
New Molybdenum(II) Complexes with α-Diimine Ligands: Synthesis, Structure, and Catalytic Activity in Olefin Epoxidation


Abstract
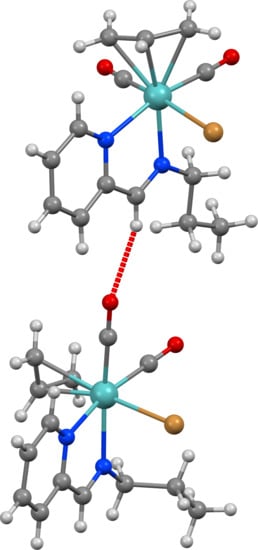
:
1. Introduction
2. Results and Discussion
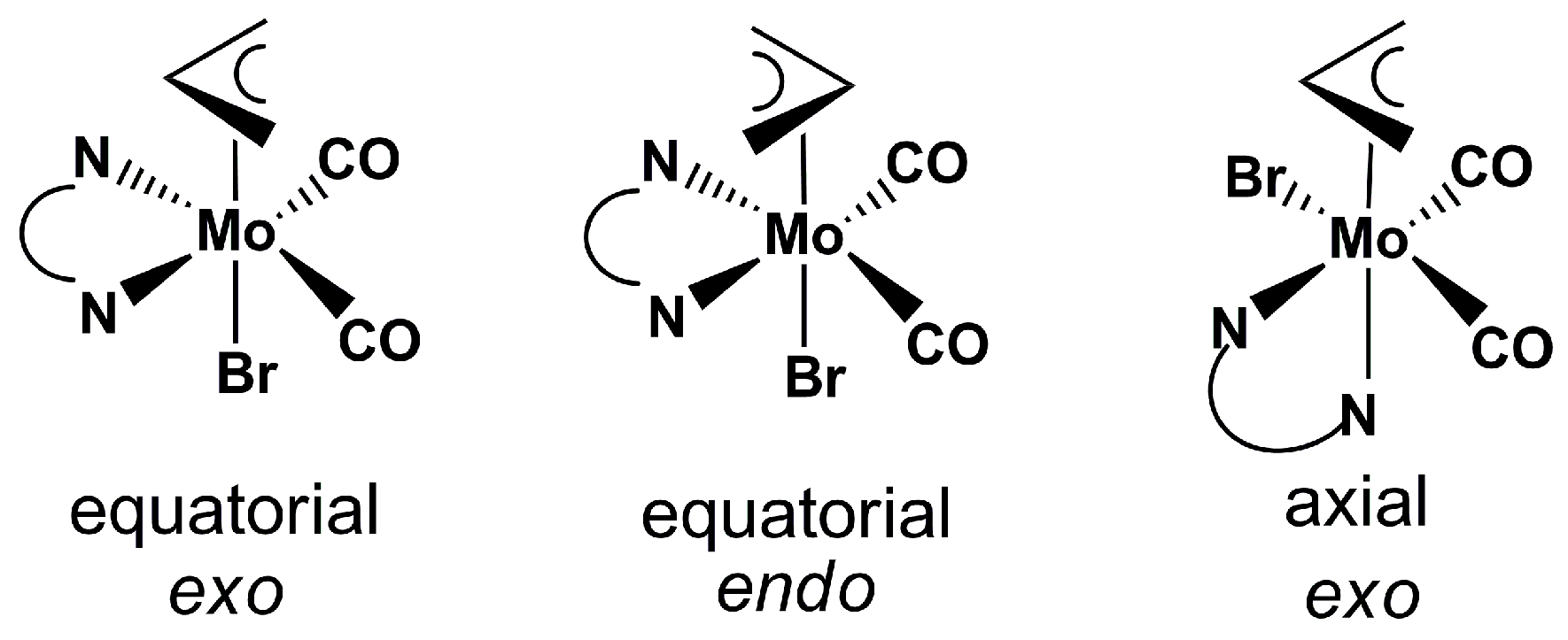
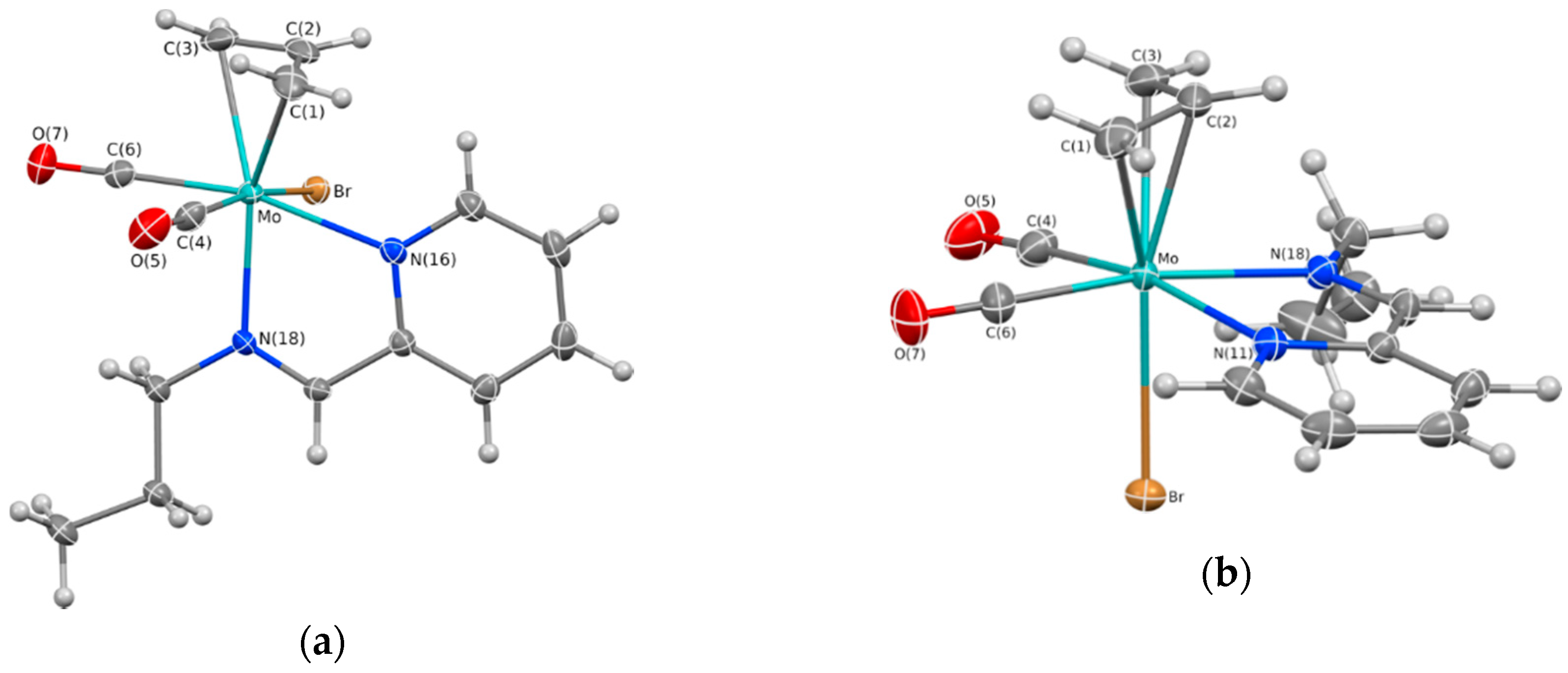
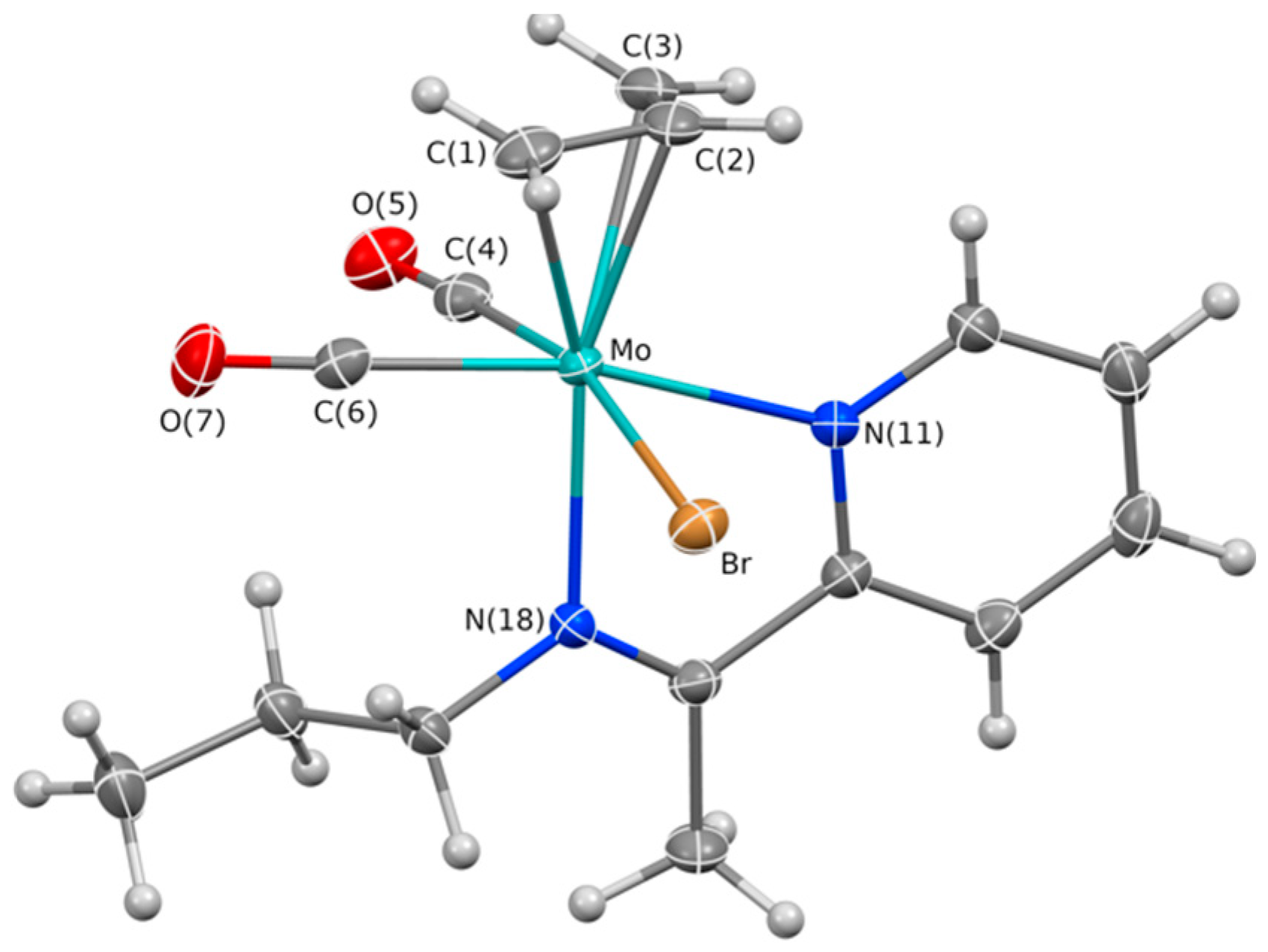
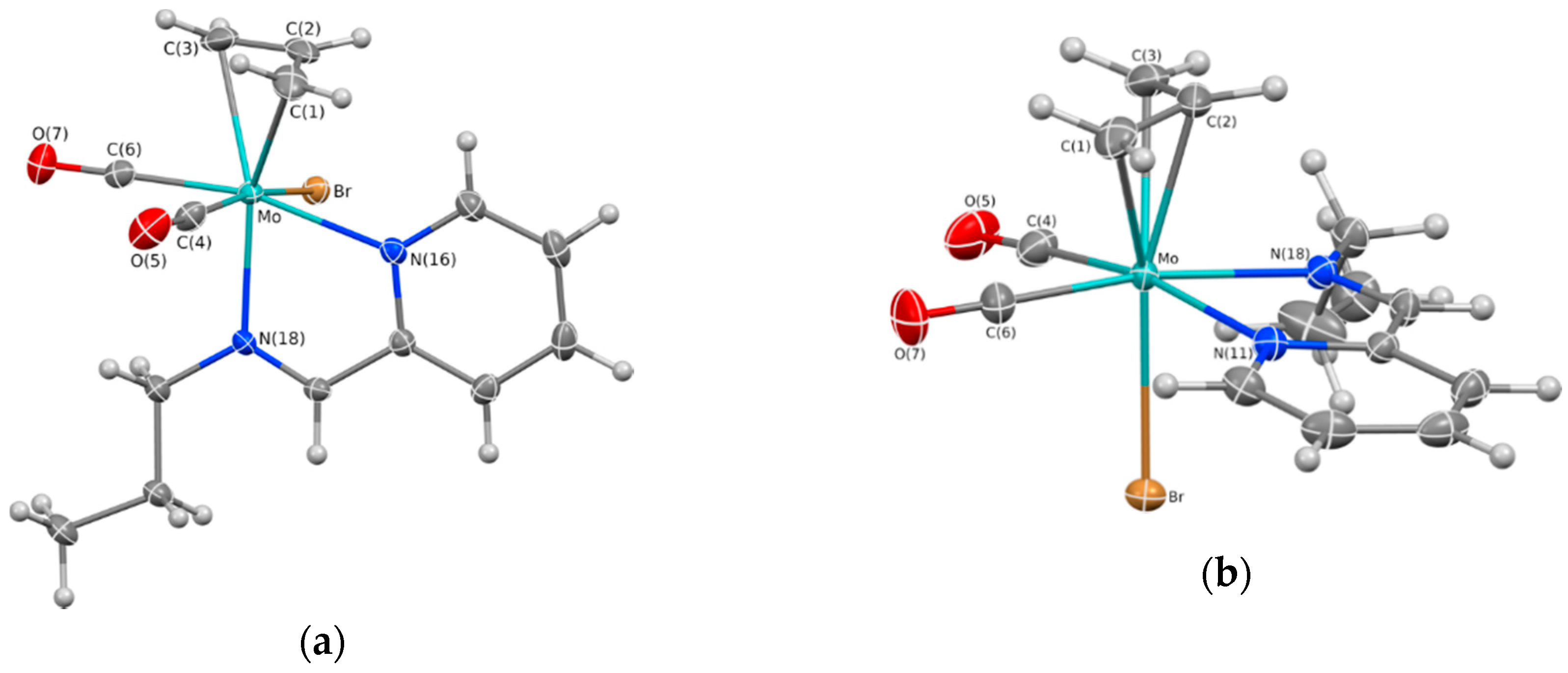
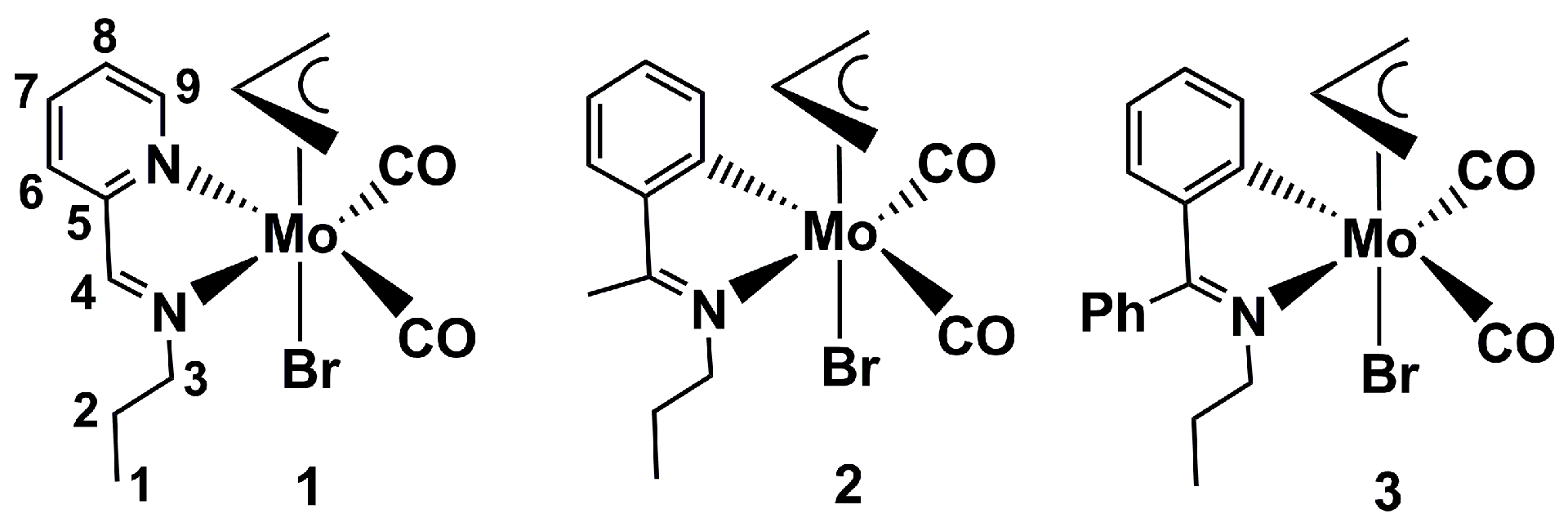
2.1. Chemical Studies: Complexes
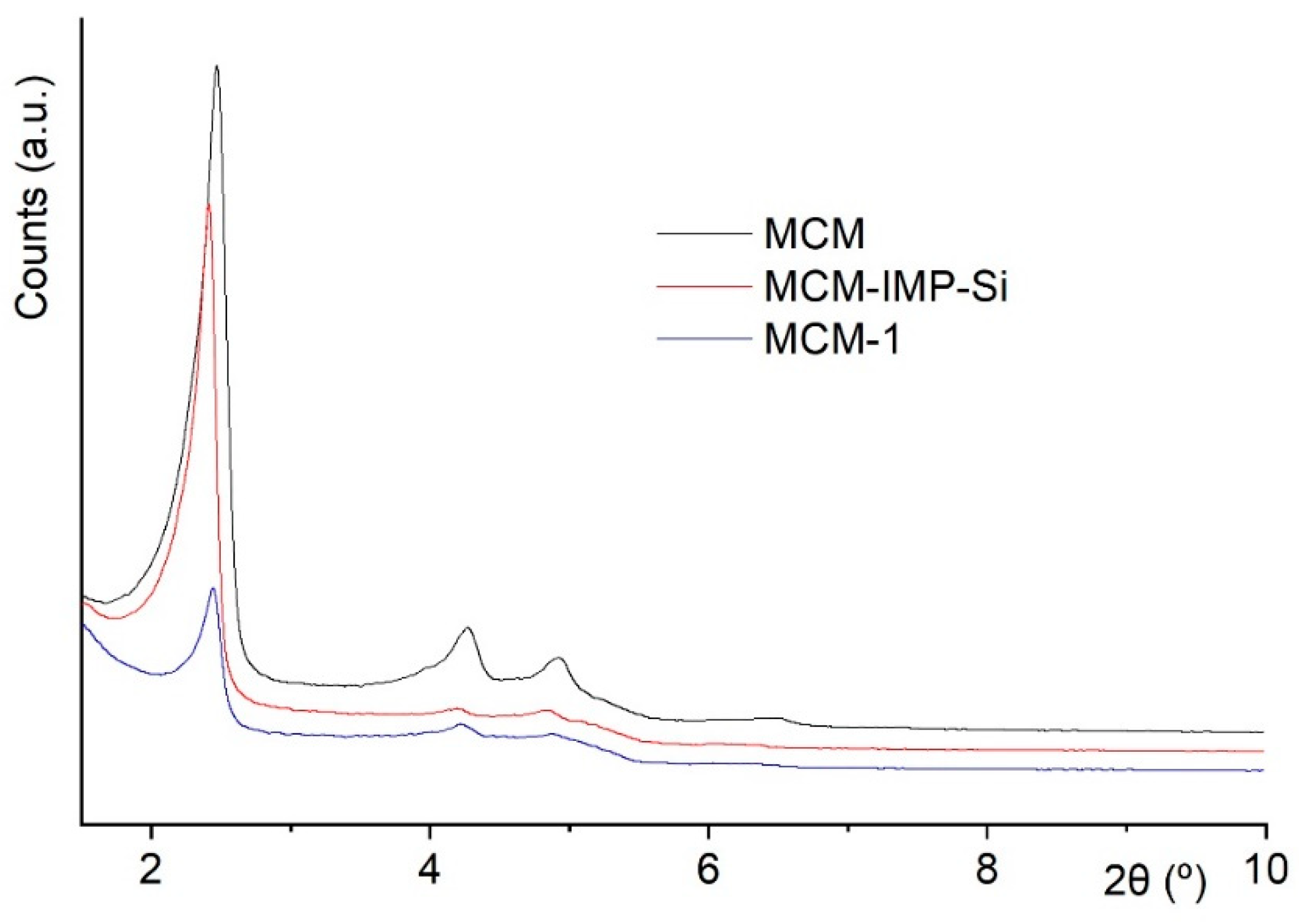
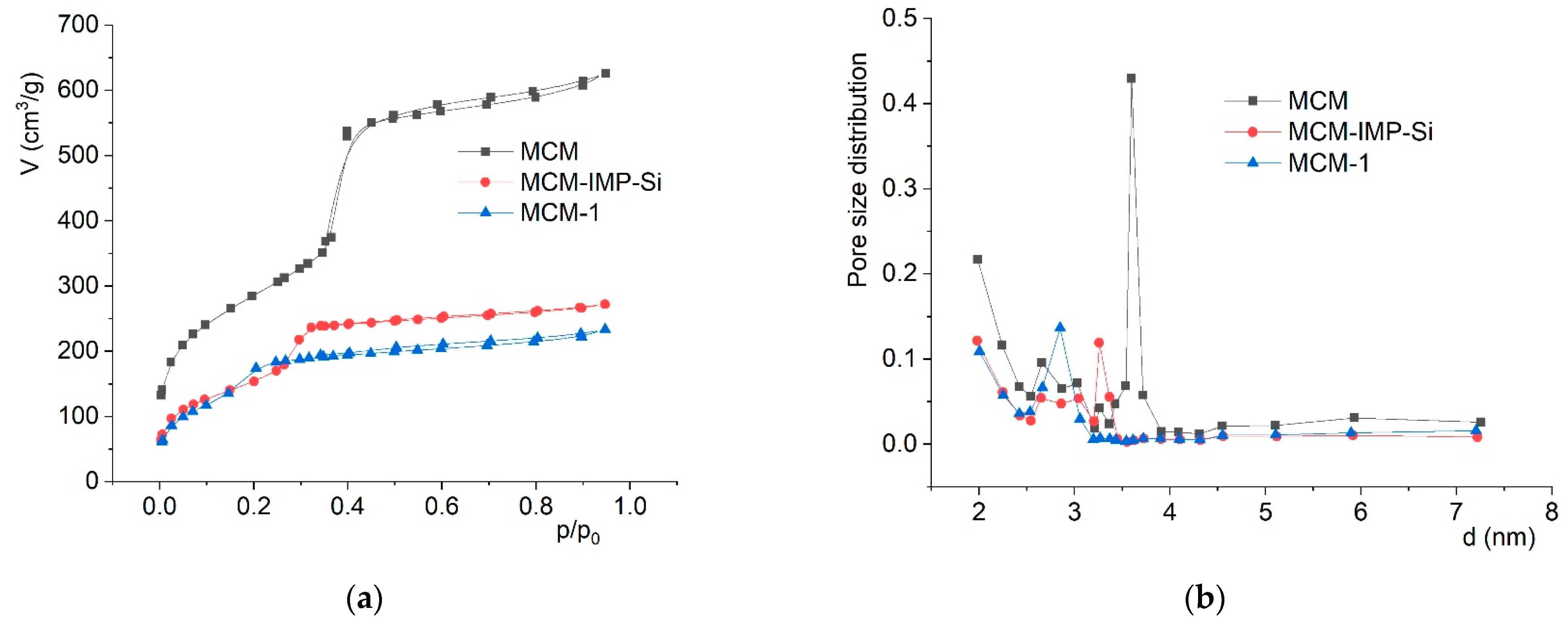
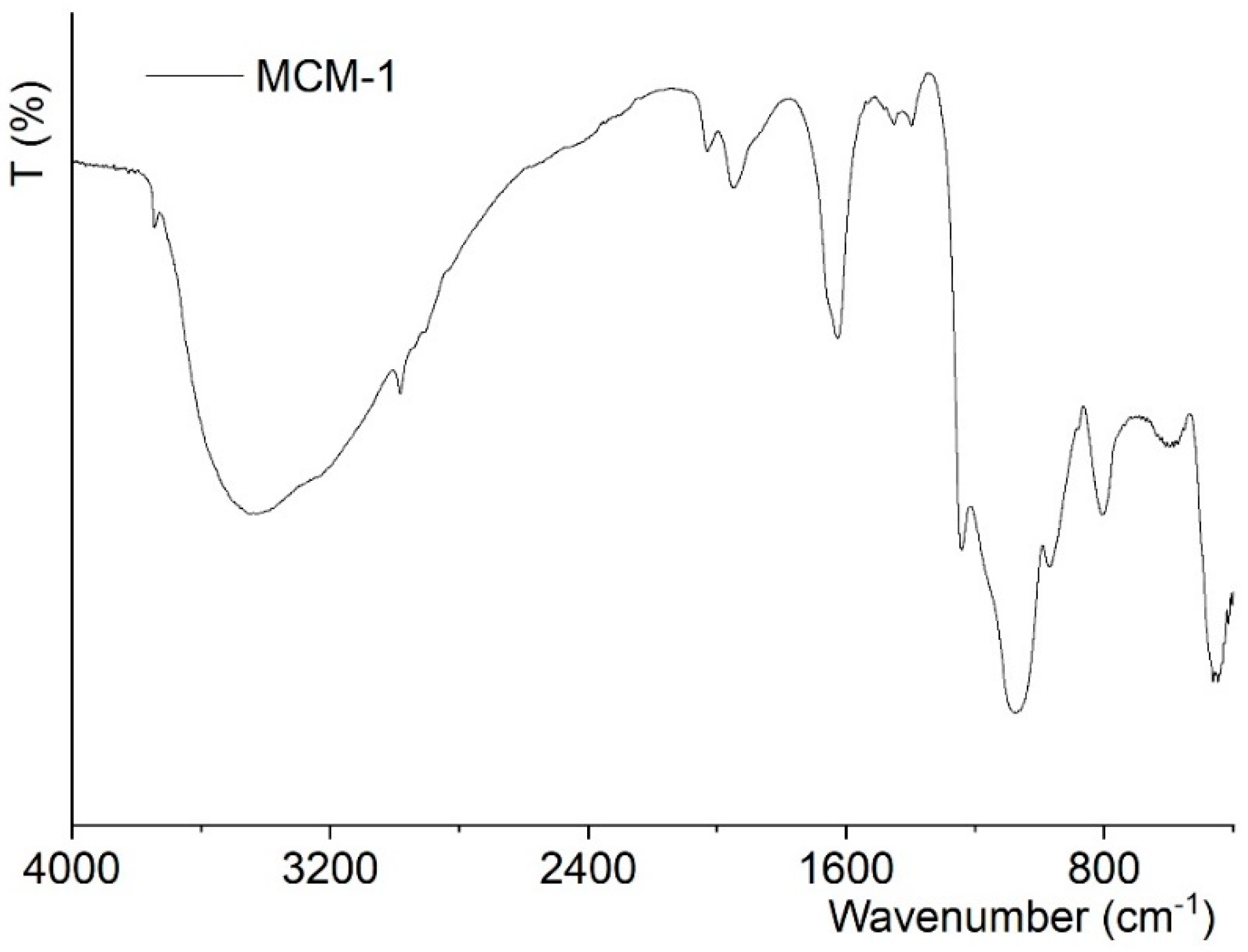
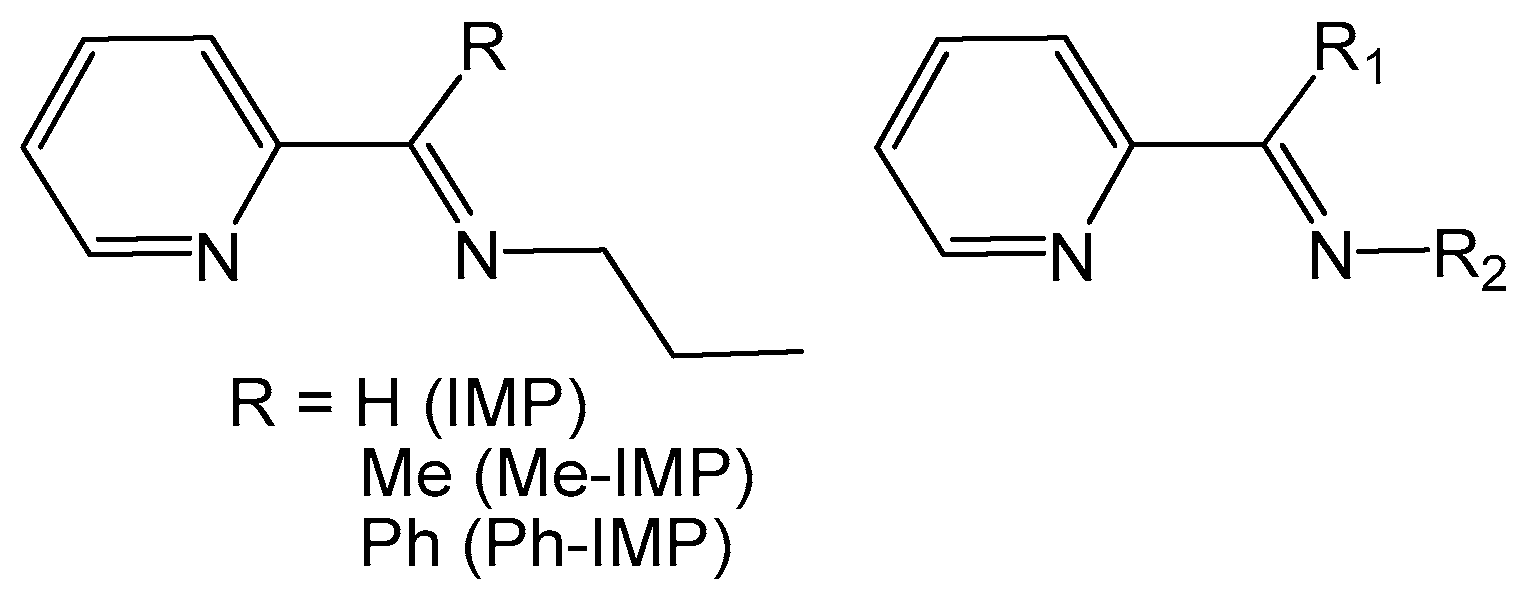
2.2. Chemical Studies: Materials
2.3. Catalytic Studies
2.4. Computational Studies
3. Materials and Methods
3.1. Synthesis of [MoBr(η3-C3H5)(CO)2C5H4NCH=N(CH2)2CH3] (Complex 1)
3.2. Synthesis of [MoBr(η3-C3H5)(CO)2C5H4NCCH3=N(CH2)2CH3] (Complex 2)
3.3. Synthesis of [MoBr(η3-C3H5)(CO)2C5H4NCPh=N(CH2)2CH3] (Complex 3)
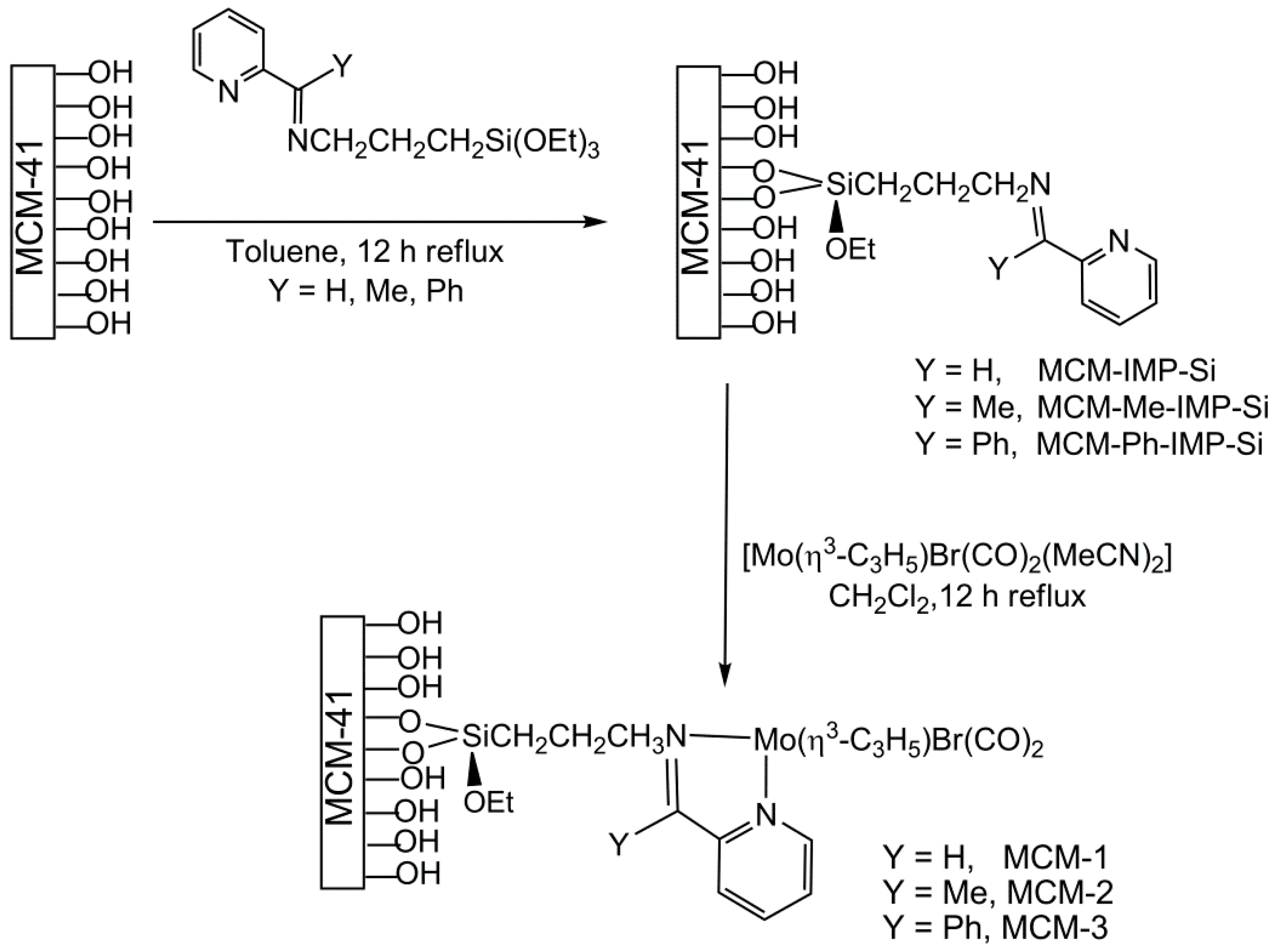
3.4. Synthesis of Materials MCM-IMP-Si, MCM-Me-IMP-Si, and MCM-Ph-IMP-Si
MCM-Ph-IMP-Si
3.5. Synthesis of Materials MCM-1, MCM-2, and MCM-3
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Balula, S.S.; Bruno, S.M.; Gomes, A.C.; Valente, A.A.; Pillinger, M.; Gonçalves, I.S.; Macquarrie, D.J.; Clark, J.H. Epoxidation of olefins using a dichlorodioxomolybdenum(VI)-pyridylimine complex as catalyst. Inorg. Chim. Acta 2012, 387, 234–239. [Google Scholar] [CrossRef]
- Mentes, A.; Kemmitt, R.D.W.; Fawcett, J.; Russell, D.R. Allyl complexes of molybdenum with Schiff base ligands. The crystal structures of [MoCl(CO)2{N(C6H4-2-OMe)=C(Me)C5H4N}(η3-C3H5)] and [MoCl(CO)2{N(Me)=C(Ph)C5H4N}(η3-C3H5)] are described. J. Organomet. Chem. 2002, 660, 91–97. [Google Scholar] [CrossRef]
- Gilani, S.R.; Mahmood, Z. Syntheses and structure determination of allyl complexes of molybdenum. J. Chem. Soc. Pak. 2003, 25, 41–43. [Google Scholar]
- Mentes, A.; Hanhan, M.E. Synthesis of water-soluble Mo(0) tetracarbonyl complexes containing nitrogen donor ligands and polymerization of methyl methacrylate. Transit. Met. Chem. 2008, 33, 91–97. [Google Scholar] [CrossRef]
- Gomes, A.C.; Bruno, S.M.; Gago, S.; Lopes, R.P.; Machado, D.A.; Carminatti, A.P.; Valente, A.A.; Pillinger, M.; Gonçalves, I.S. Epoxidation of cyclooctene using soluble or MCM-41-supported molybdenum tetracarbonyl–pyridylimine complexes as catalyst precursors. J. Organomet. Chem. 2011, 696, 3543–3550. [Google Scholar] [CrossRef]
- Matsubayashi, G.-E.; Ohara, T.; Tanaka, T. A paramagnetic 1H NMR study of tetrabromo(N-alkyl-methylpyridine-2-carbaldimine)tungsten(IV) and related complexes. Inorg. Chim. Acta 1989, 161, 67–72. [Google Scholar] [CrossRef]
- Haddleton, D.M.; Duncalf, D.J.; Kukulj, D.; Crossman, M.C.; Jackson, S.G.; Bon, S.A.F.; Clark, A.J.; Shooter, A.J. [N-Alkyl-(2-pyridyl)methanimine]copper(I) complexes: Characterisation and application as catalysts for atom-transfer polymerisation. Eur. J. Inorg. Chem. 1998, 1799–1806. [Google Scholar] [CrossRef]
- Shabbir, S.; Lee, S.; Lim, M.; Lee, H.; Ko, H.; Lee, Y.; Rhee, H. Pd nanoparticles on reverse phase silica gel as recyclable catalyst for Suzuki-Miyaura cross coupling reaction and hydrogenation in water. J. Organomet. Chem. 2017, 846, 296–304. [Google Scholar] [CrossRef]
- Kotze, H.; Mapolie, S. Cationic ruthenium(II) complexes supported on mesoporous silica as catalyst precursors in the selective oxidative cleavage of 1-octene. Appl. Organomet. Chem. 2016, 31, 1–13. [Google Scholar] [CrossRef]
- Hartl, F.; Bakker, M.J.; Santos, V.F.; Costa, P.J.; Calhorda, M.J. Accurate description of low-lying excited states in a series of photoreactive clusters [Os3(CO)10(α-diimine)] by DFT calculations. Inorg. Chem. 2018, 57, 11704–11716. [Google Scholar] [CrossRef]
- Vasconcellos-Dias, M.; Nunes, C.D.; Vaz, P.D.; Ferreira, P.; Brandão, P.; Félix, V.; Calhorda, M.J. Heptacoordinate tricarbonyl Mo(II) complexes as highly selective oxidation homogeneous and heterogeneous catalysts. J. Catal. 2008, 256, 301–311. [Google Scholar] [CrossRef]
- Ascenso, J.R.; de Azevedo, C.G.; Calhorda, M.J.; Carrondo, M.A.A.F.; Costa, P.; Dias, A.R.; Drew, M.G.B.; Galvão, A.M.; Romão, C.C.; Félix, V. Synthesis, bonding and dynamic behavior of fac-[Mo(II)(CO)2(η3-allyl)] derivatives. J. Organomet. Chem. 2001, 632, 197–208. [Google Scholar] [CrossRef]
- Costa, P.M.F.J.; Mora, M.; Calhorda, M.J.; Félix, V.; Ferreira, P.; Drew, M.G.B.; Wadepohl, H. Mono- and binuclear bipyridyl derivatives of the Mo(η3-C3H5)(CO)2 fragment: Structural studies and fluxionality in solution. J. Organomet. Chem. 2003, 687, 57–68. [Google Scholar] [CrossRef]
- Saraiva, M.S.; Nunes, C.D.; Félix, V.; Ribeiro, A.P.C.; Nieto de Castro, C.; Calhorda, M.J. Molybdenum(II) complexes with α-diimines: Catalytic activity in organic and ionic liquid solvents. Eur. J. Inorg. Chem. 2018, 3922–3932. [Google Scholar] [CrossRef]
- Alonso, J.C.; Neves, P.; Pires da Silva, M.J.; Quintal, S.; Vaz, P.D.; Silva, C.; Valente, A.A.; Ferreira, P.; Calhorda, M.J.; Félix, V.; et al. Molybdenum η3-allyl dicarbonyl complexes as a new class of precursors for highly reactive epoxidation catalysts with tert-butyl hydroperoxide. Organometallics 2007, 26, 5548–5556. [Google Scholar] [CrossRef]
- Tory, J.; Gobaille-Shaw, G.; Chippindale, A.M.; Hartl, F. Spectroelectrochemical study of complexes [Mo(CO)2(η3-allyl)(α-diimine)(NCS)] (α-diimine = bis(2,6-dimethylphenyl)-acenaphthenequinonediimine and 2,2′-bipyridine) exhibiting different molecular structure and redox reactivity. J. Organomet. Chem. 2014, 760, 30–41. [Google Scholar] [CrossRef]
- Nunes, C.D.; Valente, A.A.; Pillinger, M.; Fernandes, A.C.; Romão, C.C.; Rocha, J.; Gonçalves, I.S. MCM-41 functionalized with bipyridyl groups and its use as a support for oxomolybdenum(VI) catalysts. J. Mater. Chem. 2002, 12, 1735–1742. [Google Scholar] [CrossRef]
- Beck, J.S.; Vartuli, J.C.; Roth, W.J.; Leonowicz, M.E.; Kresge, C.T.; Schmitt, K.D.; Chu, C.T.-W.; Olson, D.H.; Sheppard, E.W.; McCullen, S.B.; et al. A new family of mesoporous molecular sieves prepared with liquid crystal templates. J. Am. Chem. Soc. 1992, 114, 10834–10843. [Google Scholar] [CrossRef]
- Marler, B.; Oberhagemann, U.; Voltmann, S.; Gies, H. Influence of the sorbate type on the XRD peak intensities of loaded MCM-41. Microporous Mater. 1996, 6, 375–383. [Google Scholar] [CrossRef]
- Hammond, W.; Prouzet, E.; Mahanti, S.D.; Pinnavaia, T.J. Structure factor for the periodic walls of mesoporous MCM-41 molecular sieves. Microporous Mesoporous Mater. 1999, 27, 19–25. [Google Scholar] [CrossRef]
- Gregg, S.J.; Sing, K.S.W. Adsorption, Surface Area and Porosity, 2nd ed.; Academic Press: London, UK, 1982. [Google Scholar]
- Alba, M.D.; Becerro, A.; Klinowski, J. Pore structure analysis of the mesoporous titanosilicate molecular sieve MCM-41 by 1H NMR and N2 sorption. J. Chem. Soc. Faraday Trans. 1996, 92, 849–854. [Google Scholar] [CrossRef]
- Romero, A.A.; Alba, M.D.; Zhou, W.; Klinowski, J. Synthesis and Characterization of the Mesoporous Silicate Molecular Sieve MCM-48. J. Phys. Chem. B 1997, 101, 5294–5300. [Google Scholar] [CrossRef]
- Kruk, M.; Jaroniec, M.; Olivier, J.P. Standard Nitrogen Adsorption Data for Characterization of Nanoporous Silicas. Langmuir 1999, 15, 5410–5413. [Google Scholar] [CrossRef]
- Vaz, P.D.; Nunes, C.D.; Vasconcellos-Dias, M.; Nolasco, M.M.; Ribeiro-Claro, P.J.A.; Calhorda, M.J. Vibrational study on the local structure of post-synthesis and hybrid mesoporous materials: Are there fundamental distinctions? Chem. Eur. J. 2007, 13, 7874–7882. [Google Scholar] [CrossRef]
- Bett, W.; Cradoc, S. Preparation of some silyl esters and study of their vibration spectra in gas and condensed phases. Monatshefte Chem. 1980, 111, 193–198. [Google Scholar] [CrossRef]
- Nunes, C.D.; Calhorda, M.J. Molybdenum(II) catalyst precursors in olefin oxidation reactions. Inorg. Chim. Acta 2015, 431, 122–131. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- SCM. Theoretical Chemistry; Vrije Universiteit: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Tom Dieck, H.; Friedel, H. Über π-Allyl-Komplexe des Molybdäns II. Die bildung von π-Allyldicarbonylmolybdän-Komplexen aus Molybdän-hexacarbonyl und seinen Derivaten. J. Organomet. Chem. 1968, 14, 375–385. [Google Scholar] [CrossRef]
- Baker, P.K.; Fraser, S.G.; Keys, E.M. The synthesis and spectral properties of some highly reactive new seven-coordinate molybdenum(II) and tungsten(II) bisacetonitrile dihalogenotricarbonyl complexes. J. Organomet. Chem. 1986, 309, 319–321. [Google Scholar] [CrossRef]
- Hayter, R.G. A new route to π-allyl complexes of molybdenum and tungsten. J. Organomet. Chem. 1968, 13, P1–P3. [Google Scholar] [CrossRef]
- Ferreira, P.; Nunes, C.D.; Vaz, P.D.; Bion, N.; Brandão, P.; Rocha, J. Hybrid mesoporous MCM-41 type material containing 1,4-diazobutadiene chelate ligand in the walls. Prog. Solid State Chem. 2005, 33, 163–170. [Google Scholar] [CrossRef]
- Saraiva, M.S.; Dias Filho, N.L.; Nunes, C.D.; Vaz, P.D.; Nunes, T.G.; Calhorda, M.J. Activity of Mo(II) allylic complexes supported in MCM-41 as oxidation catalysts precursors. Microporous Mesoporous Mater. 2009, 117, 670–677. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Becke, A.D. A new inhomogeneity parameter in density-functional theory. J. Chem. Phys. 1998, 109, 2092–2098. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Perdew, J.P. Erratum: Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 34, 7406. [Google Scholar] [CrossRef]
- van Lenthe, E.; Ehlers, A.; Baerends, E.-J. Geometry optimizations in the zero order regular approximation for relativistic effects. J. Chem. Phys. 1999, 110, 8943–8953. [Google Scholar] [CrossRef] [Green Version]
- Chemcraft Program. Available online: http://www.chemcraftprog.com/index.html (accessed on 23 November 2018).
- Calhorda, M.J.; Costa, P.J. Structure, bonding and reactivity of seven-coordinate allylic Mo(II) complexes. Coord. Chem. Rev. 2017, 344, 83–100. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex Isomer | 1 (Equatorial) | 1 (Axial) | 2 |
|---|---|---|---|
| Bonds | |||
| Mo-C(1) | 2.2995(16) | 2.3266(16) | 2.3442(14) |
| Mo-C(2) | 2.2072(15) | 2.2136(14) | 2.2148(14) |
| Mo-C(3) | 2.3273(16) | 2.3182(16) | 2.3163(15) |
| Mo-Br | 2.6961(2) | 2.6408(2) | 2.71031(19) |
| Mo-N(11) | 2.2833(12) | 2.2371(12) | 2.2652(12) |
| Mo-N(18) | 2.2031(12) | 2.2636(12) | 2.2049(11) |
| Mo-C(4) | 1.9398(16) | 1.9680(16) | 1.9561(15) |
| Mo-C(6) | 1.9695(16) | 1.9541(16) | 1.9468(15) |
| N(11)-Mo-N(18) | 73.36(4) | 72.74(4) | 71.89(4) |
| N(11)-Mo-Br | 81.22(3) | 81.49(3) | 85.15(3) |
| Angles | |||
| N(18)-Mo-Br | 83.46(3) | 82.59(3) | 80.67(3) |
| C(4)-Mo-C(6) | 80.84(7) | 78.35(6) | 76.26(6) |
| C(4)-Mo-Br | 86.62(5) | 168.52(5) | 166.23(4) |
| C(6)-Mo-Br | 89.00(5) | 96.79(4) | 96.86(4) |
| C(4)-Mo-N(11) | 167.69(5) | 100.68(5) | 98.56(5) |
| C(4)-Mo-N(18) | 103.47(6) | 87.28(6) | 87.86(5) |
| C(6)-Mo-N(11) | 100.74(6) | 166.00(5) | 165.34(5) |
| C(6)-Mo-N(18) | 171.03(6) | 93.26(5) | 94.04(5) |
| Materials | d100 (Å) | a (Å) | SBET/m2·g−1 | ΔSBET/% | Vp/cm3·g−1 | ΔVp b/% | dBJH/(Å) |
|---|---|---|---|---|---|---|---|
| MCM | 35.6 | 41.1 | 1037 | - | 0.97 | - | 36.0 |
| MCM-IMP-Si | 36.5 | 42.1 | 558 | −46 | 0.42 | −57 | 32.6 |
| MCM-1 | 36.2 | 41.8 | 553 | −47 | 0.36 | −63 | 28.5 |
| MCM | 35.6 | 41.1 | 1037 | - | 0.97 | - | 36.0 |
| MCM-Me-IMP-Si | 36.2 | 41.8 | 674 | −35 | 0.42 | −57 | 28.7 |
| MCM-2 | 36.0 | 41.6 | 523 | −50 | 0.25 | −74 | 26.7 |
| MCM | 36.0 | 41.6 | 1031 | - | 0.88 | - | 32.8 |
| MCM-Ph-IMP-Si | 36.7 | 42.4 | 683 | −33.8 | 0.46 | −48 | 28.5 |
| MCM-3 | 37.2 | 42.9 | 547 | −47.0 | 0.31 | −65 | 26.6 |
| Entry | Reaction a | Catalyst | Conv. % b | Select.% c | Yield % |
|---|---|---|---|---|---|
| 1 |  | 1 | 99 | 100 | 99 |
| 2 | 2 | 99 | 100 | 99 | |
| 3 | 3 | 100 | 100 | 100 | |
| 4 | 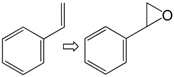 | 1 | 29 | 58 | 17/12 d |
| 5 | 2 | 33 | 41 | 20/13 d | |
| 6 | 3 | 40 | 36 | 26/14 d | |
| 7 | 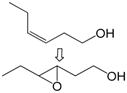 | 1 | 99 | 100 | 99 |
| 8 | 2 | 99 | 100 | 99 | |
| 9 | 3 | 99 | 100 | 99 | |
| 10 | 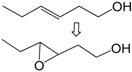 | 1 | 90 | 89 | 80/10 e |
| 11 | 2 | 82 | 86 | 70/12 e | |
| 12 | 3 | 50 | 89 | 45/5 e | |
| 13 |  | 1 | 98 | 49 | 50/48 f |
| 14 | 2 | 98 | 50 | 49/49 f | |
| 15 | 3 | 98 | 49 | 50/48 f | |
| 16 | 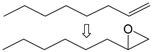 | 1 | 55 | 98 | 54/1 g |
| 17 | 2 | 43 | 96 | 41/2 g | |
| 18 | 3 | 72 | 100 | 72 |
| Entry | Reaction a | Catalyst | Conv. % b | Select. % c | Yield % |
|---|---|---|---|---|---|
| 1 |  | MCM-1 | 100/79/60 | 100/100/100 | 100 |
| 2 | MCM-2 | 100 | 100 | 100 | |
| 3 | MCM-3 | 100 | 100 | 100 | |
| 4 | 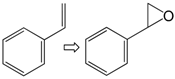 | MCM-1 | 40 | 82 | 33/7 d |
| 5 | MCM-2 | 45 | 80 | 36/9 d | |
| 6 | MCM-3 | 85 | 12 | 10/75 | |
| 7 | 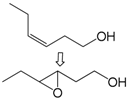 | MCM-1 | 73 | 100 | 73 |
| 8 | MCM-2 | 76 | 100 | 76 | |
| 9 | MCM-3 | 53 | 100 | 53 | |
| 10 |  | MCM-1 | 75 | 0 | 0 e |
| 11 | MCM-2 | 80 | 0 | 0 e | |
| 12 | MCM-3 | 75 | 0 | 0 e | |
| 13 | 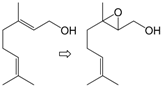 | MCM-1 | 56 | 59 | 23/33 f |
| 14 | MCM-2 | 66 | 58 | 28/38 f | |
| 15 | MCM-3 | 23 | 50 | 12/11 f | |
| 16 | 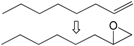 | MCM-1 | 35 | 38 | 13/22 h |
| 17 | MCM-2 | 42 | 45 | 19/23 h | |
| 18 | MCM-3 | 14 | 34 | 5/9 h | |
| 19 | 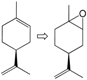 | MCM-1 | 83 | 23 | 3/16/30/34 g |
| 20 | MCM-2 | 87 | 24 | 4/17/30/36 g | |
| 21 | MCM-3 | 89 | 37 | 7/26/28/28 g |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasconcellos-Dias, M.; Marreiros, J.; Sales, R.; Félix, V.; Brandão, P.; Nunes, C.D.; Calhorda, M.J. New Molybdenum(II) Complexes with α-Diimine Ligands: Synthesis, Structure, and Catalytic Activity in Olefin Epoxidation. Molecules 2019, 24, 578. https://doi.org/10.3390/molecules24030578
Vasconcellos-Dias M, Marreiros J, Sales R, Félix V, Brandão P, Nunes CD, Calhorda MJ. New Molybdenum(II) Complexes with α-Diimine Ligands: Synthesis, Structure, and Catalytic Activity in Olefin Epoxidation. Molecules. 2019; 24(3):578. https://doi.org/10.3390/molecules24030578
Chicago/Turabian StyleVasconcellos-Dias, Maria, João Marreiros, Rita Sales, Vitor Félix, Paula Brandão, Carla D. Nunes, and Maria José Calhorda. 2019. "New Molybdenum(II) Complexes with α-Diimine Ligands: Synthesis, Structure, and Catalytic Activity in Olefin Epoxidation" Molecules 24, no. 3: 578. https://doi.org/10.3390/molecules24030578