva VLM, Silva AMS. Palladium-Catalysed Synthesis and Transformation of Quinolones. Molecules. 2019; 24(2):228. https://doi.org/10.3390/molecules24020228
Chicago/Turabian StyleSilva, Vera L. M., and Artur M. S. Silva. 2019. "Palladium-Catalysed Synthesis and Transformation of Quinolones" Molecules 24, no. 2: 228. https://doi.org/10.3390/molecules24020228