Bioluminescence Resonance Energy Transfer as a Method to Study Protein-Protein Interactions: Application to G Protein Coupled Receptor Biology


Abstract
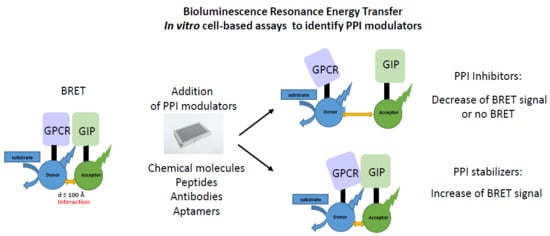
:
1. Introduction
2. BRET: An Overview of Developed Systems
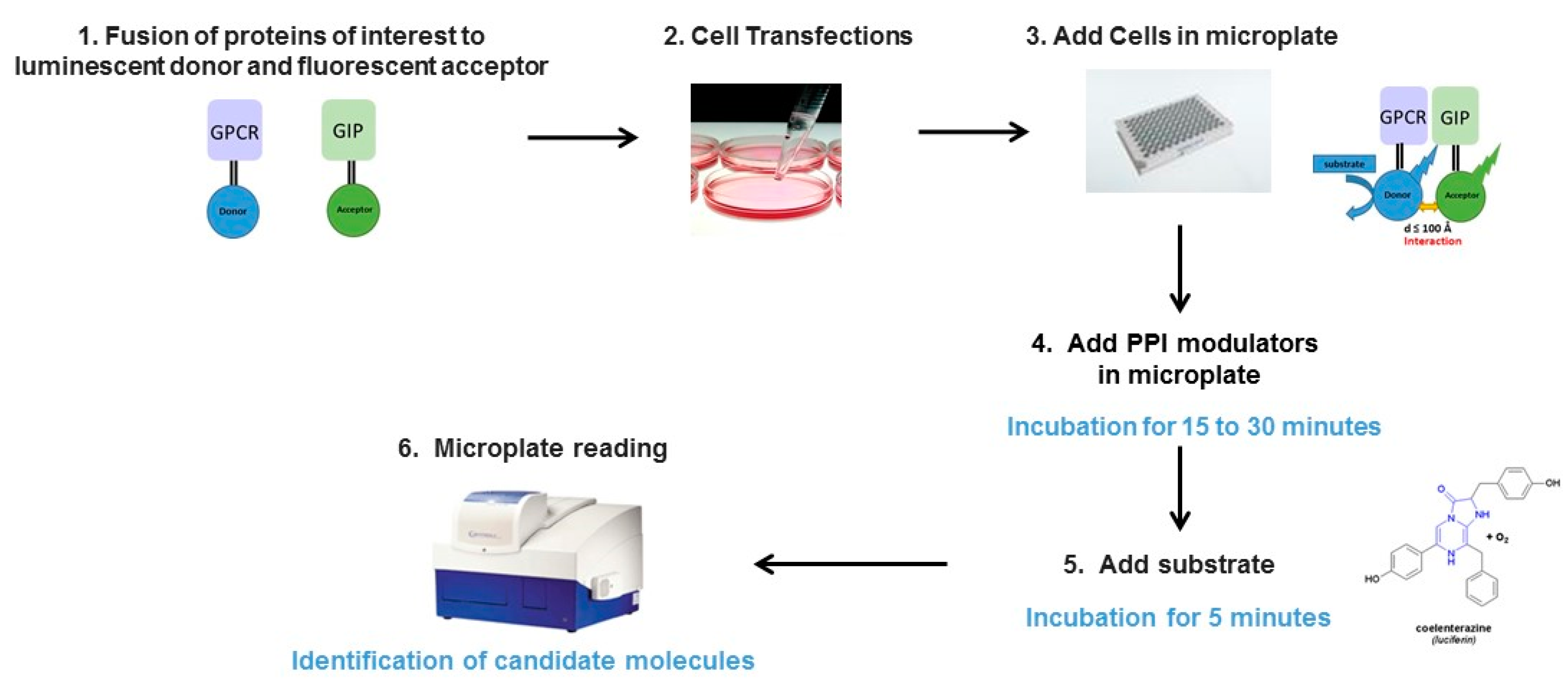
2.1. Principle of the Method
2.2. Developed BRET Systems
2.2.1. BRET 1
2.2.2. BRET 2
2.2.3. BRET 3
2.2.4. NanoBRET
2.2.5. Quantum Dot-Based BRET (QD-BRET)
3. G protein Coupled Receptors Form Molecular Complexes
3.1. GPCR Form Dimers/Oligomers
3.2. GPCR Signaling through G Proteins
3.3. GPCR Interact with Multiprotein Complexex
4. Tools to Target GPCR/Protein Interaction
4.1. Small Molecules Libraries
4.2. Peptides/Peptidomimetics
4.3. Antibodies and Nanobodies
4.4. Nuclei Acid Aptamers
5. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Bader, S.; Kuhner, S.; Gavin, A.C. Interaction networks for systems biology. FEBS Lett. 2008, 582, 1220–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janin, J.; Bahadur, R.P.; Chakrabarti, P. Protein-protein interaction and quaternary structure. Q. Rev. Biophys. 2008, 41, 133–180. [Google Scholar] [CrossRef] [PubMed]
- Arkin, M.R.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing towards the dream. Nat. Rev. Drug Discov. 2004, 3, 301–317. [Google Scholar] [PubMed]
- Keskin, O.; Gursoy, A.; Ma, B.; Nussinov, R. Principles of protein-protein interactions: What are the preferred ways for proteins to interact? Chem. Rev. 2008, 108, 1225–1244. [Google Scholar] [CrossRef] [PubMed]
- Arkin, M.R.; Tang, Y.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing toward the reality. Chem. Biol. 2014, 21, 1102–1114. [Google Scholar] [CrossRef]
- De Souza, E.B.; Cload, S.T.; Pendergrast, P.S.; Sah, D.W. Novel therapeutic modalities to address nondrugable protein interaction targets. Neuropsychopharmacology 2009, 34, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Sigalov, A.B. Targeting Intramembrane Protein-Protein Interactions: Novel Therapeutic Strategy of Millions Years Old. Adv. Protein Chem. Struct. Biol. 2018, 111, 61–99. [Google Scholar]
- Borroto-Escuela, D.O.; Flajolet, M.; Agnati, L.F.; Greengard, P.; Fuxe, K. Bioluminescence resonance energy transfer methods to study G protein-coupled receptor-receptor tyrosine kinase heteroreceptor complexes. Methods Cell Biol. 2013, 117, 141–164. [Google Scholar]
- Couturier, C.; Deprez, B. Setting Up a Bioluminescence Resonance Energy Transfer High throughput Screening Assay to Search for Protein/Protein Interaction Inhibitors in Mammalian Cells. Front. Endocrinol. 2012, 3, 100. [Google Scholar] [CrossRef]
- Mild, J.G.; Fernandez, L.R.; Gayet, O.; Iovanna, J.; Dusetti, N.; Edreira, M.M. Optimization of a Bioluminescence Resonance Energy Transfer-Based Assay for Screening of Trypanosoma cruzi Protein/Protein Interaction Inhibitors. Mol. Biotechnol. 2018, 60, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Bacart, J.; Corbel, C.; Jockers, R.; Bach, S.; Couturier, C. The BRET technology and its application to screening assays. Biotechnol. J. 2008, 3, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Dacres, H.; Michie, M.; Wang, J.; Pfleger, K.D.; Trowell, S.C. Effect of enhanced Renilla luciferase and fluorescent protein variants on the Forster distance of Bioluminescence resonance energy transfer (BRET). Biochem. Biophys. Res. Commun. 2012, 425, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Griesbeck, O.; Baird, G.S.; Campbell, R.E.; Zacharias, D.A.; Tsien, R.Y. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J. Biol. Chem. 2001, 276, 29188–29194. [Google Scholar] [CrossRef] [PubMed]
- Kocan, M.; See, H.B.; Seeber, R.M.; Eidne, K.A.; Pfleger, K.D. Demonstration of improvements to the bioluminescence resonance energy transfer (BRET) technology for the monitoring of G protein-coupled receptors in live cells. J. Biomol. Screen 2008, 13, 888–898. [Google Scholar] [CrossRef]
- Pfleger, K.D.; Dromey, J.R.; Dalrymple, M.B.; Lim, E.M.; Thomas, W.G.; Eidne, K.A. Extended bioluminescence resonance energy transfer (eBRET) for monitoring prolonged protein-protein interactions in live cells. Cell Signal 2006, 18, 1664–1670. [Google Scholar] [CrossRef]
- De, A.; Ray, P.; Loening, A.M.; Gambhir, S.S. BRET3: A red-shifted bioluminescence resonance energy transfer (BRET)-based integrated platform for imaging protein-protein interactions from single live cells and living animals. FASEB J. 2009, 23, 2702–2709. [Google Scholar] [CrossRef]
- Dragulescu-Andrasi, A.; Chan, C.T.; De, A.; Massoud, T.F.; Gambhir, S.S. Bioluminescence resonance energy transfer (BRET) imaging of protein-protein interactions within deep tissues of living subjects. Proc. Natl. Acad. Sci. USA 2011, 108, 12060–12065. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Yu, J.; Zhang, Z.; Cui, Z.; Wang, D.; Wei, H.; Zhang, X.E. Buffer enhanced bioluminescence resonance energy transfer sensor based on Gaussia luciferase for in vitro detection of protease. Anal. Chim. Acta 2012, 724, 104–110. [Google Scholar] [CrossRef]
- Kimura, T.; Hiraoka, K.; Kasahara, N.; Logg, C.R. Optimization of enzyme-substrate pairing for bioluminescence imaging of gene transfer using Renilla and Gaussia luciferases. J. Gene Med. 2010, 12, 528–537. [Google Scholar] [CrossRef]
- Li, F.; Yu, J.; Zhang, Z.; Cui, Z.; Wang, D.; Wei, H.; Zhang, X.E. Use of hGluc/tdTomato pair for sensitive BRET sensing of protease with high solution media tolerance. Talanta 2013, 109, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Arai, R.; Nakagawa, H.; Kitayama, A.; Ueda, H.; Nagamune, T. Detection of protein-protein interaction by bioluminescence resonance energy transfer from firefly luciferase to red fluorescent protein. J. Biosci. Bioeng. 2002, 94, 362–364. [Google Scholar] [CrossRef]
- Hamdan, F.F.; Audet, M.; Garneau, P.; Pelletier, J.; Bouvier, M. High-throughput screening of G protein-coupled receptor antagonists using a bioluminescence resonance energy transfer 1-based beta-arrestin2 recruitment assay. J. Biomol. Screen 2005, 10, 463–475. [Google Scholar] [CrossRef] [PubMed]
- De, A.; Loening, A.M.; Gambhir, S.S. An improved bioluminescence resonance energy transfer strategy for imaging intracellular events in single cells and living subjects. Cancer Res. 2007, 67, 7175–7183. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, Y.; Ueda, H.; Kitayama, A.; Nagamune, T. Rapid homogeneous immunoassay of peptides based on bioluminescence resonance energy transfer from firefly luciferase. J. Biosci. Bioeng. 2002, 93, 537–542. [Google Scholar] [CrossRef]
- Machleidt, T.; Woodroofe, C.C.; Schwinn, M.K.; Mendez, J.; Robers, M.B.; Zimmerman, K.; Otto, P.; Daniels, D.L.; Kirkland, T.A.; Wood, K.V. NanoBRET—A Novel BRET Platform for the Analysis of Protein-Protein Interactions. ACS Chem. Biol. 2015, 10, 1797–1804. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wen, K.; Wang, Z.; Zhang, X.; Li, C.; Zhang, S.; Shen, J. General Bioluminescence Resonance Energy Transfer Homogeneous Immunoassay for Small Molecules Based on Quantum Dots. Anal. Chem. 2016, 88, 3512–3520. [Google Scholar] [CrossRef] [PubMed]
- So, M.K.; Xu, C.; Loening, A.M.; Gambhir, S.S.; Rao, J. Self-illuminating quantum dot conjugates for in vivo imaging. Nat. Biotechnol. 2006, 24, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, N.; Mitsunaga, M.; Bhattacharyya, S.; Miller, S.C.; Choyke, P.L.; Kobayashi, H. Self-illuminating in vivo lymphatic imaging using a bioluminescence resonance energy transfer quantum dot nano-particle. Contrast. Media Mol. Imaging 2011, 6, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Alam, R.; Fontaine, D.M.; Branchini, B.R.; Maye, M.M. Designing quantum rods for optimized energy transfer with firefly luciferase enzymes. Nano Lett. 2012, 12, 3251–3256. [Google Scholar] [CrossRef] [PubMed]
- Alam, R.; Zylstra, J.; Fontaine, D.M.; Branchini, B.R.; Maye, M.M. Novel multistep BRET-FRET energy transfer using nanoconjugates of firefly proteins, quantum dots, and red fluorescent proteins. Nanoscale 2013, 5, 5303–5306. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Piston, D.W.; Johnson, C.H. A bioluminescence resonance energy transfer (BRET) system: application to interacting circadian clock proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Ciruela, F. Fluorescence-based methods in the study of protein-protein interactions in living cells. Curr. Opin. Biotechnol. 2008, 19, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Kamal, M.; Marquez, M.; Vauthier, V.; Leloire, A.; Froguel, P.; Jockers, R.; Couturier, C. Improved donor/acceptor BRET couples for monitoring beta-arrestin recruitment to G protein-coupled receptors. Biotechnol. J. 2009, 4, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Ibata, K.; Park, E.S.; Kubota, M.; Mikoshiba, K.; Miyawaki, A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 2002, 20, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Kocan, M.; Dalrymple, M.B.; Seeber, R.M.; Feldman, B.J.; Pfleger, K.D. Enhanced BRET Technology for the Monitoring of Agonist-Induced and Agonist-Independent Interactions between GPCRs and beta-Arrestins. Front. Endocrinol. 2010, 1, 12. [Google Scholar]
- Inouye, S.; Watanabe, K.; Nakamura, H.; Shimomura, O. Secretional luciferase of the luminous shrimp Oplophorus gracilirostris: cDNA cloning of a novel imidazopyrazinone luciferase(1). FEBS Lett. 2000, 481, 19–25. [Google Scholar] [CrossRef]
- Hall, M.P.; Unch, J.; Binkowski, B.F.; Valley, M.P.; Butler, B.L.; Wood, M.G.; Otto, P.; Zimmerman, K.; Vidugiris, G.; Machleidt, T.; et al. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 2012, 7, 1848–1857. [Google Scholar] [CrossRef]
- England, C.G.; Ehlerding, E.B.; Cai, W. NanoLuc: A Small Luciferase Is Brightening Up the Field of Bioluminescence. Bioconjug. Chem. 2016, 27, 1175–1187. [Google Scholar] [CrossRef] [Green Version]
- Wan, Q.; Okashah, N.; Inoue, A.; Nehme, R.; Carpenter, B.; Tate, C.G.; Lambert, N.A. Mini G protein probes for active G protein-coupled receptors (GPCRs) in live cells. J. Biol. Chem. 2018, 293, 7466–7473. [Google Scholar] [CrossRef]
- Verhoef, L.G.; Mattioli, M.; Ricci, F.; Li, Y.C.; Wade, M. Multiplex detection of protein-protein interactions using a next generation luciferase reporter. Biochim. Biophys. Acta 2016, 1863, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Soave, M.; Stoddart, L.A.; Brown, A.; Woolard, J.; Hill, S.J. Use of a new proximity assay (NanoBRET) to investigate the ligand-binding characteristics of three fluorescent ligands to the human beta1-adrenoceptor expressed in HEK-293 cells. Pharmacol. Res. Perspect. 2016, 4, e00250. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, L.A.; Johnstone, E.K.M.; Wheal, A.J.; Goulding, J.; Robers, M.B.; Machleidt, T.; Wood, K.V.; Hill, S.J.; Pfleger, K.D.G. Application of BRET to monitor ligand binding to GPCRs. Nat. Methods 2015, 12, 661–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoddart, L.A.; White, C.W.; Nguyen, K.; Hill, S.J.; Pfleger, K.D. Fluorescence- and bioluminescence-based approaches to study GPCR ligand binding. Br. J. Pharmacol. 2016, 173, 3028–3037. [Google Scholar] [PubMed]
- Stoddart, L.A.; Vernall, A.J.; Bouzo-Lorenzo, M.; Bosma, R.; Kooistra, A.J.; de Graaf, C.; Vischer, H.F.; Leurs, R.; Briddon, S.J.; Kellam, B.; et al. Development of novel fluorescent histamine H1-receptor antagonists to study ligand-binding kinetics in living cells. Sci. Rep. 2018, 8, 1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, E.; Canet, J.; Gracia, E.; Lluis, C.; Mallol, J.; Canela, E.I.; Cortes, A.; Casado, V. Molecular Evidence of Adenosine Deaminase Linking Adenosine A2A Receptor and CD26 Proteins. Front. Pharmacol. 2018, 9, 106. [Google Scholar] [CrossRef] [PubMed]
- Cobret, L.; De Tauzia, M.L.; Ferent, J.; Traiffort, E.; Henaoui, I.; Godin, F.; Kellenberger, E.; Rognan, D.; Pantel, J.; Benedetti, H.; et al. Targeting the cis-dimerization of LINGO-1 with low MW compounds affects its downstream signalling. Br. J. Pharmacol. 2015, 172, 841–856. [Google Scholar]
- Yao, J.; Li, P.; Li, L.; Yang, M. Biochemistry and biomedicine of quantum dots: From biodetection to bioimaging, drug discovery, diagnostics, and therapy. Acta Biomater. 2018, 74, 36–55. [Google Scholar] [CrossRef]
- Xiong, L.; Shuhendler, A.J.; Rao, J. Self-luminescing BRET-FRET near-infrared dots for in vivo lymph-node mapping and tumour imaging. Nat. Commun. 2012, 3, 1193. [Google Scholar] [CrossRef] [Green Version]
- Kamkaew, A.; Sun, H.; England, C.G.; Cheng, L.; Liu, Z.; Cai, W. Quantum dot-NanoLuc bioluminescence resonance energy transfer enables tumor imaging and lymph node mapping in vivo. Chem. Commun. 2016, 52, 6997–7000. [Google Scholar]
- Michalet, X.; Pinaud, F.F.; Bentolila, L.A.; Tsay, J.M.; Doose, S.; Li, J.J.; Sundaresan, G.; Wu, A.M.; Gambhir, S.S.; Weiss, S. Quantum dots for live cells, in vivo imaging, and diagnostics. Science 2005, 307, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, M. Dysfunction of various organelles provokes multiple cell death after quantum dot exposure. Int. J. Nanomed. 2018, 13, 2729–2742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luttrell, L.M. Reviews in molecular biology and biotechnology: Transmembrane signaling by G protein-coupled receptors. Mol. Biotechnol. 2008, 39, 239–264. [Google Scholar] [CrossRef] [PubMed]
- Lagerstrom, M.C.; Schioth, H.B. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discov. 2008, 7, 339–357. [Google Scholar] [CrossRef] [PubMed]
- Ferre, S.; Casado, V.; Devi, L.A.; Filizola, M.; Jockers, R.; Lohse, M.J.; Milligan, G.; Pin, J.P.; Guitart, X. G protein-coupled receptor oligomerization revisited: Functional and pharmacological perspectives. Pharmacol. Rev. 2014, 66, 413–434. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Casado, V.; Cortes, A.; Mallol, J.; Ciruela, F.; Ferre, S.; Lluis, C.; Canela, E.I. G-protein-coupled receptor heteromers: Function and ligand pharmacology. Br. J. Pharmacol. 2008, 153 (Suppl. S1), S90–S98. [Google Scholar]
- Milligan, G. A day in the life of a G protein-coupled receptor: The contribution to function of G protein-coupled receptor dimerization. Br. J. Pharmacol. 2008, 153 (Suppl. S1), S216–S229. [Google Scholar]
- Jones, K.A.; Borowsky, B.; Tamm, J.A.; Craig, D.A.; Durkin, M.M.; Dai, M.; Yao, W.J.; Johnson, M.; Gunwaldsen, C.; Huang, L.Y.; et al. GABA(B) receptors function as a heteromeric assembly of the subunits GABA(B)R1 and GABA(B)R2. Nature 1998, 396, 674–679. [Google Scholar] [CrossRef] [PubMed]
- White, J.H.; Wise, A.; Main, M.J.; Green, A.; Fraser, N.J.; Disney, G.H.; Barnes, A.A.; Emson, P.; Foord, S.M.; Marshall, F.H. Heterodimerization is required for the formation of a functional GABA(B) receptor. Nature 1998, 396, 679–682. [Google Scholar] [CrossRef]
- Galvez, T.; Duthey, B.; Kniazeff, J.; Blahos, J.; Rovelli, G.; Bettler, B.; Prezeau, L.; Pin, J.P. Allosteric interactions between GB1 and GB2 subunits are required for optimal GABA(B) receptor function. EMBO J. 2001, 20, 2152–2159. [Google Scholar] [CrossRef]
- Duthey, B.; Caudron, S.; Perroy, J.; Bettler, B.; Fagni, L.; Pin, J.P.; Prezeau, L. A single subunit (GB2) is required for G-protein activation by the heterodimeric GABA(B) receptor. J. Biol. Chem. 2002, 277, 3236–3241. [Google Scholar] [CrossRef] [PubMed]
- Moreno Delgado, D.; Moller, T.C.; Ster, J.; Giraldo, J.; Maurel, D.; Rovira, X.; Scholler, P.; Zwier, J.M.; Perroy, J.; Durroux, T.; et al. Pharmacological evidence for a metabotropic glutamate receptor heterodimer in neuronal cells. Elife 2017, 6, e25233. [Google Scholar] [CrossRef] [PubMed]
- Albizu, L.; Cottet, M.; Kralikova, M.; Stoev, S.; Seyer, R.; Brabet, I.; Roux, T.; Bazin, H.; Bourrier, E.; Lamarque, L.; et al. Time-resolved FRET between GPCR ligands reveals oligomers in native tissues. Nat. Chem. Biol. 2010, 6, 587–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khelashvili, G.; Dorff, K.; Shan, J.; Camacho-Artacho, M.; Skrabanek, L.; Vroling, B.; Bouvier, M.; Devi, L.A.; George, S.R.; Javitch, J.A.; et al. GPCR-OKB: The G Protein Coupled Receptor Oligomer Knowledge Base. Bioinformatics 2010, 26, 1804–1805. [Google Scholar] [CrossRef] [PubMed]
- Farran, B. An update on the physiological and therapeutic relevance of GPCR oligomers. Pharmacol. Res. 2017, 117, 303–327. [Google Scholar] [CrossRef] [PubMed]
- Kent, T.; McAlpine, C.; Sabetnia, S.; Presland, J. G-protein-coupled receptor heterodimerization: Assay technologies to clinical significance. Curr. Opin. Drug Discov. Dev. 2007, 10, 580–589. [Google Scholar]
- Angers, S.; Salahpour, A.; Joly, E.; Hilairet, S.; Chelsky, D.; Dennis, M.; Bouvier, M. Detection of beta 2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET). Proc. Natl. Acad. Sci. USA 2000, 97, 3684–3689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayoub, M.A. Resonance Energy Transfer-Based Approaches to Study GPCRs. Methods Cell Biol. 2016, 132, 255–292. [Google Scholar]
- Kaczor, A.A.; Makarska-Bialokoz, M.; Selent, J.; de la Fuente, R.A.; Marti-Solano, M.; Castro, M. Application of BRET for studying G protein-coupled receptors. Mini Rev. Med. Chem. 2014, 14, 411–425. [Google Scholar] [CrossRef]
- Milligan, G.; Ramsay, D.; Pascal, G.; Carrillo, J.J. GPCR dimerisation. Life Sci. 2003, 74, 181–188. [Google Scholar] [CrossRef]
- Schiedel, A.C.; Kose, M.; Barreto, C.; Bueschbell, B.; Morra, G.; Sensoy, O.; Moreira, I.S. Prediction and Targeting of Interaction Interfaces in G-protein Coupled Receptor Oligomers. Curr. Top. Med. Chem. 2018, 18, 714–746. [Google Scholar] [CrossRef] [PubMed]
- Berthouze, M.; Rivail, L.; Lucas, A.; Ayoub, M.A.; Russo, O.; Sicsic, S.; Fischmeister, R.; Berque-Bestel, I.; Jockers, R.; Lezoualc’h, F. Two transmembrane Cys residues are involved in 5-HT4 receptor dimerization. Biochem. Biophys. Res. Commun. 2007, 356, 642–647. [Google Scholar] [CrossRef] [PubMed]
- McMillin, S.M.; Heusel, M.; Liu, T.; Costanzi, S.; Wess, J. Structural basis of M3 muscarinic receptor dimer/oligomer formation. J. Biol. Chem. 2011, 286, 28584–28598. [Google Scholar] [CrossRef] [PubMed]
- Felce, J.H.; Latty, S.L.; Knox, R.G.; Mattick, S.R.; Lui, Y.; Lee, S.F.; Klenerman, D.; Davis, S.J. Receptor Quaternary Organization Explains G Protein-Coupled Receptor Family Structure. Cell Rep. 2017, 20, 2654–2665. [Google Scholar] [CrossRef] [PubMed]
- James, J.R.; Oliveira, M.I.; Carmo, A.M.; Iaboni, A.; Davis, S.J. A rigorous experimental framework for detecting protein oligomerization using bioluminescence resonance energy transfer. Nat. Methods 2006, 3, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.H.; Liu, Q.; Li, C.; Wu, G.; Steyaert, J.; Lambert, N.A. BRET evidence that beta2 adrenergic receptors do not oligomerize in cells. Sci. Rep. 2015, 5, 10166. [Google Scholar] [CrossRef] [PubMed]
- Szalai, B.; Hoffmann, P.; Prokop, S.; Erdelyi, L.; Varnai, P.; Hunyady, L. Improved methodical approach for quantitative BRET analysis of G Protein Coupled Receptor dimerization. PLoS ONE 2014, 9, e109503. [Google Scholar] [CrossRef]
- Hamm, H.E. The Many Faces of G Protein Signaling. J. Biol. Chem. 1998, 273, 669–672. [Google Scholar] [CrossRef] [Green Version]
- Ayoub, M.A.; Maurel, D.; Binet, V.; Fink, M.; Prezeau, L.; Ansanay, H.; Pin, J.P. Real-time analysis of agonist-induced activation of protease-activated receptor 1/Galphai1 protein complex measured by bioluminescence resonance energy transfer in living cells. Mol. Pharmacol. 2007, 71, 1329–1340. [Google Scholar] [CrossRef]
- Gales, C.; Rebois, R.V.; Hogue, M.; Trieu, P.; Breit, A.; Hebert, T.E.; Bouvier, M. Real-time monitoring of receptor and G-protein interactions in living cells. Nat. Methods 2005, 2, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Gales, C.; Van Durm, J.J.; Schaak, S.; Pontier, S.; Percherancier, Y.; Audet, M.; Paris, H.; Bouvier, M. Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nat. Struct. Mol. Biol. 2006, 13, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Hollins, B.; Kuravi, S.; Digby, G.J.; Lambert, N.A. The c-terminus of GRK3 indicates rapid dissociation of G protein heterotrimers. Cell Signal 2009, 21, 1015–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuho, I.; Ostrovskaya, O.; Kramer, G.M.; Jones, C.D.; Xie, K.; Martemyanov, K.A. Distinct profiles of functional discrimination among G proteins determine the actions of G protein-coupled receptors. Sci. Signal 2015, 8, ra123. [Google Scholar] [CrossRef] [PubMed]
- Bockaert, J.; Roussignol, G.; Becamel, C.; Gavarini, S.; Joubert, L.; Dumuis, A.; Fagni, L.; Marin, P. GPCR-interacting proteins (GIPs): Nature and functions. Biochem. Soc. Trans. 2004, 32, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Maurice, P.; Guillaume, J.L.; Benleulmi-Chaachoua, A.; Daulat, A.M.; Kamal, M.; Jockers, R. GPCR-interacting proteins, major players of GPCR function. Adv. Pharmacol. 2011, 62, 349–380. [Google Scholar] [PubMed]
- Sexton, P.M.; Poyner, D.R.; Simms, J.; Christopoulos, A.; Hay, D.L. Modulating receptor function through RAMPs: Can they represent drug targets in themselves? Drug Discov. Today 2009, 14, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Pitcher, J.A.; Freedman, N.J.; Lefkowitz, R.J. G protein-coupled receptor kinases. Annu. Rev. Biochem. 1998, 67, 653–692. [Google Scholar] [CrossRef] [PubMed]
- McDonald, P.H.; Lefkowitz, R.J. Beta-Arrestins: new roles in regulating heptahelical receptors’ functions. Cell Signal 2001, 13, 683–689. [Google Scholar] [CrossRef]
- Bockaert, J.; Fagni, L.; Dumuis, A.; Marin, P. GPCR interacting proteins (GIP). Pharmacol. Ther. 2004, 103, 203–221. [Google Scholar] [CrossRef]
- Hall, R.A.; Premont, R.T.; Lefkowitz, R.J. Heptahelical Receptor Signaling: Beyond the G Protein Paradigm. J. Cell Biol. 1999, 145, 6. [Google Scholar] [CrossRef]
- Milligan, G.; White, J.H. Protein–protein interactions at G-protein-coupled receptors. Trends Pharmacol. Sci. 2001, 22, 6. [Google Scholar] [CrossRef]
- Dunn, H.A.; Ferguson, S.S. PDZ Protein Regulation of G Protein-Coupled Receptor Trafficking and Signaling Pathways. Mol. Pharmacol. 2015, 88, 624–639. [Google Scholar] [CrossRef] [PubMed]
- Bologna, Z.; Teoh, J.P.; Bayoumi, A.S.; Tang, Y.; Kim, I.M. Biased G Protein-Coupled Receptor Signaling: New Player in Modulating Physiology and Pathology. Biomol. Ther. 2017, 25, 12–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whalen, E.J.; Rajagopal, S.; Lefkowitz, R.J. Therapeutic potential of beta-arrestin- and G protein-biased agonists. Trends Mol. Med. 2011, 17, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Duhr, F.; Deleris, P.; Raynaud, F.; Seveno, M.; Lopez, S.M.; la Cour, C.M.; Millan, M.J.; Bockaert, J.; Marin, P.; Chaumont-Dubel, S. Cdk5 induces constitutive activation of 5-HT6 receptors to promote neurite growth. Nat. Chem. Biol. 2014, 10, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Deraredj Nadim, W.; Chaumont-Dubel, S.; Madouri, F.; Cobret, L.; De Tauzia, M.; Pawel, Z.; Bénédetti, H.; Marin, P.; Morisset-Lopez, S. A Physical Interaction between Neurofibromin and Serotonin 5-HT6 Receptor Promotes Receptor Constitutive Activity. Proc. Natl. Acad. Sci. USA 2016, 113, 12310–12315. [Google Scholar] [CrossRef]
- Sheppard, D.W.; Lipkin, M.J.; Harris, C.J.; Catana, C.; Stouten, P.F. Strategies for small molecule library design. Curr. Pharm. Des. 2014, 20, 3314–3322. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small molecules, big targets: Drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discov. 2016, 15, 533–550. [Google Scholar] [CrossRef]
- Villar, E.A.; Beglov, D.; Chennamadhavuni, S.; Porco, J.A., Jr.; Kozakov, D.; Vajda, S.; Whitty, A. How proteins bind macrocycles. Nat. Chem. Biol. 2014, 10, 723–731. [Google Scholar] [CrossRef] [Green Version]
- Dougherty, P.G.; Qian, Z.; Pei, D. Macrocycles as protein-protein interaction inhibitors. Biochem. J. 2017, 474, 1109–1125. [Google Scholar] [CrossRef]
- Ayoub, M.A.; Pfleger, K.D. Recent advances in bioluminescence resonance energy transfer technologies to study GPCR heteromerization. Curr. Opin. Pharmacol. 2010, 10, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, M.; Schilling, J.; Beautrait, A.; Bouvier, M.; Benovic, J.L.; Shukla, A.K. Emerging Paradigm of Intracellular Targeting of G Protein-Coupled Receptors. Trends Biochem. Sci. 2018, 43, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.P.; Zhang, Y.; Van Cleemput, J.; Jiang, W.; Liao, M.; Li, L.; Wan, Q.; Backstrom, J.R.; Zhang, X. Disruption of PTEN coupling with 5-HT2C receptors suppresses behavioral responses induced by drugs of abuse. Nat. Med. 2006, 12, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Pichon, X.; Wattiez, A.S.; Becamel, C.; Ehrlich, I.; Bockaert, J.; Eschalier, A.; Marin, P.; Courteix, C. Disrupting 5-HT(2A) receptor/PDZ protein interactions reduces hyperalgesia and enhances SSRI efficacy in neuropathic pain. Mol. Ther. 2010, 18, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.J.; Kania, R.S.; Zuckermann, R.N.; Huebner, V.D.; Jewell, D.A.; Banville, S.; Ng, S.; Wang, L.; Rosenberg, S.; Marlowe, C.K.; et al. Peptoids: A modular approach to drug discovery. Proc. Natl. Acad. Sci. USA 1992, 89, 9367–9371. [Google Scholar] [CrossRef] [PubMed]
- Kirshenbaum, K.; Barron, A.E.; Goldsmith, R.A.; Armand, P.; Bradley, E.K.; Truong, K.T.; Dill, K.A.; Cohen, F.E.; Zuckermann, R.N. Sequence-specific polypeptoids: A diverse family of heteropolymers with stable secondary structure. Proc. Natl. Acad. Sci. USA 1998, 95, 4303–4308. [Google Scholar] [CrossRef] [Green Version]
- Zuckermann, R.N.; Martin, E.J.; Spellmeyer, D.C.; Stauber, G.B.; Shoemaker, K.R.; Kerr, J.M.; Figliozzi, G.M.; Goff, D.A.; Siani, M.A.; Simon, R.J.; et al. Discovery of nanomolar ligands for 7-transmembrane G-protein-coupled receptors from a diverse N-(substituted)glycine peptoid library. J. Med. Chem. 1994, 37, 2678–2685. [Google Scholar] [CrossRef]
- Stone, T.A.; Deber, C.M. Therapeutic design of peptide modulators of protein-protein interactions in membranes. Biochim. Biophys. Acta 2017, 1859, 577–585. [Google Scholar] [CrossRef]
- O’Callaghan, K.; Kuliopulos, A.; Covic, L. Turning receptors on and off with intracellular pepducins: New insights into G-protein-coupled receptor drug development. J. Biol. Chem. 2012, 287, 12787–12796. [Google Scholar] [CrossRef]
- Tepper, S.; Ashina, M.; Reuter, U.; Brandes, J.L.; Dolezil, D.; Silberstein, S.; Winner, P.; Leonardi, D.; Mikol, D.; Lenz, R. Safety and efficacy of erenumab for preventive treatment of chronic migraine: A randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2017, 16, 425–434. [Google Scholar] [CrossRef]
- Ueda, R. Clinical Application of Anti-CCR4 Monoclonal Antibody. Oncology 2015, 89 (Suppl. S1), 16–21. [Google Scholar] [CrossRef]
- Maeda, S.; Koehl, A.; Matile, H.; Hu, H.; Hilger, D.; Schertler, G.F.X.; Manglik, A.; Skiniotis, G.; Dawson, R.J.P.; Kobilka, B.K. Development of an antibody fragment that stabilizes GPCR/G-protein complexes. Nat. Commun. 2018, 9, 3712. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [PubMed]
- De Genst, E.; Silence, K.; Decanniere, K.; Conrath, K.; Loris, R.; Kinne, J.; Muyldermans, S.; Wyns, L. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. USA 2006, 103, 4586–4591. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S.; Baral, T.N.; Retamozzo, V.C.; De Baetselier, P.; De Genst, E.; Kinne, J.; Leonhardt, H.; Magez, S.; Nguyen, V.K.; Revets, H.; et al. Camelid immunoglobulins and nanobody technology. Vet. Immunol. Immunopathol. 2009, 128, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.O.; Sherwood, L.J.; Collazo, M.T.; Garza, J.A.; Hayhurst, A. Llama single domain antibodies specific for the 7 botulinum neurotoxin serotypes as heptaplex immunoreagents. PLoS ONE 2010, 5, e8818. [Google Scholar] [CrossRef] [PubMed]
- Ladenson, R.C.; Crimmins, D.L.; Landt, Y.; Ladenson, J.H. Isolation and characterization of a thermally stable recombinant anti-caffeine heavy-chain antibody fragment. Anal. Chem. 2006, 78, 4501–4508. [Google Scholar] [CrossRef]
- Li, T.; Bourgeois, J.P.; Celli, S.; Glacial, F.; Le Sourd, A.M.; Mecheri, S.; Weksler, B.; Romero, I.; Couraud, P.O.; Rougeon, F.; et al. Cell-penetrating anti-GFAP VHH and corresponding fluorescent fusion protein VHH-GFP spontaneously cross the blood-brain barrier and specifically recognize astrocytes: Application to brain imaging. FASEB J. 2012, 26, 3969–3979. [Google Scholar] [CrossRef]
- Li, T.; Vandesquille, M.; Koukouli, F.; Dudeffant, C.; Youssef, I.; Lenormand, P.; Ganneau, C.; Maskos, U.; Czech, C.; Grueninger, F.; et al. Camelid single-domain antibodies: A versatile tool for in vivo imaging of extracellular and intracellular brain targets. J. Control. Release 2016, 243, 1–10. [Google Scholar] [CrossRef]
- Jahnichen, S.; Blanchetot, C.; Maussang, D.; Gonzalez-Pajuelo, M.; Chow, K.Y.; Bosch, L.; De Vrieze, S.; Serruys, B.; Ulrichts, H.; Vandevelde, W.; et al. CXCR4 nanobodies (VHH-based single variable domains) potently inhibit chemotaxis and HIV-1 replication and mobilize stem cells. Proc. Natl. Acad. Sci. USA 2010, 107, 20565–20570. [Google Scholar] [CrossRef] [Green Version]
- Bradley, M.E.; Dombrecht, B.; Manini, J.; Willis, J.; Vlerick, D.; De Taeye, S.; Van den Heede, K.; Roobrouck, A.; Grot, E.; Kent, T.C.; et al. Potent and efficacious inhibition of CXCR2 signaling by biparatopic nanobodies combining two distinct modes of action. Mol. Pharmacol. 2015, 87, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Maussang, D.; Mujic-Delic, A.; Descamps, F.J.; Stortelers, C.; Vanlandschoot, P.; Stigter-van Walsum, M.; Vischer, H.F.; van Roy, M.; Vosjan, M.; Gonzalez-Pajuelo, M.; et al. Llama-derived single variable domains (nanobodies) directed against chemokine receptor CXCR7 reduce head and neck cancer cell growth in vivo. J. Biol. Chem. 2013, 288, 29562–29572. [Google Scholar] [CrossRef] [PubMed]
- Staus, D.P.; Strachan, R.T.; Manglik, A.; Pani, B.; Kahsai, A.W.; Kim, T.H.; Wingler, L.M.; Ahn, S.; Chatterjee, A.; Masoudi, A.; et al. Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation. Nature 2016, 535, 448–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staus, D.P.; Wingler, L.M.; Strachan, R.T.; Rasmussen, S.G.; Pardon, E.; Ahn, S.; Steyaert, J.; Kobilka, B.K.; Lefkowitz, R.J. Regulation of beta2-adrenergic receptor function by conformationally selective single-domain intrabodies. Mol. Pharmacol. 2014, 85, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Scholler, P.; Nevoltris, D.; de Bundel, D.; Bossi, S.; Moreno-Delgado, D.; Rovira, X.; Moller, T.C.; El Moustaine, D.; Mathieu, M.; Blanc, E.; et al. Allosteric nanobodies uncover a role of hippocampal mGlu2 receptor homodimers in contextual fear consolidation. Nat. Commun. 2017, 8, 1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Parashar, A. Aptamers in Therapeutics. J. Clin. Diagn. Res. 2016, 10, BE01–BE06. [Google Scholar] [CrossRef] [PubMed]
- Ng, E.W.; Shima, D.T.; Calias, P.; Cunningham, E.T., Jr.; Guyer, D.R.; Adamis, A.P. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug Discov. 2006, 5, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Kotula, J.W.; Sun, J.; Li, M.; Pratico, E.D.; Fereshteh, M.P.; Ahrens, D.P.; Sullenger, B.A.; Kovacs, J.J. Targeted disruption of beta-arrestin 2-mediated signaling pathways by aptamer chimeras leads to inhibition of leukemic cell growth. PLoS ONE 2014, 9, e93441. [Google Scholar] [CrossRef] [PubMed]
- Kahsai, A.W.; Wisler, J.W.; Lee, J.; Ahn, S.; Cahill Iii, T.J.; Dennison, S.M.; Staus, D.P.; Thomsen, A.R.; Anasti, K.M.; Pani, B.; et al. Conformationally selective RNA aptamers allosterically modulate the beta2-adrenoceptor. Nat. Chem. Biol. 2016, 12, 709–716. [Google Scholar] [CrossRef]




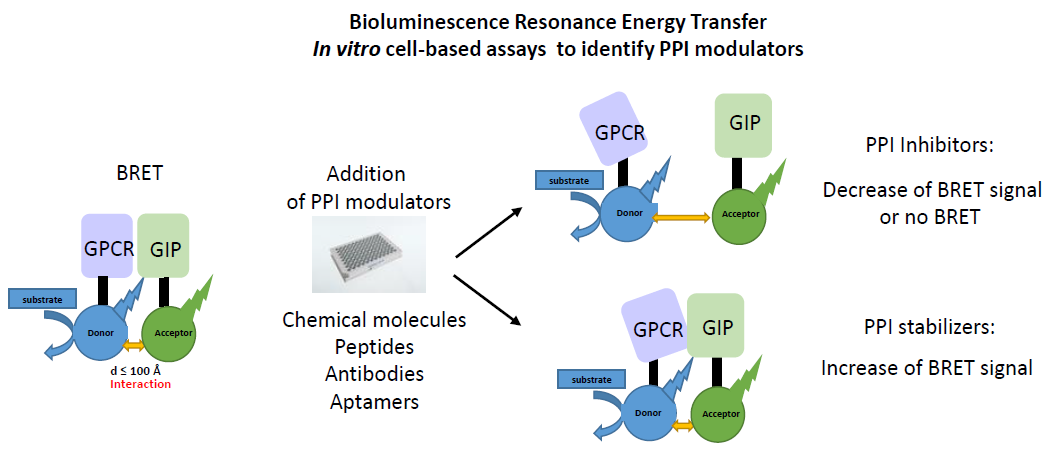{kind=link}
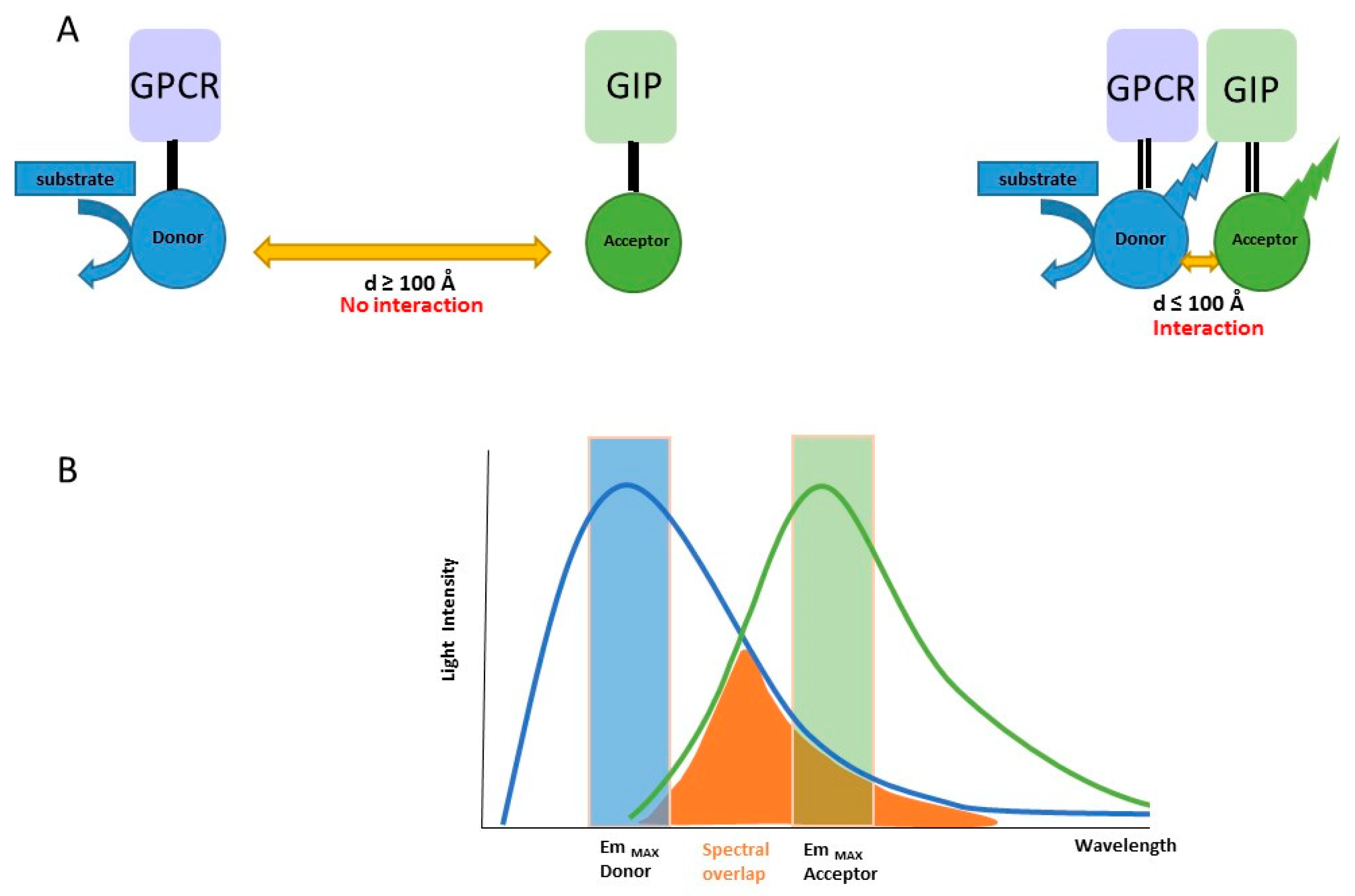{kind=link}
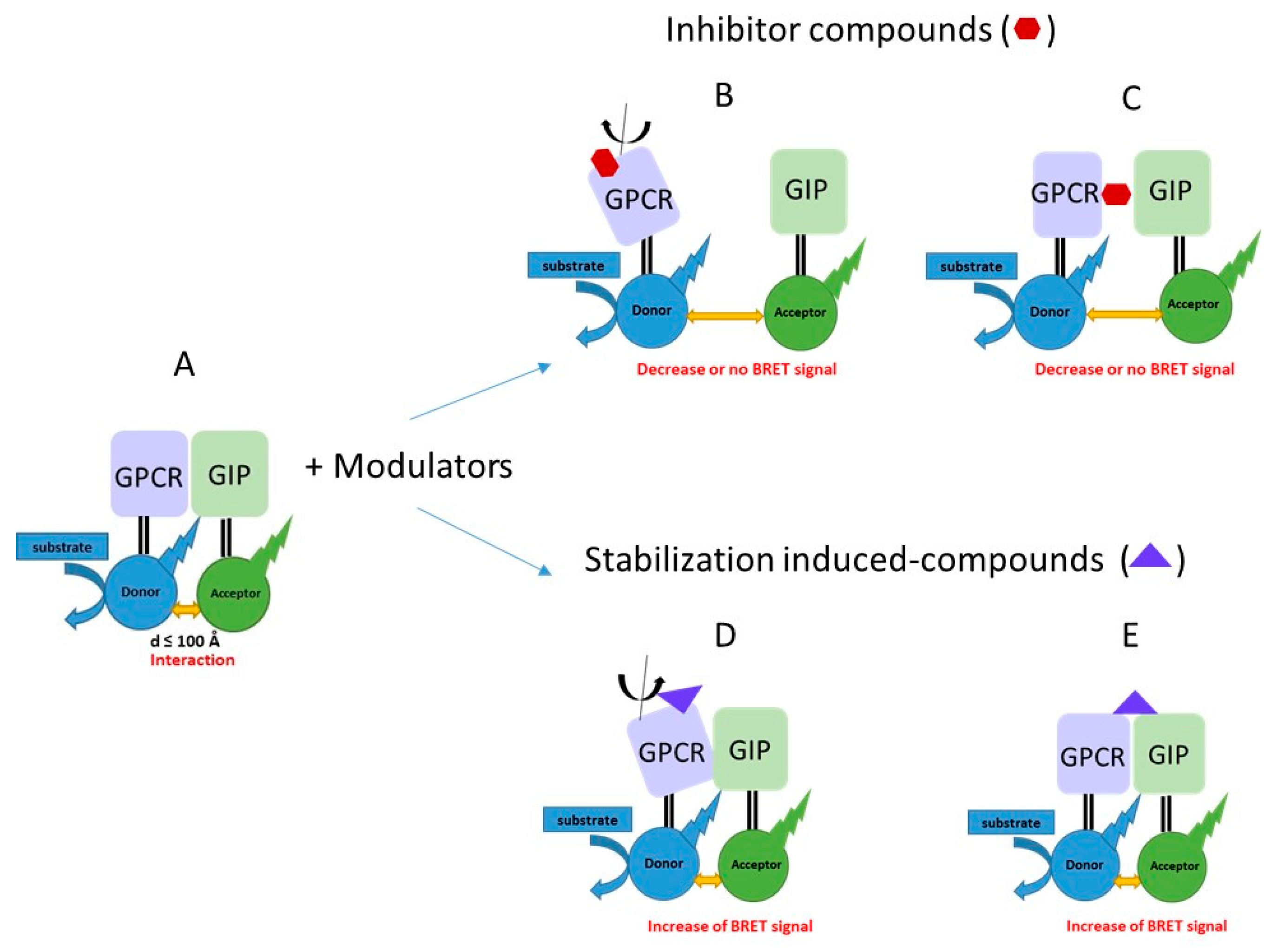{kind=link}
{kind=link}
| Name * | Donor | λem § | Acceptor | λem § | Substrate | Advantages | Drawbacks | Refs |
|---|---|---|---|---|---|---|---|---|
| BRET 1 | RLuc/RLuc8 | 480 | eYFP | 530 | CLZN h | Monitor PPI at endogenous expression levels of protein RLuc8 more stable than RLuc | Sensitive to solvent polarity, serum and pH | [13,14] |
| BRET 1 1.1 | RLuc/RLuc8 | 480 | Venus | 530 | CLZN h | Venus has faster and more efficient maturation compared to YFP Working distance range increased (2.7–8.3 nm) compared to BRET 1 (2.2–6.6 nm) | [13,15] | |
| BRET 1 2 | RLuc | 480 | eYFP | 530 | Enduren | Monitoring of PPI several hours in real-time under near-physiological conditions | Requires expensive Enduren | [15,16] |
| BRET 1 3 | RLuc8 | 480 | mOrange | 564 | CLZN h | Application for BRET imaging Wide spectral separation Δλ: 84 nm | mOrange: slow maturation processes (t1/2: 2 h) | [17] |
| BRET 1 3.1 | RLuc8 | 515 | mOrange | 564 | CLZN v | CLZN v increases the spectral overlap between donor emission and acceptor excitation | Low spectral separation Δλ: 50 nm | [18] |
| BRET 1 4.1 | RLuc8 | 515 | TagRFP | 584 | CLZN v | Low spectral separation Δλ: 70 nm | [18] | |
| BRET 1 5 | RLuc8.6 | 535 | TagRFP | 584 | CLZN h | Increased stability and enhanced enzymatic activity of RLuc8.6 compared to RLuc8 | Low spectral separation Δλ: 50 nm | [18] |
| BRET 1 6 | RLuc8.6 | 535 | TurboFP | 635 | CLZN h | High spectral separation Δλ: 100 nm Application for BRET in living animals | [18] | |
| BRET 1 7 | Gluc | 470 | eYFP | 530 | CLZN h | Gluc smaller and brighter luciferase | Glu activity depends on pH and NaCl concentration Secreted luciferase | [19,20] |
| BRET 1 7.1 | hGluc | 470 | TdTomato | 580 | CLZN h | Large spectral separation compared to Gluc/eYFP pair Δλ: 110 nm High tolerance toward the solution components (serum) and pH. | TdTomato: slow maturation processes compared to GFP Low stokes shift | [21] |
| BRET 1 7.2 | hGluc | 470 | DsRed | 583 | CLZN h | Large spectral separation: Δλ: 110 | DsRed: slow maturation processes, fluorescent intensity lower compared to GFP | [22] |
| BRET 2 | RLuc | 395 | GFP2 | 510 | DeepBlueC | Large spectral separation: Δλ 115 for BRET2 vs. 50 for BRET1 1 | DeepBlue C: weak and short lasting light emission Necessity high expression of BRET partners | [23] |
| BRET 2 | RLuc2 | 420 | GFP2 | 510 | DeepBlueC | Working distance range increased (3.8–11.5 nm) compared to BRET 1 (2.2–6.6 nm) | [13] | |
| BRET 2 | RLucM/RLuc8 | 400 | GFP2 | 510 | DeepBlueC | RLuc8 increased stability and even higher quantum yield BRET signal 30 fold higher than RLuc/GFP2 pair Application for BRET in single live cells and living animals | [24] | |
| BRET 3 | FLuc | 565 | DsRed | 583 | D luciferin | DsRed: high photostability and resistance to pH; Application for in vivo imaging | Overlap of donor/acceptor emission Low signal/noise | [17,22] |
| BRET 3 | FLuc | 565 | Cy3/Cy3.5 | 570/596 | D luciferin | Overlap of donor/acceptor emission Low signal/noise | [25] | |
| NanoBRET | Nluc | 462 | haloTag | 618 | Furimazine | NanoLuc is 100 fold brighter than RLuc. Furimazine permits longer observation (2 h compared to 25 min with coelenterazine) | Not red shifted version available Requires expensive Furimazine | [26] |
| NanoBRET | Nluc | 462 | Venus DsRed | 535 | Furimazine | Improved sensitivity and dynamic range Used as biosensor Single cell BRET imaging | Not red shifted version available Requires expensive Furimazine | [26] |
| QD-BRET 1 | RLuc | 480 | Qdot | 620 | CLZN h | Used as biosensor Larger stokes shift Resistance to photobeaching Strong fluorescence | [27] | |
| QD-BRET 2 | RLuc8 | 480 | Qdot | 655 | CLZN h | Real time in vivo imaging | Size of Qdot | [28,29] |
| QD-BRET 3 | FLuc | 565 | Qdot | 613/628 675 | CLZN h | Working distance range increased | Problem for Coupling to proteins; cellular toxicity | [30,31] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Khamlichi, C.; Reverchon-Assadi, F.; Hervouet-Coste, N.; Blot, L.; Reiter, E.; Morisset-Lopez, S. Bioluminescence Resonance Energy Transfer as a Method to Study Protein-Protein Interactions: Application to G Protein Coupled Receptor Biology. Molecules 2019, 24, 537. https://doi.org/10.3390/molecules24030537
El Khamlichi C, Reverchon-Assadi F, Hervouet-Coste N, Blot L, Reiter E, Morisset-Lopez S. Bioluminescence Resonance Energy Transfer as a Method to Study Protein-Protein Interactions: Application to G Protein Coupled Receptor Biology. Molecules. 2019; 24(3):537. https://doi.org/10.3390/molecules24030537
Chicago/Turabian StyleEl Khamlichi, Chayma, Flora Reverchon-Assadi, Nadège Hervouet-Coste, Lauren Blot, Eric Reiter, and Séverine Morisset-Lopez. 2019. "Bioluminescence Resonance Energy Transfer as a Method to Study Protein-Protein Interactions: Application to G Protein Coupled Receptor Biology" Molecules 24, no. 3: 537. https://doi.org/10.3390/molecules24030537