
Synthesis and Thermophysical Characterization of Fatty Amides for Thermal Energy Storage

 ,
,  , ,
, ,  and
and
Abstract
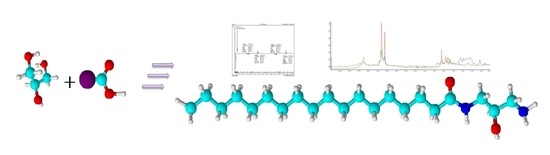
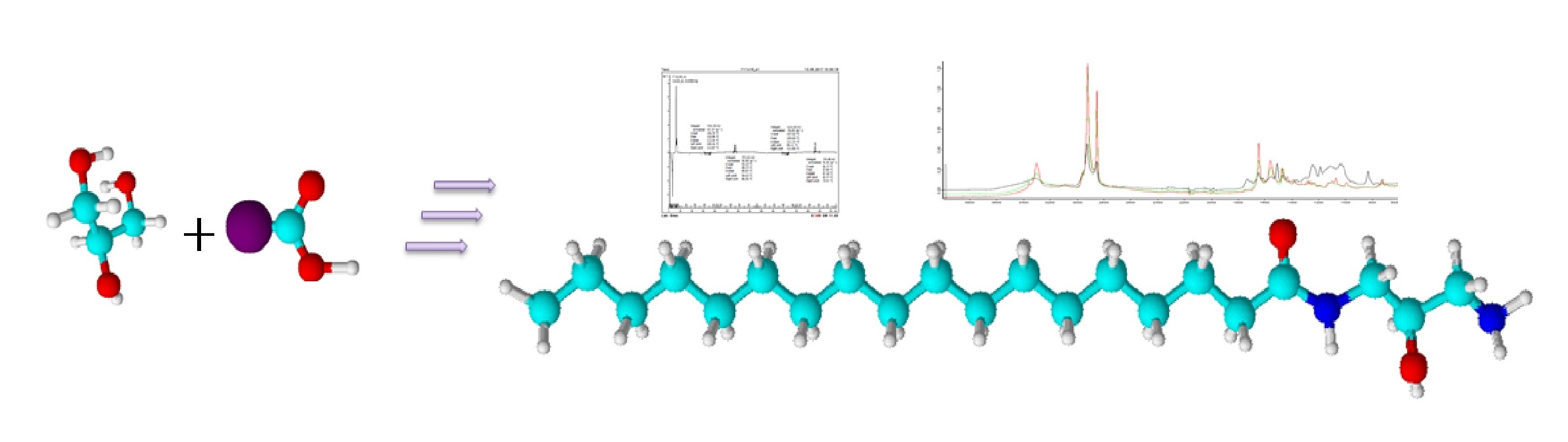
:
1. Introduction
2. Results and Discussions
3. Materials and Methods
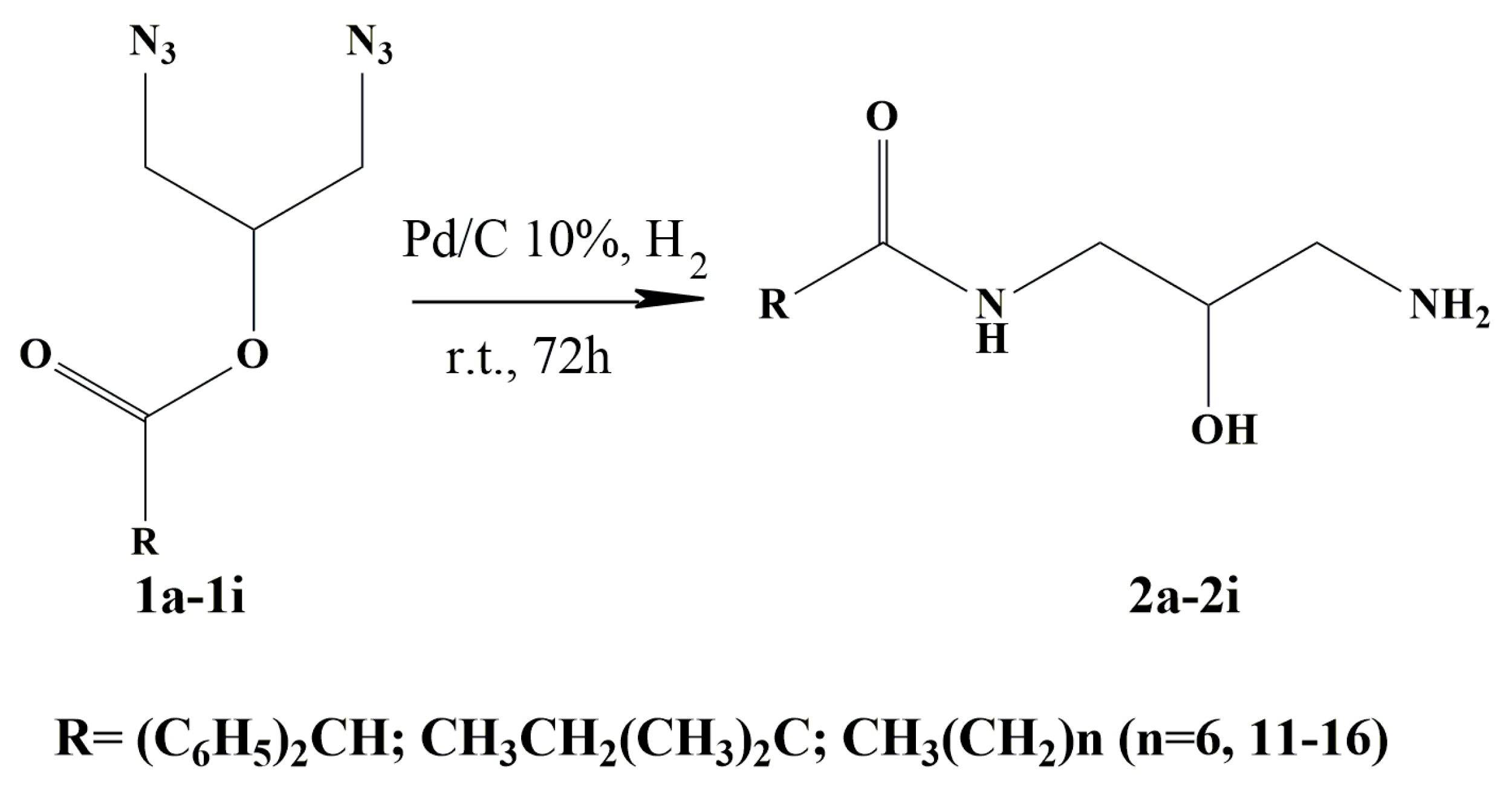
3.1. General Procedure for the Synthesis of N-(3-amino-2-hydroxypropyl)amides 2a to 2i
3.2. Thermoanalytical Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chung, O.; Jeong, S.-G.; Yu, S.; Kim, S. Thermal performance of organic PCMs/micronized silica composite for latent heat thermal energy storage. Energy Build. 2014, 70, 180–185. [Google Scholar] [CrossRef]
- Pasupathy, A.; Velraj, R.; Seeniraj, R. Phase change material-based building architecture for thermal management in residential and commercial establishments. Renew. Sustain. Energy Rev. 2008, 12, 39–64. [Google Scholar] [CrossRef]
- Oró, E.; Barreneche, C.; Farid, M.M.; Cabeza, L.F. Experimental study on the selection of phase change materials for low temperature applications. Renew. Energy 2013, 57, 130–136. [Google Scholar] [CrossRef]
- Waqas, A.; Ud, D.Z. Phase change material (PCM) storage for free cooling of buildings—A review. Renew. Sustain. Energy Rev. 2013, 18, 607–625. [Google Scholar] [CrossRef]
- Gil, A.; Barreneche, C.; Moreno, P.; Solé, C.; Fernández, A.I.; Cabeza, L.F. Thermal behaviour of d-mannitol when used as PCM: Comparison of results obtained by DSC and in a thermal energy storage unit at pilot plant scale. Appl. Energy 2013, 111, 1107–1113. [Google Scholar] [CrossRef]
- Liu, M.; Saman, W.; Bruno, F. Review on storage materials and thermal performance enhancement techniques for high temperature phase change thermal storage systems. Renew. Sustain. Energy Rev. 2012, 16, 2118–2132. [Google Scholar] [CrossRef]
- Stamatiou, A.; Obermeyer, M.; Fischer, L.J.; Schuetz, P.; Worlitschek, J. Investigation of unbranched, saturated, carboxylic esters as phase change materials. Renew. Energy 2017, 108, 401–409. [Google Scholar] [CrossRef]
- Ravotti, R.; Fellmann, O.; Lardon, N.; Fischer, L.; Stamatiou, A.; Worlitschek, J. Synthesis and Investigation of Thermal Properties of Highly Pure Carboxylic Fatty Esters to Be Used as PCM. Appl. Sci. 2018, 8, 1069. [Google Scholar] [CrossRef]
- Floros, M.C.; Kaller, K.L.C.; Poopalam, K.D.; Narine, S.S. Lipid derived diamide phase change materials for high temperature thermal energy storage. Sol. Energy 2016, 139, 23–28. [Google Scholar] [CrossRef]
- Canik, G.; Alkan, C. Hexamethylene dilauroyl, dimyristoyl, and dipalmytoyl amides as phase change materials for thermal energy storage. Sol. Energy 2010, 84, 666–672. [Google Scholar] [CrossRef]
- Zhang, Z.; Salih, A.A.M.; Li, M.; Yang, B. Synthesis and Characterization of Functionalized Ionic Liquids for Thermal Storage. Energy Fuels 2014, 28, 2802–2810. [Google Scholar] [CrossRef]
- Pielichowski, K.; Flejtuch, K. Recent developments in polymeric phase change materials for energy storage: Poly(ethylene oxide)/stearic acid blends. Polym. Adv. Technol. 2005, 16, 127–132. [Google Scholar] [CrossRef]
- Spanget-Larsen, P.E.H.J. NMR and IR Investigations of Strong Intramolecular Hydrogen Bonds. Molecules 2017, 22, 552. [Google Scholar] [Green Version]
- Lučić, B.; Stepanić, V.; Plavšić, D.; Amić, A.; Amić, D. Correlation between 13C NMR chemical shifts and antiradical activity of flavonoids. Monatsh. Chem. 2014, 145, 457–463. [Google Scholar] [CrossRef]
- Shu, I.; Scian, M.; Stewart, J.M.; Kier, B.L.; Andersen, N.H. 13C structuring shifts for the analysis of model β-hairpins and β-sheets in proteins: Diagnostic shifts appear only at the cross-strand H-bonded residues. J. Biomol. NMR 2013, 56, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Chitra, R.; Roussel, P.; Capet, F.; Murli, C.; Choudhury, R.R. 2-Aminopyridinium maleate: A structural study. J. Mol. Struct. 2008, 891, 103–109. [Google Scholar] [CrossRef]
- Beko, S.L.; Alig, E.; Schmidt, M.U.; van de Streek, J. On the correlation between hydrogen bonding and melting points in the inositols. IUCrJ. 2014, 1, 61–73. [Google Scholar] [CrossRef]
- Escribà, M.; Barreneche, C.; Yara-Varón, E.; Eras, J.; Solé, A.; Tomàs, A.; Cabeza, L.F.; Canela-Garayoa, R. Ionic compounds derived from crude glycerol: Thermal energy storage capability evaluation. Renew. Energy 2017, 114, 629–637. [Google Scholar] [CrossRef] [Green Version]
- Gallart-Sirvent, P.; Martín, M.; Villorbina, G.; Balcells, M.; Solé, A.; Barrenche, C.; Cabeza, L.F.; Canela-Garayoa, R. Fatty acid eutectic mixtures and derivatives from non-edible animal fat as phase change materials. RSC Adv. 2017, 7, 24133–24139. [Google Scholar] [CrossRef] [Green Version]
- Gallart-Sirvent, P.; Martín, M.; Solé, A.; Villorbina, G.; Balcells, M.; Cabeza, L.F.; Canela-Garayoa, R. Combining biocatalysts to achieve new phase change materials. Application to non-edible animal fat. Mol. Catal. 2018, 444, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Qu, S.; Li, F.; Wang, H.; Bai, B.; Xu, C.; Zhao, L.; Long, B.; Li, M. Twin-tapered molecules containing bi-dihydrazine units: Self-assembly through intermolecular quadruple hydrogen bonding and liquid crystalline behavior. Chem. Mater. 2007, 19, 4839–4846. [Google Scholar] [CrossRef]
- He, Y.; Zhu, B.; Inoue, Y. Hydrogen bonds in polymer blends. Prog. Polym. Sci. 2004, 29, 1021–1051. [Google Scholar] [CrossRef]
- da Silva, A.M.; Ghosh, A.; Chaudhuri, P. Effect of Hydrogen Bond Formation on the NMR Properties of Glycine–HCN Complexes. J. Phys. Chem. A 2013, 117, 10274–10285. [Google Scholar] [CrossRef] [PubMed]
- Solarte, C.; Escribà, M.; Eras, J.; Villorbina, G.; Canela, R.; Balcells, M. From Symmetric Glycerol Derivatives to Dissymmetric Chlorohydrins. Molecules 2011, 16, 2065–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Organic azides: An exploding diversity of a unique class of compounds. Angew. Chem. Int. Ed. 2005, 44, 5188–5240. [Google Scholar] [CrossRef]
- Mori, A.; Mizusaki, T.; Miyakawa, Y.; Ohashi, E.; Haga, T.; Maegawa, T.; Monguchi, Y.; Sajiki, H. Chemoselective hydrogenation method catalyzed by Pd/C using diphenylsulfide as a reasonable catalyst poison. Tetrahedron 2006, 62, 11925–11932. [Google Scholar] [CrossRef]
- Bisol, T.B.; Bortoluzzi, A.J.; Sa, M.M. Nucleophilic Ring-Opening of Epoxide and Aziridine Acetates for the Stereodivergent Synthesis of β-Hydroxy and β-Amino γ-Lactams. J. Org. Chem. 2011, 76, 948–962. [Google Scholar] [CrossRef]
- Zaffalon, P.-L.; D’Anna, V.; Hagemann, H.; Zumbuehl, A. Study of surfactant alcohols with various chemical moieties at the hydrophilic-hydrophobic interface. RSC Adv. 2013, 3, 7237–7244. [Google Scholar] [CrossRef]
- Hibino, M.; Sumi, A.; Tsuchiya, H.; Hatta, I. Odd-Even Effect in Monolayer of Fatty Acids Formed on a Graphite Surface by scanning Tunneling Microscopy. J. Phys. Chem. B. 1998, 102, 4544–4547. [Google Scholar] [CrossRef]
- Bhate, M.P.; Woodard, J.C.; Mehta, M.A. Solvation and hydrogen bonding in alanine-and glycine-containing dipeptides probed using solution-and solid-state NMR spectroscopy. J. Am. Chem. Soc. 2009, 131, 9579–9589. [Google Scholar] [CrossRef]
- Hiraga, Y.; Chaki, S.; Niwayama, S. 13C NMR spectroscopic studies of the behaviors of carbonyl compounds in various solutions. Tetrahedron Lett. 2017, 58, 4677–4681. [Google Scholar] [CrossRef]
- Skrovanek, D.J.; Howe, S.E.; Painter, P.C.; Coleman, M.M. Hydrogen bonding in polymers: Infrared temperature studies of an amorphous polyamide. Macromolecules 1985, 18, 1676–1683. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
{kind=link}
| R | Yield (%) a | Melting Point (°C) | |
|---|---|---|---|
| 2a | CH3CH2C(CH3)2 | 93.1 | – |
| 2b | (C6H5)2CH | 59.9 | 99.7–103.8 |
| 2c | CH3(CH2)6 | 68.2 | 114.8–119.0 |
| 2d | CH3(CH2)11 | 62.9 | 62.1–64.3 |
| 2e | CH3(CH2)12 | 56.6 | 112.9–115.5 |
| 2f | CH3(CH2)13 | 88.3 | 115.0–117.4 |
| 2g | CH3(CH2)14 | 74.7 | 112.1–114.9 |
| 2h | CH3(CH2)15 | 85.7 | 97.1–98.5 |
| 2i | CH3(CH2)16 | 69.1 | 110.9–113.1 |
| R | Cycle | ∆Hmelting (kJ/kg) | ∆Hsolidification (kJ/kg) | Tmelting (°C) | Tsolidification (°C) | |
|---|---|---|---|---|---|---|
| 2d | CH3(CH2)11 | 2 | 25.8 | 26.0 | 62.2 | 58.0 |
| 3 | 31.3 | 27.8 | 61.7 | 57.8 | ||
| 2e | CH3(CH2)12 | 2 | 107.2 | 101.8 | 116.4 | 101.0 |
| 3 | 98.3 | 85.8 | 112.1 | 95.9 | ||
| 2g | CH3(CH2)14 | 2 | 149.7 | 118.1 | 111.5 | 105.7 |
| 3 | 107.3 | 103.9 | 113.9 | 103.9 | ||
| 2h | CH3(CH2)15 | 2 | 145.4 | 129.3 | 96.6 | 85.3 |
| 3 | 125.1 | 122.0 | 95.2 | 82.2 | ||
| 2i | CH3(CH2)16 | 2 | 67.3 | 96.0 | 111.0 | 90.8 |
| 3 | 76.0 | 96.5 | 109.8 | 87.9 |
| R | Cα | C=O | CNH | COH | CNH2 | Solvent | [Sample] (M) | |
|---|---|---|---|---|---|---|---|---|
| 2d | CH3(CH2)11 | −0.5 | 0 | −0.5 | −0.3 | 0 | CD3OD | 0.07 |
| 2e | CH3(CH2)12 | 0 | 0 | 0 | −0.2 | 0 | CDCl3 | 0.07 |
| 2g | CH3(CH2)14 | 35.6 | 175.4 | 44.2 | 70.8 | 42.4 | CDCl3 | 0.06 |
| 2h | CH3(CH2)15 | 0 | 0.1 | 4 | −1.5 | 4.7 | CD3OD | 0.06 |
| 2i | CH3(CH2)16 | −4 | 0 | −1.7 | −0.3 | −6.7 | CD3OD | 0.06 |
| R | T (°C) | NH/OH (cm−1) | CH2 (cm−1) | CON (cm−1) | |
|---|---|---|---|---|---|
| 2d | CH3(CH2)11 | 30 | 3338 (w)/3299 (s) | 2920 (s)/2850 (s) | 1642 (s) |
| 125 | 3319 (m/b) | 2917 (m)/2853 (m) | 1729 (w)/1703 (w)/1646 (m) | ||
| 2g | CH3(CH2)14 | 30 | 3360 (w)/3299 (s) | 2920 (s)/2853 (s) | 1639 (s) |
| 125 | 3321 (m/b) | 2920 (m)/2853 (m) | 1732 (w)/1703 (w)/1639 (w) | ||
| 2h | CH3(CH2)15 | 30 | 3360 (w)/3299 (s) | 2917 (s)/2853 (s) | 1649 (s) |
| 135 | 3350 (w/b)/3309 (s) | 2917(s)/2853 (s) | 1729 (w)/1649 (s) | ||
| 2i | CH3(CH2)16 | 30 | 3357 (w)/3303 (s) | 2917 (s)/2853 (s) | 1642 (s) |
| 125 | 3305 (m/b) | 2917 (m)/2853 (m) | 1729 (w)/1703 (w)/1646 (w) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canela-Xandri, A.; Villorbina, G.; Balcells, M.; Fernández-Francos, X.; Cabeza, L.F.; Canela-Garayoa, R. Synthesis and Thermophysical Characterization of Fatty Amides for Thermal Energy Storage. Molecules 2019, 24, 3777. https://doi.org/10.3390/molecules24203777
Canela-Xandri A, Villorbina G, Balcells M, Fernández-Francos X, Cabeza LF, Canela-Garayoa R. Synthesis and Thermophysical Characterization of Fatty Amides for Thermal Energy Storage. Molecules. 2019; 24(20):3777. https://doi.org/10.3390/molecules24203777
Chicago/Turabian StyleCanela-Xandri, Anna, Gemma Villorbina, Mercè Balcells, Xavier Fernández-Francos, Luisa F. Cabeza, and Ramon Canela-Garayoa. 2019. "Synthesis and Thermophysical Characterization of Fatty Amides for Thermal Energy Storage" Molecules 24, no. 20: 3777. https://doi.org/10.3390/molecules24203777