
Early Celastrol Administration Prevents Ketamine-Induced Psychotic-Like Behavioral Dysfunctions, Oxidative Stress and IL-10 Reduction in The Cerebellum of Adult Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
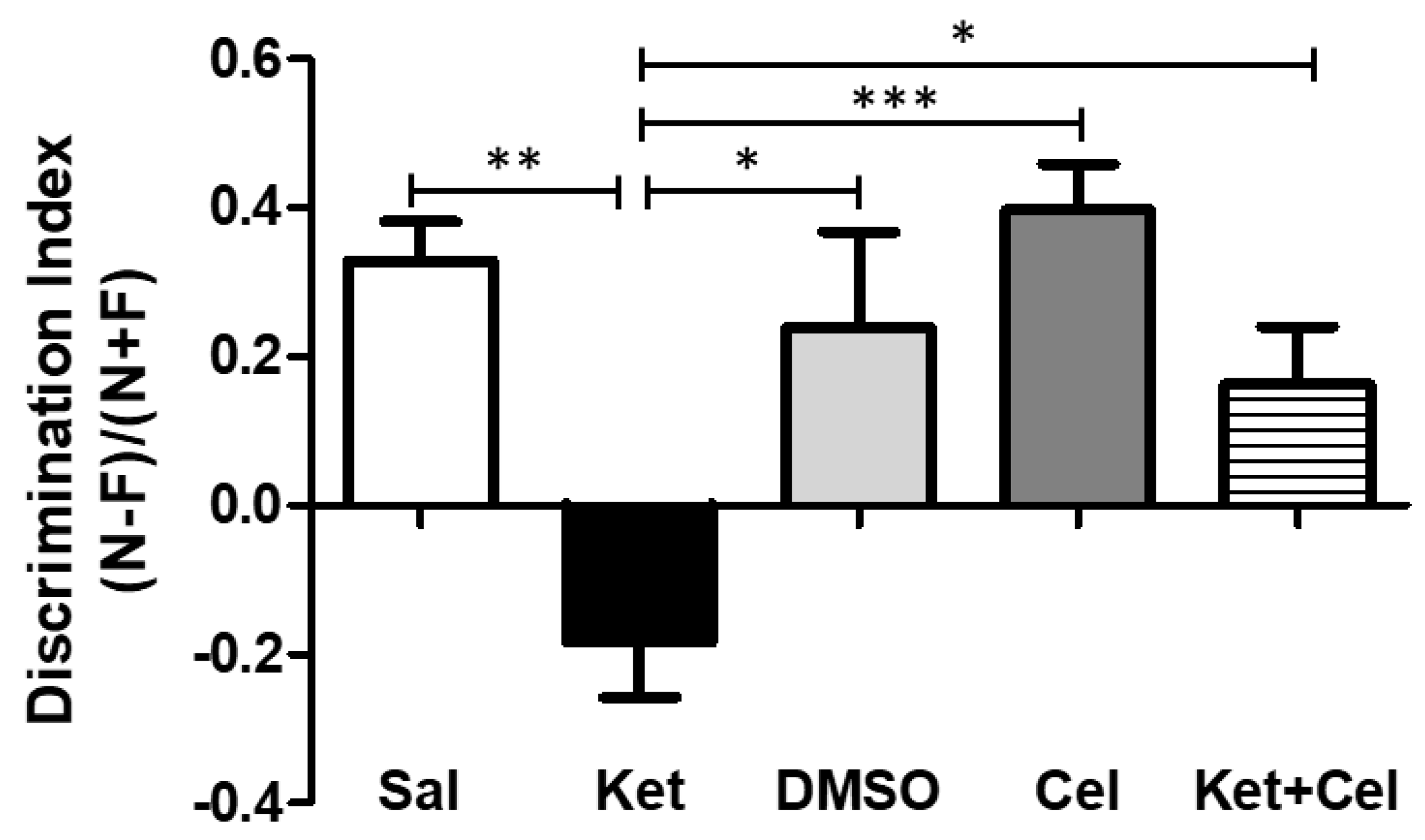
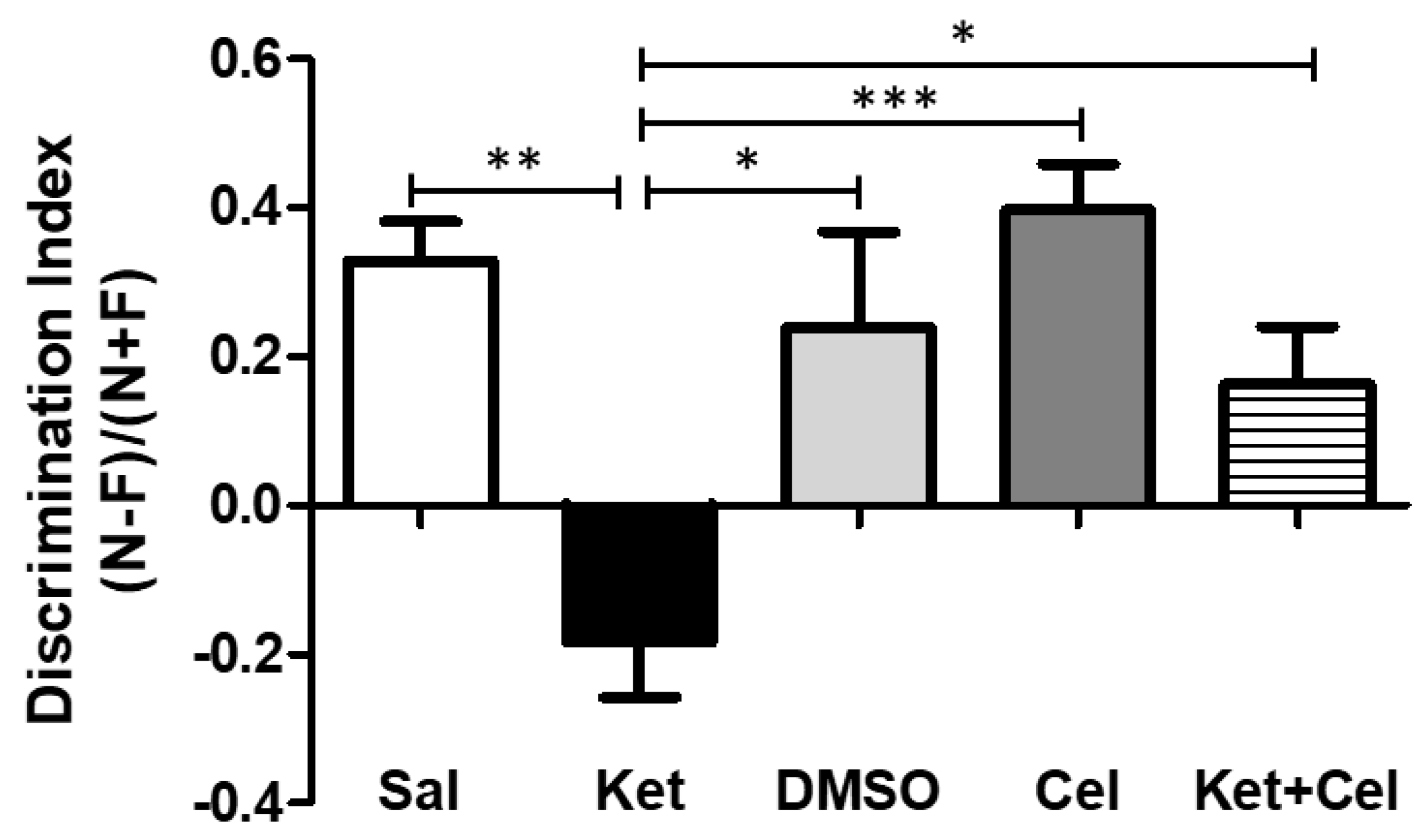
2.1. Early Celastrol Administration Prevented Cognitive Dysfunctions in Adult Mice Exposed to Ketamine in Postnatal Life
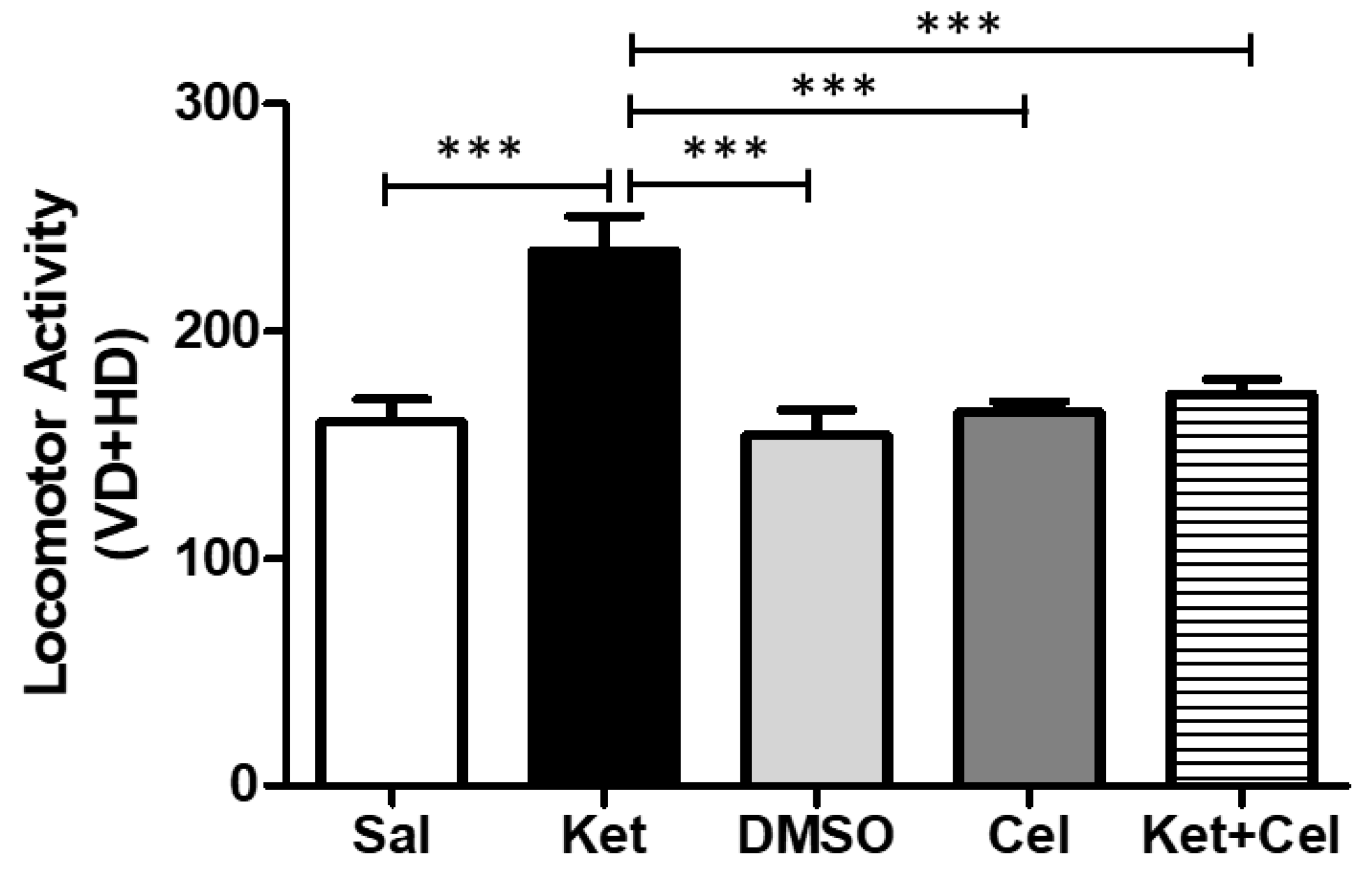
2.2. Early Celastrol Administration Prevented Locomotor Dysfunctions in Adult Mice Exposed to Ketamine in Postnatal Life
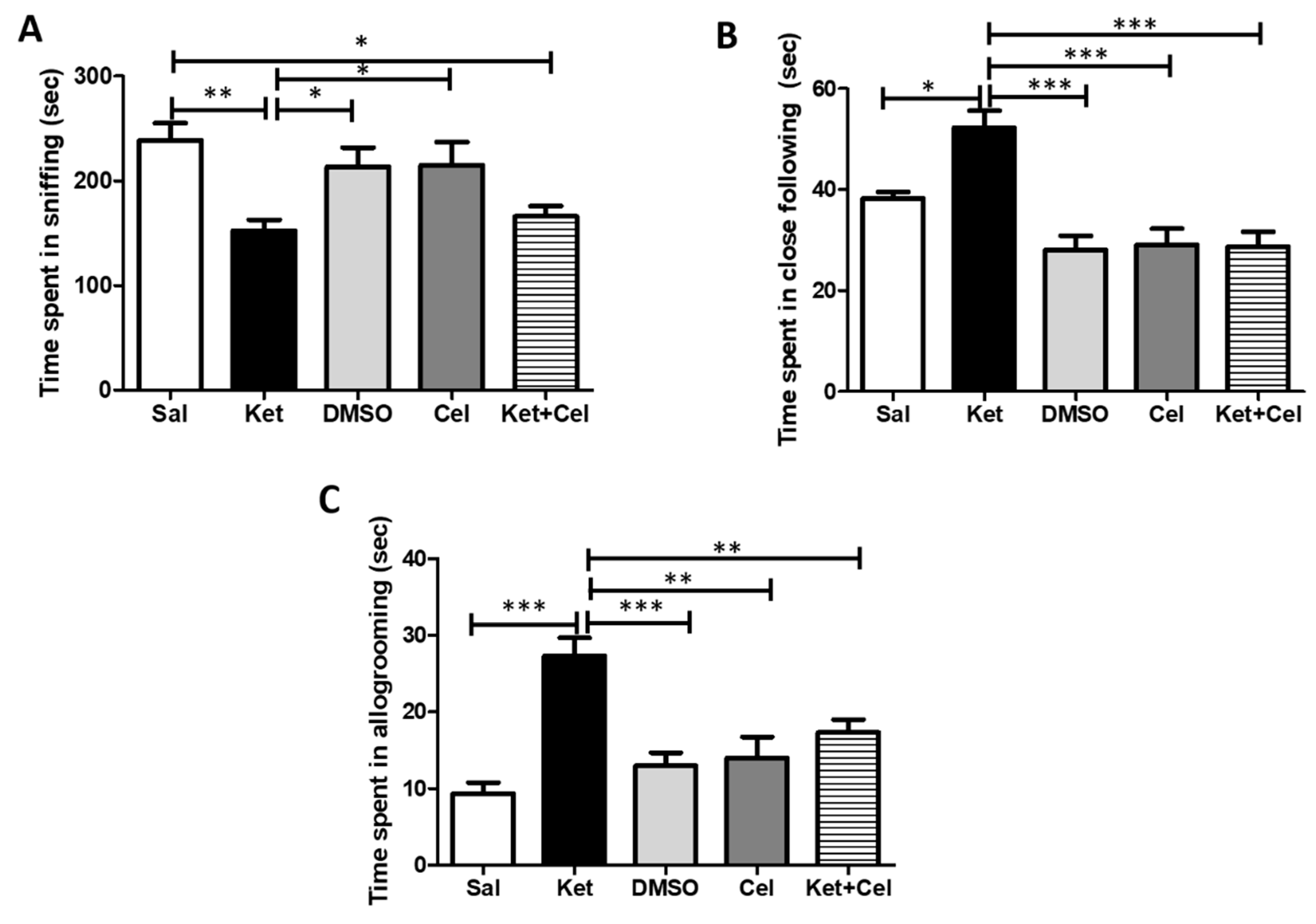
2.3. Early Celastrol Administration Prevented Social Behavior Dysfunctions in Adult Mice Exposed to Ketamine in Postnatal Life
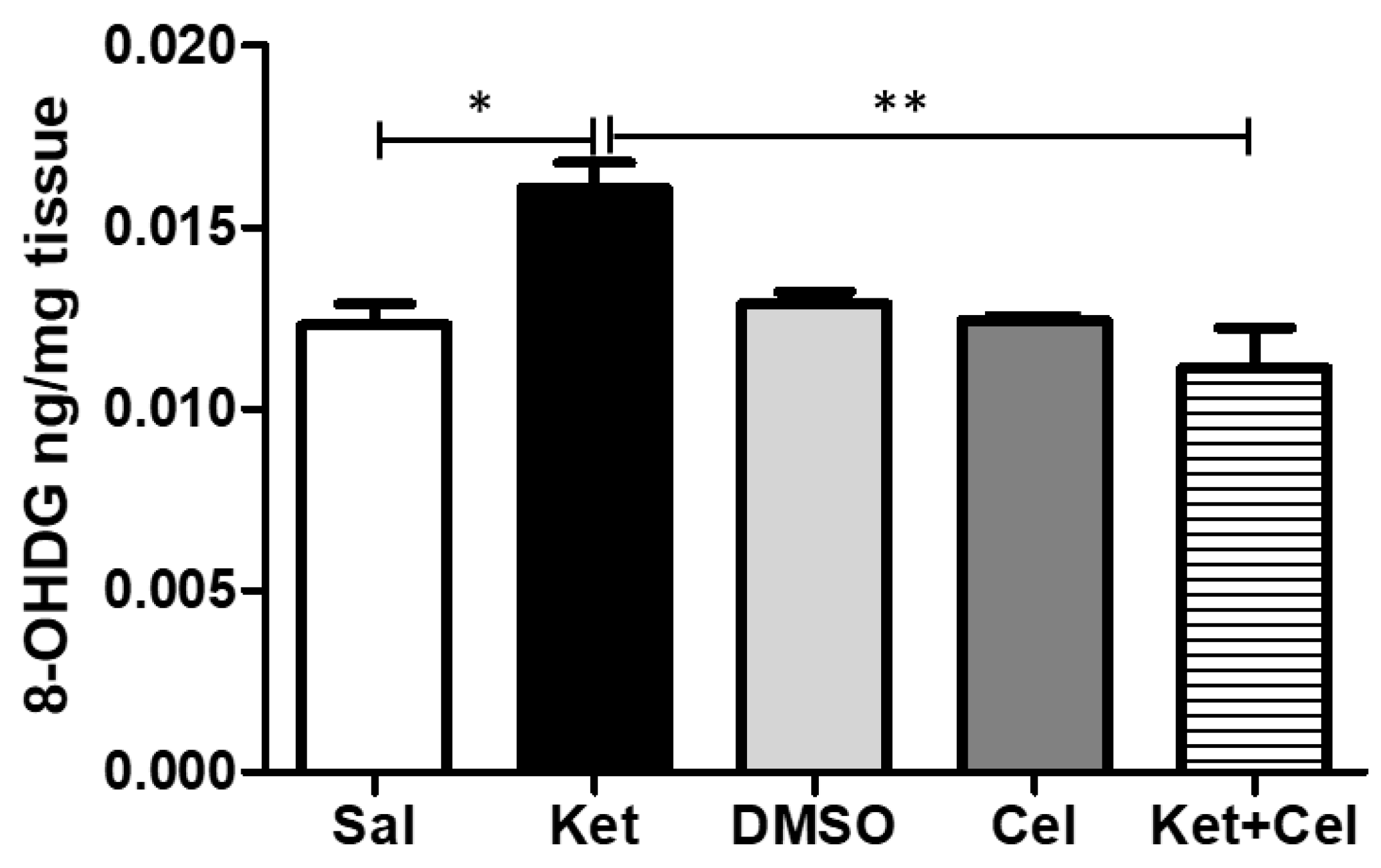
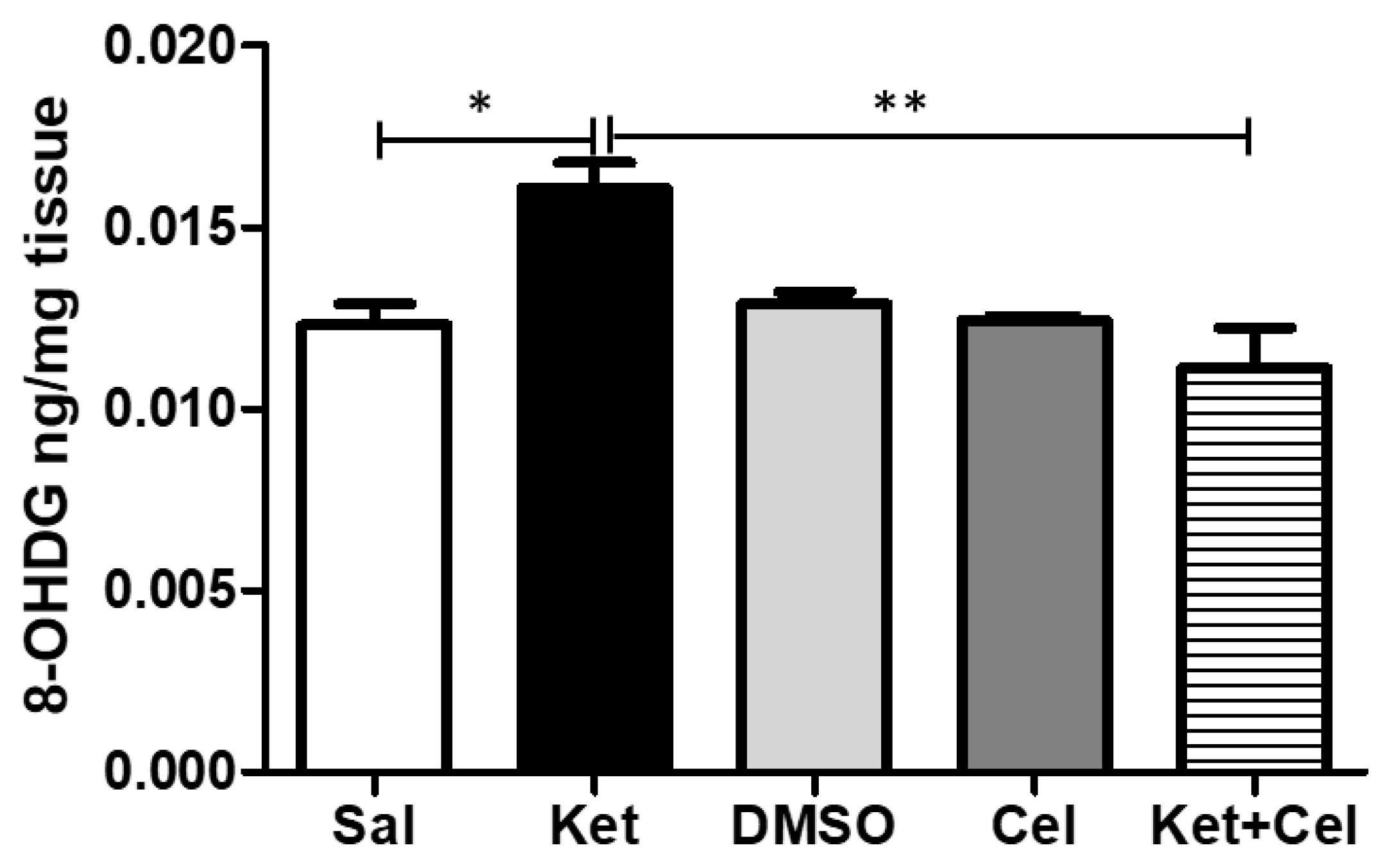
2.4. Early Celastrol Administration Prevented Oxidative Stress Increase in the Cerebellum of Adult Mice Exposed to Ketamine in Postnatal Life
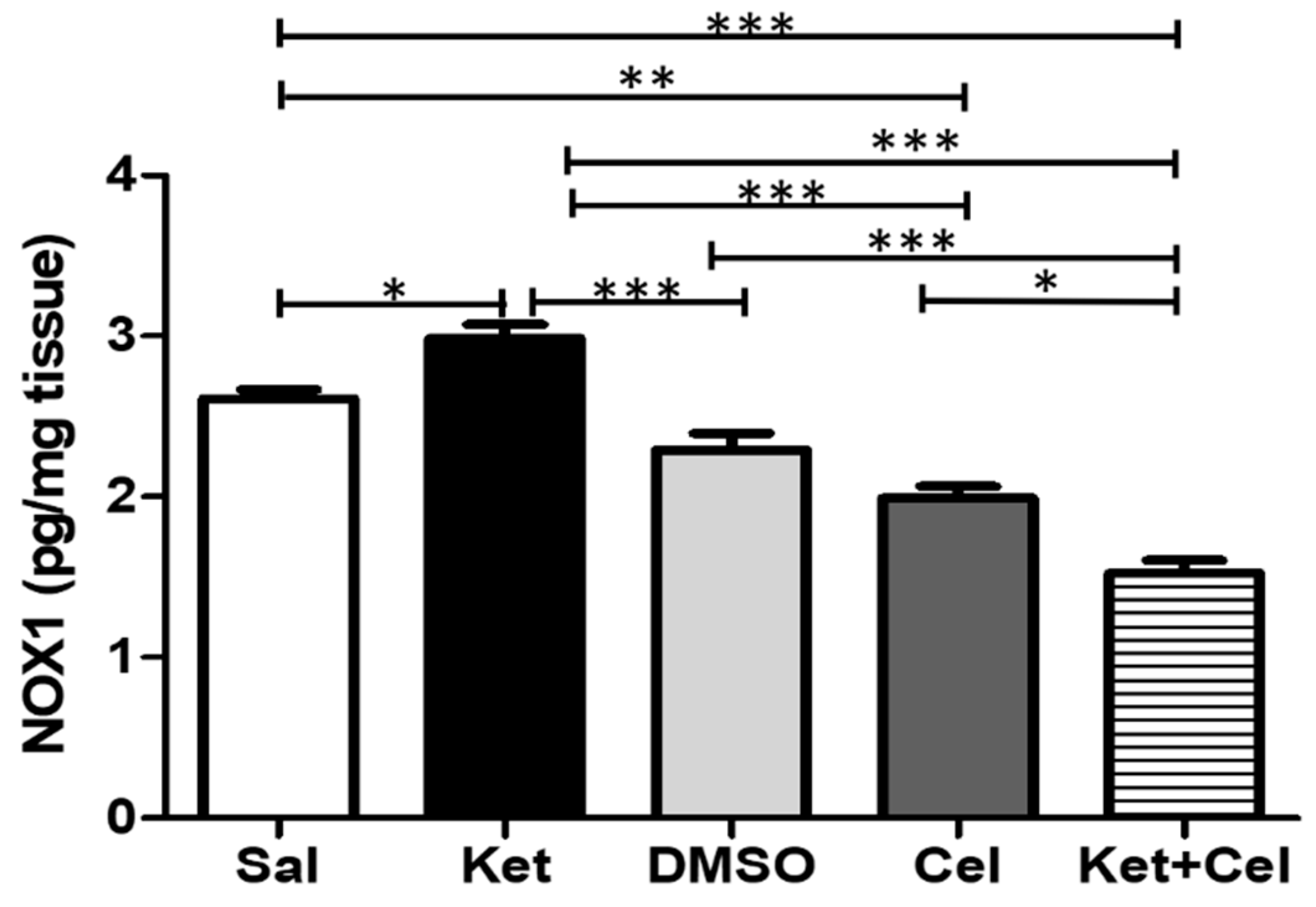
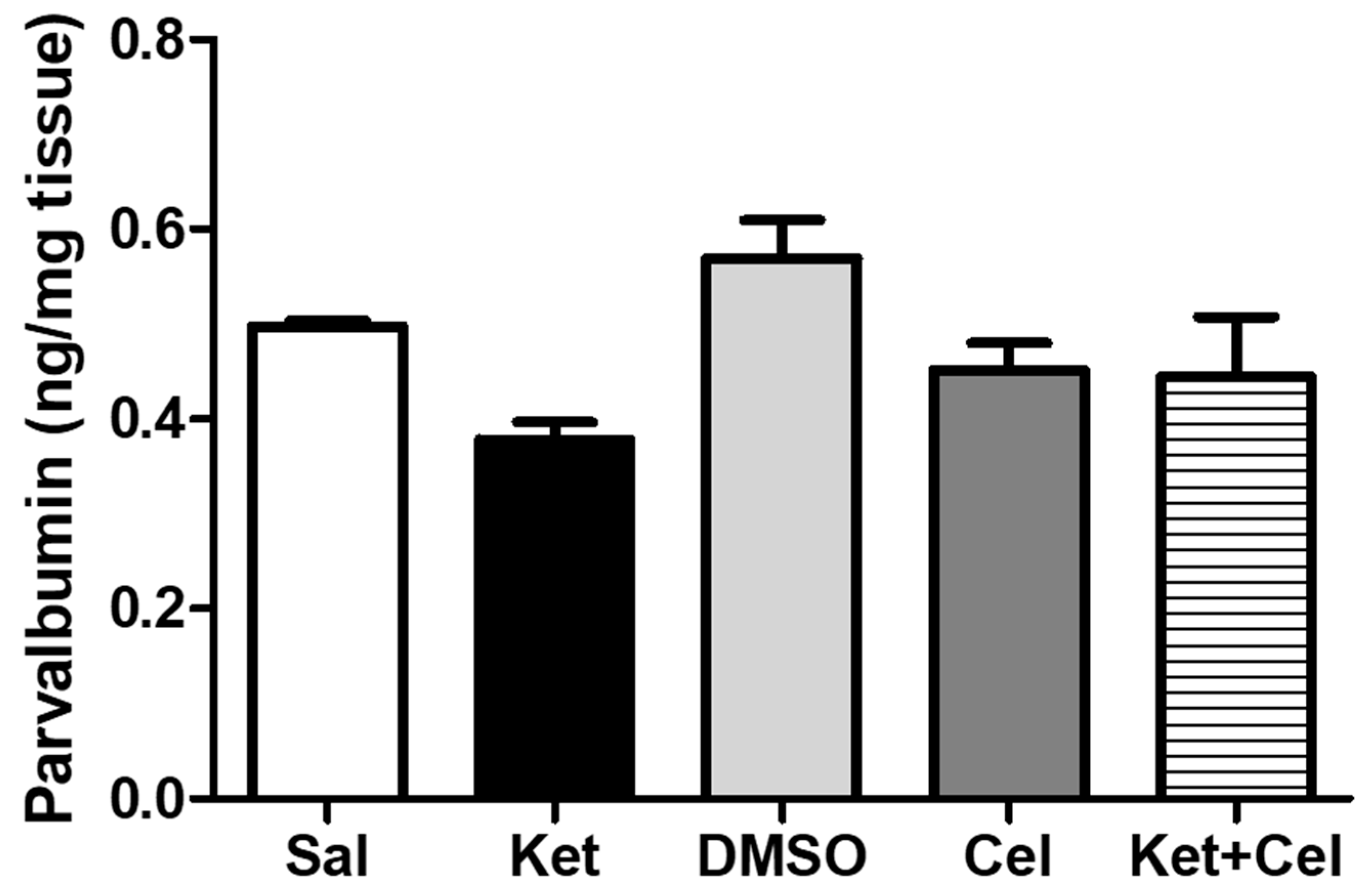
2.5. Early Celastrol Administration Decreased NOX1 Levels in the Cerebellum of Adult Mice Per Se and Following Ketamine Exposure
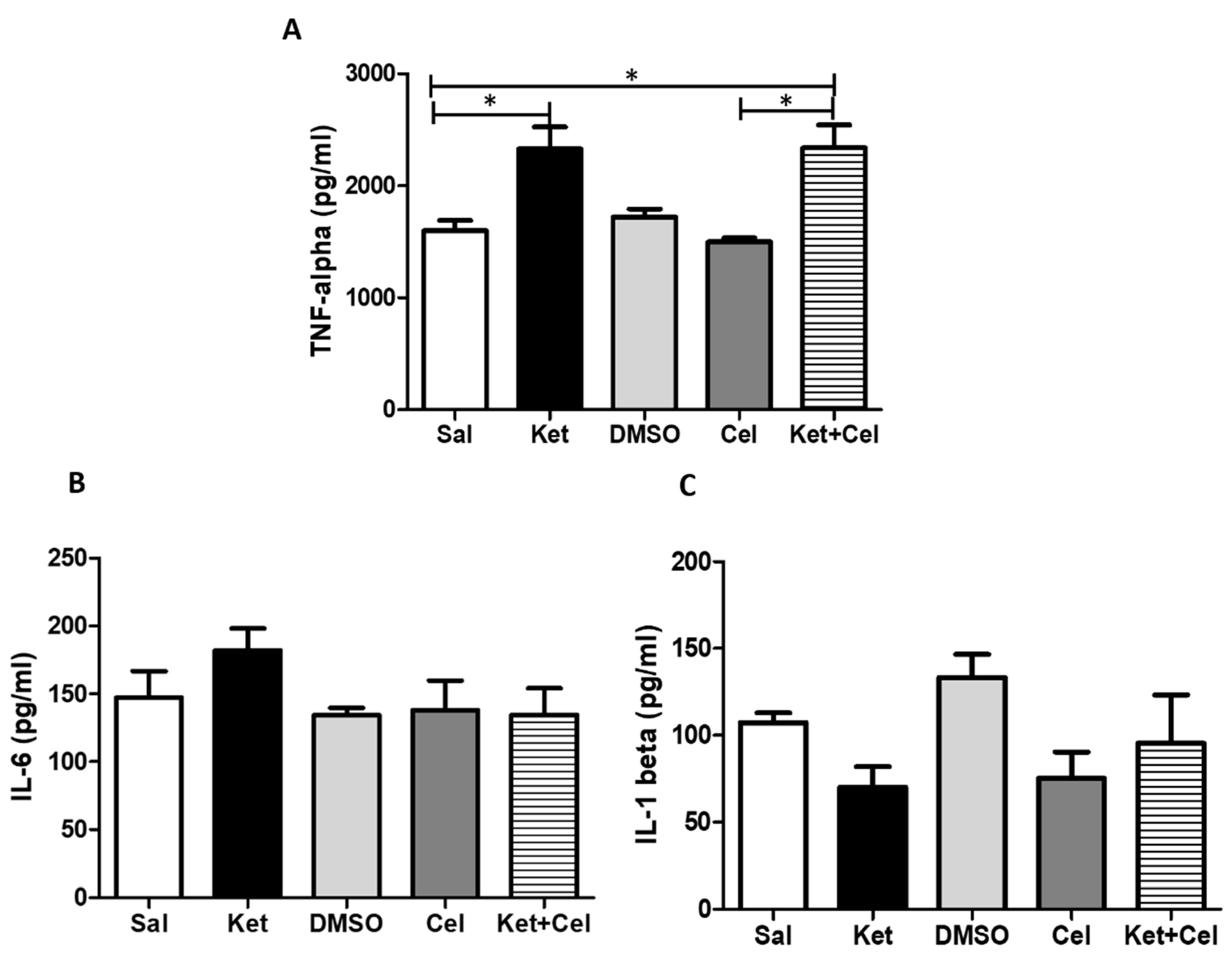
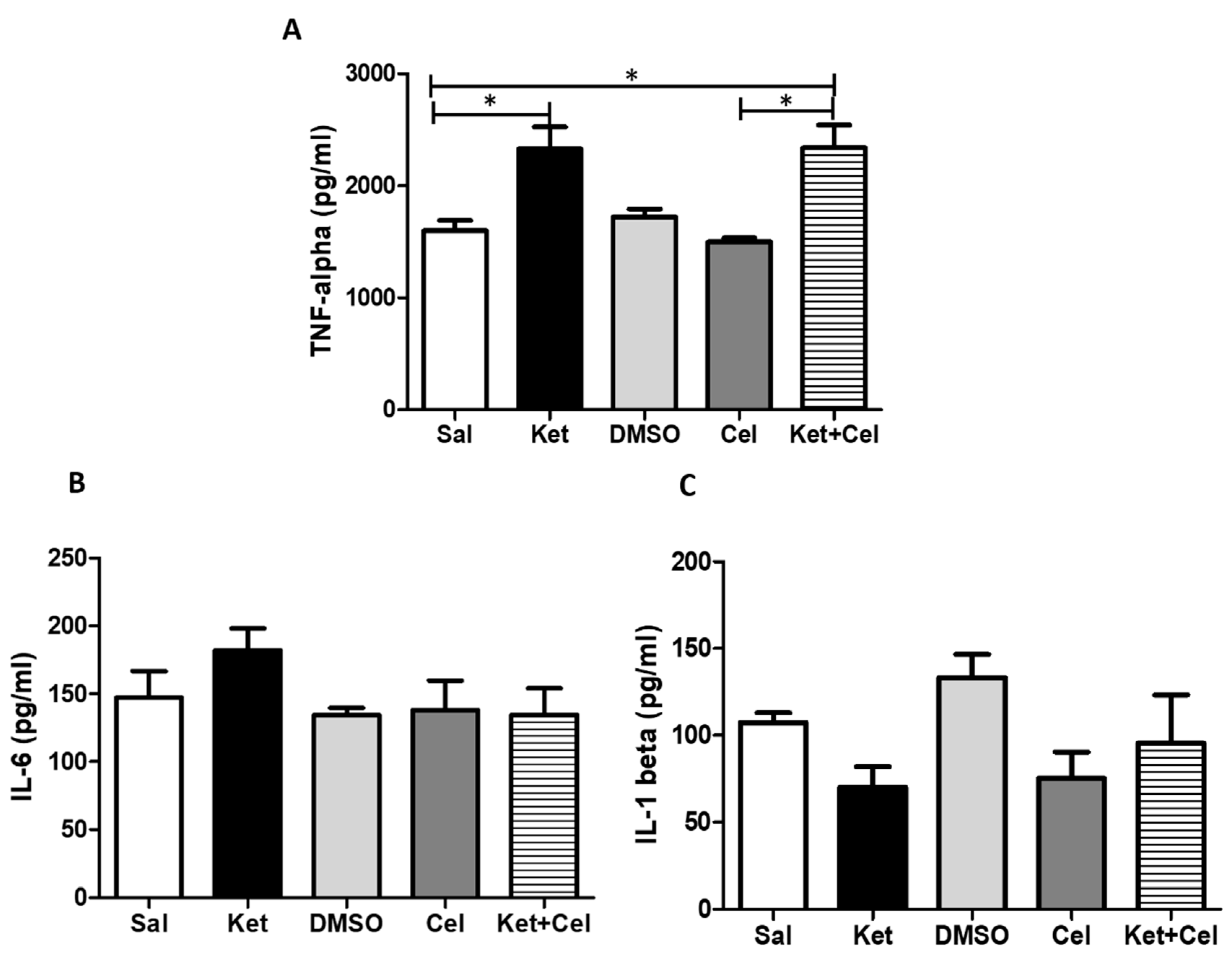
2.6. Early Celastrol Administration Did not Prevent TNF-α Increase in the Cerebellum of Adult Mice Exposed to Ketamine in Postnatal Life
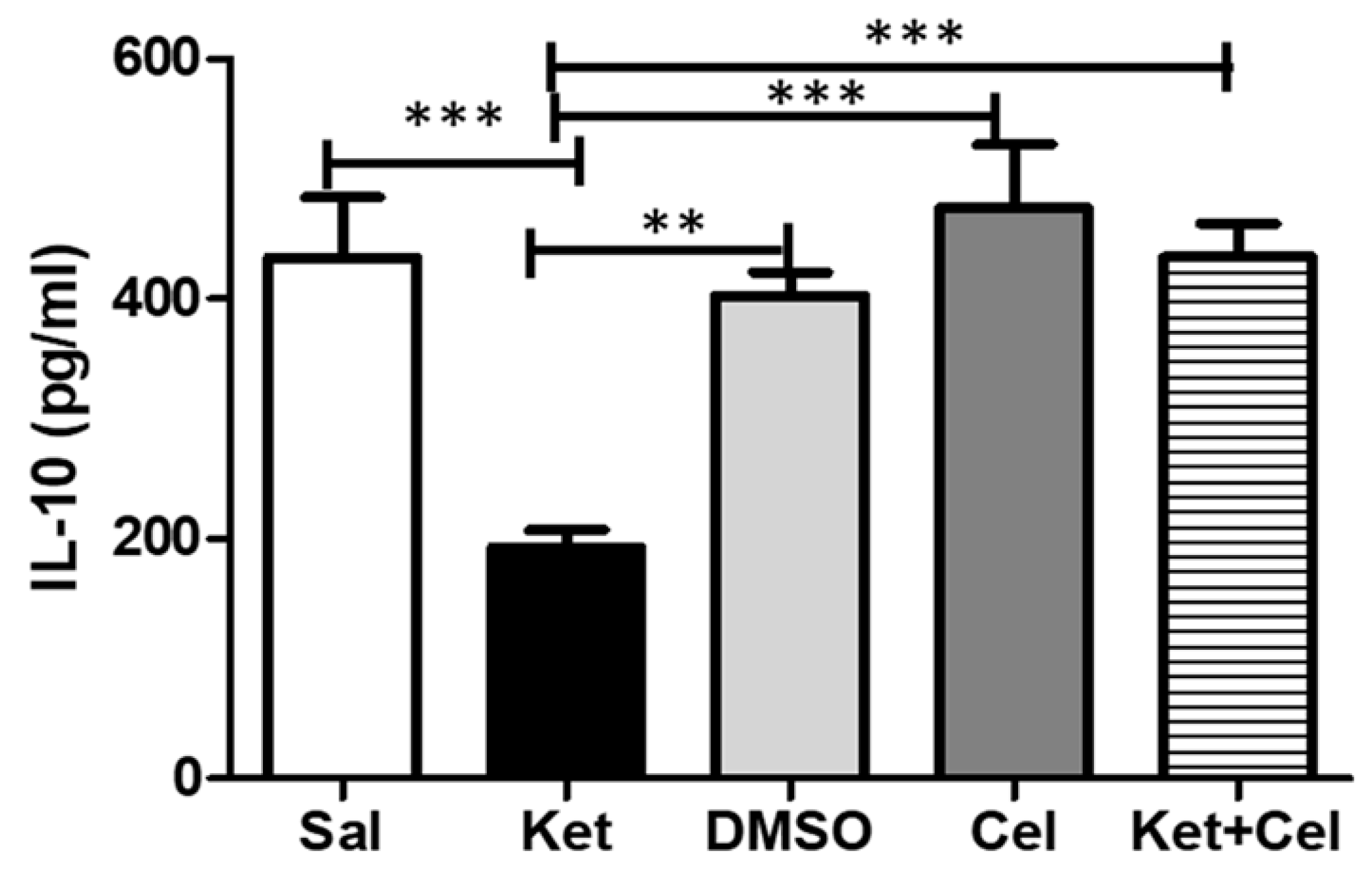
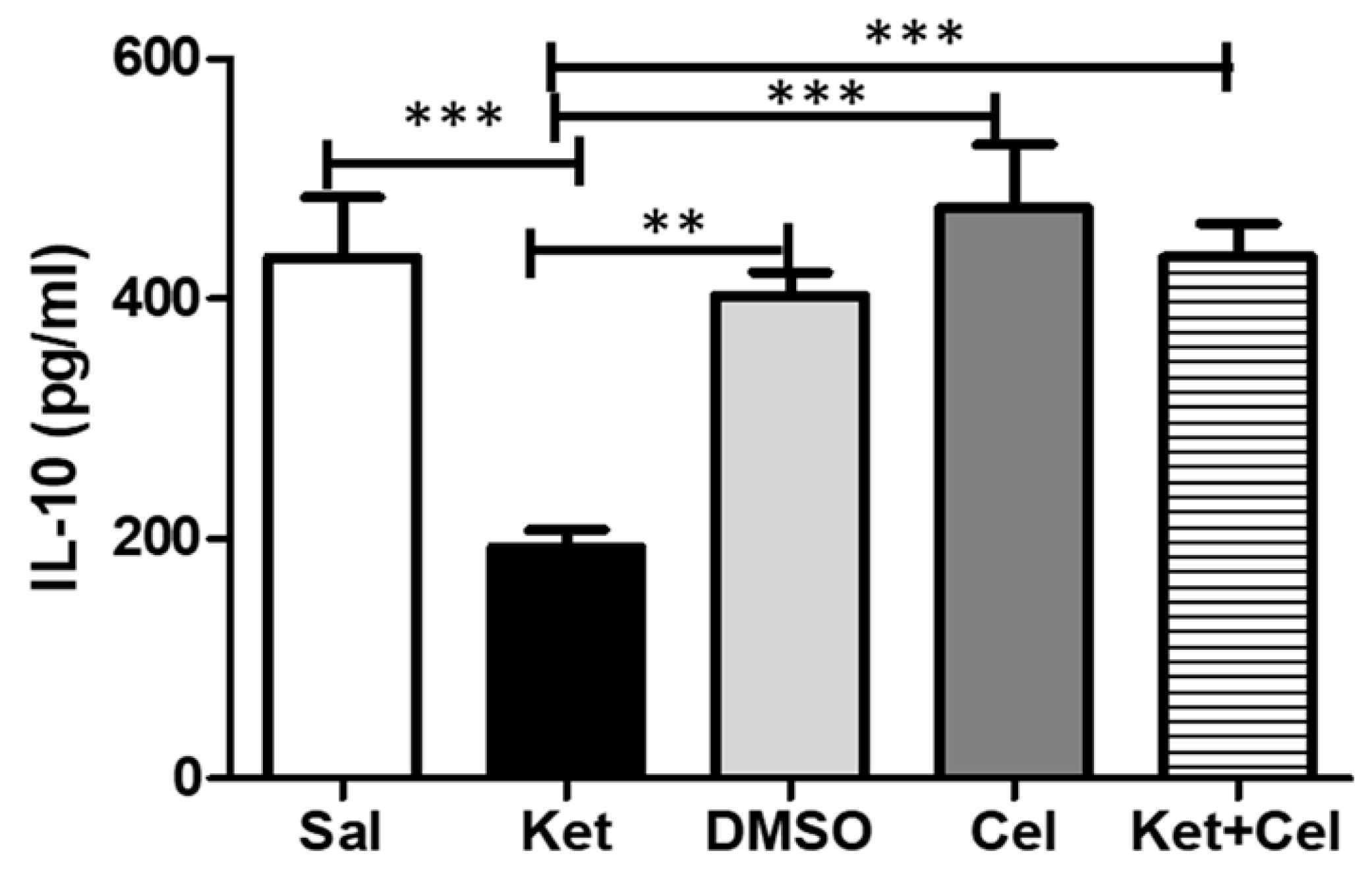
2.7. Early Celastrol Administration Prevented IL-10 Decrease in the Cerebellum of Adult Mice Exposed to Ketamine in Postnatal Life
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Experimental Design
- pups administered with saline (10 mL/kg i.p.);
- pups administered with celastrol (Sigma Aldrich, Milano, Italy; 1 mg/kg i.p., dissolved in 50% DMSO/PBS) [43];
- pups administered with a 50% DMSO/PBS solution (5 mL/kg i.p.)—we have referred to this treatment throughout the text as “DMSO”;
- pups administered with ketamine (30 mg/kg i.p., dissolved in saline, injected in the right side of the peritoneum) and celastrol (1 mg/kg i.p., dissolved in 50% DMSO/PBS, injected in the left side of the peritoneum)—we have referred to this treatment throughout the text as “ketamine + celastrol”.
4.3. Behavioral Tests
4.3.1. NOR Test
4.3.2. OF Test
4.3.3. SI Test
4.4. Enzyme-Linked Immunosorbent Assays (ELISAs)
4.5. Blindness of the Study
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Curran, H.V.; Nutt, D.; de Wit, H. Psychedelics and related drugs: Therapeutic possibilities, mechanisms and regulation. Psychopharmacology 2018, 235, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Vlisides, P.E.; Bel-Bahar, T.; Nelson, A.; Chilton, K.; Smith, E.; Janke, E.; Tarnal, V.; Picton, P.; Harris, R.E.; Mashour, G.A. Subanaesthetic ketamine and altered states of consciousness in humans. Br. J. Anaesth. 2018, 121, 249–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomarol-Clotet, E.; Honey, G.D.; Murray, G.K.; Corlett, P.R.; Absalom, A.R.; Lee, M.; McKenna, P.J.; Bullmore, E.T.; Fletcher, P.C. Psychological effects of ketamine in healthy volunteers. Phenomenological study. Br. J. Psychiatry J. Ment. Sci. 2006, 189, 173–179. [Google Scholar] [CrossRef]
- Frohlich, J.; Van Horn, J.D. Reviewing the ketamine model for schizophrenia. J. Psychopharmacol. 2014, 28, 287–302. [Google Scholar] [CrossRef]
- Powell, S.B.; Zhou, X.; Geyer, M.A. Prepulse inhibition and genetic mouse models of schizophrenia. Behav. Brain Res. 2009, 204, 282–294. [Google Scholar] [CrossRef] [Green Version]
- Watson, D.J.; Marsden, C.A.; Millan, M.J.; Fone, K.C. Blockade of dopamine D(3) but not D(2) receptors reverses the novel object discrimination impairment produced by post-weaning social isolation: Implications for schizophrenia and its treatment. Int. J. Neuropsychopharmacol. Off. Sci. J. Coll. Int. Neuropsychopharmacol. 2012, 15, 471–484. [Google Scholar] [CrossRef]
- Forrest, A.D.; Coto, C.A.; Siegel, S.J. Animal models of psychosis: Current state and future directions. Curr. Behav. Neurosci. Rep. 2014, 1, 100–116. [Google Scholar] [CrossRef]
- Mattei, D.; Schweibold, R.; Wolf, S.A. Brain in flames—Animal models of psychosis: Utility and limitations. Neuropsychiatr. Dis. Treat. 2015, 11, 1313–1329. [Google Scholar]
- Owen, M.J.; O’Donovan, M.C.; Thapar, A.; Craddock, N. Neurodevelopmental hypothesis of schizophrenia. Br. J. Psychiatry J. Ment. Sci. 2011, 198, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Jeevakumar, V.; Driskill, C.; Paine, A.; Sobhanian, M.; Vakil, H.; Morris, B.; Ramos, J.; Kroener, S. Ketamine administration during the second postnatal week induces enduring schizophrenia-like behavioral symptoms and reduces parvalbumin expression in the medial prefrontal cortex of adult mice. Behav. Brain Res. 2015, 282, 165–175. [Google Scholar] [CrossRef]
- Yeganeh-Doost, P.; Gruber, O.; Falkai, P.; Schmitt, A. The role of the cerebellum in schizophrenia: From cognition to molecular pathways. Clinics 2011, 66 (Suppl. S1), 71–77. [Google Scholar] [CrossRef] [PubMed]
- Bernard, J.A.; Orr, J.M.; Mittal, V.A. Cerebello-thalamo-cortical networks predict positive symptom progression in individuals at ultra-high risk for psychosis. Neuroimage. Clin. 2017, 14, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, R. The cerebellum and neuropsychiatric disorders. Psychiatry Res. 2012, 198, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Bernard, J.A.; Orr, J.M.; Dean, D.J.; Mittal, V.A. The cerebellum and learning of non-motor associations in individuals at clinical-high risk for psychosis. Neuroimage Clin. 2018, 19, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Schmahmann, J.D. The cerebellum and cognition. Neurosci. Lett. 2019, 688, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, N.C.; Pierson, R. The role of the cerebellum in schizophrenia. Biol. Psychiatry 2008, 64, 81–88. [Google Scholar] [CrossRef]
- Arasappa, R.; Rao, N.; Venkatasubramanian, G.; Jayakumar, P.; Gangadhar, B. Structural cerebellar abnormalities in antipsychotic-naive schizophrenia: Evidence for cognitive dysmetria. Indian J. Psychol. Med. 2008, 30, 83–89. [Google Scholar] [CrossRef]
- Bielawski, M.; Bondurant, H. Psychosis following a stroke to the cerebellum and midbrain: A case report. Cerebellum Ataxias 2015, 2, 17. [Google Scholar] [CrossRef]
- Schiavone, S.; Sorce, S.; Dubois-Dauphin, M.; Jaquet, V.; Colaianna, M.; Zotti, M.; Cuomo, V.; Trabace, L.; Krause, K.H. Involvement of NOX2 in the development of behavioral and pathologic alterations in isolated rats. Biol. Psychiatry 2009, 66, 384–392. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, X.R.; Wang, J.; Zhang, Z.Z.; Zhao, H.T.; Li, H.H.; Ji, M.H.; Li, K.Y.; Yang, J.J. Reactive oxygen species-mediated loss of phenotype of parvalbumin interneurons contributes to long-term cognitive impairments after repeated neonatal ketamine exposures. Neurotox. Res. 2016, 30, 593–605. [Google Scholar] [CrossRef]
- Sabbagh, J.J.; Murtishaw, A.S.; Bolton, M.M.; Heaney, C.F.; Langhardt, M.; Kinney, J.W. Chronic ketamine produces altered distribution of parvalbumin-positive cells in the hippocampus of adult rats. Neurosci. Lett. 2013, 550, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiavone, S.; Morgese, M.G.; Bove, M.; Colia, A.L.; Maffione, A.B.; Tucci, P.; Trabace, L.; Cuomo, V. Ketamine administration induces early and persistent neurochemical imbalance and altered NADPH oxidase in mice. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 96, 109750. [Google Scholar] [CrossRef] [PubMed]
- Schwaller, B.; Meyer, M.; Schiffmann, S. ‘New’ functions for ‘old’ proteins: The role of the calcium-binding proteins calbindin D-28k, calretinin and parvalbumin, in cerebellar physiology. Studies with knockout mice. Cerebellum 2002, 1, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Bastianelli, E. Distribution of calcium-binding proteins in the cerebellum. Cerebellum 2003, 2, 242–262. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.F.; Yang, L.D.; Sun, X.R.; Zhang, H.; Pan, W.; Wang, X.M.; Yang, J.J.; Ji, M.H.; Yuan, H.M. NOX2 mediated-parvalbumin interneuron loss might contribute to anxiety-like and enhanced fear learning behavior in a rat model of post-traumatic stress disorder. Mol. Neurobiol. 2016, 53, 6680–6689. [Google Scholar] [CrossRef]
- Schiavone, S.; Trabace, L. Pharmacological targeting of redox regulation systems as new therapeutic approach for psychiatric disorders: A literature overview. Pharmacol. Res. 2016, 107, 195–204. [Google Scholar] [CrossRef]
- Barron, H.; Hafizi, S.; Andreazza, A.C.; Mizrahi, R. Neuroinflammation and oxidative stress in psychosis and psychosis risk. Int. J. Mol. Sci. 2017, 18, 651. [Google Scholar] [CrossRef]
- Sorce, S.; Krause, K.H. NOX enzymes in the central nervous system: From signaling to disease. Antioxid. Redox Signal. 2009, 11, 2481–2504. [Google Scholar] [CrossRef]
- Ibi, M.; Liu, J.; Arakawa, N.; Kitaoka, S.; Kawaji, A.; Matsuda, K.I.; Iwata, K.; Matsumoto, M.; Katsuyama, M.; Zhu, K.; et al. Depressive-like behaviors are regulated by NOX1/NADPH oxidase by redox modification of NMDA receptor 1. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 4200–4212. [Google Scholar] [CrossRef]
- Ma, M.W.; Wang, J.; Zhang, Q.; Wang, R.; Dhandapani, K.M.; Vadlamudi, R.K.; Brann, D.W. NADPH oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegener. 2017, 12, 7. [Google Scholar] [CrossRef]
- Schiavone, S.; Jaquet, V.; Sorce, S.; Dubois-Dauphin, M.; Hultqvist, M.; Backdahl, L.; Holmdahl, R.; Colaianna, M.; Cuomo, V.; Trabace, L.; et al. NADPH oxidase elevations in pyramidal neurons drive psychosocial stress-induced neuropathology. Transl. Psychiatry 2012, 2, e111. [Google Scholar] [CrossRef] [PubMed]
- Schiavone, S.; Mhillaj, E.; Neri, M.; Morgese, M.G.; Tucci, P.; Bove, M.; Valentino, M.; Di Giovanni, G.; Pomara, C.; Turillazzi, E.; et al. Early loss of blood-brain barrier integrity precedes NOX2 elevation in the prefrontal cortex of an animal model of psychosis. Mol. Neurobiol. 2017, 54, 2031–2044. [Google Scholar] [CrossRef] [PubMed]
- Sorce, S.; Schiavone, S.; Tucci, P.; Colaianna, M.; Jaquet, V.; Cuomo, V.; Dubois-Dauphin, M.; Trabace, L.; Krause, K.H. The NADPH oxidase NOX2 controls glutamate release: A novel mechanism involved in psychosis-like ketamine responses. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 11317–11325. [Google Scholar] [CrossRef] [PubMed]
- Behrens, M.M.; Ali, S.S.; Dao, D.N.; Lucero, J.; Shekhtman, G.; Quick, K.L.; Dugan, L.L. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science 2007, 318, 1645–1647. [Google Scholar] [CrossRef] [PubMed]
- Behrens, M.M.; Ali, S.S.; Dugan, L.L. Interleukin-6 mediates the increase in NADPH-oxidase in the ketamine model of schizophrenia. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 13957–13966. [Google Scholar] [CrossRef]
- Coyoy, A.; Olguin-Albuerne, M.; Martinez-Briseno, P.; Moran, J. Role of reactive oxygen species and NADPH-oxidase in the development of rat cerebellum. Neurochem. Int. 2013, 62, 998–1011. [Google Scholar] [CrossRef]
- Fraguas, D.; Diaz-Caneja, C.M.; Rodriguez-Quiroga, A.; Arango, C. Oxidative stress and inflammation in early onset first episode psychosis: A systematic review and meta-Analysis. Int. J. Neuropsychopharmacol. Off. Sci. J. Coll. Int. Neuropsychopharmacol. 2017, 20, 435–444. [Google Scholar] [CrossRef]
- Khandaker, G.M.; Cousins, L.; Deakin, J.; Lennox, B.R.; Yolken, R.; Jones, P.B. Inflammation and immunity in schizophrenia: Implications for pathophysiology and treatment. Lancet. Psychiatry 2015, 2, 258–270. [Google Scholar] [CrossRef]
- Kirkpatrick, B.; Miller, B.J. Inflammation and schizophrenia. Schizophr. Bull. 2013, 39, 1174–1179. [Google Scholar] [CrossRef]
- Hagberg, H.; Mallard, C. Effect of inflammation on central nervous system development and vulnerability. Curr. Opin. Neurol. 2005, 18, 117–123. [Google Scholar] [CrossRef]
- Ng, S.W.; Chan, Y.; Chellappan, D.K.; Madheswaran, T.; Zeeshan, F.; Chan, Y.L.; Collet, T.; Gupta, G.; Oliver, B.G.; Wark, P.; et al. Molecular modulators of celastrol as the keystones for its diverse pharmacological activities. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 109, 1785–1792. [Google Scholar] [CrossRef] [PubMed]
- Kiaei, M.; Kipiani, K.; Petri, S.; Chen, J.; Calingasan, N.Y.; Beal, M.F. Celastrol blocks neuronal cell death and extends life in transgenic mouse model of amyotrophic lateral sclerosis. Neuro-Degener. Dis. 2005, 2, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Paris, D.; Ganey, N.J.; Laporte, V.; Patel, N.S.; Beaulieu-Abdelahad, D.; Bachmeier, C.; March, A.; Ait-Ghezala, G.; Mullan, M.J. Reduction of beta-amyloid pathology by celastrol in a transgenic mouse model of Alzheimer’s disease. J. Neuroinflamm. 2010, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.S.; Kim, H.; Lee, H.J.; Sapkota, K.; Park, S.E.; Kim, S.; Kim, S.J. Celastrol from ’Thunder God Vine’ protects SH-SY5Y cells through the preservation of mitochondrial function and inhibition of p38 MAPK in a rotenone model of Parkinson’s disease. Neurochem. Res. 2014, 39, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Tarafdar, A.; Pula, G. The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders. Int. J. Mol. Sci. 2018, 19, E3824. [Google Scholar] [CrossRef]
- Jaquet, V.; Marcoux, J.; Forest, E.; Leidal, K.G.; McCormick, S.; Westermaier, Y.; Perozzo, R.; Plastre, O.; Fioraso-Cartier, L.; Diebold, B.; et al. NADPH oxidase (NOX) isoforms are inhibited by celastrol with a dual mode of action. Br. J. Pharmacol. 2011, 164, 507–520. [Google Scholar] [CrossRef] [Green Version]
- Schiavone, S.; Jaquet, V.; Trabace, L.; Krause, K.H. Severe life stress and oxidative stress in the brain: From animal models to human pathology. Antioxid. Redox Signal. 2013, 18, 1475–1490. [Google Scholar] [CrossRef]
- Kawanishi, S.; Oikawa, S. Mechanism of telomere shortening by oxidative stress. Ann. N. Y. Acad. Sci. 2004, 1019, 278–284. [Google Scholar] [CrossRef]
- Konieczny, J.; Jantas, D.; Lenda, T.; Domin, H.; Czarnecka, A.; Kuter, K.; Smialowska, M.; Lason, W.; Lorenc-Koci, E. Lack of neuroprotective effect of celastrol under conditions of proteasome inhibition by lactacystin in in vitro and in vivo studies: Implications for Parkinson’s disease. Neurotox. Res. 2014, 26, 255–273. [Google Scholar] [CrossRef]
- Cleren, C.; Calingasan, N.Y.; Chen, J.; Beal, M.F. Celastrol protects against MPTP- and 3-nitropropionic acid-induced neurotoxicity. J. Neurochem. 2005, 94, 995–1004. [Google Scholar] [CrossRef]
- Faust, K.; Gehrke, S.; Yang, Y.; Yang, L.; Beal, M.F.; Lu, B. Neuroprotective effects of compounds with antioxidant and anti-inflammatory properties in a Drosophila model of Parkinson’s disease. BMC Neurosci. 2009, 10, 109. [Google Scholar] [CrossRef] [PubMed]
- Brown, I.R. Heat shock proteins and protection of the nervous system. Ann. N. Y. Acad. Sci. 2007, 1113, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Abdin, A.A.; Hasby, E.A. Modulatory effect of celastrol on Th1/Th2 cytokines profile, TLR2 and CD3 + T-lymphocyte expression in a relapsing-remitting model of multiple sclerosis in rats. Eur. J. Pharmacol. 2014, 742, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cao, L.; Xu, L.M.; Cao, F.F.; Peng, B.; Zhang, X.; Shen, Y.F.; Uzan, G.; Zhang, D.H. Celastrol Ameliorates EAE Induction by Suppressing Pathogenic T Cell Responses in the Peripheral and Central Nervous Systems. J. Neuroimmune Pharmacol. Off. J. Soc. Neuroimmune Pharmacol. 2015, 10, 506–516. [Google Scholar] [CrossRef]
- Malkov, A.; Ivanov, A.I.; Latyshkova, A.; Bregestovski, P.; Zilberter, M.; Zilberter, Y. Activation of nicotinamide adenine dinucleotide phosphate oxidase is the primary trigger of epileptic seizures in rodent models. Ann. Neurol. 2019, 85, 907–920. [Google Scholar] [CrossRef]
- Von Ruden, E.L.; Wolf, F.; Gualtieri, F.; Keck, M.; Hunt, C.R.; Pandita, T.K.; Potschka, H. Genetic and pharmacological targeting of heat shock protein 70 in the mouse amygdala-kindling model. ACS Chem. Neurosci. 2019, 10, 1434–1444. [Google Scholar] [CrossRef]
- Jiang, M.; Liu, X.; Zhang, D.; Wang, Y.; Hu, X.; Xu, F.; Jin, M.; Cao, F.; Xu, L. Celastrol treatment protects against acute ischemic stroke-induced brain injury by promoting an IL-33/ST2 axis-mediated microglia/macrophage M2 polarization. J. Neuroinflamm. 2018, 15, 78. [Google Scholar] [CrossRef]
- Li, Y.; He, D.; Zhang, X.; Liu, Z.; Zhang, X.; Dong, L.; Xing, Y.; Wang, C.; Qiao, H.; Zhu, C.; et al. Protective effect of celastrol in rat cerebral ischemia model: Down-regulating p-JNK, p-c-Jun and NF-kappaB. Brain Res. 2012, 1464, 8–13. [Google Scholar] [CrossRef]
- Zhu, F.; Li, C.; Jin, X.P.; Weng, S.X.; Fan, L.L.; Zheng, Z.; Li, W.L.; Wang, F.; Wang, W.F.; Hu, X.F.; et al. Celastrol may have an anti-atherosclerosis effect in a rabbit experimental carotid atherosclerosis model. Int. J. Clin. Exp. Med. 2014, 7, 1684–1691. [Google Scholar]
- Kim, J.Y.; Kim, N.; Zheng, Z.; Lee, J.E.; Yenari, M.A. The 70 kDa heat shock protein protects against experimental traumatic brain injury. Neurobiol. Dis. 2013, 58, 289–295. [Google Scholar] [CrossRef] [Green Version]
- Eroglu, B.; Kimbler, D.E.; Pang, J.; Choi, J.; Moskophidis, D.; Yanasak, N.; Dhandapani, K.M.; Mivechi, N.F. Therapeutic inducers of the HSP70/HSP110 protect mice against traumatic brain injury. J. Neurochem. 2014, 130, 626–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Readhead, B.; Hartley, B.J.; Eastwood, B.J.; Collier, D.A.; Evans, D.; Farias, R.; He, C.; Hoffman, G.; Sklar, P.; Dudley, J.T.; et al. Expression-based drug screening of neural progenitor cells from individuals with schizophrenia. Nat. Commun. 2018, 9, 4412. [Google Scholar] [CrossRef] [PubMed]
- Cheung, H.M.; Yew, D.T.W. Effects of perinatal exposure to ketamine on the developing brain. Front. Neurosci. 2019, 13, 138. [Google Scholar] [CrossRef] [PubMed]
- Coronel-Oliveros, C.M.; Pacheco-Calderon, R. Prenatal exposure to ketamine in rats: Implications on animal models of schizophrenia. Dev. Psychobiol. 2018, 60, 30–42. [Google Scholar] [CrossRef]
- Liao, W.T.; Xiao, X.Y.; Zhu, Y.; Zhou, S.P. The effect of celastrol on learning and memory in diabetic rats after sevoflurane inhalation. Arch. Med Sci. AMS 2018, 14, 370–380. [Google Scholar] [CrossRef] [Green Version]
- Hooper, P.L.; Durham, H.D.; Torok, Z.; Hooper, P.L.; Crul, T.; Vigh, L. The central role of heat shock factor 1 in synaptic fidelity and memory consolidation. Cell Stress Chaperones 2016, 21, 745–753. [Google Scholar] [CrossRef] [Green Version]
- Barker-Haliski, M.L.; Loscher, W.; White, H.S.; Galanopoulou, A.S. Neuroinflammation in epileptogenesis: Insights and translational perspectives from new models of epilepsy. Epilepsia 2017, 58 (Suppl. S3), 39–47. [Google Scholar] [CrossRef] [Green Version]
- Wesson, D.W. Sniffing behavior communicates social hierarchy. Curr. Biol. 2013, 23, 575–580. [Google Scholar] [CrossRef]
- Lee, P.R.; Brady, D.L.; Shapiro, R.A.; Dorsa, D.M.; Koenig, J.I. Social interaction deficits caused by chronic phencyclidine administration are reversed by oxytocin. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2005, 30, 1883–1894. [Google Scholar] [CrossRef]
- Snigdha, S.; Neill, J.C. Efficacy of antipsychotics to reverse phencyclidine-induced social interaction deficits in female rats--a preliminary investigation. Behav. Brain Res. 2008, 187, 489–494. [Google Scholar] [CrossRef]
- Bozdagi, O.; Sakurai, T.; Papapetrou, D.; Wang, X.; Dickstein, D.L.; Takahashi, N.; Kajiwara, Y.; Yang, M.; Katz, A.M.; Scattoni, M.L.; et al. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol. Autism 2010, 1, 15. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Peters, B.; Schroeder, H.; Mann, T.; Huether, G.; Grecksch, G. Ketamine-induced changes in rat behaviour: A possible animal model of schizophrenia. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2003, 27, 687–700. [Google Scholar] [CrossRef]
- Schweinfurth, M.K.; Stieger, B.; Taborsky, M. Experimental evidence for reciprocity in allogrooming among wild-type Norway rats. Sci. Rep. 2017, 7, 4010. [Google Scholar] [CrossRef] [PubMed]
- Alleva, E. 7—Assessment of Aggressive Behavior in Rodents. In Methods in Neurosciences; Conn, P.M., Ed.; Academic Press: Cambridge, MA, USA, 1993; Volume 14, pp. 111–137. [Google Scholar]
- Shin, S.Y.; Baek, N.J.; Han, S.H.; Min, S.S. Chronic administration of ketamine ameliorates the anxiety- and aggressive-like behavior in adolescent mice induced by neonatal maternal separation. Korean J. Physiol. Pharmacol. Off. J. Korean Physiol. Soc. Korean Soc. Pharmacol. 2019, 23, 81–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hira, S.; Saleem, U.; Anwar, F.; Ahmad, B. Antioxidants Attenuate Isolation- and L-DOPA-Induced Aggression in Mice. Front. Pharmacol. 2017, 8, 945. [Google Scholar] [CrossRef] [PubMed]
- Garratt, M.; Brooks, R.C. A genetic reduction in antioxidant function causes elevated aggression in mice. J. Exp. Biol. 2015, 218 Pt 2, 223–227. [Google Scholar] [CrossRef]
- Kim, T.; Lee, K.H.; Oh, H.; Lee, T.Y.; Cho, K.I.K.; Lee, J.; Kwon, J.S. Cerebellar structural abnormalities associated with cognitive function in patients with first-episode psychosis. Front. Psychiatry 2018, 9, 286. [Google Scholar] [CrossRef]
- Moberget, T.; Ivry, R.B. Prediction, Psychosis, and the Cerebellum. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2019, 4, 820–831. [Google Scholar] [CrossRef]
- Jones, C.A.; Watson, D.J.; Fone, K.C. Animal models of schizophrenia. Br. J. Pharmacol. 2011, 164, 1162–1194. [Google Scholar] [CrossRef]
- Shinn, A.K.; Roh, Y.S.; Ravichandran, C.T.; Baker, J.T.; Ongur, D.; Cohen, B.M. Aberrant cerebellar connectivity in bipolar disorder with psychosis. Biol. Psychiatry. Cogn. Neurosci. Neuroimaging 2017, 2, 438–448. [Google Scholar] [CrossRef]
- Yadav, M.; Parle, M.; Jindal, D.K.; Dhingra, S. Protective effects of stigmasterol against ketamine-induced psychotic symptoms: Possible behavioral, biochemical and histopathological changes in mice. Pharmacol. Rep. Pr 2018, 70, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.; Jindal, D.K.; Dhingra, M.S.; Kumar, A.; Parle, M.; Dhingra, S. Protective effect of gallic acid in experimental model of ketamine-induced psychosis: Possible behaviour, biochemical, neurochemical and cellular alterations. Inflammopharmacology 2018, 26, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Brambrink, A.M.; Evers, A.S.; Avidan, M.S.; Farber, N.B.; Smith, D.J.; Martin, L.D.; Dissen, G.A.; Creeley, C.E.; Olney, J.W. Ketamine-induced neuroapoptosis in the fetal and neonatal rhesus macaque brain. Anesthesiology 2012, 116, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Tobe, E.H. Mitochondrial dysfunction, oxidative stress, and major depressive disorder. Neuropsychiatr. Dis. Treat. 2013, 9, 567–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Cheng, L.; Craig, D.W.; Redman, M.; Liu, C. Cerebellar telomere length and psychiatric disorders. Behav. Genet. 2010, 40, 250–254. [Google Scholar] [CrossRef]
- Filiou, M.D.; Teplytska, L.; Otte, D.M.; Zimmer, A.; Turck, C.W. Myelination and oxidative stress alterations in the cerebellum of the G72/G30 transgenic schizophrenia mouse model. J. Psychiatr. Res. 2012, 46, 1359–1365. [Google Scholar] [CrossRef]
- Streck, E.L.; Rezin, G.T.; Barbosa, L.M.; Assis, L.C.; Grandi, E.; Quevedo, J. Effect of antipsychotics on succinate dehydrogenase and cytochrome oxidase activities in rat brain. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2007, 376, 127–133. [Google Scholar] [CrossRef]
- Assis, L.C.; Scaini, G.; Di-Pietro, P.B.; Castro, A.A.; Comim, C.M.; Streck, E.L.; Quevedo, J. Effect of antipsychotics on creatine kinase activity in rat brain. Basic Clin. Pharmacol. Toxicol. 2007, 101, 315–319. [Google Scholar] [CrossRef]
- Cao, H.; Chen, O.Y.; Chung, Y.; Forsyth, J.K.; McEwen, S.C.; Gee, D.G.; Bearden, C.E.; Addington, J.; Goodyear, B.; Cadenhead, K.S.; et al. Cerebello-thalamo-cortical hyperconnectivity as a state-independent functional neural signature for psychosis prediction and characterization. Nat. Commun. 2018, 9, 3836. [Google Scholar] [CrossRef]
- Gu, F.; Chauhan, V.; Chauhan, A. Impaired synthesis and antioxidant defense of glutathione in the cerebellum of autistic subjects: Alterations in the activities and protein expression of glutathione-related enzymes. Free Radic. Biol. Med. 2013, 65, 488–496. [Google Scholar] [CrossRef]
- Olguin-Albuerne, M.; Moran, J. ROS produced by NOX2 control in vitro development of cerebellar granule neurons development. ASN Neuro 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Coyoy, A.; Valencia, A.; Guemez-Gamboa, A.; Moran, J. Role of NADPH oxidase in the apoptotic death of cultured cerebellar granule neurons. Free Radic. Biol. Med. 2008, 45, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Sorce, S.; Nuvolone, M.; Keller, A.; Falsig, J.; Varol, A.; Schwarz, P.; Bieri, M.; Budka, H.; Aguzzi, A. The role of the NADPH oxidase NOX2 in prion pathogenesis. PLoS Pathog. 2014, 10, e1004531. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, A.; Ahmad, S.F.; Al-Harbi, N.O.; Attia, S.M.; Alshammari, M.A.; Alzahrani, K.S.; Bakheet, S.A. Increased oxidative stress in the cerebellum and peripheral immune cells leads to exaggerated autism-like repetitive behavior due to deficiency of antioxidant response in BTBR T + tf/J mice. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 89, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Cristovao, A.C.; Guhathakurta, S.; Bok, E.; Je, G.; Yoo, S.D.; Choi, D.H.; Kim, Y.S. NADPH oxidase 1 mediates alpha-synucleinopathy in Parkinson’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 14465–14477. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Sun, Q.; Chen, S. Oxidative stress: A major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog. Neurobiol. 2016, 147, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Belarbi, K.; Cuvelier, E.; Destée, A.; Gressier, B.; Chartier-Harlin, M.C. NADPH oxidases in Parkinson’s disease: A systematic review. Mol. Neurodegener. 2017, 12, 84. [Google Scholar] [CrossRef]
- Cheret, C.; Gervais, A.; Lelli, A.; Colin, C.; Amar, L.; Ravassard, P.; Mallet, J.; Cumano, A.; Krause, K.H.; Mallat, M. Neurotoxic activation of microglia is promoted by a nox1-dependent NADPH oxidase. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 12039–12051. [Google Scholar] [CrossRef]
- Nakki, R.; Nickolenko, J.; Chang, J.; Sagar, S.M.; Sharp, F.R. Haloperidol prevents ketamine- and phencyclidine-induced HSP70 protein expression but not microglial activation. Exp. Neurol. 1996, 137, 234–241. [Google Scholar] [CrossRef]
- Vasconcellos, L.R.; Dutra, F.F.; Siqueira, M.S.; Paula-Neto, H.A.; Dahan, J.; Kiarely, E.; Carneiro, L.A.; Bozza, M.T.; Travassos, L.H. Protein aggregation as a cellular response to oxidative stress induced by heme and iron. Proc. Natl. Acad. Sci. USA 2016, 113, E7474–E7482. [Google Scholar] [CrossRef]
- Chen, F.; Pandey, D.; Chadli, A.; Catravas, J.D.; Chen, T.; Fulton, D.J. Hsp90 regulates NADPH oxidase activity and is necessary for superoxide but not hydrogen peroxide production. Antioxid. Redox Signal. 2011, 14, 2107–2119. [Google Scholar] [CrossRef] [PubMed]
- Troyanova, N.I.; Shevchenko, M.A.; Boyko, A.A.; Mirzoyev, R.R.; Pertseva, M.A.; Kovalenko, E.I.; Sapozhnikov, A.M. Modulating effect of extracellular HSP70 on generation of reactive oxigen species in populations of phagocytes. Bioorganicheskaia Khimiia 2015, 41, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Boczek, T.; Lisek, M.; Ferenc, B.; Wiktorska, M.; Ivchevska, I.; Zylinska, L. Region-specific effects of repeated ketamine administration on the presynaptic GABAergic neurochemistry in rat brain. Neurochem. Int. 2015, 91, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Shi, C.; Yang, X.; Yang, M.; Sun, H.; Wang, C. Celastrol suppresses obesity process via increasing antioxidant capacity and improving lipid metabolism. Eur. J. Pharmacol. 2014, 744, 52–58. [Google Scholar] [CrossRef]
- Divya, T.; Dineshbabu, V.; Soumyakrishnan, S.; Sureshkumar, A.; Sudhandiran, G. Celastrol enhances Nrf2 mediated antioxidant enzymes and exhibits anti-fibrotic effect through regulation of collagen production against bleomycin-induced pulmonary fibrosis. Chem. Biol. Interact. 2016, 246, 52–62. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, X.; Zhao, M.; Wang, Y.; Cheng, X.; Wang, D.; Xu, Y.; Du, Z.; Yu, X. Celastrol targets mitochondrial respiratory chain complex I to induce reactive oxygen species-dependent cytotoxicity in tumor cells. BMC Cancer 2011, 11, 170. [Google Scholar] [CrossRef]
- Braun, I.; Genius, J.; Grunze, H.; Bender, A.; Möller, H.J.; Rujescu, D. Alterations of hippocampal and prefrontal GABAergic interneurons in an animal model of psychosis induced by NMDA receptor antagonism. Schizophr. Res. 2007, 97, 254–263. [Google Scholar] [CrossRef]
- Schiavone, S.; Neri, M.; Trabace, L.; Turillazzi, E. The NADPH oxidase NOX2 mediates loss of parvalbumin interneurons in traumatic brain injury: Human autoptic immunohistochemical evidence. Sci. Rep. 2017, 7, 8752. [Google Scholar] [CrossRef]
- Meyer, U.; Feldon, J.; Dammann, O. Schizophrenia and autism: Both shared and disorder-specific pathogenesis via perinatal inflammation? Pediatric Res. 2011, 69, 26–33. [Google Scholar] [CrossRef]
- Wang, H.; Guo, W.; Liu, F.; Wang, G.; Lyu, H.; Wu, R.; Chen, J.; Wang, S.; Li, L.; Zhao, J. Patients with first-episode, drug-naive schizophrenia and subjects at ultra-high risk of psychosis shared increased cerebellar-default mode network connectivity at rest. Sci. Rep. 2016, 6, 26124. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Zhou, J.; Xia, Y. The role of TNF-α in regulating ketamine-induced hippocampal neurotoxicity. Arch. Med. Sci. AMS 2015, 11, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shen, R.; Wen, G.; Ding, R.; Du, A.; Zhou, J.; Dong, Z.; Ren, X.; Yao, H.; Zhao, R.; et al. Effects of ketamine on levels of inflammatory cytokines IL-6, IL-1beta, and TNF-alpha in the hippocampus of mice following acute or chronic administration. Front. Pharmacol. 2017, 8, 139. [Google Scholar] [PubMed]
- Onuki, Y.; Van Someren, E.J.W.; De Zeeuw, C.I.; Van der Werf, Y.D. Hippocampal–cerebellar interaction during spatio-temporal prediction. Cereb. Cortex 2013, 25, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Babayan, B.M.; Watilliaux, A.; Viejo, G.; Paradis, A.-L.; Girard, B.; Rondi-Reig, L. A hippocampo-cerebellar centred network for the learning and execution of sequence-based navigation. Sci. Rep. 2017, 7, 17812. [Google Scholar] [CrossRef] [PubMed]
- Decourt, B.; Lahiri, D.K.; Sabbagh, M.N. Targeting tumor necrosis factor alpha for Alzheimer’s disease. Curr. Alzheimer Res. 2017, 14, 412–425. [Google Scholar] [CrossRef]
- Allison, A.; Cacabelos, R.; Lombardi, V.; Alvarez, X.; Vigo, C. Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer’s disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2001, 25, 1341–1357. [Google Scholar] [CrossRef]
- Müller, N. Inflammation in Schizophrenia: Pathogenetic Aspects and Therapeutic Considerations. Schizophr. Bull. 2018, 44, 973–982. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-K.; Jung, H.-G.; Myint, A.-M.; Kim, H.; Park, S.-H. Imbalance between pro-inflammatory and anti-inflammatory cytokines in bipolar disorder. J. Affect. Disord. 2007, 104, 91–95. [Google Scholar] [CrossRef]
- Garcia, J.M.; Stillings, S.A.; Leclerc, J.L.; Phillips, H.; Edwards, N.J.; Robicsek, S.A.; Hoh, B.L.; Blackburn, S.; Dore, S. Role of Interleukin-10 in Acute Brain Injuries. Front. Neurol. 2017, 8, 244. [Google Scholar] [CrossRef]
- Stoll, G.; Jander, S.; Schroeter, M. Cytokines in CNS Disorders: Neurotoxicity versus Neuroprotection; Advances in Dementia Research, Vienna, 2000//; Jellinger, K., Schmidt, R., Windisch, M., Eds.; Springer Vienna: Vienna, Austria, 2000; pp. 81–89. [Google Scholar]
- Zhu, Y.; Chen, X.; Liu, Z.; Peng, Y.P.; Qiu, Y.H. Interleukin-10 Protection against lipopolysaccharide-induced neuro-inflammation and neurotoxicity in ventral mesencephalic cultures. Int. J. Mol. Sci. 2015, 17, 25. [Google Scholar] [CrossRef]
- Bachis, A.; Colangelo, A.M.; Vicini, S.; Doe, P.P.; De Bernardi, M.A.; Brooker, G.; Mocchetti, I. Interleukin-10 prevents glutamate-mediated cerebellar granule cell death by blocking caspase-3-like activity. J. Neurosci. 2001, 21, 3104–3112. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.H.; Park, E.; You, B.; Jung, Y.; Park, A.R.; Park, S.G.; Lee, J.R. Neuronal synapse formation induced by microglia and interleukin 10. PLoS ONE 2013, 8, e81218. [Google Scholar] [CrossRef] [PubMed]
- Lueptow, L.M. Novel object recognition test for the investigation of learning and memory in mice. J. Vis. Exp. Jove 2017, 126, e55718. [Google Scholar] [CrossRef] [PubMed]
- Trabace, L.; Cassano, T.; Colaianna, M.; Castrignano, S.; Giustino, A.; Amoroso, S.; Steardo, L.; Cuomo, V. Neurochemical and neurobehavioral effects of ganstigmine (CHF2819), a novel acetylcholinesterase inhibitor, in rat prefrontal cortex: An in vivo study. Pharmacol. Res. 2007, 56, 288–294. [Google Scholar] [CrossRef]
- Carratu, M.R.; Borracci, P.; Coluccia, A.; Giustino, A.; Renna, G.; Tomasini, M.C.; Raisi, E.; Antonelli, T.; Cuomo, V.; Mazzoni, E.; et al. Acute exposure to methylmercury at two developmental windows: Focus on neurobehavioral and neurochemical effects in rat offspring. Neuroscience 2006, 141, 1619–1629. [Google Scholar] [CrossRef]
- Nogueira Neto, J.D.; de Almeida, A.A.; da Silva Oliveira, J.; Dos Santos, P.S.; de Sousa, D.P.; de Freitas, R.M. Antioxidant effects of nerolidol in mice hippocampus after open field test. Neurochem. Res. 2013, 38, 1861–1870. [Google Scholar] [CrossRef]
- Fortes, A.C.; Almeida, A.A.; Mendonca-Junior, F.J.; Freitas, R.M.; Soares-Sobrinho, J.L.; de La Roca Soares, M.F. Anxiolytic properties of new chemical entity, 5TIO1. Neurochem. Res. 2013, 38, 726–731. [Google Scholar] [CrossRef]
- Crawley, J.N.; Chen, T.; Puri, A.; Washburn, R.; Sullivan, T.L.; Hill, J.M.; Young, N.B.; Nadler, J.J.; Moy, S.S.; Young, L.J.; et al. Social approach behaviors in oxytocin knockout mice: Comparison of two independent lines tested in different laboratory environments. Neuropeptides 2007, 41, 145–163. [Google Scholar] [CrossRef]
- Silverman, J.L.; Turner, S.M.; Barkan, C.L.; Tolu, S.S.; Saxena, R.; Hung, A.Y.; Sheng, M.; Crawley, J.N. Sociability and motor functions in Shank1 mutant mice. Brain Res. 2011, 1380, 120–137. [Google Scholar] [CrossRef] [Green Version]
- Kaidanovich-Beilin, O.; Lipina, T.; Vukobradovic, I.; Roder, J.; Woodgett, J.R. Assessment of social interaction behaviors. J. Vis. Exp. Jove 2011, 25, e2473. [Google Scholar] [CrossRef]
- Schiavone, S.; Tucci, P.; Mhillaj, E.; Bove, M.; Trabace, L.; Morgese, M.G. Antidepressant drugs for beta amyloid-induced depression: A new standpoint? Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 78, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Morgese, M.G.; Tucci, P.; Mhillaj, E.; Bove, M.; Schiavone, S.; Trabace, L.; Cuomo, V. Lifelong nutritional omega-3 deficiency evokes depressive-like state through soluble beta amyloid. Mol. Neurobiol. 2017, 54, 2079–2089. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: All the compounds used in this work are commercially available. Details about selling companies are provided in Materials and Methods. |




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schiavone, S.; Tucci, P.; Trabace, L.; Morgese, M.G. Early Celastrol Administration Prevents Ketamine-Induced Psychotic-Like Behavioral Dysfunctions, Oxidative Stress and IL-10 Reduction in The Cerebellum of Adult Mice. Molecules 2019, 24, 3993. https://doi.org/10.3390/molecules24213993
Schiavone S, Tucci P, Trabace L, Morgese MG. Early Celastrol Administration Prevents Ketamine-Induced Psychotic-Like Behavioral Dysfunctions, Oxidative Stress and IL-10 Reduction in The Cerebellum of Adult Mice. Molecules. 2019; 24(21):3993. https://doi.org/10.3390/molecules24213993
Chicago/Turabian StyleSchiavone, Stefania, Paolo Tucci, Luigia Trabace, and Maria Grazia Morgese. 2019. "Early Celastrol Administration Prevents Ketamine-Induced Psychotic-Like Behavioral Dysfunctions, Oxidative Stress and IL-10 Reduction in The Cerebellum of Adult Mice" Molecules 24, no. 21: 3993. https://doi.org/10.3390/molecules24213993