Resonance Raman Spectro-Electrochemistry to Illuminate Photo-Induced Molecular Reaction Pathways


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
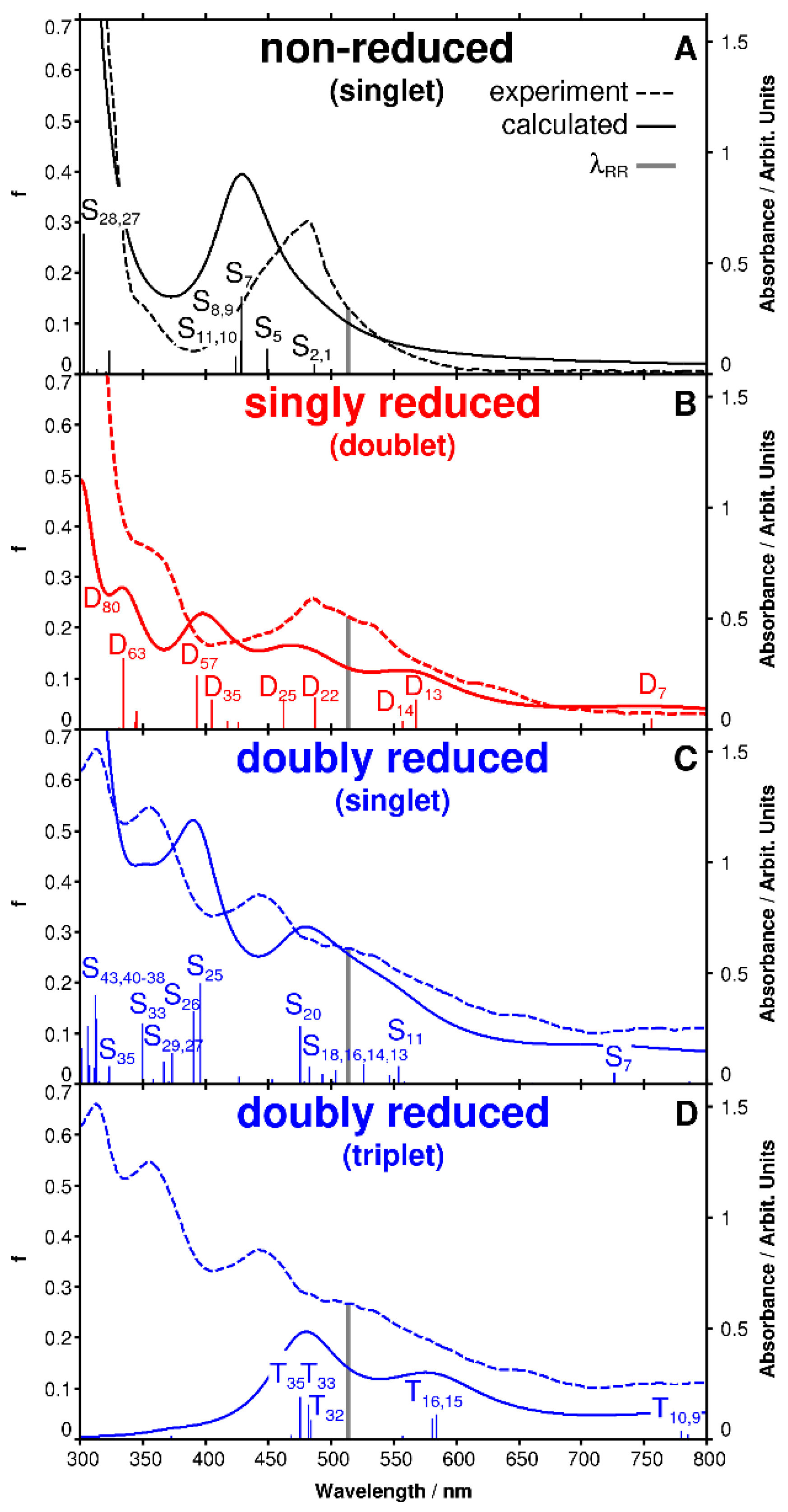
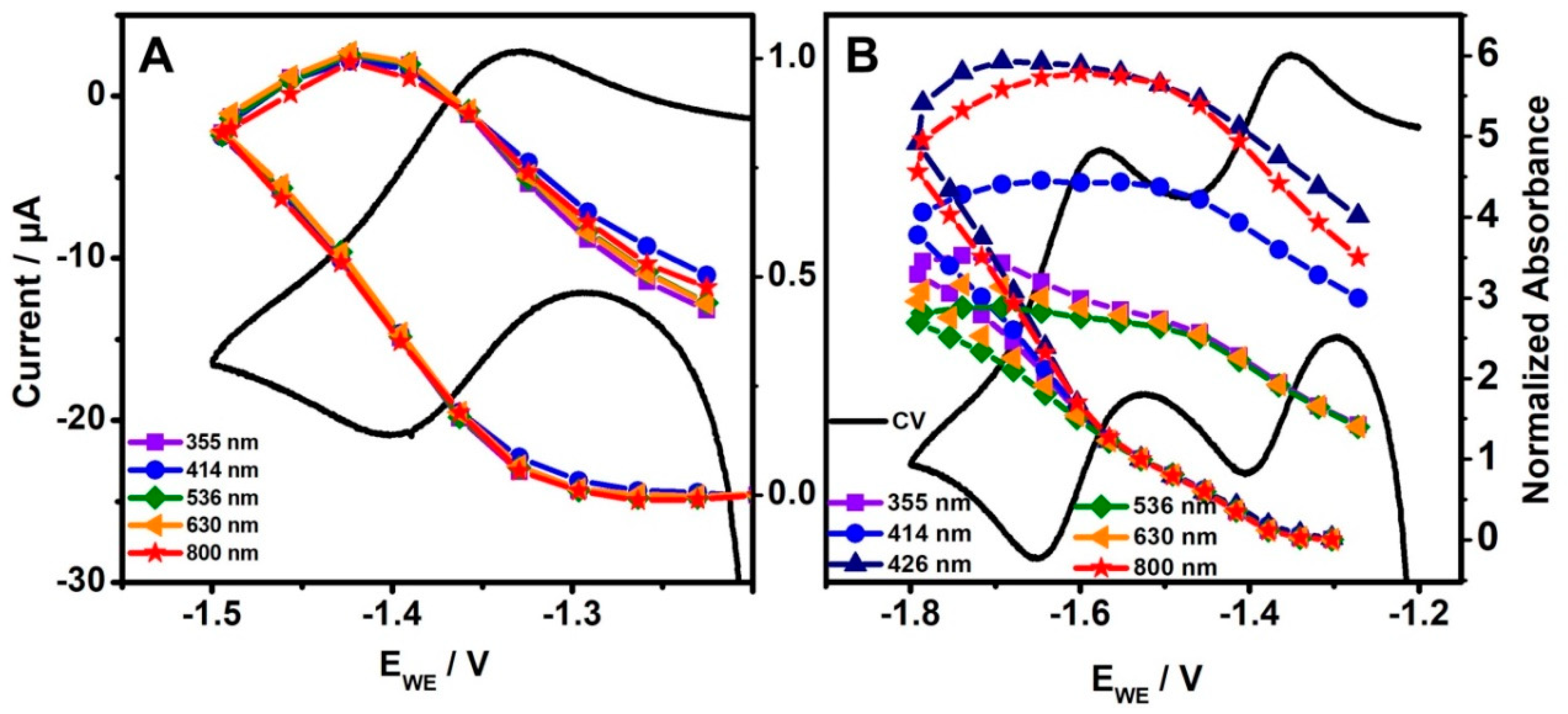
2.1. UV-Vis Spectro-Electrochemistry
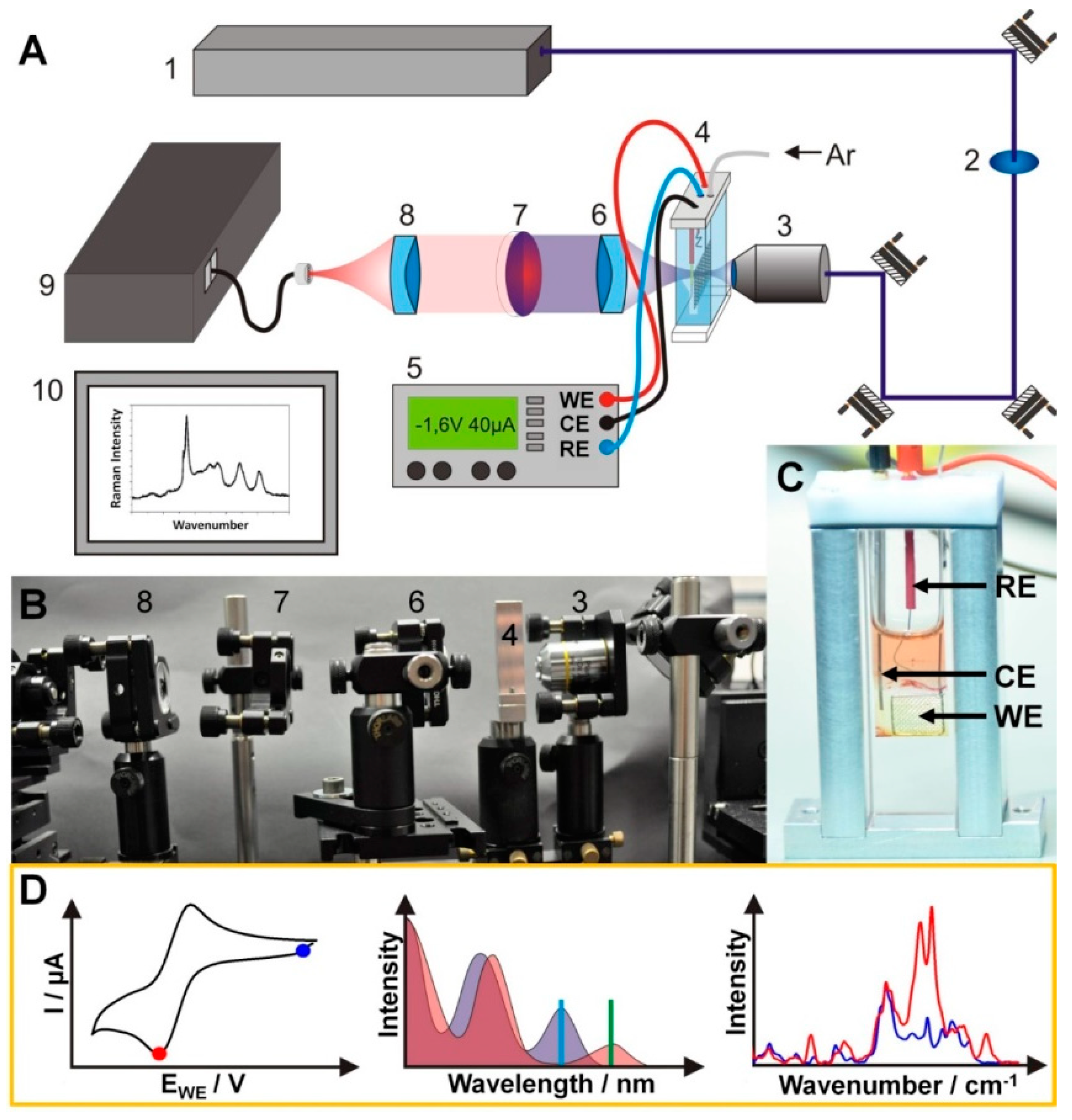
2.2. Resonance Raman Spectro-Electrochemistry
3. Methods
3.1. Experimental
3.2. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Berardi, S.; Drouet, S.; Francàs, L.; Gimbert-Suriñach, C.; Guttentag, M.; Richmond, C.; Stoll, T.; Llobet, A. Molecular artificial photosynthesis. Chem. Soc. Rev. 2014, 43, 7501–7519. [Google Scholar] [CrossRef] [PubMed]
- Hammarström, L. Accumulative charge separation for solar fuels production: Coupling light-induced single electron transfer to multielectron catalysis. Acc. Chem. Res. 2015, 48, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Zedler, L.; Guthmuller, J.; Rabelo de Moraes, I.; Krieck, S.; Schmitt, M.; Popp, J.; Dietzek, B. Redox state sensitive spectroscopy of the model compound [(H-dcbpy)2RuII(NCS)2]2– (dcbpy = 2,2′-bipyridine-4,4′-dicarboxylato). J. Phys. Chem. C 2013, 117, 6669–6677. [Google Scholar] [CrossRef]
- Zedler, L.; Guthmuller, J.; Rabelo de Moraes, I.; Kupfer, S.; Krieck, S.; Schmitt, M.; Popp, J.; Rau, S.; Dietzek, B. Resonance-Raman spectro-electrochemistry of intermediates in molecular artificial photosynthesis of bimetallic complexes. Chem. Comm. 2014, 50, 5227–5229. [Google Scholar] [CrossRef] [PubMed]
- Zedler, L.; Kupfer, S.; de Moraes, I.R.; Wächtler, M.; Beckert, R.; Schmitt, M.; Popp, J.; Rau, S.; Dietzek, B. Trapped in imidazole: How to accumulate multiple photoelectrons on a black-absorbing ruthenium complex. Chemistry 2014, 20, 3793–3799. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kupfer, S.; Zedler, L.; Schindler, J.; Bocklitz, T.; Guthmuller, J.; Rau, S.; Dietzek, B. In situ spectroelectrochemical and theoretical study on the oxidation of a 4H-imidazole-ruthenium dye adsorbed on nanocrystalline TiO2 thin film electrodes. Phys. Chem. Chem. Phys. 2015, 17, 29637–29646. [Google Scholar] [CrossRef] [PubMed]
- Harriman, A. Artificial light-harvesting arrays for solar energy conversion. Chem. Comm. 2015, 51, 11745–11756. [Google Scholar] [CrossRef]
- Esswein, A.J.; Nocera, D.G. Hydrogen production by molecular photocatalysis. Chem. Rev. 2007, 107, 4022–4047. [Google Scholar] [CrossRef]
- El-Khouly, M.E.; El-Mohsnawy, E.; Fukuzumi, S. Solar energy conversion: From natural to artificial photosynthesis. J. Photochem. Photobiol. C Photochem. Rev. 2017, 31, 36–83. [Google Scholar] [CrossRef]
- Wächtler, M.; Kübel, J.; Barthelmes, K.; Winter, A.; Schmiedel, A.; Pascher, T.; Lambert, C.; Schubert, U.S.; Dietzek, B. Energy transfer and formation of long-lived 3MLCT states in multimetallic complexes with extended highly conjugated bis-terpyridyl ligands. Phys. Chem. Chem. Phys. 2016, 18, 2350–2360. [Google Scholar] [CrossRef]
- Pfeffer, M.G.; Kowacs, T.; Wachtler, M.; Guthmuller, J.; Dietzek, B.; Vos, J.G.; Rau, S. Optimization of hydrogen-evolving photochemical molecular devices. Angew. Chem. Int. Ed. Engl. 2015, 54, 6627–6631. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wächtler, M.; Barthelmes, K.; Winter, A.; Schubert, U.S.; Dietzek, B. Direct detection of the photoinduced charge-separated state in a Ru(II) bis(terpyridine)–polyoxometalate molecular dyad. Chem. Comm. 2018, 54, 2970–2973. [Google Scholar] [CrossRef] [PubMed]
- Constable, E.C.; Cargill Thompson, A.M.W.; Armaroli, N.; Balzani, V.; Maestri, M. Ligand substitution patterns control photophysical properties of ruthenium(II)-2,2′:6′,2″-terpyridine complexes—room temperature emission from [Ru(tpy)2]2+ analogues. Polyhedron 1992, 11, 2707–2709. [Google Scholar] [CrossRef]
- Klein, J.; Stuckmann, A.; Sobottka, S.; Suntrup, L.; van der Meer, M.; Hommes, P.; Reissig, H.-U.; Sarkar, B. Ruthenium complexes with strongly electron-donating terpyridine ligands: Effect of the working electrode on electrochemical and spectroelectrochemical properties. Chem. A Eur. J. 2017, 23, 12314–12325. [Google Scholar] [CrossRef] [PubMed]
- Schubert, U.S.; Winter, A.; Newkome, G.R. Terpyridine-Based Materials: For Catalytic, Optoelectronic and Life Science Applications; John Wiley & Sons: Hoboken, NJ, USA, 2012; p. 881. [Google Scholar]
- Sauvage, J.P.; Collin, J.P.; Chambron, J.C.; Guillerez, S.; Coudret, C.; Balzani, V.; Barigelletti, F.; De Cola, L.; Flamigni, L. Ruthenium(II) and Osmium(II) bis(terpyridine) complexes in covalently-linked multicomponent systems: Synthesis, electrochemical behavior, absorption spectra, and photochemical and photophysical properties. Chem. Rev. 1994, 94, 993–1019. [Google Scholar] [CrossRef]
- Beley, M.; Collin, J.-P.; Sauvage, J.-P.; Sugihara, H.; Heisel, F.; Miehé, A. Photophysical and photochemical properties of ruthenium and osmium complexes with substituted terpyridines. J. Chem. Soc. Dalton Trans. 1991, 0, 3157–3159. [Google Scholar] [CrossRef]
- Harriman, A.; Ziessel, R. Making photoactive molecular-scale wires. Chem. Comm. 1996, 0, 1707–1716. [Google Scholar] [CrossRef]
- Maestri, M.; Armaroli, N.; Balzani, V.; Constable, E.C.; Thompson, A.M.W.C. Complexes of the ruthenium(II)-2,2’:6’,2’’-terpyridine family. Effect of electron-accepting and -donating substituents on the photophysical and electrochemical properties. Inorg. Chem. 1995, 34, 2759–2767. [Google Scholar] [CrossRef]
- Stone, M.L.; Crosby, G.A. Charge-transfer luminescence from ruthenium(II) complexes containing tridentate ligands. Chem. Phys. Lett. 1981, 79, 169–173. [Google Scholar] [CrossRef]
- Zhang, Y.-M.; Wu, S.-H.; Yao, C.-J.; Nie, H.-J.; Zhong, Y.-W. A Bis(terpyridine)ruthenium complex with three redox-active amine sites: Electrochemical, optical, and computational studies. Inorg. Chem. 2012, 51, 11387–11395. [Google Scholar] [CrossRef]
- Benniston, A.C.; Grosshenny, V.; Harriman, A.; Ziessel, R. Electron delocalization in ethynyl-bridged binuclear ruthenium(II) polypyridine complexes. Angew. Chem. Int. Ed. Eng. 1994, 33, 1884–1885. [Google Scholar] [CrossRef]
- Preiß, J.; Jäger, M.; Rau, S.; Dietzek, B.; Popp, J.; Martínez, T.; Presselt, M. How does peripheral functionalization of ruthenium(II)–terpyridine complexes affect spatial charge redistribution after photoexcitation at the Franck–Condon point? ChemPhysChem 2015, 16, 1395–1404. [Google Scholar] [CrossRef] [PubMed]
- Siebert, R.; Winter, A.; Dietzek, B.; Schubert, U.S.; Popp, J. Dual emission from highly conjugated 2,2′:6′:2″-terpyridine complexes—a potential route to white emitters. Macromol. Rapid Comm. 2010, 31, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Siebert, R.; Winter, A.; Schubert, U.S.; Dietzek, B.; Popp, J. The molecular mechanism of dual emission in terpyridine transition metal complexes—ultrafast investigations of photoinduced dynamics. Phys. Chem. Chem. Phys. 2011, 13, 1606–1617. [Google Scholar] [CrossRef] [PubMed]
- Wood, C.J.; Robson, K.C.D.; Elliott, P.I.P.; Berlinguette, C.P.; Gibson, E.A. Novel triphenylamine-modified ruthenium(II) terpyridine complexes for nickel oxide-based cathodic dye-sensitized solar cells. RSC Adv. 2014, 4, 5782–5791. [Google Scholar] [CrossRef]
- Yang, S.-H.; Wu, K.-L.; Chi, Y.; Cheng, Y.-M.; Chou, P.-T. Tris(thiocyanate) ruthenium(II) sensitizers with functionalized dicarboxyterpyridine for dye-sensitized solar cells. Angew. Chem. Int. Ed. 2011, 50, 8270–8274. [Google Scholar] [CrossRef] [PubMed]
- Schulze, B.; Escudero, D.; Friebe, C.; Siebert, R.; Görls, H.; Köhn, U.; Altuntas, E.; Baumgaertel, A.; Hager, M.D.; Winter, A.; et al. A Heteroleptic bis(tridentate) ruthenium(II) complex of a click-derived abnormal carbene pincer ligand with potential for photosensitzer application. Chem. A Eur. J. 2011, 17, 5494–5498. [Google Scholar] [CrossRef] [PubMed]
- Schulze, B.; Brown, D.G.; Robson, K.C.; Friebe, C.; Jäger, M.; Birckner, E.; Berlinguette, C.P.; Schubert, U.S. Cyclometalated ruthenium(II) complexes featuring tridentate click-derived ligands for dye-sensitized solar cell applications. Chem. A Eur. J. 2013, 19, 14171–14180. [Google Scholar] [CrossRef] [PubMed]
- Berger, R.M.; McMillin, D.R. Localized states in reduced and excited-state ruthenium(II) terpyridyls. Inorg. Chem. 1988, 27, 4245–4249. [Google Scholar] [CrossRef]
- Das, A.; Ghosh, T.K.; Dutta Chowdhury, A.; Mobin, S.M.; Lahiri, G.K. Electronic structure and catalytic aspects of [(trpy)(Cl)Ru(L)]n incorporating potential non-innocent ligands, L−: 9-Oxidophenalenone and trpy: 2,2′:6′,2″-terpyridine. Polyhedron 2013, 52, 1130–1137. [Google Scholar] [CrossRef]
- England, J.; Scarborough, C.C.; Weyhermüller, T.; Sproules, S.; Wieghardt, K. Electronic structures of the electron transfer series [M(bpy)3]n, [M(tpy)2]n, and [Fe(tbpy)3]n (M = Fe, Ru; n = 3+, 2+, 1+, 0, 1–): A mössbauer spectroscopic and DFT study. Eur. J. Inorg. Chem. 2012, 2012, 4605–4621. [Google Scholar] [CrossRef]
- DeArmond, M.K.; Hanck, K.W.; Wertz, D.W. Spatially ioslated redox orbitals—an update. Coord. Chem. Rev. 1985, 64, 65–81. [Google Scholar] [CrossRef]
- Kaim, W.; Fiedler, J. Spectroelectrochemistry: the best of two worlds. Chem. Soc. Rev. 2009, 38, 3373–3382. [Google Scholar] [CrossRef] [PubMed]
- Wadman, S.H.; Lutz, M.; Tooke, D.M.; Spek, A.L.; Hartl, F.; Havenith, R.W.A.; van Klink, G.P.M.; van Koten, G. Consequences of N,C,N′- and C,N,N′-coordination modes on electronic and photophysical properties of cyclometalated aryl ruthenium(II) complexes. Inorg. Chem. 2009, 48, 1887–1900. [Google Scholar] [CrossRef] [PubMed]
- Heinze, K.; Hempel, K.; Tschierlei, S.; Schmitt, M.; Popp, J.; Rau, S. Resonance Raman studies of bis(terpyridine)ruthenium(II) amino acid esters and diesters. Eur. J. Inorg. Chem. 2009, 2009, 3119–3126. [Google Scholar] [CrossRef]
- Kupfer, S.; Guthmuller, J.; Wächtler, M.; Losse, S.; Rau, S.; Dietzek, B.; Popp, J.; Gonzalez, L. Protonation effects on the resonance Raman properties of a novel (terpyridine)Ru(4H-imidazole) complex: an experimental and theoretical case study. Phys. Chem. Chem. Phys. 2011, 13, 15580–15588. [Google Scholar] [CrossRef] [PubMed]
- Latouche, C.; Skouteris, D.; Palazzetti, F.; Barone, V. TD-DFT Benchmark on inorganic Pt(II) and Ir(III) complexes. J. Chem. Theory Comput. 2015, 11, 3281–3289. [Google Scholar] [CrossRef]
- Hewitt, J.T.; Vallett, P.J.; Damrauer, N.H. Dynamics of the 3MLCT in Ru(II) terpyridyl complexes probed by ultrafast spectroscopy: Evidence of excited-state equilibration and interligand electron transfer. J. Phys. Chem. A 2012, 116, 11536–11547. [Google Scholar] [CrossRef] [PubMed]
- Braterman, P.S.; Song, J.I.; Peacock, R.D. Electronic absorption spectra of the iron(II) complexes of 2,2’-bipyridine, 2,2’-bipyrimidine, 1,10-phenanthroline, and 2,2’:6’,2’’-terpyridine and their reduction products. Inorg. Chem. 1992, 31, 555–559. [Google Scholar] [CrossRef]
- Tokel-Takvoryan, N.E.; Hemingway, R.E.; Bard, A.J. Electrogenerated chemiluminescence. XIII. electrochemical and electrogenerated chemiluminescence studies of ruthenium chelates. J. Am. Chem. Soc. 1973, 95, 6582–6589. [Google Scholar] [CrossRef]
- Morris, D.E.; Hanck, K.W.; DeArmond, M.K. Electrochemical and ESR studies of [Fe(terpy)2]2+ and [Ru(terpy)2]2+ and their reduction products. J. Electroanal. Chem. Inter. Electrochem. 1983, 149, 115–130. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.1, Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Becke, A.D. Density-functional thermochemistry III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Kupfer, S.; Wächtler, M.; Guthmuller, J.; Popp, J.; Dietzek, B.; González, L. A novel Ru(II) polypyridine black dye investigated by resonance Raman spectroscopy and TDDFT calculations. J. Phys. Chem. C 2012, 116, 19968–19977. [Google Scholar] [CrossRef]
- Wächtler, M.; Kupfer, S.; Guthmuller, J.; Rau, S.; González, L.; Dietzek, B. Structural control of photoinduced dynamics in 4H-imidazole-ruthenium dyes. J. Phys. Chem. C 2012, 116, 25664–25676. [Google Scholar] [CrossRef]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chimica Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Merrick, J.P.; Moran, D.; Radom, L. An evaluation of harmonic vibrational frequency scale factors. J. Phys. Chem. A 2007, 111, 11683–11700. [Google Scholar] [CrossRef]
- Mennucci, B.; Cappelli, C.; Guido, C.A.; Cammi, R.; Tomasi, J. Structures and properties of electronically excited chromophores in solution from the polarizable continuum model coupled to the time-dependent density functional theory. J. Phys. Chem. A 2009, 113, 3009–3020. [Google Scholar] [CrossRef]
- Wächtler, M.; Guthmuller, J.; González, L.; Dietzek, B. Analysis and characterization of coordination compounds by resonance Raman spectroscopy. Coord. Chem. Rev. 2012, 256, 1479–1508. [Google Scholar] [CrossRef]
- Guthmuller, J. Comparison of simplified sum-over-state expressions to calculate resonance Raman intensities including Franck-Condon and Herzberg-Teller effects. J. Chem. Phys. 2016, 144. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compound [Ru(tbtpy)2](PF6)2 are available from the authors. |



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zedler, L.; Krieck, S.; Kupfer, S.; Dietzek, B. Resonance Raman Spectro-Electrochemistry to Illuminate Photo-Induced Molecular Reaction Pathways. Molecules 2019, 24, 245. https://doi.org/10.3390/molecules24020245
Zedler L, Krieck S, Kupfer S, Dietzek B. Resonance Raman Spectro-Electrochemistry to Illuminate Photo-Induced Molecular Reaction Pathways. Molecules. 2019; 24(2):245. https://doi.org/10.3390/molecules24020245
Chicago/Turabian StyleZedler, Linda, Sven Krieck, Stephan Kupfer, and Benjamin Dietzek. 2019. "Resonance Raman Spectro-Electrochemistry to Illuminate Photo-Induced Molecular Reaction Pathways" Molecules 24, no. 2: 245. https://doi.org/10.3390/molecules24020245