Small Molecule Docking of DNA Repair Proteins Associated with Cancer Survival Following PCNA Metagene Adjustment: A Potential Novel Class of Repair Inhibitors

Abstract
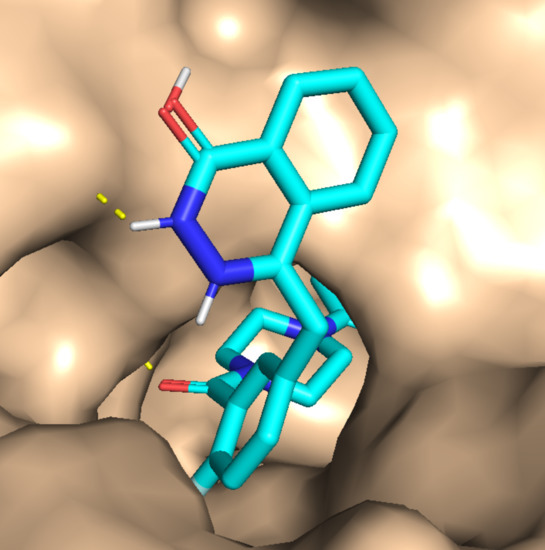
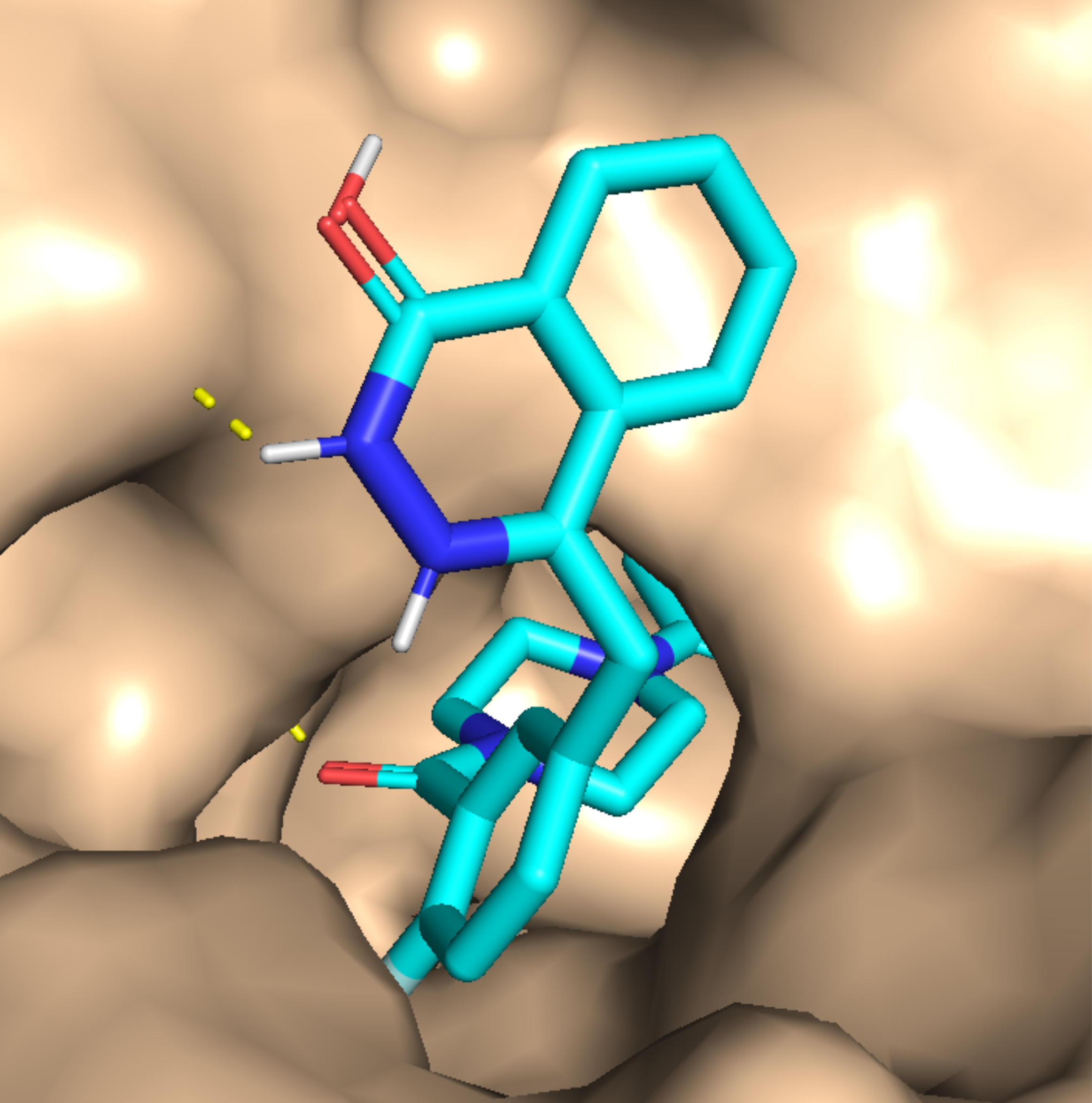
1. Introduction
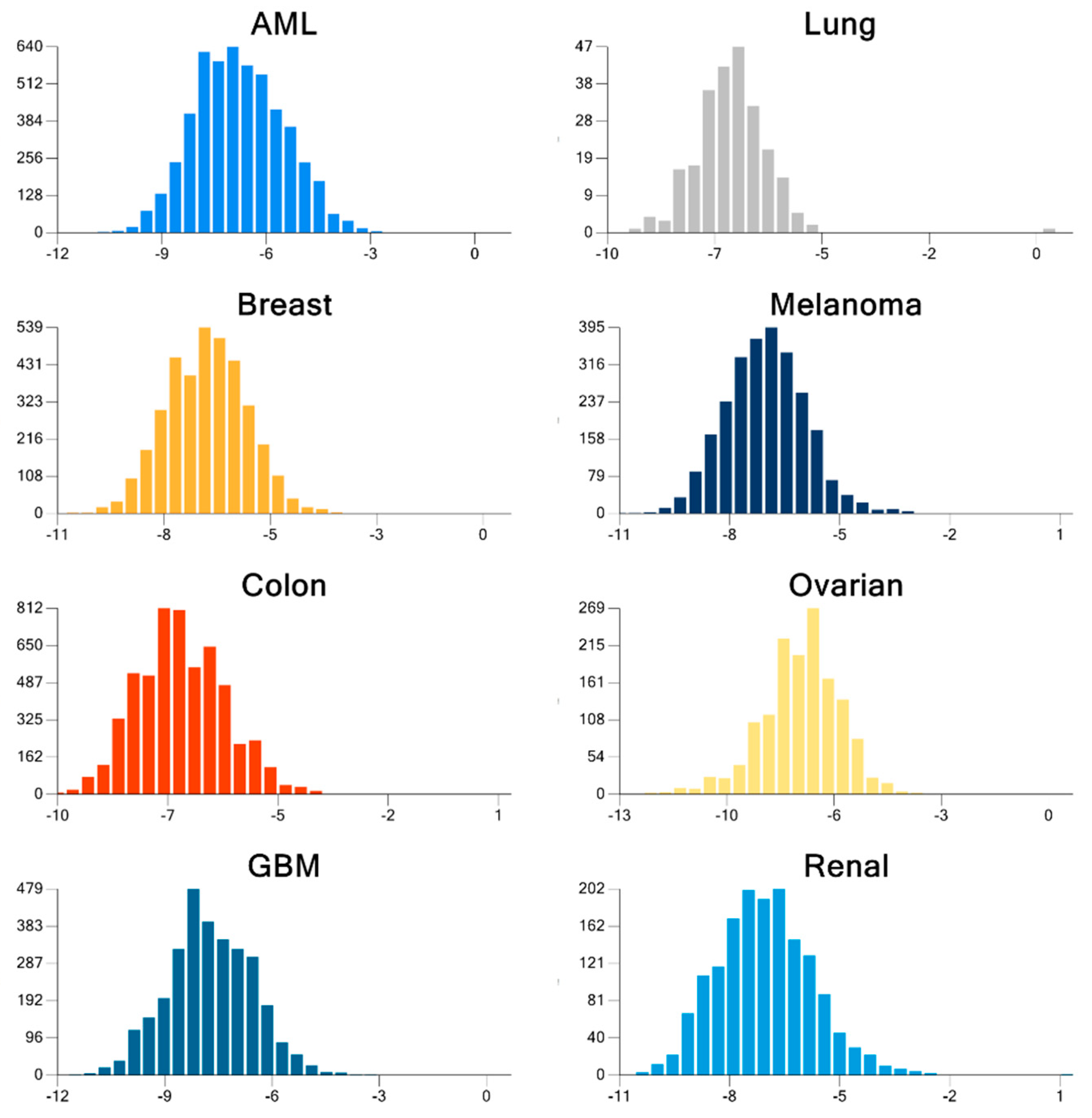
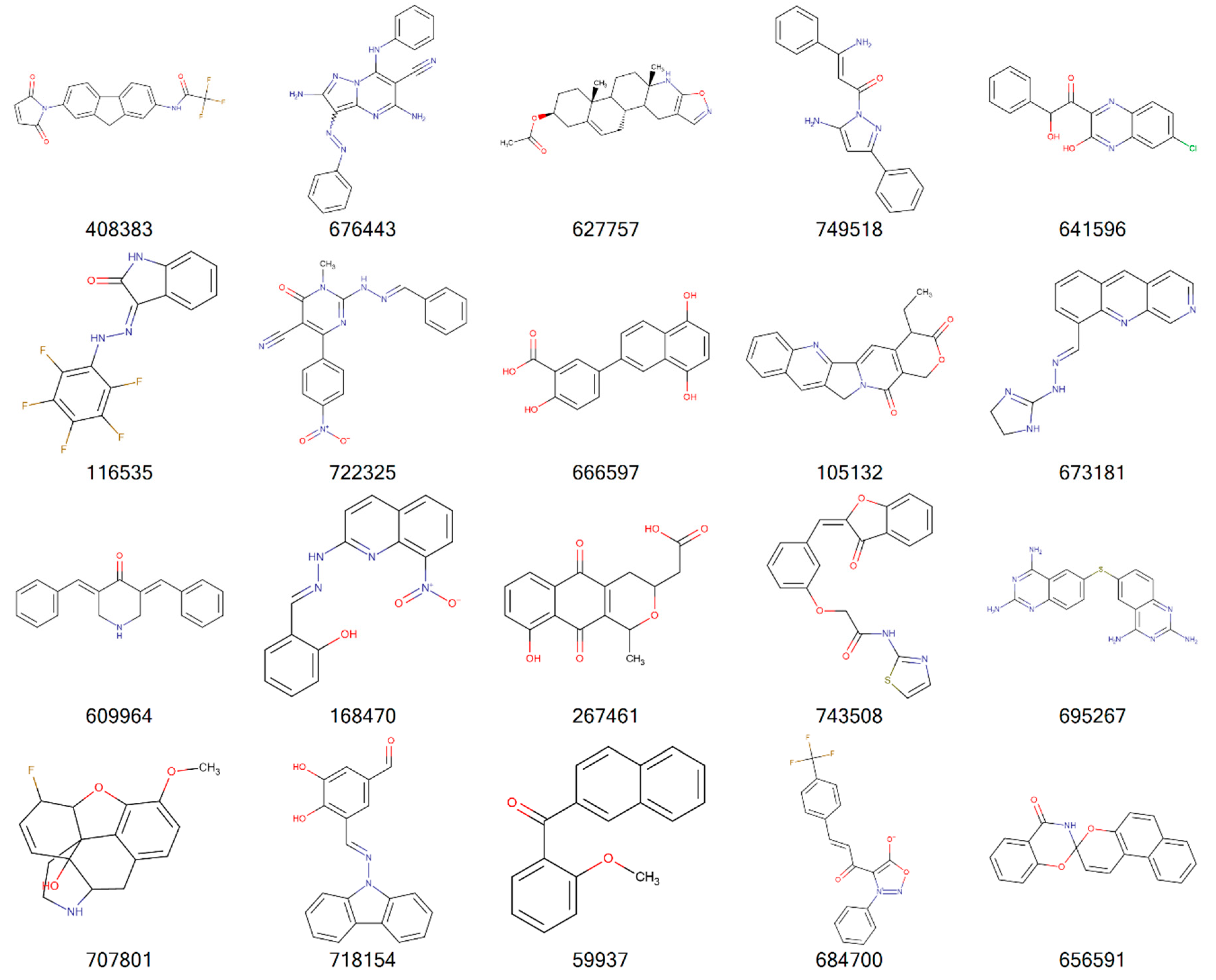
2. Results
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Funding
Conflicts of Interest
References
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Cannan, W.J.; Pederson, D.S. Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin. J. Cell. Physiol. 2016, 231, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Forment, J.V.; Kaidi, A.; Jackson, S.P. Chromothripsis and cancer: Causes and consequences of chromosome shattering. Nat. Rev. Cancer 2012, 12, 663–670. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, M.; Allen, C.; Nickoloff, J.A.; Hromas, R. Synthetic lethality: Exploiting the addiction of cancer to DNA repair. Blood 2011, 117, 6074–6082. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Ashley, A.K.; Hromas, R.; Nickoloff, J.A. More forks on the road to replication stress recovery. J. Mol. Cell Biol. 2011, 3, 4–12. [Google Scholar] [CrossRef]
- Budzowska, M.; Kanaar, R. Mechanisms of dealing with DNA damage-induced replication problems. Cell Biochem. Biophys. 2009, 53, 17–31. [Google Scholar] [CrossRef]
- Nickoloff, J.A.; Jones, D.; Lee, S.H.; Williamson, E.A.; Hromas, R. Drugging the Cancers Addicted to DNA Repair. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Ashworth, A.; Lord, C.J. Synthetic lethal therapies for cancer: what’s next after PARP inhibitors? Nat. Rev. Clin. Oncol. 2018, 15, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Dedes, K.J.; Wilkerson, P.M.; Wetterskog, D.; Weigelt, B.; Ashworth, A.; Reis-Filho, J.S. Synthetic lethality of PARP inhibition in cancers lacking BRCA1 and BRCA2 mutations. Cell Cycle 2011, 10, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Gavande, N.S.; VanderVere-Carozza, P.S.; Hinshaw, H.D.; Jalal, S.I.; Sears, C.R.; Pawelczak, K.S.; Turchi, J.J. DNA repair targeted therapy: The past or future of cancer treatment? Pharmacol. Ther. 2016, 160, 65–83. [Google Scholar] [CrossRef]
- Rehman, F.L.; Lord, C.J.; Ashworth, A. Synthetic lethal approaches to breast cancer therapy. Nat. Rev. Clin. Oncol. 2010, 7, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Aoki, D.; Chiyoda, T. PARP inhibitors and quality of life in ovarian cancer. Lancet Oncol. 2018, 19, 1012–1014. [Google Scholar] [CrossRef]
- Jiang, Y.; Dai, H.; Li, Y.; Yin, J.; Guo, S.; Lin, S.Y.; McGrail, D.J. PARP inhibitors synergize with gemcitabine by potentiating DNA damage in non-small-cell lung cancer. Int. J. Cancer 2018, 144, 1092–1103. [Google Scholar] [CrossRef]
- Lyons, T.G.; Robson, M.E. Resurrection of PARP Inhibitors in Breast Cancer. J. Natl. Compr. Cancer Netw. 2018, 16, 1150–1156. [Google Scholar] [CrossRef]
- O’Cearbhaill, R.E. Using PARP Inhibitors in Advanced Ovarian Cancer. Oncology 2018, 32, 339–343. [Google Scholar]
- Paradiso, A.; Andreopoulou, E.; Conte, P.; Eniu, A.; Saghatchian, M. PARP Inhibitors in Breast Cancer: Why, How, and When? Breast Care 2018, 13, 216–219. [Google Scholar] [CrossRef]
- Robert, M.; Patsouris, A.; Frenel, J.S.; Gourmelon, C.; Augereau, P.; Campone, M. Emerging PARP inhibitors for treating breast cancer. Expert Opin. Emerg. Drugs 2018, 23, 211–221. [Google Scholar] [CrossRef]
- Szalat, R.; Samur, M.K.; Fulciniti, M.; Lopez, M.; Nanjappa, P.; Cleynen, A.; Wen, K.; Kumar, S.; Perini, T.; Calkins, A.S.; et al. Nucleotide excision repair is a potential therapeutic target in multiple myeloma. Leukemia 2018, 32, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Andrews, B.J.; Turchi, J.J. Development of a high-throughput screen for inhibitors of replication protein A and its role in nucleotide excision repair. Mol. Cancer Ther. 2004, 3, 385–391. [Google Scholar] [PubMed]
- Gentile, F.; Tuszynski, J.A.; Barakat, K.H. New design of nucleotide excision repair (NER) inhibitors for combination cancer therapy. J. Mol. Graph. Model. 2016, 65, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Rocha, J.C.; Busatto, F.F.; Guecheva, T.N.; Saffi, J. Role of nucleotide excision repair proteins in response to DNA damage induced by topoisomerase II inhibitors. Mutat. Res. Rev. Mutat. Res. 2016, 768, 8–77. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Myint, M.; Ang, W.H.; Song, L.; Lippard, S.J. Monofunctional platinum-DNA adducts are strong inhibitors of transcription and substrates for nucleotide excision repair in live mammalian cells. Cancer Res. 2012, 72, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Davidson, D.; Amrein, L.; Panasci, L.; Aloyz, R. Small Molecules, Inhibitors of DNA-PK, Targeting DNA Repair, and Beyond. Front. Pharmacol. 2013, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Mould, E.; Berry, P.; Jamieson, D.; Hill, C.; Cano, C.; Tan, N.; Elliott, S.; Durkacz, B.; Newell, D.; Willmore, E. Identification of dual DNA-PK MDR1 inhibitors for the potentiation of cytotoxic drug activity. Biochem. Pharmacol. 2014, 88, 58–65. [Google Scholar] [CrossRef]
- Olsen, B.B.; Fritz, G.; Issinger, O.G. Characterization of ATM and DNA-PK wild-type and mutant cell lines upon DSB induction in the presence and absence of CK2 inhibitors. Int. J. Oncol. 2012, 40, 592–598. [Google Scholar]
- Pospisilova, M.; Seifrtova, M.; Rezacova, M. Small molecule inhibitors of DNA-PK for tumor sensitization to anticancer therapy. J. Physiol. Pharmacol. 2017, 68, 337–344. [Google Scholar]
- Singh, S.K.; Wu, W.; Zhang, L.; Klammer, H.; Wang, M.; Iliakis, G. Widespread dependence of backup NHEJ on growth state: Ramifications for the use of DNA-PK inhibitors. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 540–548. [Google Scholar] [CrossRef]
- Ronco, C.; Martin, A.R.; Demange, L.; Benhida, R. ATM, ATR, CHK1, CHK2 and WEE1 inhibitors in cancer and cancer stem cells. Medchemcomm 2017, 8, 295–319. [Google Scholar] [CrossRef] [PubMed]
- Sarkaria, J.N. Identifying inhibitors of ATM and ATR kinase activities. Methods Mol. Med. 2003, 85, 49–56. [Google Scholar] [PubMed]
- Arnaudeau, C.; Rozier, L.; Cazaux, C.; Defais, M.; Jenssen, D.; Helleday, T. RAD51 supports spontaneous non-homologous recombination in mammalian cells, but not the corresponding process induced by topoisomerase inhibitors. Nucleic Acids Res. 2001, 29, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Motlekar, N.A.; Burgwin, C.M.; Napper, A.D.; Diamond, S.L.; Mazin, A.V. Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem. Biol. 2011, 6, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Nomme, J.; Renodon-Corniere, A.; Asanomi, Y.; Sakaguchi, K.; Stasiak, A.Z.; Stasiak, A.; Norden, B.; Tran, V.; Takahashi, M. Design of potent inhibitors of human RAD51 recombinase based on BRC motifs of BRCA2 protein: Modeling and experimental validation of a chimera peptide. J. Med. Chem. 2010, 53, 5782–5791. [Google Scholar] [CrossRef] [PubMed]
- Normand, A.; Riviere, E.; Renodon-Corniere, A. Identification and characterization of human Rad51 inhibitors by screening of an existing drug library. Biochem. Pharmacol. 2014, 91, 293–300. [Google Scholar] [CrossRef]
- Reed, A.M.; Fishel, M.L.; Kelley, M.R. Small-molecule inhibitors of proteins involved in base excision repair potentiate the anti-tumorigenic effect of existing chemotherapeutics and irradiation. Future Oncol. 2009, 5, 713–726. [Google Scholar] [CrossRef]
- Wilson, S.H.; Beard, W.A.; Shock, D.D.; Batra, V.K.; Cavanaugh, N.A.; Prasad, R.; Hou, E.W.; Liu, Y.; Asagoshi, K.; Horton, J.K.; et al. Base excision repair and design of small molecule inhibitors of human DNA polymerase beta. Cell. Mol. Life Sci. 2010, 67, 3633–3647. [Google Scholar] [CrossRef]
- Peterson, L.; Kovyrshina, T. DNA Repair Gene Expression Adjusted by the PCNA Metagene Predicts Survival in Multiple Cancers. BioRxiv 2018. [Google Scholar] [CrossRef]
- Venet, D.; Dumont, J.E.; Detours, V. Most random gene expression signatures are significantly associated with breast cancer outcome. PLoS Comput. Biol. 2011, 7, e1002240. [Google Scholar] [CrossRef]
- Sun, H. A universal molecular descriptor system for prediction of logP, logS, logBB, and absorption. J. Chem. Inf. Comput. Sci. 2004, 44, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Bunin, B.A. Increasing the efficiency of small-molecule drug discovery. Drug Discov. Today 2003, 8, 823–826. [Google Scholar] [CrossRef]
- Driscoll, J.S. The preclinical new drug research program of the National Cancer Institute. Cancer Treat. Rep. 1984, 68, 63–76. [Google Scholar]
- Rubinstein, L.V.; Shoemaker, R.H.; Paull, K.D.; Simon, R.M.; Tosini, S.; Skehan, P.; Scudiero, D.A.; Monks, A.; Boyd, M.R. Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J. Natl. Cancer Inst. 1990, 82, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Reinhold, W.C.; Sunshine, M.; Liu, H.; Varma, S.; Kohn, K.W.; Morris, J.; Doroshow, J.; Pommier, Y. CellMiner: A web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set. Cancer Res. 2012, 72, 3499–3511. [Google Scholar] [CrossRef] [PubMed]
- Cellminer hdnngc: 2018, 2.1; US National Cancer Institute: Bethesda, MD, USA, 2018.
- UniProt Consortium, T. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [Google Scholar] [CrossRef] [PubMed]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Gallo Cassarino, T.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30, S162–S173. [Google Scholar] [CrossRef] [PubMed]
- Benkert, P.; Kunzli, M.; Schwede, T. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009, 37, W510–W514. [Google Scholar] [CrossRef]
- Bertoni, M.; Kiefer, F.; Biasini, M.; Bordoli, L.; Schwede, T. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 2017, 7, 10480. [Google Scholar] [CrossRef] [PubMed]
- Boratyn, G.M.; Camacho, C.; Cooper, P.S.; Coulouris, G.; Fong, A.; Ma, N.; Madden, T.L.; Matten, W.T.; McGinnis, S.D.; Merezhuk, Y.; et al. BLAST: A more efficient report with usability improvements. Nucleic Acids Res. 2013, 41, W29–W33. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S.; Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S.; Henikoff, J.G. Amino acid substitution matrices. Adv. Protein Chem. 2000, 54, 73–97. [Google Scholar] [PubMed]
- Eyrich, V.A.; Marti-Renom, M.A.; Przybylski, D.; Madhusudhan, M.S.; Fiser, A.; Pazos, F.; Valencia, A.; Sali, A.; Rost, B. EVA: Continuous automatic evaluation of protein structure prediction servers. Bioinformatics 2001, 17, 1242–1243. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.; Roth, S.; Arnold, K.; Kiefer, F.; Schmidt, T.; Bordoli, L.; Schwede, T. The Protein Model Portal—A comprehensive resource for protein structure and model information. Database 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Gilson, M.K.; Given, J.A.; Bush, B.L.; McCammon, J.A. The statistical-thermodynamic basis for computation of binding affinities: A critical review. Biophys. J. 1997, 72, 1047–1069. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck Molecular Force Field. I Basis, Form, Scope, Parameterization, and Performance of MMFF94. J. Comp. Chem. 1996, 17, 490–515. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
- De Winter, H. SILICOS-IT Filter-It. Available online: http://silicos-itbes3-website-eu-west-1amazonawscom/ (accessed on 11 July 2018).
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Shen, J.; Li, W.; Lee, P.W.; Tang, Y. In silico prediction of terrestrial and aquatic toxicities for organic chemicals. Chin. J. Pestic. Sci. 2010, 12, 477–488. [Google Scholar]
- EPA. US EPA ECOTOX Database. Available online: https://cfpub.epa.gov/ecotox/ (accessed on 11 July 2018).
- Cheng, F.; Shen, J.; Li, W.; Yu, Y.; Liu, G.; Lee, P.W.; Tang, Y. In silico prediction of Tetrahymena Pyriformis toxicity for diverse industrial chemicals with substructure pattern recognition and machine learning methods. Chemosphere 2011, 82, 1636–1643. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Li, H.; Ung, C.Y.; Yap, C.W.; Chen, Y.Z. Classification of a diverse set of Tetrahymena pyriformis toxicity chemical compounds from molecular descriptors by statistical learning methods. Chem. Res. Toxicol. 2006, 19, 1030–1039. [Google Scholar] [CrossRef]
- Shen, J.; Cheng, F.; Xu, Y.; Li, W.; Tang, Y. Estimation of ADME properties with substructure pattern recognition. J. Chem. Inf. Model. 2010, 50, 1034–1041. [Google Scholar] [CrossRef]
- NCBI. NCBI PubChem Database AID-1851. Cytochrome Panel Assay with Activity Outcomes. Available online: https://pubchem.ncbi.nlm.nih.gov/bioassay/1851/ (accessed on 11 July 2018).
- Zaretzki, J.; Boehm, K.M.; Swamidass, S.J. Improved Prediction of CYP-Mediated Metabolism with Chemical Fingerprints. J. Chem. Inf. Model. 2015, 55, 972–982. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the NCI’s DTP program. |







{kind=link}
{kind=link}
{kind=link}
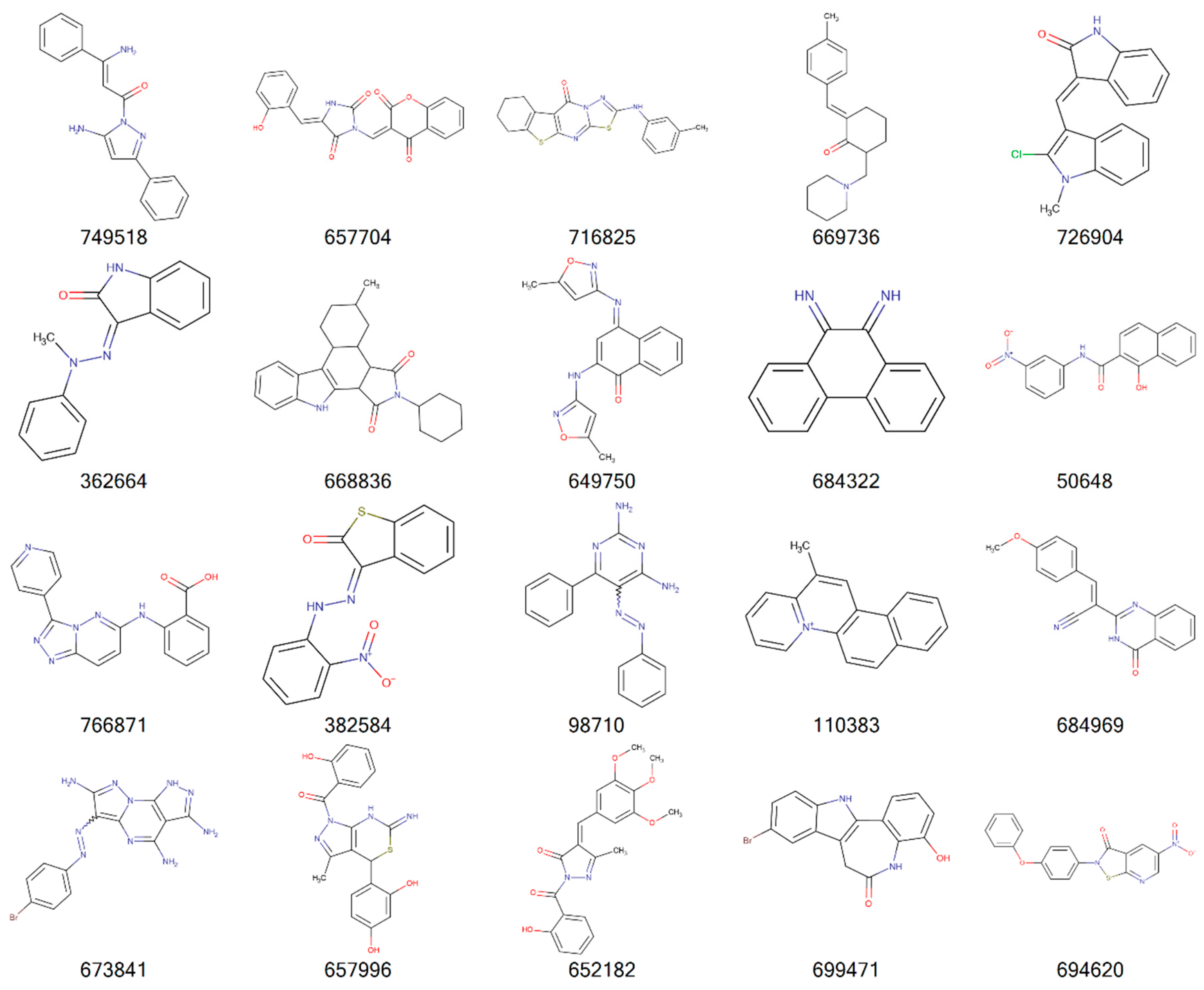{kind=link}
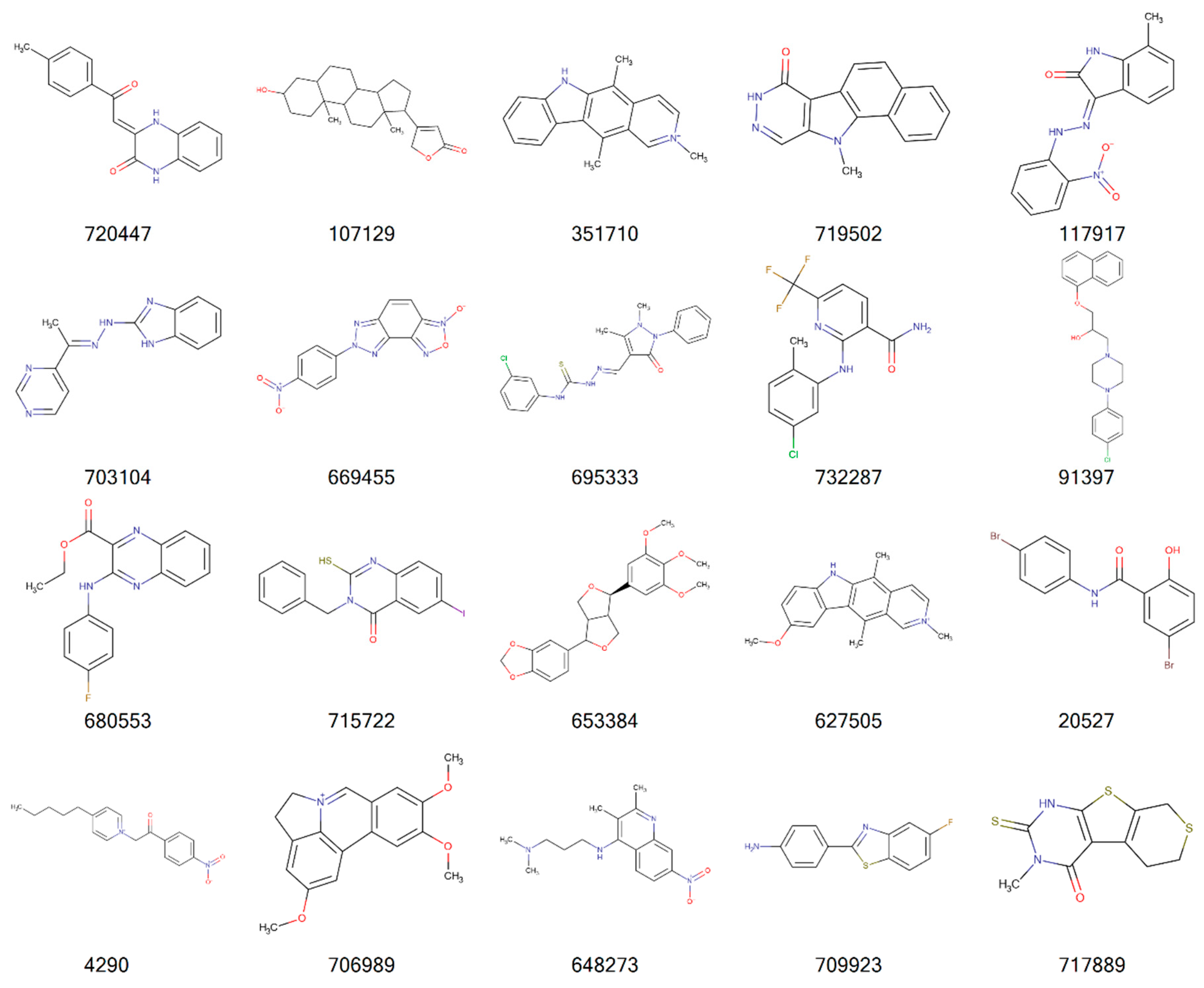{kind=link}
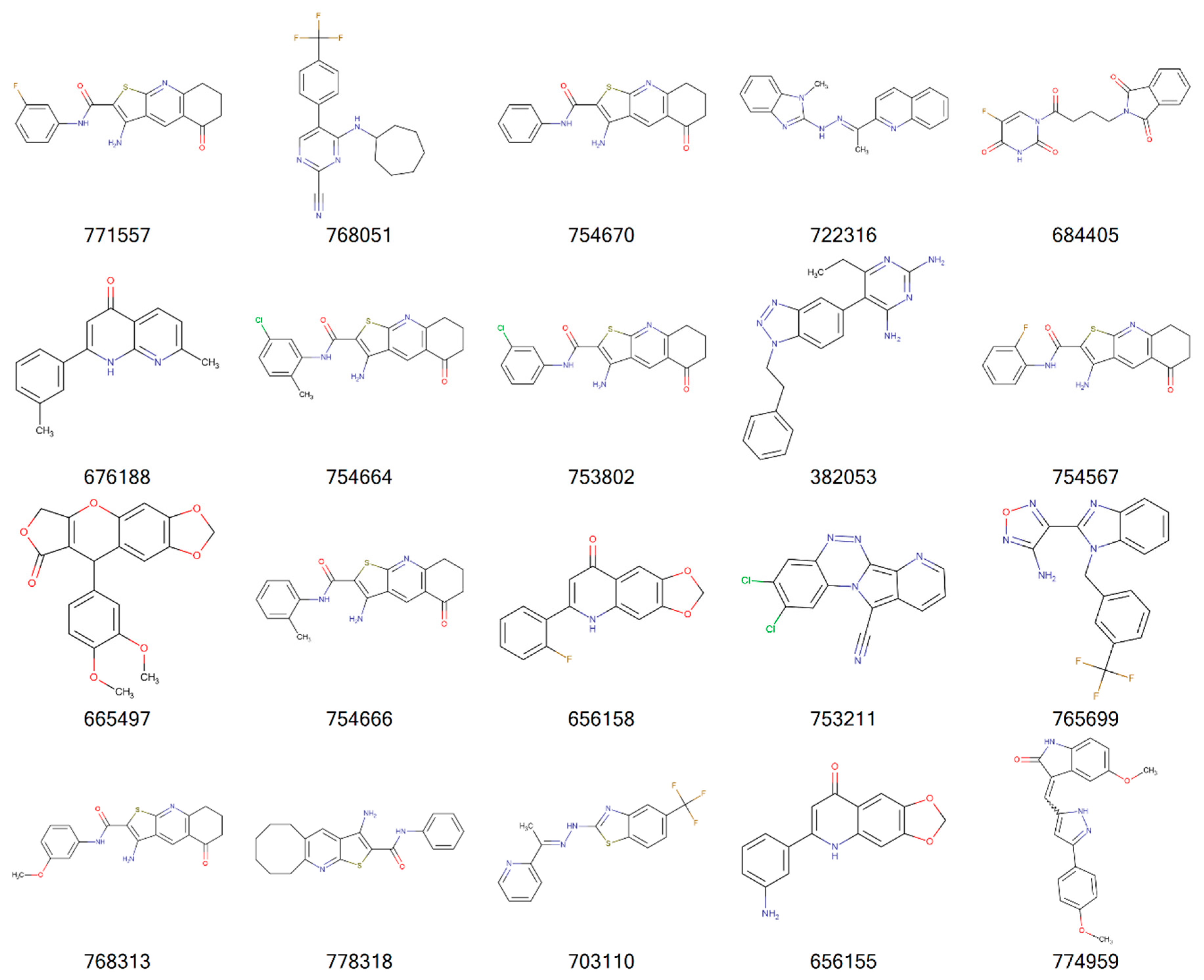{kind=link}
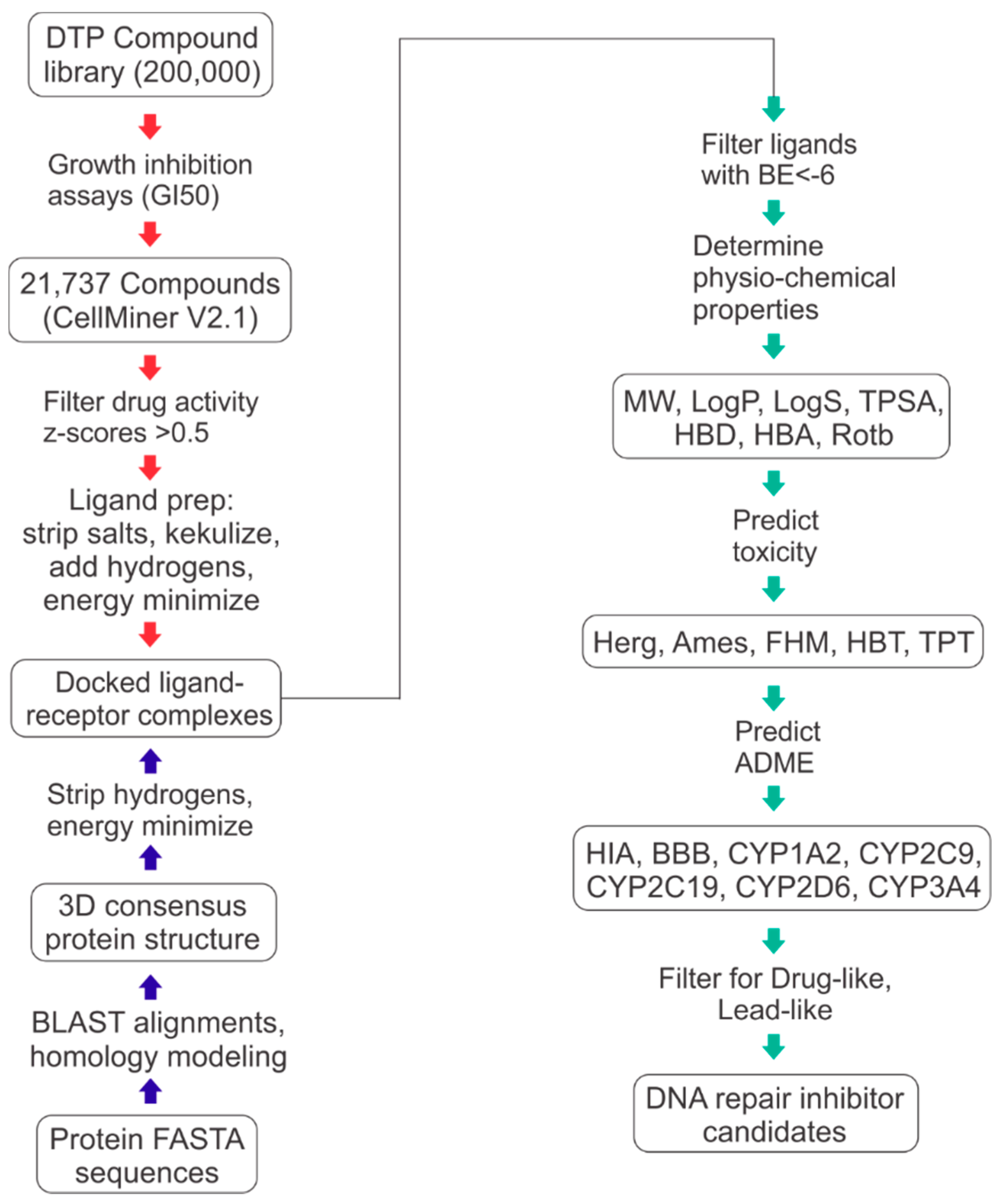{kind=link}
| Cancer | DNA Repair Proteins Whose Downregulation is Associated with Prolonged OS |
|---|---|
| AML | RAD23A, EME2, APEX2 |
| Breast | ATRIP, FANCC, RAD1, RFC3, NEIL3, EXO1, FANCB, FANCD2, FANCI, RAD51, XRCC4 |
| Colon | RAD23A, RFC2, POLL, MLH3, FANCL |
| GBM | XRCC5, NBN, DDB1, GTF2H2, ERCC4, ALKBH2, APEX1, PRKDC, PMS1, REV1 |
| Renal papillary | BLM, RAD1, FEN1, LIG1, EXO1, MSH6, BRCA2, EME1, FANCB, LIG4 |
| Lung | BRCA1, NBN, RAD1, NEIL3, MMS19, FANCI, XRCC1, XRCC5 |
| Melanoma | MDC1, NBN, MUTYH, POLE, UNG, FANCE, FANCI, POLI, POLK |
| Ovarian | PARP2, GTF2H4, SMUG1, DCLRE1B, GPS1, FANCL, APEX2, PMS1, XRCC6, MSH6, UNG, RAD51, RAD23A, EXO1, MUS81 |
| Cancer | Docked | Binding Energy (kcal/mol) < −6 | Drug-Like | Lead-Like |
|---|---|---|---|---|
| AML | 3835 | 1533 | 1181 | 173 |
| Breast | 684 | 336 | 237 | 47 |
| GBM | 343 | 334 | 238 | 38 |
| Colon | 1123 | 1049 | 751 | 106 |
| Lung | 31 | 30 | 20 | 7 |
| Melanoma | 291 | 276 | 224 | 52 |
| Ovarian | 105 | 102 | 75 | 8 |
| Renal papillary | 161 | 149 | 103 | 24 |
| Total | 3669 | 3392 |




© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peterson, L.E. Small Molecule Docking of DNA Repair Proteins Associated with Cancer Survival Following PCNA Metagene Adjustment: A Potential Novel Class of Repair Inhibitors. Molecules 2019, 24, 645. https://doi.org/10.3390/molecules24030645
Peterson LE. Small Molecule Docking of DNA Repair Proteins Associated with Cancer Survival Following PCNA Metagene Adjustment: A Potential Novel Class of Repair Inhibitors. Molecules. 2019; 24(3):645. https://doi.org/10.3390/molecules24030645
Chicago/Turabian StylePeterson, Leif E. 2019. "Small Molecule Docking of DNA Repair Proteins Associated with Cancer Survival Following PCNA Metagene Adjustment: A Potential Novel Class of Repair Inhibitors" Molecules 24, no. 3: 645. https://doi.org/10.3390/molecules24030645
APA StylePeterson, L. E. (2019). Small Molecule Docking of DNA Repair Proteins Associated with Cancer Survival Following PCNA Metagene Adjustment: A Potential Novel Class of Repair Inhibitors. Molecules, 24(3), 645. https://doi.org/10.3390/molecules24030645