
Retro-1-Oligonucleotide Conjugates. Synthesis and Biological Evaluation
by
 ,
,
Jordi Agramunt
1 ,
,
Enrique Pedroso
1,
Silvia M. Kreda
2,
Rudolph L. Juliano
3,* and
Anna Grandas
1,* 1
Departament de Química Inorgànica i Orgànica (Secció de Química Orgànica) and IBUB, Facultat de Química, Universitat de Barcelona, Martí i Franquès 1-11, 08028 Barcelona, Spain
2
UNC Cystic Fibrosis Center, School of Medicine, University of North Carolina, Chapel Hill, NC 27516, USA
3
UNC Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, NC 27599, USA
*
Authors to whom correspondence should be addressed.
Molecules 2019, 24(3), 579; https://doi.org/10.3390/molecules24030579
Submission received: 19 December 2018
/
Revised: 28 January 2019
/
Accepted: 1 February 2019
/
Published: 6 February 2019
(This article belongs to the Special Issue Molecular Tools for Nucleic Acid Manipulation for Biological Intervention)
Abstract
:Addition of small molecule Retro-1 has been described to enhance antisense and splice switching oligonucleotides. With the aim of assessing the effect of covalently linking Retro-1 to the biologically active oligonucleotide, three different derivatives of Retro-1 were prepared that incorporated a phosphoramidite group, a thiol or a 1,3-diene, respectively. Retro-1–oligonucleotide conjugates were assembled both on-resin (coupling of the phosphoramidite) and from reactions in solution (Michael-type thiol-maleimide reaction and Diels-Alder cycloaddition). Splice switching assays with the resulting conjugates showed that they were active but that they provided little advantage over the unconjugated oligonucleotide in the well-known HeLa Luc705 reporter system.
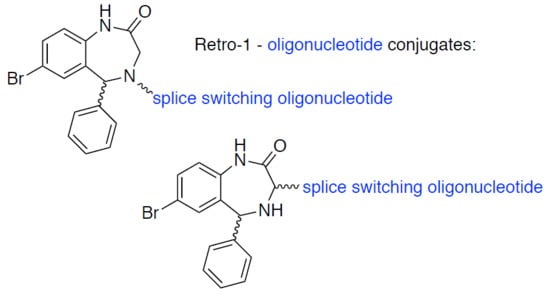
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Forty years after the therapeutic potential of synthetic oligonucleotides was first perceived [1], delivery is the one problem that remains not well solved [2,3]. Oligonucleotides can easily be modified to favor hybridization and ameliorate their bioavailability [4,5], but there is no gold standard methodology for effective and complete internalization. Most forms of oligonucleotides are thought to be taken up by endocytosis and accumulate in intracellular endomembrane compartments [6,7]. Only a tiny fraction of the accumulated oligonucleotide spontaneously leaks from the endosomes, although this is sometimes enough to provide a pharmacological effect. There have been many efforts to increase both the cellular uptake and intracellular release of oligonucleotides including various targeting ligands and endomembrane destabilizing polymers or nanoparticles [2,8].
A few years ago, Juliano and cols. described that the compound named Retro-1 enhanced the pharmaceutical potency of antisense and siRNA oligonucleotides [9]. Retro-1 is a member of a group of compounds able to reduce the action of bacterial toxins [10] by interfering with their intracellular trafficking. Subsequently, high throughput screening has allowed identification of additional molecules capable of enhancing the effectiveness of synthetic oligonucleotides [11,12,13]. Some of the hits developed in these studies have been modified to assess their effect on oligonucleotide delivery [14,15]. However, to our knowledge the impact of covalently linking one of these molecules to the oligonucleotide chain has not been evaluated.
Retro-1, the first compound identified to ameliorate both antisense and siRNA oligonucleotides effect, was chosen for this evaluation. In this manuscript we wish to describe the preparation of three Retro-1 derivatives, their conjugation reactions to a splice switching oligonucleotide, and the results of the biological evaluation. These studies indicate that the efficacy of covalently linking the two moieties is inferior to that achieved by administering the two separate molecules.
2. Results and Discussion
2.1. Modification of Retro-1 for Conjugation
One alternative to prepare oligonucleotide conjugates, that is, to covalently link oligonucleotides to other molecules, involves the use of a phosphoramidite derivative that can be incorporated into the chain using standard oligonucleotide elongation methodologies. Another possibility is making use of a click reaction [16] that chemoselectively links the two components of the conjugate. Irrespective of the synthesis approach, Retro-1 had to be derivatized so as to incorporate an additional functional group.
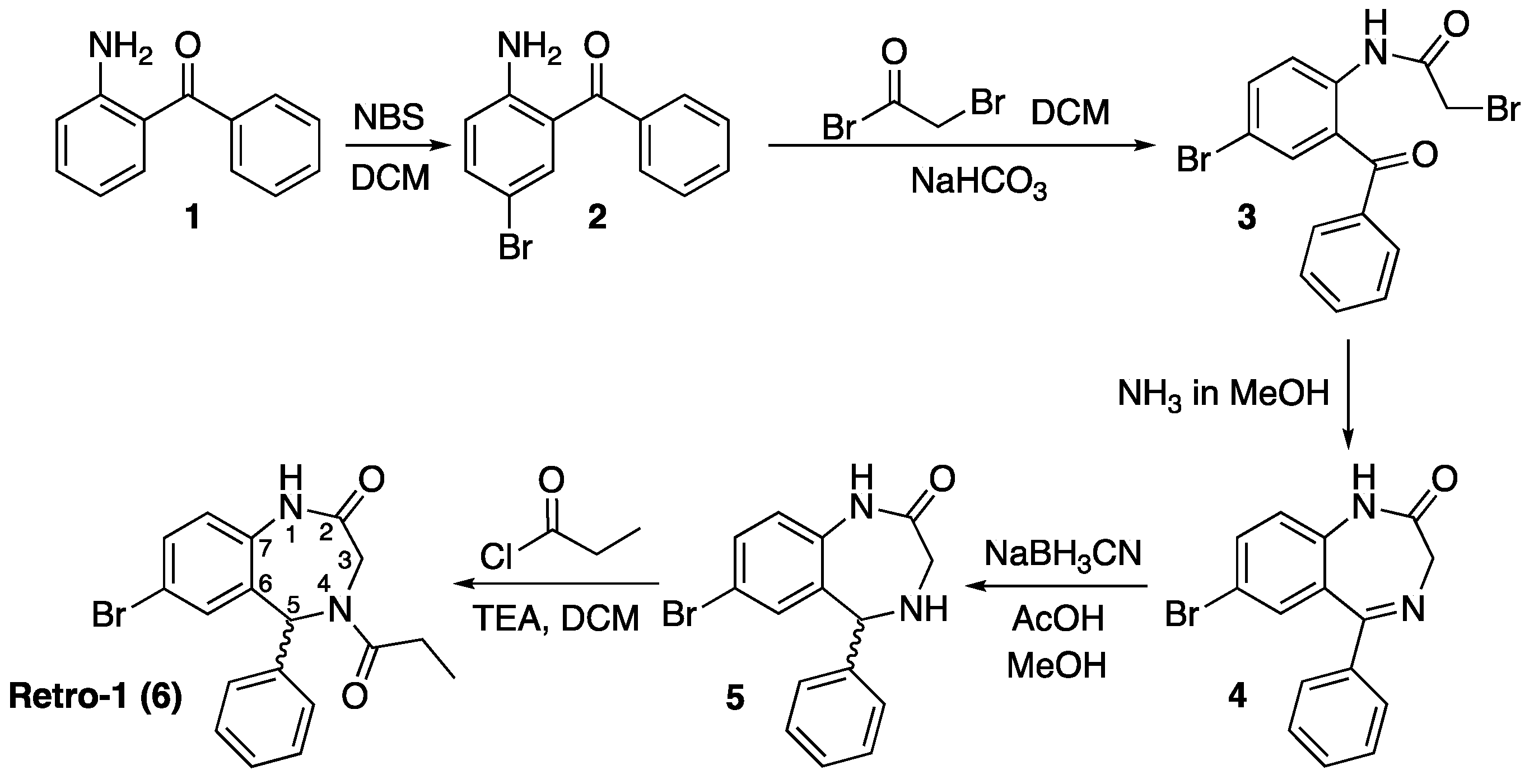
The structure of Retro-1 (6), which is shown on Scheme 1, has many points in common with that of benzodiazepine psychoactive drugs. It is also composed of two fused rings, where two atoms of the benzene ring and carbons 6–7 of the diazepine function as bridgehead, and the 1,4-diazepine ring contains a carbonyl group at the 2 position and an aromatic ring appending from carbon 5. The main difference is that atoms 4 and 5 of the diazepine are not linked by a double bond but by a single one. As a result, carbon 5 is a stereocenter.
The synthesis of racemic Retro-1 has been described [17], as well as the separation of the two enantiomers using a chiral HPLC column (conformers, which are observed at the NMR spectra, could not be separated). Both enantiomers were shown to be biologically active, and the difference between their EC50 values was small (the S isomer was a bit more active than the R one, but both were within the same 1–10 μM range as the racemic mixture). Therefore, we have worked with the racemic mixture.
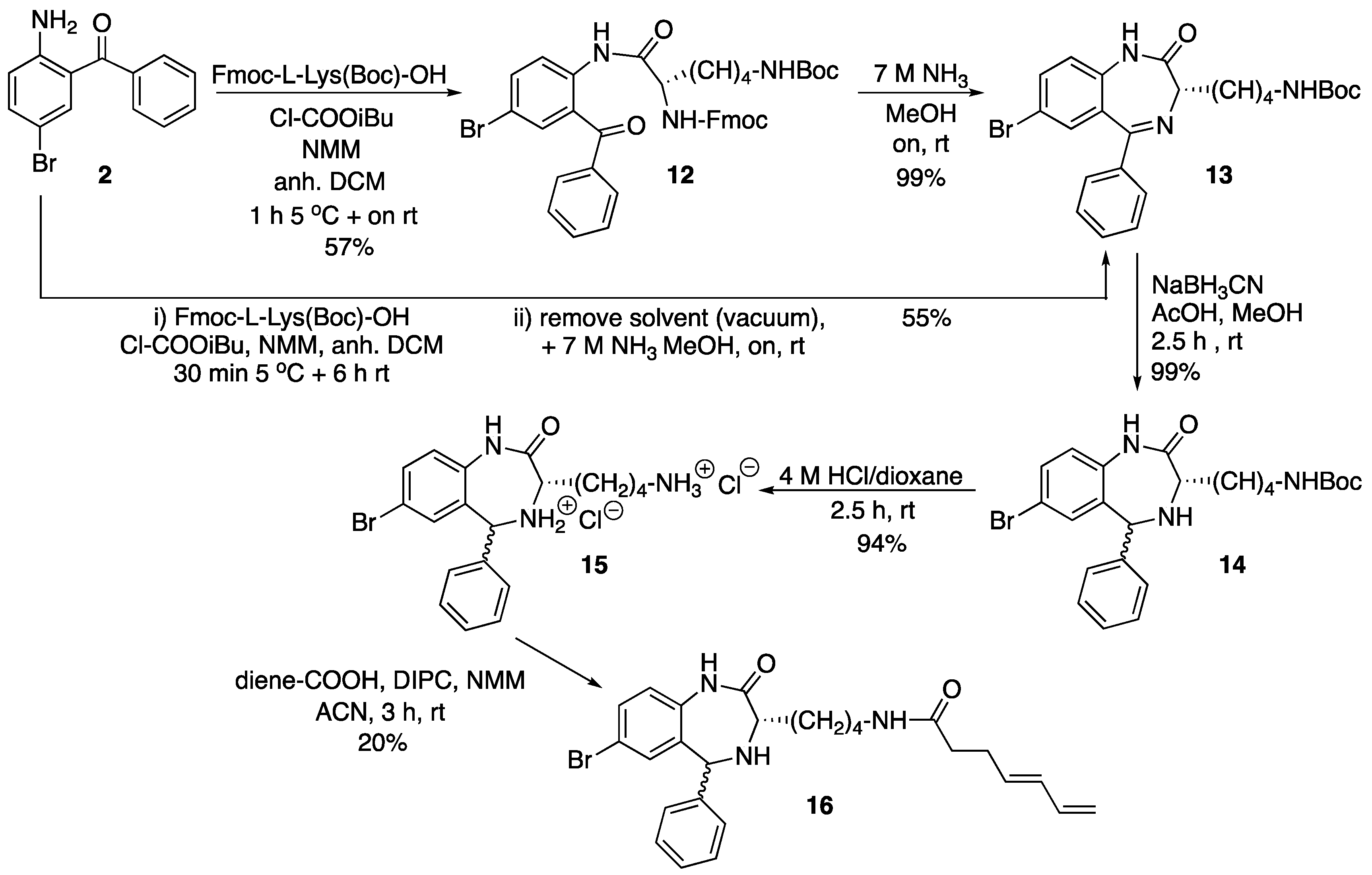
The only information available on the biological activity of Retro-1 analogs [14] indicates that compounds with no substituent at the 4 position (see Scheme 1) favor oligonucleotide activity, whilst N-alkylation seems best suited for reducing bacterial toxins effect. Since Retro-1, in which N-4 is acylated, exhibits both effects, we decided to prepare two N-4 acylated analogs (Scheme 2) and an additional one modified at the 3 position (Scheme 3).
As shown in Scheme 1, the diazepine ring is made from three building blocks: the aminobenzophenone, the acylating reagent and ammonia. Acylation of the aromatic amine links the two main components and imine formation closes the cycle.
Based on this scheme, which we could perfectly reproduce, changing the reagents of the two acylation reactions appeared to be a simple option to adapt Retro-1 for conjugation. Replacement of bromoacetyl bromide with an activated amino acid would modify position 3 of the diazepine ring, and use of a suitably protected trifunctional amino acid would incorporate an additional group suitable for further derivatization. Alternatively, substitution of propionyl chloride with another acylation reagent would allow position 4 to be modified.
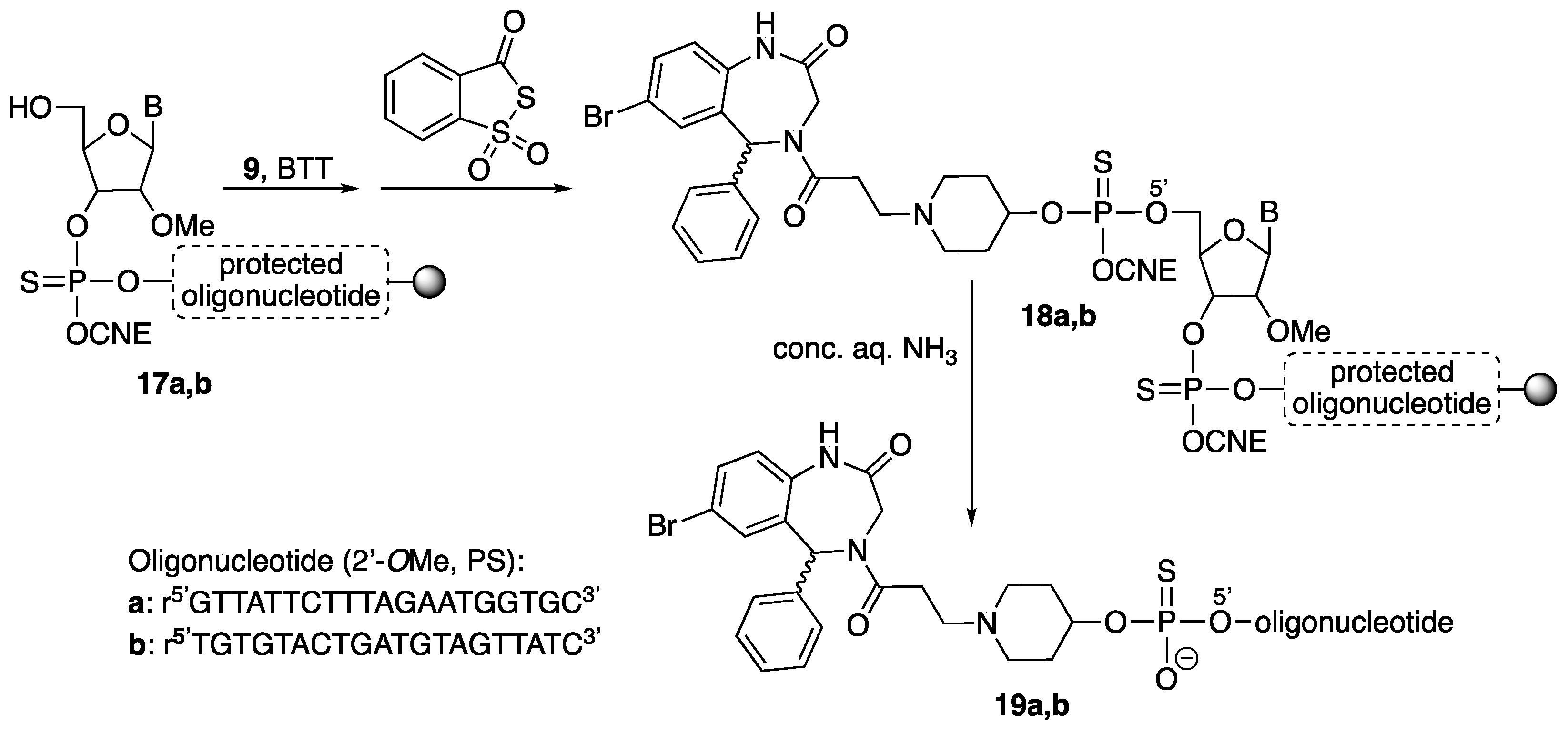
We decided that the three Retro-1 analogs would be differently derivatized for conjugation. One would include a phosphitylatable hydroxyl group, and could be attached to the 5’ end of a resin-linked chain. The other two would contain functional groups suitable for click conjugations in solution. For this purpose we chose a thiol and a 1,3-diene, which were intended to react with a maleimide moiety. Maleimido-oligonucleotides can be easily assembled on a solid support making use of the appropriate maleimide protection [18].
2.1.1. Retro-1 Derivatives Modified at the 4 Position
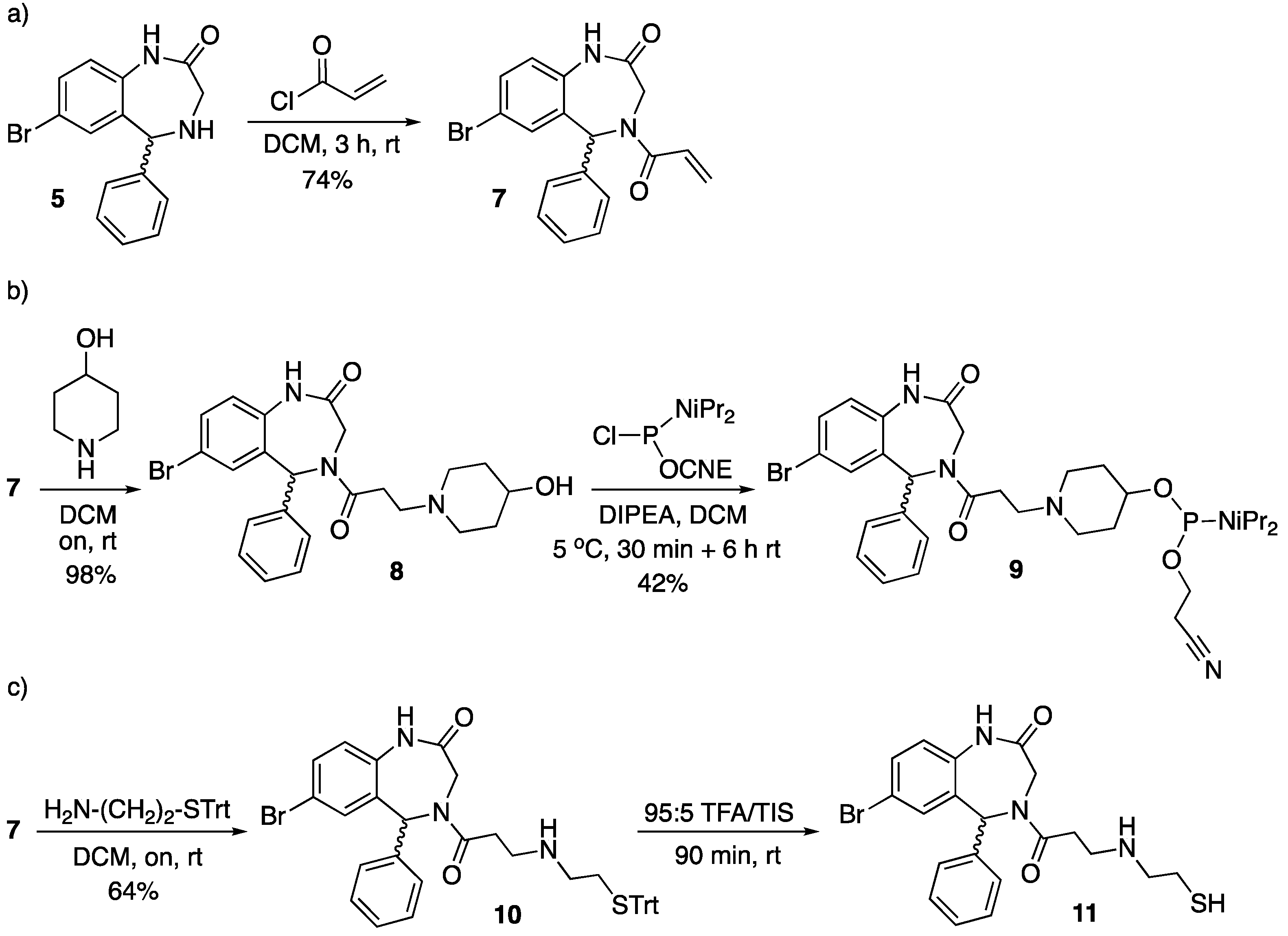
Two of the three analogs were synthesized from the amine precursor of Retro-1 5 (see Scheme 1). Replacement of propionyl chloride with acryloyl chloride gave 7 (Scheme 2a), which could be easily modified by means of Michael-type reactions.
Reaction between 7 and 4-hydroxypiperidine afforded 8, from which phosphoramidite 9 was prepared by reaction with a chlorophosphine and a base (Scheme 2b). The other aza-Michael reaction was carried out with S-trityl cysteamine, which furnished 10, and removal of the trityl group under acidic conditions gave 11 (Scheme 2c). To minimize the extent of thiol oxidation to disulfide the thiol-containing Retro-1 analog was kept protected, and thiol deprotection was carried out not long before the conjugation reaction.
2.1.2. Modification of the 3 Position of Retro-1
The starting material for the preparation of the third Retro-1 derivative was the first intermediate in the synthesis of Retro-1, 2 (see Scheme 1). Acylation of the amine with a suitably protected lysine derivative was found not to be straightforward. Attempts to activate the carboxyl group with either a combination of a carbodiimide and 1-hydroxybenzotriazole, 1-[(1-cyano-2-ethoxy-2-oxoethylideneaminooxy)-dimethylaminomorpholinomethylene)] methan- aminium hexafluorophosphate (COMU), or mesitylenesulfonyl 3-nitro-1,2,4-triazole (MSNT) in the presence of N-methylimidazole were unsuccessful. Gratifyingly, the mixed carboxylic carbonic anhydride methodology did work (Scheme 3). Carboxyl group activation with isobutyl chloroformate in the presence of N-methylmorpholine afforded 12, and overnight treatment of 12 with 7 M ammonia in methanol quantitatively removed the Fmoc group and promoted cyclization to 13. These two steps were also carried out without isolating 12, and 13 was obtained in a similar yield.
Diene-derivatized 16 was finally obtained after the following series of steps: (i) Reduction of the imine of compound 13 with NaBH3CN as previously reported, which gave 14; (ii) Removal of the Boc group with an acidic treatment, which yielded 15; (iii) Carbodiimide-promoted acylation of the primary amine with (4E)-4,6-heptadienoic acid. We were initially concerned with the possible acylation of the secondary amine (N-4 of the diazepine ring), but we found that reaction took place exclusively on the lysine ε-amine, furnishing 16. In fact, all attempts carried out to acylate N-4 failed. Therefore, derivative 16 differed from the other Retro-1 analogs in the presence of a substituent appending from carbon 3 and no acyl group on nitrogen 4.
2.2. Preparation of the Retro-1 Conjugates
The three Retro-1 derivatives (9, 11, 16) were linked to oligonucleotide r5’GTTATTCTTTAGAATGGTGC3’ (all 2’-O-methyl and phosphorothioate, T = ribothymidine). This sequence is complementary to that of a mutated intron from thalassemic hemoglobin that is introduced into the firefly luciferase gene used for the splice switching experiments [19]. Compounds 9, 11 and 16 were also linked to a control oligonucleotide with a scrambled sequence, namely r5’TGTGTACTGATGTAGTTATC3’ (all 2’-O-methyl and phosphorothioate; T = ribothymidine).
After elongation of the two oligonucleotide chains using standard methodology and BTT as activator (BTT = 5-benzylthio-1H-tetrazole), removal of the DMT group (DMT = 4,4’-dimethoxytrityl) on the 5’ end furnished oligonucleotide-resins 17. This was followed by coupling of phosphoramidite 9 (2 × 10 min coupling, activation with BTT) and sulfurization (Beaucage reagent). The resulting Retro-1-oligonucleotide-resins (18) were treated with conc. aq. ammonia (3 h, rt) to yield conjugates 19a and 19b (Scheme 4).
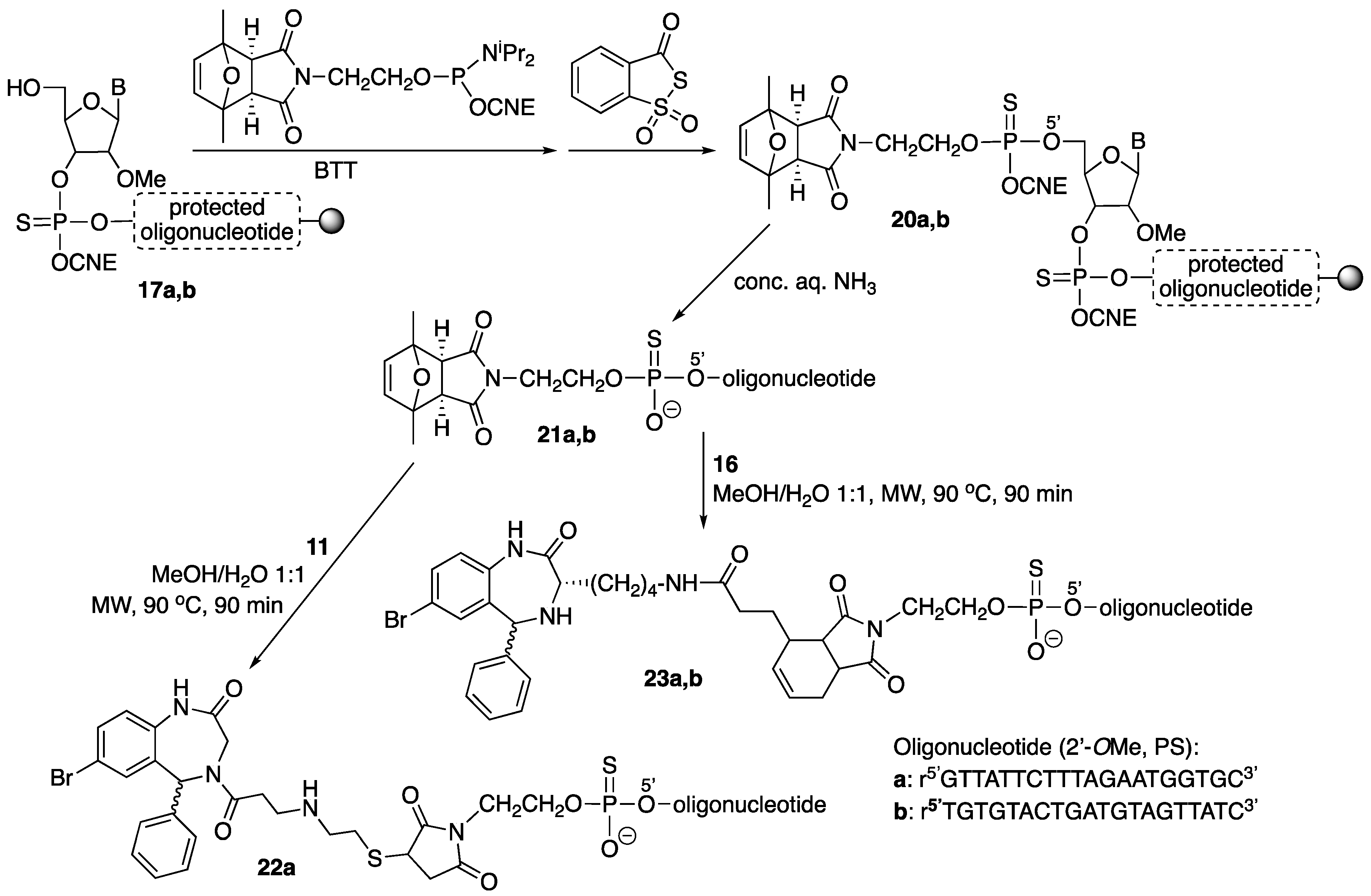
For the preparation of the other conjugates (Scheme 5), the 2,5-dimethylfuran-protected maleimide phosphoramidite was coupled to either oligonucleotide-resin 17 (2 × 10 min coupling, activation with BTT), and the resulting phosphite triester sulfurized (Beaucage reagent). Treatment of oligonucleotide-reins 20 with conc. aq. ammonia afforded [protected maleimido]-oligonucleotides 21, which were purified. Conjugates 22 and 23 were obtained by simultaneously carrying out maleimide deprotection (retro Diels-Alder reaction) and reaction with either the thiol-containing derivative 11 (Michael-type reaction) or the diene-derivatized compound 16 (Diels-Alder cycloaddition) in a microwave (MW) oven.
All conjugates were purified by reversed phase HPLC and characterized by MALDI-TOF MS.
2.3. Splice-Switching Assays
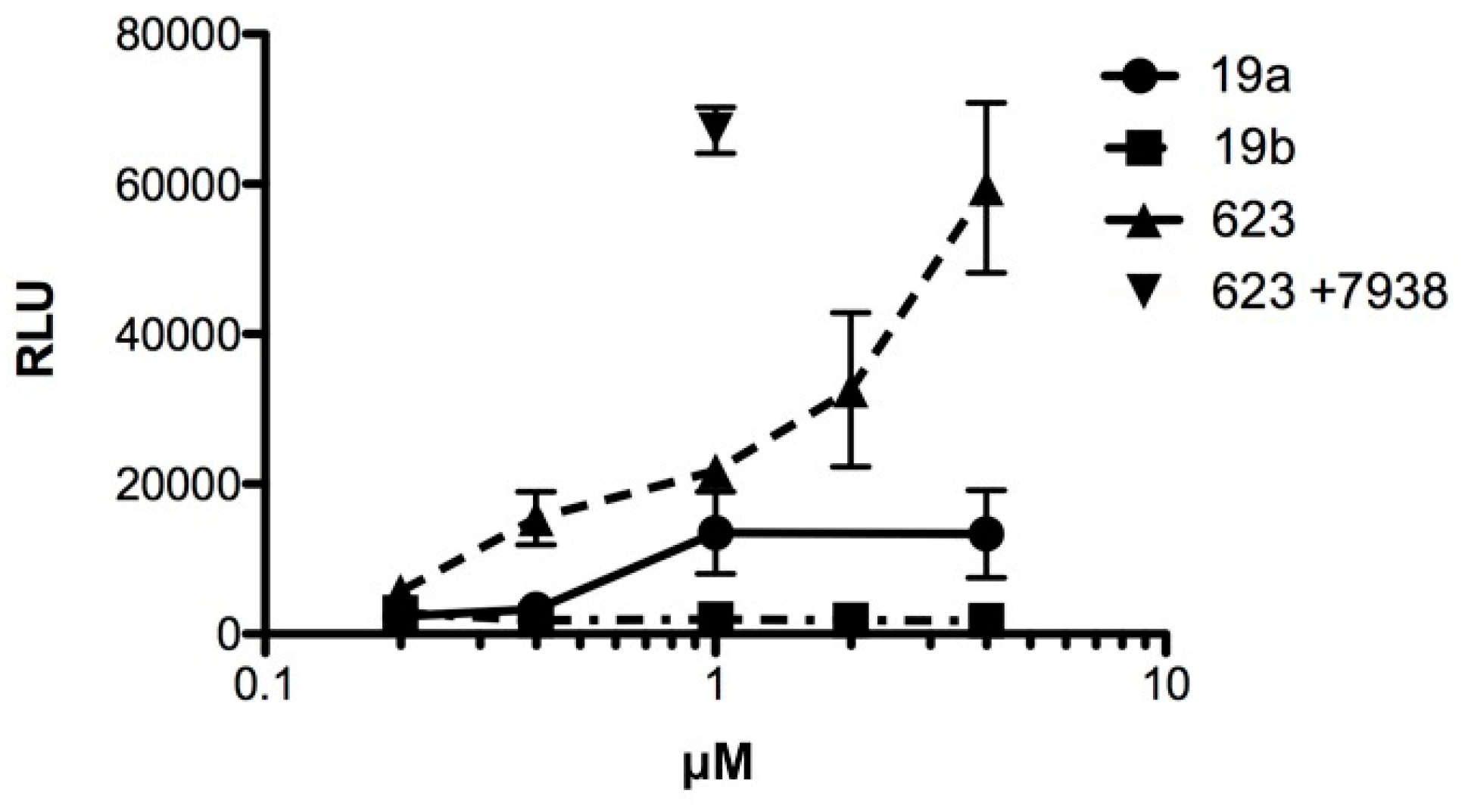
Oligonucleotides conjugated to Retro-1 derivatives were examined for splice correction activity. The experiments utilized the HeLa Luc 705 cell line [19], as described in Materials and Methods. Oligonucleotides having the known splice correcting sequence r5’GTTATTCTTTAGAATGGTGC3’, previously termed SSO623 [19], were used in conjugated or unconjugated form and were compared to control oligonucleotides with the inactive sequence r5’TGTGTACTGATGTAGTTATC3’. In Figure 1 oligonucleotide 19a, which is the immediate conjugate of Retro-1, was compared to its control conjugate 19b. Furthermore, we examined unmodified SSO623, as well as SSO623 followed by treatment with the small molecule UNC7938 as a positive control. As seen in the figure, oligonucleotide 19a produced a dose-dependent luciferase induction while the control oligonucleotide 19b did not. However, SSO623 itself also gave rise to a progressive induction of luciferase that was somewhat greater than that produced by 19a. The dual use of SSO623 and UNC7938 provided the largest degree of splice correction and luciferase induction as expected. This set of experiments demonstrated that coupling of the Retro moiety did not prevent the splice correcting activity of the active oligonucleotide. However, conjugate 19a did not provide an advantage, in terms of potency or efficacy, over SSO623 itself. We also examined two additional conjugates 22a and 23a. However, neither of these conjugates displayed an advantage over SSO623 in the HeLa Luc 705 induction assay.
In summary, conjugation of the Retro moiety to a splice switching oligonucleotide did not provide a major enhancement of splice correction activity in the widely used HeLa Luc705 reporter system. At this point it is unclear why the conjugates displayed slightly lower activity than the unmodified oligonucleotide. One could hypothesize that the presence of the bulky Retro group might affect either cell uptake of the oligo or might affect the interaction of the oligo with the splicing machinery. However, detailed investigation of these possibilities, especially the splicing aspect, would involve a very substantial amount of new biological investigation and is beyond the scope of this work.
3. Experimental Section
3.1. Materials and Methods
3.1.1. Materials for Solution Organic Synthesis
(E)-Hepta-4,6-dienoic acid was prepared as described by Baillie et al. [20], and S-trityl cysteamine as reported by Naumiec et al. [21]. Fmoc-l-Lys(Boc)-OH was from Novabiochem (Zaragoza, Spain); all of the other chemicals were either from Sigma-Aldrich (Zaragoza, Spain) or Across Organics (Zaragoza, Spain) (7 M ammonia in methanol), and were used without further purification. Water was obtained from a MilliQ system (Zaragoza, Spain).
3.1.2. Analysis and Characterization Techniques for Small Organic Molecules (2–16)
TLC was carried out on silica gel plates 60 F254 from Merck (Zaragoza, Spain). IR spectra were recorded in a Nicolet 6700 FT-IR spectrometer (Thermo Scientific, Zaragoza, Spain). 1H and 13C NMR spectra were recorded on either Varian Mercury 400 MHz or Brucker 400 MHz spectrometers (reference: TMS or residual solvent signals). HR-ESI mass spectra were obtained using an LC/MSD-TOF Instrument (Agilent Technologies, Zaragoza, Spain). HPLC/MS analyses were recorded in an Alliance Waters 2690 separation module with a Waters micromass ZQ4000 MS detector (Waters, Zaragoza, Spain). Aqueous solutions were lyophilized in either Labconco (Vertex, Zaragoza, Spain) or Christ freeze dryers (Inycom, Zaragoza, Spain).
3.1.3. Materials for Oligonucleotide Assembly
Nucleoside phosphoramidites (2′-OMe APac, CAc, GiPrPac, and 5-MeU), 5′-O-DMT-2′-OMe-CAc-succinyl-derivatized glass beads, and oligonucleotide synthesis reagents were from Link Technologies (Manchester, UK). [Protected maleimido]-phosphoramidite (see structure in Scheme 5) was synthesized as previously described [18].
3.1.4. Oligonucleotide Assembly
Oligonucleotide chains were elongated in a 3400 ABI automatic synthesizer at the 1 μmol scale, using standard phosphite triester methodology. The activator used at the coupling step was 5-benzylthio-1H-tetrazole (0.3 M solution in anh. acetonitrile, 10 min coupling time), and the Beaucage reagent was used for sulfurization (0.05 M in anh. acetonitrile; 2 × 4 min). All phosphoramidites were dissolved in anh. acetonitrile (0.1 M solutions), except that of 2′-OMe-5-MeU (which was dissolved in anh. DCM).
Final deprotection conditions: conc. aq. ammonia, 3 h, room temperature. After this treatment the resin was filtered and washed with water (3×), the filtrates pooled and concentrated in a SpeedVac apparatus (Inycom, Zaragoza, Spain) (removal of ammonia), and the resulting crude lyophilized.
3.1.5. RP-HPLC Analysis and Purification of Oligonucleotides and Conjugates
Reversed-phase HPLC analysis and purification was performed using a Shimadzu system (Izasa, Zaragoza, Spain). Linear gradients were always used.
Analysis conditions for oligonucleotides and conjugates were Jupiter C18 column (10 μm, 300 Å, 250 × 4.6 mm) from Phenomenex (Zaragoza, Spain), solvent A: 0.1 M aq. triethylammonium acetate, solvent B: acetonitrile, flow: 1 mL/min, detection wavelength: 254 nm. Semipreparative purification conditions: Jupiter C18 column (10 μm, 300 Å, 250 × 10.0 mm) from Phenomenex, solvent A: 0.1 M aq. triethylammonium acetate, solvent B: acetonitrile, flow: 3 mL/min, detection wavelength: 254 nm.
3.1.6. MALDI-TOF MS of Oligonucleotides and Conjugates
MALDI-TOF mass spectra were recorded on a 4800 Plus instrument (AB Sciex, Millipore, Spain), not using the reflector mode unless otherwise indicated. Typical analysis conditions: 1:1 (v/v) 2,4,6-trihydroxyacetophenone/ammonium citrate (THAP/CA), negative mode.
3.2. Synthesis of Small Molecules (Compounds 8–16)
Compounds 1–6 were Synthesized as Previously Described [17].
7-Bromo-1,3,4,5-tetrahydro-4-(acryloyl)-5-phenyl-2H-1,4-benzodiazepin-2-one (7). To a suspension of 5 (150 mg, 0.47 mmol) in DCM (5 mL) acryloyl chloride (50 µL, 0.62 mmol) was added, and the mixture stirred at room temperature for 3 h. Afterwards, additional DCM (30 mL) was added, and the mixture was transferred to a separatory funnel and washed with H2O (3 × 20 mL). The organic layer was dried over anh. MgSO4, filtered, and the solvent removed under low pressure. The resulting crude was purified by silica gel flash column chromatography eluting with DCM/EtOAc mixtures from 100:0 to 75:25. The title compound (7) was obtained as a white foam (130 mg, 74%).
TLC (DCM/EtOAc 80:20): Rf = 0.50; IR (ATR, solid): 3221, 2920, 2359, 2340, 1682, 1647, 1515, 1416, 665 cm−1; 1H NMR (CDCl3, 400 MHz): δ 8.88 and 8.85 (s, 1H), 7.45 (s, 1H), 7.46 and 7.40 (d, J = 8.5 Hz, 1H), 7.32–7.24 (m, 3H), 7.05 (s, 1H), 7.04 and 6.16 (s, 1H), 6.93 and 6.89 (d, J = 8.5 Hz, 1H), 6.61 and 6.54 (dd, J = 10.4, 16.4 Hz, 1H), 6.45 and 6.41 (s, 1H), 5.84–5.78 (m, 1H), 4.34–4.02 (m, 2H) ppm; diastereomer 1, 13C NMR (CDCl3, 101 MHz): δ 170.39, 166.04, 138.44, 134.30, 132.09, 130.61, 129.02, 127.93, 126.67, 122.91, 117.43, 59.46, 48.72 ppm; diastereomer 2, 13C NMR (CDCl3, 101 MHz): δ 170.39, 166.63, 137.52, 135.52, 134.71, 133.15, 132.73, 130.86, 130.00, 129.06, 128.39, 128.21, 127.08, 126.79, 123.67, 117.55, 63.49, 46.30 ppm; ESI-HRMS (positive mode): m/z 371.0391/373.0368 (81Br) [M + H]+, M calcd for C18H16BrN2O2 371.0390.
7-Bromo-1,3,4,5-tetrahydro-4-[1-oxo-3-(4-hydroxypiperidin-1-yl)propyl]-5-phenyl-2H-1,4-benzodiazep-in-2-one (8). 7 (100 mg, 0.27 mmol) and 4-hydroxypiperidine (273.1 mg, 2.70 mmol) were dissolved in DCM (5 mL) and reacted overnight at room temperature. Afterwards, the solvent was removed under low pressure, the resulting crude dissolved in EtOAc (40 mL) and washed with H2O (3 × 20 mL). The organic phase was dried over anh. MgSO4, filtered, and the solvent removed under vacuum. The title compound (8) was obtained as a white solid (125 mg, 98%).
TLC (DCM:EtOAc 50:50): Rf = 0.20; IR (ATR, solid): 3214, 3119, 2983, 2353, 2334, 1865, 1650, 1558, 1553, 1508, 1239, 783, 666 cm−1; 1H NMR (CDCl3, 400 MHz): δ 8.70 and 8.68 (s, 1H), 7.46–7.38 (m, 2H), 7.34–7.27 (m, 3H), 7.06–7.00 (m, 2H), 6.95 and 6.20 (s, 1H), 6.92–6.87 (m, 1H), 4.45–3.98 (m, 2H), 3.65 (p, J = 4.5 Hz, 1H), 2.80–2.54 (m, 6H), 2.20–2.12 (m, 2H), 1.87–1.79 (m, 4H), 1.57–1.47 (m, 1H) ppm; diastereomer 1, 13C NMR (CDCl3, 101 MHz): δ 171.64, 170.40, 138.33, 134.86, 134.25, 132.05, 130.34, 129.14, 128.96, 127.74, 122.87, 117.39, 67.49, 63.07, 59.29, 53.75, 51.19, 34.18, 31.60 ppm; diastereomer 2, 13C NMR (CDCl3, 101 MHz): δ 171.96, 170.94, 137.60, 135.50, 133.17, 132.60, 130.37, 128.44, 128.10, 127.05, 123.45, 117.19, 67.44, 60.42, 53.82, 51.30, 46.10, 34.19, 31.30 ppm; ESI-HRMS (positive mode): m/z 472.1222/474.1205 (81Br) [M + H]+, M calcd for C23H27BrN3O3 472.1230.
7-Bromo-1,3,4,5-tetrahydro-4-[1-oxo-3-(4-hydroxypiperidin-1-yl)propyl]-5-phenyl-2H-1,4-benz-odiazepin-2-one, 2-cyanoethyl N,N-diisopropylphosphoramidite (9). 8 (453.5 mg, 0.96 mmol) was dissolved in anh. DCM (15 mL). Subsequently, anh. N,N-diisopropylethylamine (310 µL, 2.41 mmol) was added and the mixture was stirred in an ice bath for 5 min under an argon atmosphere. Afterwards, a solution of chloro(2-cyanoethoxy)diisopropylaminophosphine (250 mg, 1.06 mmol) in anh. DCM (1 mL) was added, and the mixture was reacted in an ice bath for 30 min. Then, it was allowed to warm up and left stirring for 6 h at room temperature until complete phosphitylation as shown by TLC. The solvent was removed under low pressure; the crude was dissolved in EtOAc (50 mL) and washed with aq. NaHCO3(sat.) (3 × 30 mL). The organic phase was dried over anh. MgSO4, filtered, and the solvent removed under low pressure. The resulting crude was further purified by silica gel flash column chromatography eluting with DCM/EtOAc/NEt3 mixtures from 98:0:2 to 48:50:2. The title compound (9) was obtained as a white foam (272.4 mg, 42%).
TLC (DCM/EtOAc/NEt3 48:50:2): Rf = 0.50; 1H NMR (CDCl3, 400 MHz): δ 7.90 and 7.86 (s, 1H), 7.48–7.40 (m, 2H), 7.33–7.27 (m, 3H), 7.05–7.01 (m, 2H), 6.95 and 6.22 (s, 1H), 6.88–6.80 (m, 1H), 4.44–4.00 (m, 2H), 3.90–3.71 (m, 3H), 3.65–3.45 (m, 2H), 2.80–2.56 (m, 8H), 2.38–2.23 (m, 2H), 1.93–1.79 (m, 2H), 1.76–1.60 (m, 2H), 1.27 and 1.17 (dd, J = 6.8, 5.5 Hz, 12H) ppm; 31P NMR (CDCl3, 162 MHz): δ 145.85 ppm; ESI-HRMS (positive mode): m/z 672.2298/674.2281 (81Br) [M + H]+, M calcd for C32H44BrN5O4P 672.2309.
7-Bromo-5-phenyl-4-{3-[(2-(tritylthio)ethyl)amino]propanoyl}-1,3,4,5-tetrahydro-2H-benzo[e][1,4]di-azepin-2-one (10). 7 (370 mg, 0.99 mmol) and S-tritylcysteamine (1.50 g, 4.49 mmol) were dissolved in DCM (5 mL) and reacted overnight at room temperature. Afterwards, the solvent was removed under low pressure, and the resulting crude was purified by silica gel flash column chromatography eluting with hexanes/EtOAc/MeOH mixtures from 30:70:0 to 0:100:3. The title compound (10) was obtained as a white solid (441 mg, 64%).
TLC (EtOAc/MeOH 97:3): Rf = 0.35; IR (ATR, solid): 3211, 2926, 1729, 1666, 1664, 1482, 1448, 1368, 1242, 1216, 1188, 1058, 821, 745, 694 cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.44–7.40 (m, 9H), 7.30–7.18 (m, 14H), 7.03–6.99 (m, 1H), 6.90 and 6.11 (s, 1H), 4.42–3.96 (m, 2H), 2.80–2.66 (m, 2H), 2.60–2.45 (m, 3H), 2.41–2.31 (m, 3H), 1.87 (br s, 1H) ppm; diastereomer 1, 13C NMR (CDCl3, 101 MHz): δ 171.49, 169.75, 145.01, 138.34, 134.80, 134.45, 133.43, 132.28, 130.38, 129.15, 128.00, 127.85, 127.13, 126.80, 122.89, 117.61, 66.71, 63.16, 48.73, 44.67, 41.11, 33.67, 31.86 ppm; diastereomer 2, 13C NMR (CDCl3, 101 MHz): δ 171.86, 170.35, 144.90, 137.43, 135.47, 133.43, 132.83, 130.41, 129.33, 128.62, 128.30, 128.04, 127.13, 126.80, 123.40, 117.61, 66.80, 63.16, 48.40, 44.89, 41.11, 33.31, 31.81 ppm; ESI-HRMS (positive mode): m/z 690.1776/692.1765 (81Br) [M + H]+, M calcd for C39H37BrN3O2S 690.1784.
7-Bromo-4-{3-[(2-mercaptoethyl)amino]propanoyl}-5-phenyl-1,3,4,5-tetrahydro-2H-benzo[e][1,4]diaze-pin-2-one (11). 10 (20 mg, 0.03 mmol) was dissolved in a mixture of TFA/TIS (95:5) and reacted at room temperature for 90 min. Afterwards, the solvent was removed under a N2 stream and the resulting crude dissolved in a mixture MeOH/H2O (1:1, 2 mL) and filtered through a hydrophilic PTFE (polytetrafluoroethylene) syringe filter (0.22 µm), lyophilized and used without further purification (quantitative thiol deprotection as assessed by HPLC, Figure 2).
HPLC-MS: Analysis conditions: 0 → 50 % B in 30 min, tR = 19.0 min. ESI-MS (positive mode) of the main peak: m/z 448.2/450.2 (81Br), M calcd for C20H23BrN3O2S 448.07.
N-(Nα-Fmoc-Nε-l-lysinyl)-2-amino-5-bromobenzophenone (12). N-Methylmorpholine (200 µL, 1.81 mmol) and isobutylchloroformate (94 µL, 0.73 mmol) were added to a solution of Fmoc-l-Lys(Boc)-OH (340.5 mg, 0.73 mmol) in anh. DCM (5 mL), and the mixture cooled in an ice bath. After 15 min, a solution of 2 (200.0 mg, 0.73 mmol) in anh. DCM (1 mL) was added and the mixture was stirred for 1 h at 5 °C and overnight at room temperature. Afterwards, further DCM was added (15 mL) and the mixture was washed with aq. HCl 10% (2 × 10 mL). The organic phase was dried over anh. MgSO4, filtered and the solvent evaporated under low vacuum. The resulting crude was purified by silica gel flash column chromatography eluting with DCM/MeOH mixtures from 100:0 to 97:3. The title compound (12) was obtained as a pale white foam (301 mg, 57%).
TLC (DCM/MeOH 95:5): Rf = 0.40; IR (ATR, solid): 2980, 2905, 2355, 1730, 1658, 1599, 1571, 1497, 1425, 1437, 1391, 1254, 1248, 1173, 1158, 1071, 1043, 757, 744, 694, 533 cm−1; 1H NMR (CDCl3, 400 MHz): δ 8.57 (d, J = 8.8 Hz, 1H), 7.85–7.52 (m, 9H), 7.46–7.23 (m, 7H), 5.95 (br. s, 1H), 4.78–4.63 (m, 1H), 4.55–4.34 (m, 2H), 4.32–4.20 (m, 2H), 3.13 (t, J = 8.0 Hz, 2H), 2.02 (m, 2H), 1.93–1.78 (m, 2H), 1.77–1.50 (m, 2H), 1.45 (s, 9H) ppm; 13C NMR (CDCl3, 101 MHz): δ 197.84, 171.21, 156.47, 156.31, 143.69, 141.23, 138.78, 137.60, 136.75, 135.51, 132.91, 129.94, 128.46, 127.73, 127.66, 127.09, 127.05, 125.47, 125.14, 120.01, 119.89, 115.13, 79.21, 77.48, 77.16, 76.84, 67.44, 56.46, 47.20, 39.77, 31.73, 29.81, 28.47, 22.42 ppm. ESI-HRMS (positive mode): m/z 726.2173/728.2161 (81Br) [M + H]+; M calcd for C39H40BrN3O6 726.2173.
Tert-Butyl (S)-[4-(7-bromo-2-oxo-5-phenyl-2,3-dihydro-1H-benzo[e][1,4]diazepin-3-yl)butyl]carbamate (13). 12 (150 mg, 0.21 mmol) was dissolved in a 7 M ammonia solution (in MeOH, 3 mL) and reacted at room temperature overnight. Afterwards, the reaction mixture was taken to dryness and the crude purified by silica gel flash column chromatography eluting with a 70:30 hexanes/EtOAc mixture. The title compound (13) was obtained as a pale yellow solid (104 mg, 99%). For characterization see below.
One-pot synthesis of compound13from2. A solution of Fmoc-l-Lys(Boc)-OH (1.87 g, 4.00 mmol) in anh. DCM (15 mL) was cooled in an ice bath. N-Methylmorpholine (1.37 mL, 9.09 mmol) and isobutyl chloroformate (0.81 mL, 3.64 mmol) were added. After 10 min, a solution of 2 (1.00 g, 3.64 mmol) in anh. DCM (2 mL) was added, and the mixture was stirred for 30 min at 5 °C and 5.5 h at room temperature. Afterwards, the solvent was removed under vacuum and the crude dissolved in 7 M NH3 (in MeOH, 40 mL), and the solution was left to react overnight at room temperature. Subsequently, the solvent was removed under vacuum and the resulting crude dissolved in EtOAc (100 mL) and washed with 10% aq. HCl (3 × 30 mL). The organic phase was dried over anh. MgSO4, filtered and the solvent evaporated under vacuum. The crude material was further purified by silica gel flash column chromatography eluting with a 70:30 hexanes/EtOAc mixture. The title compound (13) was obtained as a pale yellow solid (970 mg, 55%).
TLC (hexanes/EtOAc 1:1): Rf = 0.42; IR (ATR, solid): 3414, 3309, 2929, 2359, 2337, 1682, 1653, 1508, 1232, 1156, 669 cm−1; 1H NMR (CDCl3, 400 MHz): δ 9.45 (s, 1H), 7.60 (dd, J = 8.6, 2.3 Hz, 1H), 7.52–7.34 (m, 6H), 7.08 (d, J = 8.6 Hz, 1H), 4.63 (s br, 1H), 3.50 (dd, J = 8.2, 5.7 Hz, 1H), 3.19 (d, J = 7.6 Hz, 2H), 2.32–2.14 (m, 2H), 1.69–1.56 (m, 2H), 1.44 (s, 11H) ppm; 13C NMR (CDCl3, 101 MHz): δ 172.13, 168.09, 156.03, 138.64, 137.65, 134.57, 133.30, 130.51, 129.67, 129.10, 128.34, 123.04, 116.00, 79.01, 63.22, 40.47, 30.66, 30.00, 28.44, 23.32 ppm; ESI-HRMS (positive mode): m/z 486.1386/488.1370 (81Br) [M + H]+, M calcd for C24H29BrN3O3 486.1387.
7-Bromo-1,3,4,5-tetrahydro-4-[4-(N-Boc-amino)butyl]-5-phenyl-2H-1,4-benzodiazepin-2-one (14). To a solution of 13 (800 mg, 1.65 mmol) and NaBH3CN (155.4 mg, 2.48 mmol) in MeOH (10 mL), AcOH (500 µL, 8.24 mmol) was added, and the mixture was stirred at room temperature for 2.5 h until complete reduction of the imine as assessed by TLC. Afterwards, the solvent was removed under low pressure, the crude was dissolved in EtOAc (50 mL) and washed with aq. NaHCO3(sat) (2 × 20 mL). The organic layer was dried over anh. MgSO4, filtered and the solvent removed under low pressure. The title compound (14) was obtained as a pale yellow solid (799 mg, 99%).
TLC (hexanes/EtOAc 1:1): Rf = 0.52; IR (ATR, solid): 3290, 2967, 2929, 2866, 1663, 1479, 1365, 1245, 1159, 817, 700 cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.99 and 7.67 (s, 1H), 7.45–7.27 (m, 5H), 7.13 and 6.76 (d, J = 2.3 Hz, 1H), 6.89 and 6.80 (d, 8.4 Hz, 1H), 5.34 and 5.24 (s, 1H), 4.56 (br s, 1H), 3.57 and 3.31 (t, J = 7.2 Hz, 1H), 3.14–3.05 (m, 2H), 1.94–1.44 (m, 6H), 1.42 (s, 9H) ppm; diastereomer 1, 13C NMR (CDCl3, 101 MHz): δ 175.02, 156.00, 142.23, 139.97, 136.56, 132.23, 128.64, 128.45, 127.19, 122.69, 117.53, 79.06, 63.98, 59.94, 40.29, 31.65, 29.93, 28.41, 23.50 ppm; diastereomer 2, 13C NMR (CDCl3, 101 MHz): δ 174.27, 156.00, 142.33, 139.97, 135.88, 131.35, 128.57, 128.20, 127.54, 123.32, 118.88, 79.06, 58.97, 56.14, 40.29 31.65, 30.04, 28.42, 23.21 ppm; ESI-HRMS (positive mode): m/z 488.1559/490.1555 (81Br) [M + H]+, M calcd for C24H31BrN3O3 488.1543.
7-Bromo-1,3,4,5-tetrahydro-4-(4-aminobutyl)-5-phenyl-2H-1,4-benzodiazepin-2-one dihydrochloride (15). In a 10 mL round-bottom flask, 14 (100 mg, 0.21 mmol) was dissolved in 4 M HCl (in dioxane, 5 mL) and the reaction mixture was left stirring for 2.5 h at room temperature. Afterwards the solvent was removed under low pressure to afford the title product (15) as a pale yellow solid (94 mg, 99%).
TLC (DCM/MeOH 9:1): Rf = 0.20; IR (ATR, solid): 3474, 3199, 2911, 2711, 2524, 1704, 1590, 1568, 1479, 1448, 1375, 1315, 1270, 1245, 1131, 998, 916, 827, 697 cm−1; 1H NMR (CD3OD, 400 MHz): δ 7.75 and 7.73 (s, 1H), 7.69–7.55 (m, 4H), 7.44–7.38 (m, 1H), 7.34–7.29 (m, 2H), 7.19 (d, J = 8.5 Hz) and 7.09 (d, J = 9.2 Hz, 1H), 6.02 and 5.71 (s, 1H), 4.12 (dd, J = 9.0, 4.6 Hz) and 3.86 (dd, J = 10.5, 3.1 Hz, 1H), 2.99–2.89 (m, 2H), 2.32–2.10 (m, 1H), 2.00–1.80 (m, 1H), 1.71 (h, J = 7.3 Hz, 2H), 1.58–1.34 (m, 2H) ppm; diastereomer 1, 13C NMR (CDCl3, 101 MHz): δ 166.45, 138.28, 135.43, 135.17, 133.98, 130.67, 130.20, 129.58, 130.20, 129.58, 128.34, 125.45, 120.24, 60.83, 57.09, 40.24, 28.13, 27.96, 23.88 ppm; diastereomer 2, 13C NMR (CDCl3, 101 MHz): δ 167.82, 136.80, 135.87, 134.92, 133.34, 131.13, 130.35, 129.58, 128.34, 126.16, 119.84, 63.60, 57.38, 40.24, 29.17, 28.05, 23.66 ppm; ESI-HRMS (positive mode): m/z 388.1016/390.1000 (81Br) [M + H]+, M calcd for C19H23BrN3O 388.1019.
7-Bromo-1,3,4,5-tetrahydro-4-{4-[N-[(4E)-4,6-heptadienoyl]-amino]butyl}-5-phenyl-2H-1,4-benzodiazepin-2-one (16). N,N’-Diisopropylcarbodiimide (61 µL, 0.39 mmol) and N-methylmorpholine (2.6 µL, 0.03 mmol) were added to a solution of (4E)-4,6-heptadienoic acid (49 mg, 0.39 mmol) in HPLC-quality acetonitrile (1 mL), and the mixture was left stirring for 10 min. Afterwards, 15 (50 mg, 0.13 mmol) and an additional amount of N-methylmorpholine (2.6 µL, 0.03 mmol) were poured onto the mixture. Additional amounts of N-methylmorpholine (2.6 µL, 0.03 mmol) were added after 20 and 40 min, respectively, and the solution was stirred for up to 2 h at room temperature. Subsequently, acetonitrile was added (4 mL), and the crude was filtered using a hydrophilic PTFE syringe filter (0.22 µm), purified by RP-HPLC (Jupiter Proteo C18 (10 μm, 250 nm, 250 × 10 mm) from Phenomenex, solvent A: H2O 0.1% formic acid; solvent B: ACN 0.1% formic acid, linear gradient 40 → 80% B, 3 mL/min, detection wavelength 254 nm) and lyophilized. The title compound (16) was obtained as a white solid (10.7 mg, 20%).
TLC (EtOAc/MeOH 9:1): Rf = 0.10; IR (ATR, solid): 2980, 2905, 2355, 1730, 1658, 1599, 1571, 1497, 1425, 1437, 1391, 1254, 1248, 1173, 1158, 1071, 1043, 757, 744, 694, 533 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.65–7.53 (m, 4H), 7.42–7.28 (m, 4H), 7.16 and 7.04 (d, J = 8.5 Hz, 1H), 7.47 and 6.97 (d, J = 2.2 Hz, 1H), 6.35–6.21 (m, 1H), 6.13–6.02 (m, 1H), 5.80 and 5.68 (s, 1H), 5.70–5.61 (m, 1H), 5.06 (dd, J = 16.9, 1.9 Hz, 1H), 4.92 (dt, J = 10.2, 2.2 Hz, 1H), 4.17–3.77 (m, 1H), 3.72 (dd, J = 10.1, 3.7 Hz, 1H), 3.23–3.07 (m, 2H), 2.39–2.31 (m, 2H), 2.27–2.12 (m, 3H), 1.81–1.63 (m, 1H), 1.55–1.31 (m, 4H) ppm; 13C NMR (CDCl3, 101 MHz): δ 173.99, 173.93, 136.91, 135.65, 133.59, 132.93, 132.50, 131.86, 129.62, 129.28, 128.67, 127.77, 126.88, 123.98, 118.74, 114.45, 59.34, 55.99, 38.24, 35.25, 29.37, 28.76, 28.34, 22.80 ppm; ESI-HRMS (positive mode): m/z 496.1584/498.1573 (81Br) [M + H]+, M calcd. for C26H30BrN3O3 496.1594.
RP-HPLC analysis conditions (Figure 3): Jupiter Proteo C18 (4 μm, 254 nm, 250 × 4.6 mm) from Phenomenex, 30 → 70% B in 30 min, 1 mL/min, tR = 14.3 min and 18.1 min; purification conditions: Jupiter Proteo C18 (10 μm, 254 nm, 250 × 10 mm) from Phenomenex, 40 → 80% B in 30 min, 3 mL/min (tR = 7.5 min and 8.9 min).
3.3. Preparation of Oligonucleotides and Conjugates
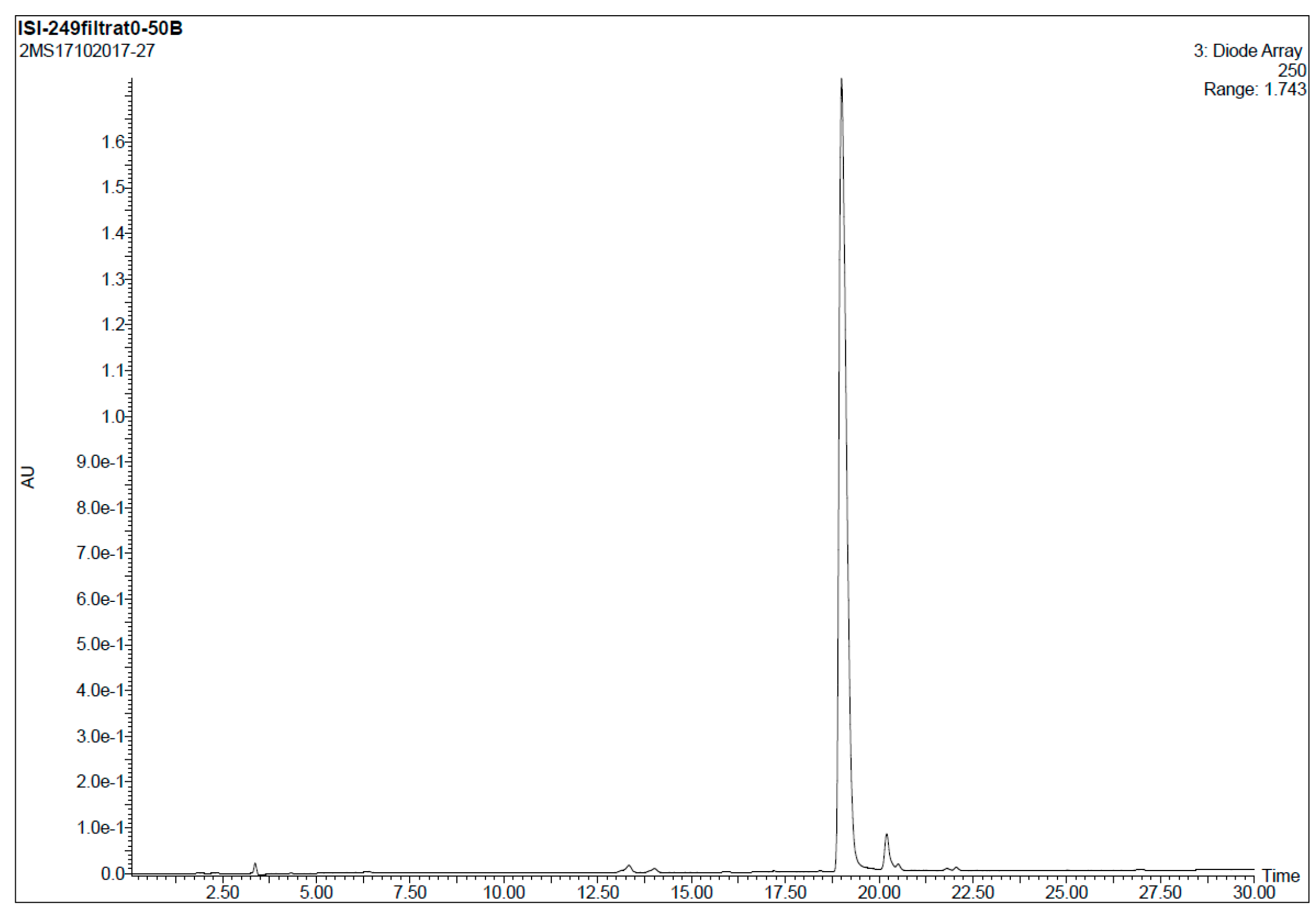
r5’GTTATTCTTTAGAATGGTGC3’ (2’-OMe, PS) (purchased from Avecia, Manchester, UK). HPLC analysis conditions (Figure 4): 0 → 50% B in 30 min, tR = 19.0 min.
MALDI-TOF MS (negative mode, THAP/CA): m/z 7060.9 [M − H]−, M calcd. for C218H290BrN69O124P19S19 7053.8.
r5’TGTGTACTGATGTAGTTATC3’ (2’-OMe, PS). HPLC: Analysis conditions, 0 → 50% B in 30 min, tR = 19.2 min (Figure 5). Purification conditions: 20 → 40% B in 30 min (tR = 9.0 min). Yield (synthesis and purification): 43%.
MALDI-TOF MS (negative mode, THAP/CA): m/z 7053.8 [M − H]−, M calcd. for C218H290N69O124P19S19 7053.8.
Conjugates19. Oligonucleotide-resins were automatically assembled at the 1 μmol-scale as stated above (Materials and Methods section). After oligonucleotide elongation was completed and the 5’ end deprotected, phosphoramidite 9 was incorporated (0.1 M solution in anh. acetonitrile, 2 × 10 min coupling) and the resulting phosphite triester sulfurized using the same procedure as in all the other synthesis cycles, which afforded 18. Final deprotection was carried out under standard conditions (see above).
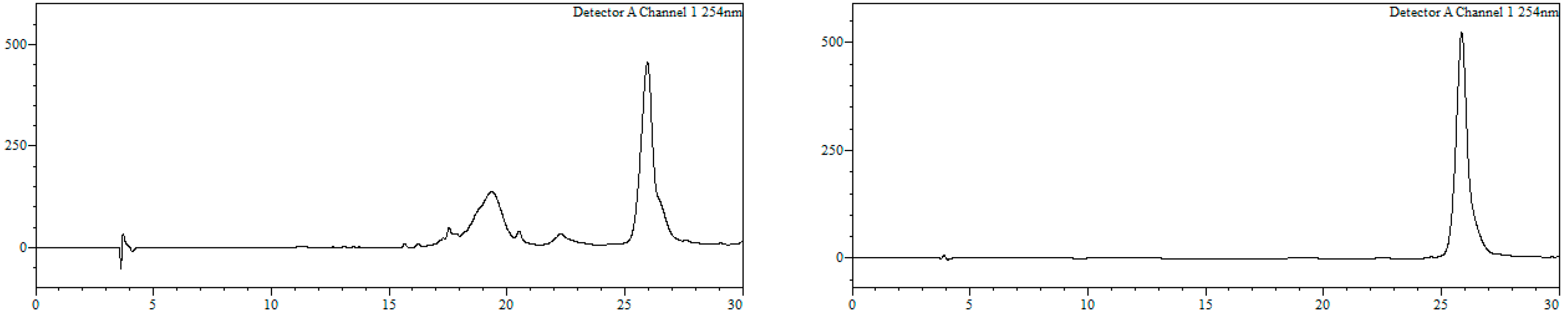
Conjugate19a. HPLC (Figure 6): Analysis conditions: 0 → 50% B in 30 min, tR = 22.0 min; purification conditions: 0 → 40% B in 30 min (tR = 19.1 min). Yield (synthesis and purification): 36%.
MALDI-TOF MS (negative mode, THAP/CA): m/z 7610.5 [M − H]−, M calcd. for C241H315BrN72O128P20S20 7602.8.
Conjugate19b. HPLC (Figure 7): Analysis conditions, 0 → 50% B in 30 min, tR = 21.7 min; purification conditions: 20 → 40% B in 30 min (tR = 13.8 min). Yield (synthesis and purification): 20%.
MALDI-TOF MS (negative mode, THAP/CA): m/z 7602.1 [M − H]−, M calcd. for C241H315BrN72O128P20S20 7602.8.
Oligonucleotides21. Oligonucleotide-resins were assembled as previously described. The [2,5-dimetylfuran-protected maleimide]-phosphoramidite (see structure in Scheme 5) was subsequently incorporated (0.1 M solution in anh. acetonitrile, 2 × 10 min coupling), which after sulfurization afforded 20. Treatment with ammonia (as described above) furnished [protected maleimido]-oligonucleotides 21.
Oligonucleotide21a. HPLC (Figure 8): Analysis conditions: 0 → 50% B in 30 min, tR = 19.7 min; purification conditions: 20 → 40% B in 30 min (tR = 10.7 min). Yield (synthesis and purification): 26%.
MALDI-TOF MS (negative mode, THAP/CA): m/z 7375.1 [M − H]−, M calcd. for C230H304N70O129P20S20 7368.8.
Oligonucleotide21b. HPLC (Figure 9): Analysis conditions, 0 → 50% B in 30 min, tR = 20.2 min; purification conditions: 20→ 40% B in 30 min (tR = 11.3 min). Yield (synthesis and purification): 20%.
MALDI-TOF MS (negative mode, THAP/CA): m/z 7369.0 [M − H]−, M calcd. for C230H304N70O129P20S20 7368.8.
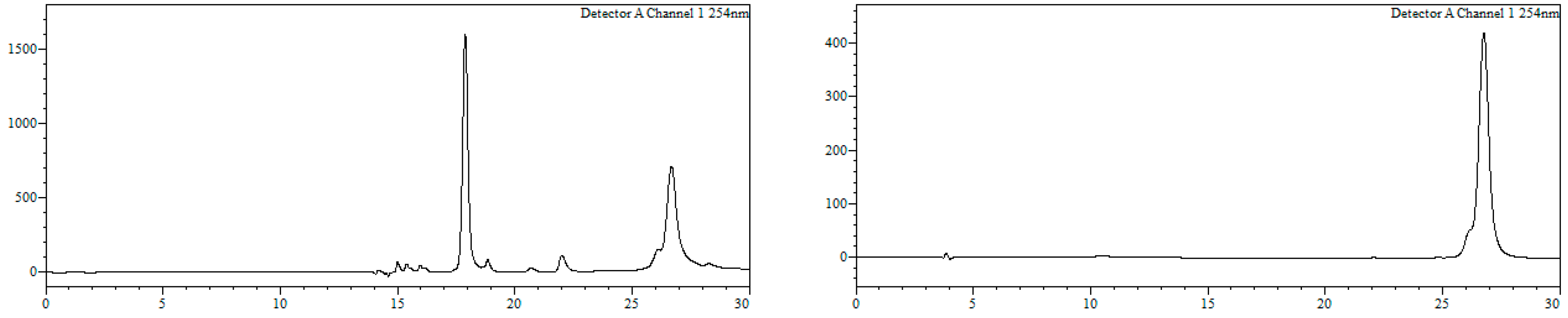
Conjugate22a. To a solution of 21a (260 nmol) in MeOH/H2O (1:1, 468 µL) a solution of 11 (520 nmol, 52 µL, 10 mM) in MeOH/H2O (1:1) was added, and the resulting mixture (final oligonucleotide concentration = 140 µM) heated at 90 °C in a microwave oven for 90 min. Afterwards, MeOH was removed under reduced pressure and the resulting crude purified by HPLC. HPLC (Figure 10): Analysis conditions, 0 → 50% B in 30 min, tR = 22.4 min, purification conditions: 20 → 40% B in 30 min (tR = 14.4 min). Yield (synthesis and purification): 11%.
MALDI-TOF MS (negative mode, THAP/CA): m/z 7720.7 [M − H]−, M calcd. for C244H318BrN73O130P20S21 7719.8.
Conjugate23a. To a solution of 21a (800 nmol) in MeOH/H2O (1:1, 18 mL) 16 was added (4000 nmol, 2000 µL, 2 mM), and the resulting mixture (final oligonucleotide concentration = 0.5 mM) heated at 90 °C in a microwave oven for 90 min. Afterwards, MeOH was removed under reduced pressure and the resulting crude purified by HPLC. HPLC (Figure 11): Analysis conditions, 0 → 50% B in 30 min, tR = 25.9 min; purification conditions: 20 → 40%B in 30 min (tR = 21.3 min). Yield (synthesis and purification): 20%.
MALDI-TOF MS (negative mode, THAP/CA): m/z 7772.5 [M − H]−, M calcd. for C250H326BrN73O130P20S20 7767.9
Conjugate23b. A solution of 21b (800 nmol) in MeOH/H2O (1:1, 19 mL) (final oligonucleotide concentration = 40 µM) was heated at 90 °C in a microwave oven for 90 min. Afterwards, a solution of 16 (4000 nmol) in H2O (1 mL) was added and the resulting mixture left stirring for 1 h. Finally, MeOH was removed under reduced pressure and the resulting crude purified by RP-HPLC. HPLC (Figure 12): Analysis conditions, 0→ 50% B in 30 min, tR = 26.7 min; purification conditions: 20 → 40% B in 30 min (tR = 8.5 min). Yield (synthesis and purification): 29%.
MALDI-TOF MS (negative mode, THAP/CA): m/z 7758.7 [M − H]−, M calcd. for C250H326BrN73O130P20S20 7767.9.
3.4. Luciferase Induction Experiments
Oligonucleotides conjugated to Retro 1 or its derivatives were tested in a splice correction assay using HeLa Luc 705 cells. These cells contain a firefly luciferase expression cassette interrupted by an abnormal intron from thalassemic beta-globin. As a result the cells fail to splice correctly and do not produce luciferase. However, successful intracellular delivery of a splice switching antisense oligonucleotide can correct splicing leading to luciferase expression. Thus this system provides a rapid and convenient assay for the intracellular delivery of oligonucleotides. In the current experiments the HeLa Luc 705 cells were incubated with various concentrations of oligonucleotide and then processed for luciferase induction as described [19]. In some cases there was a brief post-incubation with UNC7938, a small molecule known to enhance oligonucleotide effectiveness [15]. The cells were initially plated at 50,000 per well in 24 well tissue culture plates in DMEM (Dulbecco modified Eagles minimal essential medium) + 10% fetal bovine serum. After overnight incubation at 37 °C and 5% CO2, medium was removed and replaced with fresh complete medium containing various concentrations of the oligonucleotides. Cells were then further incubated for 16 h. In some samples this was followed by a 1 h exposure to UNC7938 followed by removal of the compound. After an additional incubation of 2 h, cells were rinsed twice with PBS and then lysed in 1/5 diluted luciferase lysis buffer (Promega, Madison, WI, USA) according to manufacturer’s directions. Luciferase activity was measured using a FLUOstar Omega microplate reader (BMG LABTECH, Cary, NC, USA) and luciferase reagents obtained from Promega.
Author Contributions
Conceptualization, E.P. and A.G.; funding acquisition, E.P. and A.G.; investigation, J.A., S.M.K. and R.L.J.; writing—original draft preparation, J.A., R.L.J. and A.G.; writing—review and editing, J.A., E.P., S.M.K., R.L.J. and A.G.; visualization, J.A., R.L.J. and A.G.; supervision, R.L.J. and A.G.
Funding
This research was funded by the Spanish Ministry of Economy and Competitiveness (MINECO), grant numbers CTQ2014-52658-R and CTQ2017-84779-R.
Acknowledgments
We wish to acknowledge the Generalitat de Catalunya (2014SGR187 and 2017SGR391).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.; Corey, D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018, 46, 1584–1600. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.K.; Sharma, R.K.; Singh, S.K. Antisense oligonucleotides: Modifications and clinical trials. Med. Chem. Commun. 2014, 5, 1454–1471. [Google Scholar] [CrossRef]
- Crooke, S.T.; Witzum, J.L.; Bennett, C.F.; Baker, B.F. RNA-targeted therapeutics. Cell Metab. 2018, 27, 714–739. [Google Scholar] [CrossRef] [PubMed]
- Bennet, C.F.; Baker, B.F.; Pham, N.; Swayze, E.; Geary, R.S. Pharmacology of antisense drugs. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 81–105. [Google Scholar] [CrossRef] [PubMed]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef]
- Asami, Y.; Yoshioka, K.; Nishina, K.; Nagata, T.; Yokota, T. Drug delivery system of therapeutic oligonucleotides. Drug Discov. Ther. 2016, 10, 252–262. [Google Scholar] [CrossRef]
- Ming, X.; Carver, K.; Fisher, M.; Noel, R.; Cintrat, J.-C.; Gillet, D.; Barbier, J.; Cao, C.; Bauman, J.; Juliano, R.L. The small molecule Retro-1 enhances the pharmacological actions of antisense and splice switching oligonucleotides. Nucleic Acids Res. 2013, 41, 3673–3687. [Google Scholar] [CrossRef] [Green Version]
- Stechmann, B.; Bai, S.K.; Gobbo, E.; Lopez, R.; Merer, G.; Pinchard, S.; Panigai, L.; Tenza, D.; Raposo, G.; Beaumelle, B.; et al. Inhibition of retrograde transport protects mice from lethal ricin challenge. Cell 2010, 141, 231–242. [Google Scholar] [CrossRef]
- Yang, B.; Ming, X.; Cao, C.; Laing, B.; Yuan, A.; Porter, M.A.; Hull-Ryde, E.A.; Maddry, J.; Suto, M.; Janzen, W.P.; et al. High-throughput screening identifies small molecules that enhance the pharmacological effects of oligonucleotides. Nucleic Acids Res. 2015, 43, 1987–1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborn, M.F.; Alterman, J.F.; Nikan, M.; Cao, H.; Didiot, M.C.; Hassler, M.R.; Coles, A.H.; Khvorova, A. Guanabenz (WytensinTM) selectively enhances uptake and efficacy of hydrophobically modified siRNAs. Nucleic Acids Res. 2015, 43, 8664–8672. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ariyarathna, Y.; Ming, X.; Yang, B.; James, L.I.; Kreda, S.M.; Porter, M.; Janzen, W.; Juliano, R.L. A novel family of small molecules that enhance the intracellular delivery and pharmacological effectiveness of antisense and splice switching oligonucleotides. ACS Chem. Biol. 2017, 12, 1999–2007. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Abdelkafi, H.; Pons, V.; Michau, A.; Gillet, D.; Cintrat, J.-C.; Barbier, J.; Juliano, R.L. Retro-1 analogues differentially affect oligonucleotide delivery and toxin trafficking. ChemMedChem 2016, 11, 2506–2510. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L.; Wang, L.; Tavares, F.; Brown, E.G.; James, L.; Ariyarathna, Y.; Ming, X.; Mao, C.; Suto, M. Structure-activity relationships and cellular mechanism of action of small molecules that enhance the delivery of oligonucleotides. Nucleic Acids Res. 2018, 46, 1601–1613. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Abdelkafi, H.; Michau, A.; Clerget, A.; Buisson, D.-A.; Johannes, L.; Gillet, D.; Barbier, J.; Cintrat, J.-C. Synthesis, chiral separation, absolute configuration assignment, and biological activity of enantiomers of retro-1 as potent inhibitors of Shiga toxin. ChemMedChem 2015, 10, 1153–1156. [Google Scholar] [CrossRef]
- Sánchez, A.; Pedroso, E.; Grandas, A. Maleimide-dimethylfuran exo adducts: Effective maleimide protection in the synthesis of oligonucleotide conjugates. Org. Lett. 2011, 13, 4364–4367. [Google Scholar] [CrossRef]
- Alam, M.R.; Dixit, V.; Kang, H.; Li, Z.-B.; Chen, X.; Trejo, J.A.; Fisher, M.; Juliano, R.L. Intracellular delivery of an anionic antisense oligonucleotide via receptor-mediated endocytosis. Nucleic Acids Res. 2008, 36, 2764–2776. [Google Scholar] [CrossRef] [Green Version]
- Baillie, L.C.; Batsanov, A.; Bearder, J.R.; Whiting, D.A. Synthesis of the A/E/F sections of conaconitine, napelline and related diterpenoid alkaloids of the aconitine group. J. Chem. Soc. Perkin Trans. 1 1998, 3471–3478. [Google Scholar] [CrossRef]
- Naumiec, G.R.; Lincourt, G.; Clever, J.P.; McGregor, M.A.; Kovoor, A.; DeBoef, B. Synthesis of a β-CCT-lanthanide conjugate for binding the dopamine transporter. Org. Biomol. Chem. 2015, 13, 2537–2540. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 3, 4, 5, 8 and 13 are available from the authors. |
Scheme 1.
Synthesis scheme described [17] for the preparation of Retro-1, 6. DCM = dichloromethane; NBS = N-bromosuccinimide; TEA = triethylamine. The wavy bond indicates that the stereochemistry is not defined (in other words, the compound is a mixture of isomers).
Scheme 1.
Synthesis scheme described [17] for the preparation of Retro-1, 6. DCM = dichloromethane; NBS = N-bromosuccinimide; TEA = triethylamine. The wavy bond indicates that the stereochemistry is not defined (in other words, the compound is a mixture of isomers).

Scheme 2.
Synthesis of the Retro-1 analogs modified at position 4 of the benzodiazepine ring. The first step (a) afforded the common precursor, which was subsequently modified to obtain either phosphoramidite derivative 9 (b) or the thiol-modified compound 11 (c). CNE = 2-cyanoethyl; DCM = dichloromethane; DIPEA = N,N-diisopropylethylamine; rt = room temperature; TEA = triethylamine; TFA = trifluoroacetic acid; TIS = triisopropylsilane; Trt = trityl. The wavy bond indicates a not defined stereochemistry.
Scheme 2.
Synthesis of the Retro-1 analogs modified at position 4 of the benzodiazepine ring. The first step (a) afforded the common precursor, which was subsequently modified to obtain either phosphoramidite derivative 9 (b) or the thiol-modified compound 11 (c). CNE = 2-cyanoethyl; DCM = dichloromethane; DIPEA = N,N-diisopropylethylamine; rt = room temperature; TEA = triethylamine; TFA = trifluoroacetic acid; TIS = triisopropylsilane; Trt = trityl. The wavy bond indicates a not defined stereochemistry.

Scheme 3.
Synthesis of diene-derivatized Retro-1 analog 16. ACN = acetonitrile; Boc = tert-butoxycarbonyl; DCM = dichloromethane; diene-COOH = (4E)-4,6-heptadienoic acid; DIPC = N,N’-diisopropylcarbodiimide; Fmoc = 9-fluorenylmethoxycarbonyl; NMM = N-methylmorpholine; on = overnight; rt = room temperature. The wavy bond indicates a not defined stereochemistry.
Scheme 3.
Synthesis of diene-derivatized Retro-1 analog 16. ACN = acetonitrile; Boc = tert-butoxycarbonyl; DCM = dichloromethane; diene-COOH = (4E)-4,6-heptadienoic acid; DIPC = N,N’-diisopropylcarbodiimide; Fmoc = 9-fluorenylmethoxycarbonyl; NMM = N-methylmorpholine; on = overnight; rt = room temperature. The wavy bond indicates a not defined stereochemistry.

Scheme 4.
Solid-phase assembly of conjugates 19. B = G (oligonucleotide a)/T (oligonucleotide b); BTT = 5-benzylthio-1H-tetrazole; CNE = 2-cyanoethyl; PS = phosphorothioate. The wavy bond indicates a not defined stereochemistry.
Scheme 4.
Solid-phase assembly of conjugates 19. B = G (oligonucleotide a)/T (oligonucleotide b); BTT = 5-benzylthio-1H-tetrazole; CNE = 2-cyanoethyl; PS = phosphorothioate. The wavy bond indicates a not defined stereochemistry.

Scheme 5.
Preparation of conjugates 22 and 23. B = G (oligonucleotide a)/T (oligonucleotide b); BTT = 5-benzylthio-1H-tetrazole; CNE = 2-cyanoethyl; MW = microwave; PS = phosphorothioate. The wavy bond indicates a not defined stereochemistry.
Scheme 5.
Preparation of conjugates 22 and 23. B = G (oligonucleotide a)/T (oligonucleotide b); BTT = 5-benzylthio-1H-tetrazole; CNE = 2-cyanoethyl; MW = microwave; PS = phosphorothioate. The wavy bond indicates a not defined stereochemistry.

Figure 1.
HeLa705 cells in complete growth medium were incubated for 16 h with various concentrations of conjugated or unconjugated oligonucleotide. Thereafter the cells were recovered and assayed for luciferase activity. In one case cells were post-treated with 10 μM UNC7938 for 4 h after exposure to SSO623 and then assayed. RLU = relative luminescence units; n = 3. Means and standard errors shown. SSO623: r5’GTTATTCTTTAGAATGGTGC3’, all 2’-O-Me and phosphorothioate (PS) (T = ribothymidine).
Figure 1.
HeLa705 cells in complete growth medium were incubated for 16 h with various concentrations of conjugated or unconjugated oligonucleotide. Thereafter the cells were recovered and assayed for luciferase activity. In one case cells were post-treated with 10 μM UNC7938 for 4 h after exposure to SSO623 and then assayed. RLU = relative luminescence units; n = 3. Means and standard errors shown. SSO623: r5’GTTATTCTTTAGAATGGTGC3’, all 2’-O-Me and phosphorothioate (PS) (T = ribothymidine).

Figure 2.
Analytical HPLC trace of crude 11 (detection wavelength: 250 nm).

Figure 3.
Analytical HPLC traces of crude 16 (top), purified diastereomer 1 (bottom, left) and purified diastereomer 2 (bottom, right). Detection wavelength: 250 nm. Top trace, tR ~ 2–3 min: 15 & N,N’-diisopropylurea.
Figure 3.
Analytical HPLC traces of crude 16 (top), purified diastereomer 1 (bottom, left) and purified diastereomer 2 (bottom, right). Detection wavelength: 250 nm. Top trace, tR ~ 2–3 min: 15 & N,N’-diisopropylurea.

Figure 4.
Analytical HPLC trace of oligonucleotide r5’GTTATTCTTTAGAATGGTGC3’. Detection wavelength: 250 nm.
Figure 4.
Analytical HPLC trace of oligonucleotide r5’GTTATTCTTTAGAATGGTGC3’. Detection wavelength: 250 nm.
Figure 5.
Analytical HPLC trace of oligonucleotide r5’TGTGTACTGATGTAGTTATC3’. Detection wavelength: 250 nm.
Figure 5.
Analytical HPLC trace of oligonucleotide r5’TGTGTACTGATGTAGTTATC3’. Detection wavelength: 250 nm.

Figure 6.
HPLC trace of crude (left) and purified (right) conjugate 19a. Detection wavelength: 254 nm.
Figure 6.
HPLC trace of crude (left) and purified (right) conjugate 19a. Detection wavelength: 254 nm.

Figure 7.
HPLC trace of crude (left) and purified (right) conjugate 19b. Detection wavelength: 254 nm. Left trace, tR ~ 19 min: r5’TGTGTACTGATGTAGTTATC3’.
Figure 7.
HPLC trace of crude (left) and purified (right) conjugate 19b. Detection wavelength: 254 nm. Left trace, tR ~ 19 min: r5’TGTGTACTGATGTAGTTATC3’.

Figure 8.
HPLC traces of crude (left) and purified (right) oligonucleotide 21a. Detection wavelength: 254 nm.
Figure 8.
HPLC traces of crude (left) and purified (right) oligonucleotide 21a. Detection wavelength: 254 nm.

Figure 9.
HPLC traces of crude (left) and purified (right) oligonucleotide 21b. Detection wavelength: 254 nm. Left trace, tR ~ 19 min: r5’TGTGTACTGATGTAGTTATC3’.
Figure 9.
HPLC traces of crude (left) and purified (right) oligonucleotide 21b. Detection wavelength: 254 nm. Left trace, tR ~ 19 min: r5’TGTGTACTGATGTAGTTATC3’.

Figure 10.
HPLC traces of crude (left) and purified (right) conjugate 22a. Detection wavelength: 254 nm.
Figure 10.
HPLC traces of crude (left) and purified (right) conjugate 22a. Detection wavelength: 254 nm.

Figure 11.
HPLC traces of crude (left) and purified (right) conjugate 23a. Detection wavelength: 254 nm.
Figure 11.
HPLC traces of crude (left) and purified (right) conjugate 23a. Detection wavelength: 254 nm.

Figure 12.
HPLC traces of crude (left) and purified (right) conjugate 23b. Detection wavelength: 254 nm. Left trace, tR ~ 18 min: 16.
Figure 12.
HPLC traces of crude (left) and purified (right) conjugate 23b. Detection wavelength: 254 nm. Left trace, tR ~ 18 min: 16.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Agramunt, J.; Pedroso, E.; Kreda, S.M.; Juliano, R.L.; Grandas, A. Retro-1-Oligonucleotide Conjugates. Synthesis and Biological Evaluation. Molecules 2019, 24, 579. https://doi.org/10.3390/molecules24030579
AMA Style
Agramunt J, Pedroso E, Kreda SM, Juliano RL, Grandas A. Retro-1-Oligonucleotide Conjugates. Synthesis and Biological Evaluation. Molecules. 2019; 24(3):579. https://doi.org/10.3390/molecules24030579
Chicago/Turabian StyleAgramunt, Jordi, Enrique Pedroso, Silvia M. Kreda, Rudolph L. Juliano, and Anna Grandas. 2019. "Retro-1-Oligonucleotide Conjugates. Synthesis and Biological Evaluation" Molecules 24, no. 3: 579. https://doi.org/10.3390/molecules24030579