
First Total Synthesis of Varioxiranol A
by
 and
and
Angelika Lásiková
1,*, 
Jana Doháňošová
2,
Mária Štiblariková
1,
Martin Parák
1,
Ján Moncol
3 and
Tibor Gracza
1 1
Department of Organic Chemistry, Slovak University of Technology in Bratislava, Radlinského 9, 812 37 Bratislava, Slovakia
2
Central Laboratories, Slovak University of Technology in Bratislava, Radlinského 9, 812 37 Bratislava, Slovakia
3
Department of Inorganic Chemistry, Slovak University of Technology in Bratislava, Radlinského 9, 812 37 Bratislava, Slovakia
*
Author to whom correspondence should be addressed.
Molecules 2019, 24(5), 862; https://doi.org/10.3390/molecules24050862
Submission received: 4 February 2019
/
Revised: 15 February 2019
/
Accepted: 23 February 2019
/
Published: 28 February 2019
(This article belongs to the Special Issue Synthesis, Study and Utilization of Natural Products)
Abstract
:The paper describes the first total synthesis of natural varioxiranol A by chiral pool approach and confirmation of its absolute configuration by single-crystal X-ray analysis. The target varioxiranol A and its 4-epimer were obtained after 10 steps from single and available chiral source 1,2-O-isopropylidene-d-glyceraldehyde in an overall yield of 10% and 6%, respectively. A synthetic strategy based on the Julia–Kocieński coupling reaction between aromatic sulfone and corresponding aldose derivative makes it possible to prepare other interesting polyketide derivatives (varioxiranols B-G, varioxirane, varioxiranediols).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The fungus Emericella variecolor [1] is considered as a promising source of interesting bioactive compounds. During the past several decades, the large number of natural products of this origin had been isolated and evaluated for their diversiform biological activities. First, Dunn and Johnstone [2] isolated from a static culture of a pure strain of the fungus Aspergillus variecolor (imperfect state of Emericella variecolor) 2-methoxy-6-(3,4-dihydroxyhepta-1,5-dienyl) benzyl alcohol 1 (Figure 1) along with other metabolites-6-methoxymellein, siderin, andibenin, and andilesin A–C. The structure of 1 was established by NMR spectroscopy [3], absolute configuration (3R,4S) was later determined by total synthesis [4], and the trivial name “andytriol” was kindly suggested by prof. Johnstone and used for the first time in the manuscript dealing with the synthesis [4]. In 2002, Malmstrøm et al. [5] reported the isolation of benzyl alcohols (varitriol 3, varioxirane 2), prenylxanthones (shamixanthone, varixanthone, tajixanthone), and cyclopentanones from a strain of E. variecolor derived from a Caribbean sponge. In particular, 3 showed notably increased potency toward selected renal, CNS, and breast cancer cell lines. The authors also proposed a hypothetical biogenetic relationship between these products via enzymatic intramolecular SN2 epoxide ring opening and pointed out that natural andytriol 1 could be involved in this biosynthetic pathway to 3 via epoxide 2. Recently, seven new polyketide derivatives with benzyl alcohol structural motif, namely varioxiranols A–G, isolated by chemical examination of a sponge (Cinachyrella sp.)-associated E. variecolor fungus and tested for lipid-lowering effects against oleic acid, elicited lipid accumulation in HepG2 liver cells. Among these secondary metabolites, varioxiranol A 4 exerted inhibition activity and showed no toxicity [6].
Interesting activity against drug-resistant microbial pathogens was observed by varioxiranediols 6 and 7. These metabolites of formal cyclisation of varioxirane 2 and varioxiranol A 4 were isolated from the same endophytic fungus. The structure and absolute configuration of 6 [7] and varioxiranediols A 7 and B 8 [8] were confirmed by the X-ray analysis supporting the structural relationship of the isolated natural compounds.
In the course of our long-term program directed towards the synthesis of natural compounds and secondary metabolites isolated from E. variecolor, we have developed the synthesis of varitriol 3 [9,10,11,12,13], andytriol 1 [4], and varioxirane 2 [4] and examined their antitumor activity. Herein, we describe the first total synthesis of natural varioxiranol A 4 and its 4-epimer 9 that should also be general route for the synthesis of all other varioxiranols, varioxirane, and varioxiranediols.
2. Results and Discussion
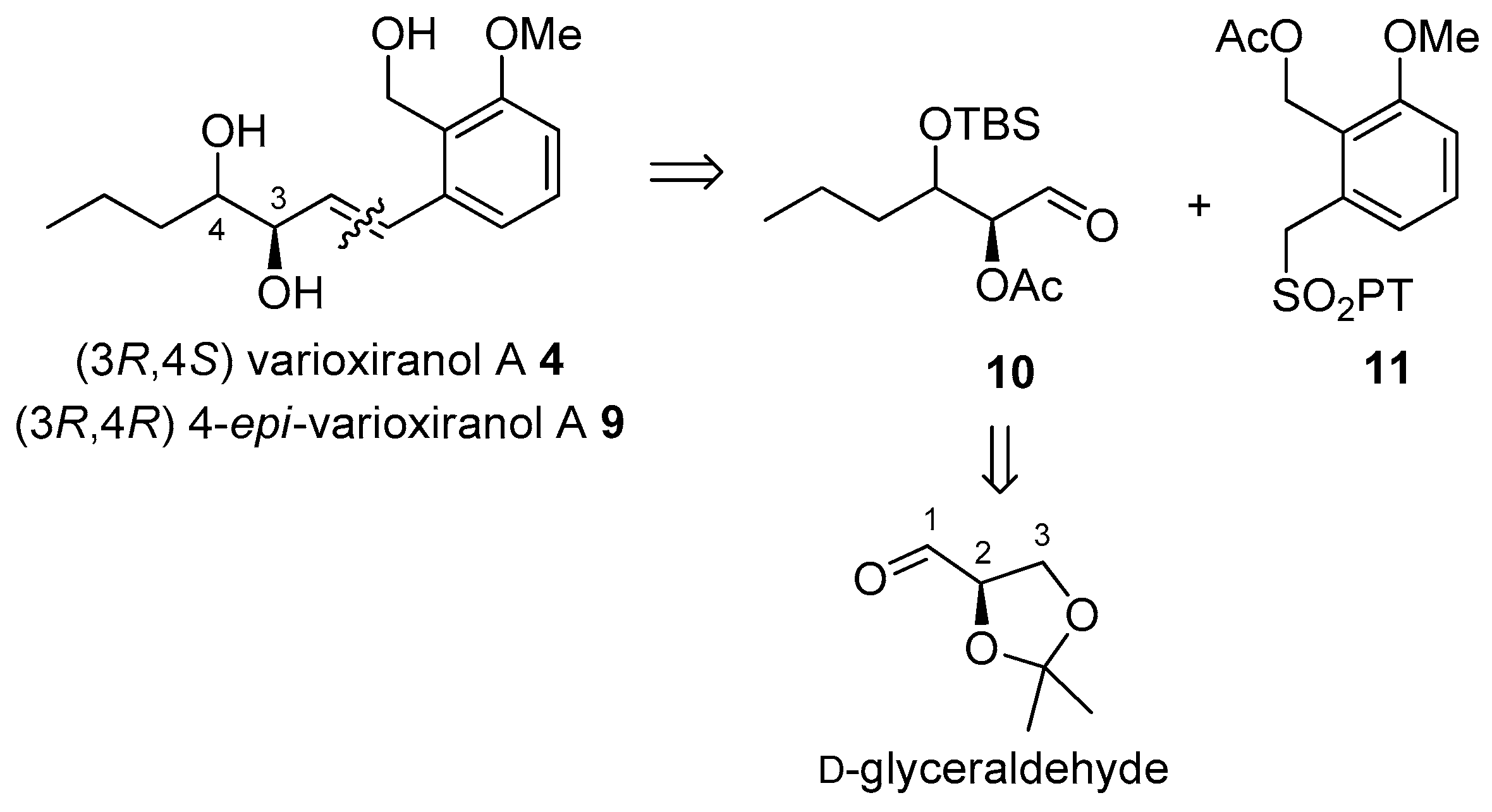
The synthetic strategy takes advantage of our previous synthesis of andytriol 1. Target compounds could be readily obtained by coupling of the known sulfone 11 [4] bearing benzyl alcohol moiety with corresponding aldehydes via Julia–Kocieński olefination (Scheme 1). The aldehydic partner for the olefination, dihydroxyhexanal 10, having the configuration of natural enantiomer 4 and its 4-epimer 9 at C-2, could be accessible from d-glyceraldehyde by introduction of the propyl group at C-1 followed by the oxidation of carbon at the other end (C-3).
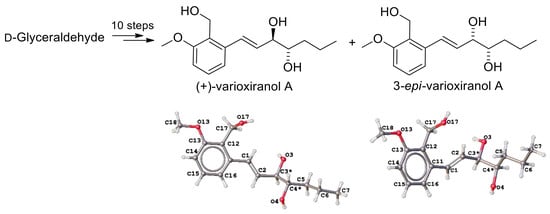
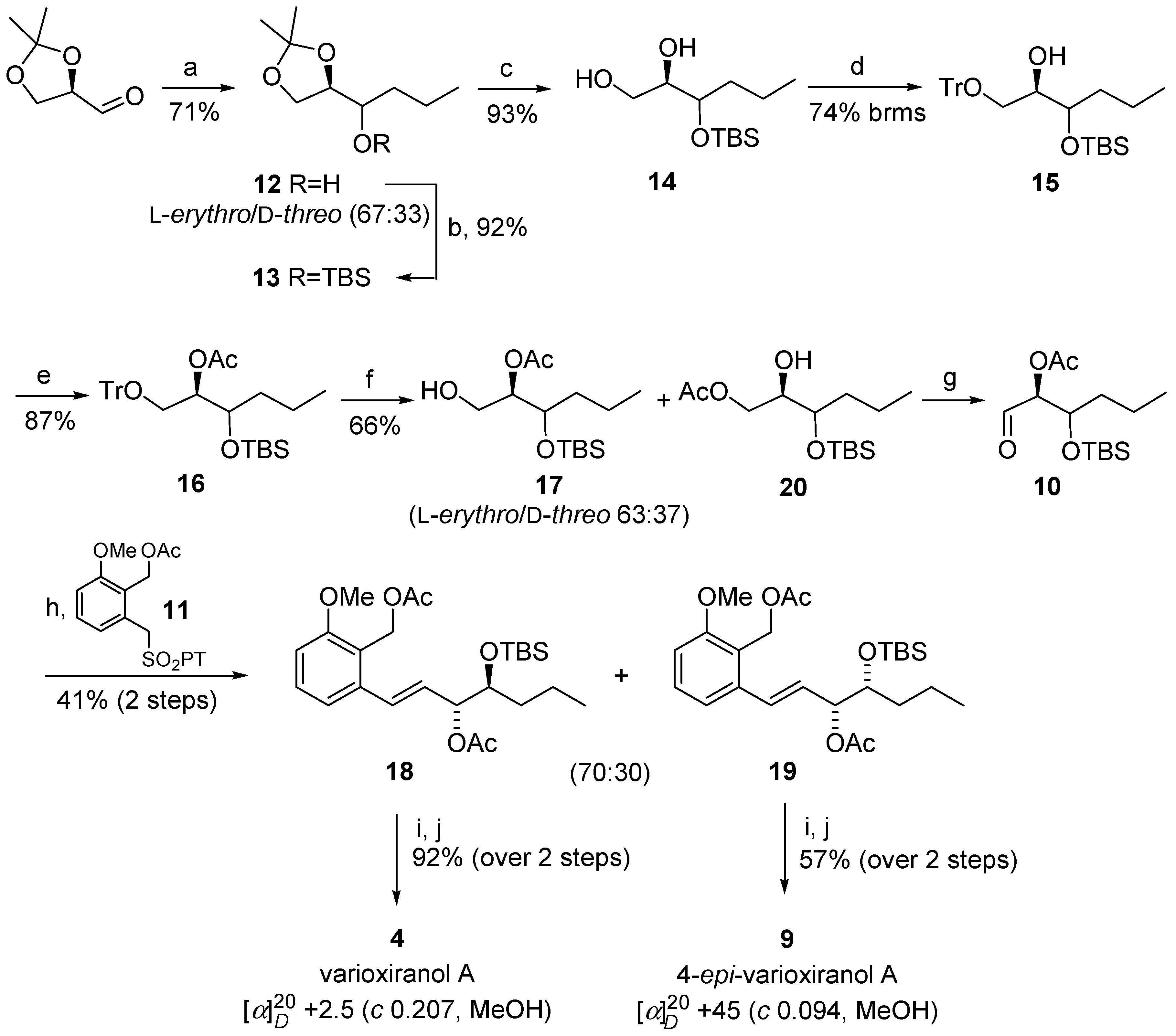
The synthesis of varioxiranols A 4 commenced from isopropylidene-d-glyceraldehyde, a commercially available starting material, or readily obtainable from d-mannitol via the route using standard carbohydrate chemistry [14]. The required six-carbon chain of the key fragment 10 [15] was obtained by the Grignard addition of propylmagnesium chloride to isopropylidene-d-glyceraldehyde in THF/Et2O [15] (Scheme 2). A diastereomeric mixture of partially protected l-erythro/d-threo hexenetriols 12 was isolated in the ratio of 67:33 with 71% yield. To prepare both epimers of varioxiranol A 4, we continued synthesis with the mixture of diastereomers 12. The aldehydic partner for the Julia–Kocieński coupling, hexenose derivative 10 was prepared using a selective protection–deprotection sequence. Firstly, free hydroxyl group of 12 was protected as tert-butyldimethylsilylether 13 with good yield (92%). Acidic hydrolysis of 13 with trifluoroacetic acid in dichloromethane afforded vicinal diol 14 (93%) which was selectively tritylated on terminal hydroxyl group to give 15. Subsequent acetylation of 15 provided fully protected triol 16 (87%). Selective deprotection of primary hydroxy group was then achieved by smooth tritylether hydrolysis using formic acid in ether furnishing alcohol 17 [16], however, as an inseparable mixture along with the product of the acetyl group migration 20 taking place even during MPLC. Swern oxidation of primary hydroxyl group under conventional reaction conditions provided desired aldehyde 10, which was used in the next step without further purification. The crude aldehyde was subjected to Julia–Kocieński coupling with 2-methoxy-6-[(1-phenyl-1H-tetrazol-5-ylsulfonyl)methyl]benzyl acetate 11, which was prepared according to the literature [4]. Thus, potassium hexamethyldisilazane was added to a nearly equimolar mixture of sulfone 11 and aldehyde 10 in dimethoxyethane at −60 °C and stirred for 40 min at room temperature affording coupling products 18/19 in 41% yield (in ratio 70:30) and with excellent E-selectivity. The resulting alkenes 18/19 could be separated by MPLC and preparative TLC. The final steps, removal of all protecting groups, were run in parallel with the pure diastereomers 18 and 19. The first, basic hydrolysis of acetyl groups with K2CO3 in MeOH, was followed by treatment of the crude mixture with TBAF in THF and finally, flash chromatography purification furnished the target compounds (+)-4 and 9 in 92% and 57% yield, respectively, over the last two steps. The 1H and 13C NMR, HRMS spectra, and the specific rotation {[α] +2.5 (c 0.207, MeOH), lit. [2], [α] +5.8 (c 0.53, MeOH)} of synthetic varioxiranol A 4 were in good agreement with the reported data for the natural product.
Definitive confirmation of absolute configuration of the target compound has been provided by single-crystal X-ray analysis. An X-ray study of both epimers confirmed 3R,4S (anti) configuration of the natural varioxiranol A 4 and 3R,4R (syn) configuration of its 4-epimer 9 (Figure 2). Interestingly, the crystal lattice of the 4-epimer 9 is composed of S-cis and S-trans conformers (two crystallographic independent molecules in cell; see Supplementary Materials).
3. Experimental Section
3.1. General Methods
Commercial reagents were used without further purification. All solvents were distilled before use. Hexanes refer to the fraction boiling at 60–65 °C. Flash column liquid chromatography (FLC) was performed on silica gel Kieselgel 60 (40–63 μm, 230–400 mesh, Merck, Darmstadt, Germany) and analytical thin-layer chromatography (TLC) was performed on aluminium plates precoated with either 0.2 mm (DC-Alufolien, Merck) or 0.25 mm silica gel 60 F254 (ALUGRAM® SIL G/UV254, Macherey-Nagel, Fisher Scientific, Loughborough, UK). The compounds were visualized by UV fluorescence and by dipping the plates in an aqueous H2SO4 solution of cerium sulphate/ammonium molybdate followed by charring with a heat gun. Melting points were obtained using a Boecius apparatus (Büchi® melting point apparatus Model B-545, BÜCHI Labortechnik AG, Flawil, Switzerland) and are uncorrected. Optical rotations were measured with a JASCO P-2000 polarimeter (JASCO, Easton, MD, USA) and are given in units of 10−1 deg.cm2.g−1. FTIR spectra were obtained on a Nicolet 5700 spectrometer (Thermo Electron, Thermo Fisher Scientific, Waltham, MA, USA) equipped with a Smart Orbit (diamond crystal ATR) accessory, using the reflectance technique (4000–400 cm−1).1H and 13C NMR spectra were recorded on either 300 (75) MHz or 600 (150) MHz Varian spectrometer (Varian Inc., Palo Alto, CA, USA). Chemical shifts (δ) are quoted in ppm and are referenced to tetramethylsilane (TMS, δ = 0 ppm) as internal standard for 1H NMR and to CDCl3 peak (δ = 77.16 ppm in case of 13C NMR). High-resolution mass spectra (HRMS) were recorded on an OrbitrapVelos mass spectrometer (Thermo Scientific, Waltham, MA, USA; Bremen, Germany) with a heated electrospray ionisation (HESI) source. The mass spectrometer was operated with full scan (50–2000 amu) in positive or negative FT mode (at a resolution of 100,000). The analyte was dissolved in methanol and infused via syringe pump at a rate of 5 mL/min. The heated capillary was maintained at 275 °C with a source heater temperature of 50 °C and the sheath, auxiliary, and sweep gases were at 10, 5, and 0 units, respectively. Source voltage was set to 3.5 kV.
Data collection and cell refinement of 4 and 9 were made on a Stoe StadiVari diffractometer (Stoe & Cie GmbH, Darmstadt, Germany) using a Pilatus3R 300K HPAD detector and the microfocus source Xenocs Genix3D Cu HF (λ = 1.54186 Å). The structures were solved using SHELXT [17] and refined by the full-matrix least-squares procedure with SHELXL (ver. 2018/3) (for 4) [18] or CRYSTALS (ver. 14.61) (9) [19]. The structures were drawn using the OLEX2 package [20]. The absolute configurations of both compounds were determined. The Flack parameter x = −0.08(5) for 9 was calculated by Parsons method [21]. The absolute structure of very small crystal of 4 is impossible to determine based on the Flack parameter, however, using Hooft parameter [22] with (Gaussian) statistics led to the conclusive value of y = 0.07(11). The deposition numbers CCDC 1892452 (4) and CCDC 1892453 (9) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK.
3.2. (2R,3S)-1,2-O-Isopropylidene-hexane-1,2,3-triol (l-erythro-12) and (2R,3R)-1,2-O-isopropylidene-hexane-1,2,3-triol (d-threo-12)
A solution of propylmagnesium chloride in diethyl ether (2.0 M solution, 5.8 mL, 11.6 mmol) was added dropwise to a stirred solution of freshly distilled 1,2-O-isopropylidene-d-glyceraldehyde (1 g, 7.68 mmol) in dry THF (62 mL) at room temperature. Following the addition, the reaction mixture was stirred for 1 h (TLC control). The reaction was quenched by pouring into a sat. aqueous NH4Cl (62 mL), the aqueous layer was extracted with diethyl ether (3 × 35 mL), and the combined organic layers were dried and concentrated. The residue was purified by MPLC (gradient AcOEt/hexanes 0/100 to 30/70) to give 12 (954 mg, 71%, l-erythro-12/d-threo-12 67:33) as colorless liquid. 1H NMR (600 MHz, CDCl3) δ (l-erythro-12) 0.95 (t, H-6, J = 7.1 Hz, 3H), 1.33–1.44 (m, H-4, H-5a, 2 × CH3, 9H), 1.51–1.59 (m, H-5b, 1H), 1.96 (d, OH, J = 2.9 Hz, 1H), 3.78–3.82 (m, H-3, 1H), 3.91 (dd, H-1a, J = 7.3, 8.0 Hz, 1H), 3.97 (dd, H-1b, J = 6.5, 8.0 Hz, 1H), 4.04 (ddd, H-2, J = 4.0, 6.5, 7.2 Hz, 1H); δ (D-threo-12) 0.94 (t, H-6, J = 7.2 Hz, 3H), 1.30–1.36 (m, H-4a, 1H), 1.37 (s, CH3, 3H), 1.37–1.44 (m, H-5a, 1H), 1.44 (s, CH3, 3H), 1.44–1.48 (m, H-4b, 1H), 1.53–1.59 (m, H-5b, 1H), 2.15 (d, OH, J = 5.2 Hz, 1H), 3.48-3.53 (m, H-3, 1H), 3.73 (dd, H-1a, J = 6.3, 7.7 Hz, 1H), 3.96–4.00 (m, H-2, 1H), 4.02 (dd, H-1b, J = 6.6, 7.8 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ (l-erythro-12) 14.2 (C-6), 19.1 (C-5), 25.5 (CH3), 26.6 (CH3), 34.8 (C-4), 64.6 (C-1), 70.5 (C-3), 78.8 (C-2), 109.0 (Cq); δ (D-threo-12) 14.2 (C-6), 18.9 (C-5), 25.5 (CH3), 26.8 (CH3), 36.0 (C-4), 66.3 (C-1), 72.2 (C-3), 79.3 (C-2), 109.5 (Cq); HRMS (ESI) calcd for C9H18O3Na+ [M + Na]+: 197.1148, found: 197.1148.
3.3. (2R,3S)-1,2-O-Isopropylidene-3-O-tert-butyldimethylsilyl-hexane-1,2,3-triol (l-erythro-13) and (2R,3R)-1,2-O-isopropylidene-3-O-tert-butyldimethylsilyl-hexane-1,2,3-triol (d-threo-13)
Imidazole (1.09 g, 16.0 mmol) was added to a solution of diastereomeric mixture 12 (928 mg, 5.33 mmol) in dry CH2Cl2 (11 mL) at room temperature. The mixture was subsequently cooled to 0 °C and tert-butyldimethylsilyl chloride (1.61 g, 10.7 mmol) was added. The reaction mixture was then stirred for 23 h at room temperature. After the dilution with CH2Cl2 (140 mL), the reaction mixture was washed with water (2 × 140 mL), the water phase was extracted with CH2Cl2 (3 × 90 mL), and combined organic layers were dried and concentrated. The residue was purified by MPLC (isocratic AcOEt/hexanes 2/98) to afford 13 (1.42 g, 92%, l-erythro-13/d-threo-13 67:33) as colorless liquid. 1H NMR (600 MHz, CDCl3) δ (l-erythro-13) 0.06 (s, CH3, 3H), 0.07 (s, CH3, 3H), 0.88 (s, tBu, 9H), 0.91 (t, H-6, J = 7.3 Hz, 3H), 1.34 (s, CH3, 3H), 1.35–1.47 (m, H-4a, H-5, CH3, 6H), 1.49–1.52 (m, H-4b, 1H), 3.73–3.76 (m, H-3, 1H), 3.80–3.84 (m, H-1a, 1H), 3.96–4.00 (m, H-1b, H-2c, 2H); δ (D-threo-13) 0.07 (s, CH3, 3H), 0.08 (s, CH3, 3H), 0.89 (s, tBu, 9H), 0.91 (t, H-6, J = 7.2 Hz, 3H), 1.30–1.39 (H-4, H-5a, CH3, 6H), 1.41 (s, CH3, 3H), 1.45-1.50 (m, H-5b, 1H), 3.69 (ddd, H-3, J = 4.2, 6.0, 7.7 Hz, 1H), 3.71 (dd, H-1a, J = 7.4, 8.2 Hz, 1H), 3.94 (dd, H-1b, J = 6.6, 8.2 Hz, 1H), 4.04 (ddd, H-2, J = 6.0, 6.6, 7.3 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ (l-erythro-13) −4.2 (CH3), −4.1 (CH3), 14.5 (C-6), 17.8 (C-5), 18.2 (tBu), 25.6 (CH3), 26.0 (tBu), 26.8 (CH3), 37.0 (C-4), 66.6 (C-1), 72.4 (C-3), 78.4 (C-2), 109.0 (Cq); δ (D-threo-13) −4.5 (CH3), −4.1 (CH3), 14.4 (C-6), 18.4 (tBu), 19.0 (C-5), 25.4 (CH3), 26.1 (tBu), 26.6 (CH3), 34.7 (C-4), 65.7 (C-1), 73.2 (C-3), 78.9 (C-2), 109.2 (Cq); HRMS (ESI) calcd for C15H32O3SiNa+ [M + Na]+: 311.2013, found: 311.2013.
3.4. (2R,3S)-3-O-tert-Butyldimethylsilyl-hexane-1,2,3-triol (l-erythro-14) and (2R,3R)-3-O-tert-butyldimethylsilyl-hexane-1,2,3-triol (d-threo-14)
Trifluoroacetic acid (50%, 3.3 mL) was added dropwise to a vigorously stirred solution of compound 13 (1.39 g, 4.81 mmol) in CH2Cl2 (120 mL) at room temperature. The reaction mixture was stirred for 55 min, then diluted with CH2Cl2 (220 mL), and washed with sat. aqueous NaHCO3 (60 mL) and water (2 × 130 mL). The organic phase was dried and concentrated to give crude 14 (1.11 g, 93%, l-erythro-14/d-threo-14 67:33) as colorless oil, which was used in the next step without further purification. 1H NMR (600 MHz, CDCl3) δ (l-erythro-14) 0.09 (s, CH3, 3H), 0.11 (s, CH3, 3H), 0.89 (s, tBu, 9H), 0.91 (t, H-6, J = 7.3 Hz, 3H), 1.26–1.44 (m, H-4a, H-5, 3H), 1.52–1.58 (m, H-4b, 1H), 2.20 (bs, 2x OH, 2H), 3.60 (ddd, H-2, J = 3.5, 3.7, 5.5 Hz, 1H), 3.66 (dd, H-1a, J = 3.5, 11.5 Hz, 1H), 3.79 (dd, H-1b, J = 5.5, 11.5 Hz, 1H), 3.83 (ddd, H-3, J = 3.7, 5.6, 6.6 Hz, 1H); δ (D-threo-14) 0.08 (s, CH3, 3H), 0.09 (s, CH3, 3H), 0.90 (s, tBu, 9H), 0.92 (t, H-6, J = 7.3 Hz, 3H), 1.28–1.44 (m, H-4a, H-5, 3H), 1.61–1.68 (m, H-4b, 1H), 2.20 (bs, 2 × OH, 2H), 3.56-3.60 (m, H-1, H-2, 3H), 3.68 (ddd, H-3, J = 2.8, 4.6, 6.9 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ (l-erythro-14) −4.5 (CH3), −4.4 (CH3), 14.4 (C-6), 18.2 (tBu), 18.7 (C-5), 26.0 (tBu), 35.8 (C-4), 63.4 (C-1), 73.2 (C-2), 75.3 (C-3); δ (D-threo-14) −4.6 (CH3), −4.0 (CH3), 14.4 (C-6), 18.2 (tBu), 18.4 (C-5), 26.0 (tBu), 36.2 (C-4), 64.7 (C-1), 72.4 (C-3), 73.1 (C-2); HRMS (ESI) calcd for C12H28O3SiNa+ [M + Na]+: 271.1700, found: 271.1700.
3.5. (2R,3S)-3-O-tert-Butyldimethylsilyl-1-O-trityl-hexane-1,2,3-triol (l-erythro-15) and (2R,3R)-3-O-tert-butyldimethylsilyl-1-O-trityl-hexane-1,2,3-triol (d-threo-15)
A solution of trityl chloride (1.01 g, 3.64 mmol) in dry CH2Cl2 (2.4 mL) was cooled to 0 °C, and triethylamine (0.95 mL, 6.84 mmol) and DMAP (46 mg, 0.37 mmol) were added. Subsequently, a solution of compound 14 (772 mg, 3.11 mmol) in CH2Cl2 (2.4 mL) was added dropwise. After warming to room temperature, the reaction mixture was stirred for 15 h, then quenched with sat. aqueous NH4Cl (25 mL), the aqueous layer was extracted with CH2Cl2 (3 × 10 mL), and the combined organic layers were dried and concentrated. The residue was purified by MPLC (gradient AcOEt/hexanes 0/100 to 5/95) to afford 15 (1.01 g, 74% brsm, l-erythro-15/d-threo-15 64:36) as colorless oil. 1H NMR (600 MHz, CDCl3) δ (l-erythro-15) −0.07 (s, CH3, 3H), 0.01 (s, CH3, 3H), 0.79 (s, tBu, 9H), 0.85 (t, H-6, J = 7.2 Hz, 3H), 1.21–1.28 (m, H-4a, H-5a, 2H), 1.36–1.47 (m, H-4b, H-5b, 2H), 2.36 (d, OH, J = 2.8 Hz, 1H), 3.17 (dd, H-1a, J = 7.7, 9.4 Hz, 1H), 3.23 (dd, H-1b, J = 4.5, 9.4 Hz, 1H), 3.69 (ddd, H-3, J = 4.1, 4.5, 6.9 Hz, 1H), 3.79-3.83 (m, H-2, 1H), 7.22–7.25 (m, Tr-Hp, 3H), 7.28–7.32 (m, Tr-Hm, 6H), 7.42–7.45 (m, Tr-Ho, 6H); δ (D-threo-15) -0.13 (s, CH3, 3H), 0.02 (s, CH3, 3H), 0.78 (s, tBu, 9H), 0.90 (t, H-6, J = 7.2 Hz, 3H), 1.30-1.35 (m, H-4a, H-5, 3H), 1.58–1.63 (m, H-4b, 1H), 2.37 (d, OH, J = 7.5 Hz, 1H), 3.03 (dd, H-1a, J = 6.1, 9.4 Hz, 1H), 3.22 (dd, H-1b, J = 6.3, 9.4 Hz, 1H), 3.65–3.69 (m, H-2, 1H), 3.79-3.82 (m, H-3, 1H), 7.22–7.25 (m, Tr-Hp, 3H), 7.27–7.31 (m, Tr-Hm, 6H), 7.42–7.45 (m, Tr-Ho, 6H); 13C NMR (150 MHz, CDCl3) δ (l-erythro-15) −4.4 (CH3), −4.3 (CH3), 14.4 (C-6), 18.2 (tBu), 18.3 (C-5), 26.0 (tBu), 34.5 (C-4), 65.1 (C-1), 73.3 (C-3), 73.5 (C-2), 86.9 (Trt-Cq), 127.2 (3 × Tr-Cp), 128.0 (6 × Tr-Cm), 128.8 (6 × Tr-Co), 144.1 (3 × Tr-Cipso); δ (D-threo-15) −4.7 (CH3), −4.1 (CH3), 14.4 (C-6), 18.2 (tBu), 18.7 (C-5), 26.0 (tBu), 36.1 (C-4), 65.3 (C-1), 71.8 (C-2), 71.9 (C-3), 86.8 (Tr-Cq), 127.1 (3 × Tr-Cp), 127.9 (6 × Tr-Cm), 128.8 (6 × Tr-Co), 144.2 (3 × Tr-Cipso); HRMS (ESI) calcd for C31H42O3SiNa+ [M + Na]+: 513.2795, found: 513.2795.
3.6. (2R,3S)-2-O-Acetyl-3-O-tert-butyldimethylsilyl-1-O-trityl-hexane-1,2,3-triol (l-erythro-16) and (2R,3R)-2-O-acetyl-3-O-tert-butyldimethylsilyl-1-O-trityl-hexane-1,2,3-triol (d-threo-16)
DMAP (771 mg, 6.32 mmol) and Ac2O (0.60 mL, 6.32 mmol) were added to a soluion of triol 15 (1.03 g, 2.11 mmol) in dry CH2Cl2 (30 mL) at room temperature. The reaction mixture was stirred for 30 min and quenched with sat. aqueous NaHCO3 (30 mL). The water phase was extracted with CH2Cl2 (3 × 40 mL), combined organic layers were dried and concentrated. The residue was purified by MPLC (gradient AcOEt/hexanes 0/100 to 5/95) to give 16 (975 mg, 87%, l-erythro-16/d-threo-16 65:35) as colorless oil. 1H NMR (600 MHz, CDCl3) δ (l-erythro-16) -0.10 (s, CH3, 3H), -0.04 (s, CH3, 3H), 0.74 (s, tBu, 9H), 0.85 (t, H-6, J = 7.1 Hz, 3H), 1.22–1.39 (m, H-4, H-5, 4H), 2.10 (s, C(O)CH3, 3H), 3.25–3.29 (m, H-1, 2H), 3.78 (ddd, H-3, J = 3.5, 5.0, 6.7 Hz, 1H), 5.15 (ddd, H-2, J = 3.5, 4.7, 6.8 Hz, 1H), 7.20–7.24 (m, Tr-Hp, 3H), 7.27–7.30 (m, Tr-Hm, 6H), 7.40–7.43 (m, Tr-Ho, 6H); δ (D-threo-16) 0.01 (s, CH3, 3H), 0.02 (s, CH3, 3H), 0.79 (s, tBu, 9H), 0.80 (t, H-6, J = 7.1 Hz, 3H), 1.16–1.30 (m, H-4a, H-5, 3H), 1.33–1.38 (m, H-4b, 1H), 2.15 (s, C(O)CH3, 3H), 3.17 (dd, H-1a, J = 6.8, 10.2 Hz, 1H), 3.25 (dd, H-1b, J = 2.7, 10.2 Hz, 1H), 3.86 (ddd, H-3, J = 4.3, 5.5, 6.8 Hz, 1H), 5.07 (ddd, H-2, J = 2.7, 5.5, 6.8 Hz, 1H), 7.20–7.24 (m, Tr-Hp, 3H), 7.27-7.30 (m, Tr-Hm, 6H), 7.40–7.43 (m, Tr-Ho, 6H); 13C NMR (150 MHz, CDCl3) δ (l-erythro-16) −4.6 (CH3), −4.4 (CH3), 14.3 (C-6), 18.1 (tBu), 18.7 (C-5), 21.4 (C(O)CH3), 25.9 (tBu), 36.1 (C-4), 62.3 (C-1), 72.5 (C-3), 76.0 (C-2), 86.8 (Tr-Cq), 127.1 (3 × Tr-Cp), 127.9 (6 × Tr-Cm), 128.8 (6 × Tr-Co), 144.1 (3 × Tr-Cipso), 170.5 (C(O)CH3); δ (D-threo-16) −4.4 (CH3), −4.4 (CH3), 14.3 (C-6), 18.1 (tBu), 18.5 (C-5), 21.5 (C(O)CH3), 25.9 (tBu), 35.1 (C-4), 62.6 (C-1), 70.9 (C-3), 75.8 (C-2), 86.5 (Tr-Cq), 127.1 (3 × Tr-Cp), 127.9 (6 × Tr-Cm), 128.8 (6 × Tr-Co), 144.1 (3 × Tr-Cipso), 170.7 (C(O)CH3); HRMS (ESI) calcd for C33H44O4SiNa+ [M + Na]+: 555.2901, found: 555.2901.
3.7. (2R,3S)-2-O-Acetyl-3-O-tert-butyldimethylsilyl-hexane-1,2,3-triol (l-erythro-17) and (2R,3R)-2-O-acetyl-3-O-tert-butyldimethylsilyl-hexane-1,2,3-triol (d-threo-17)
Formic acid (12.2 mL) was added to a solution of protected triol 16 (959 mg, 1.80 mmol) in diethyl ether (12.2 mL) at 0 °C. The reaction mixture was stirred for 50 min at room temperature, diluted with diethyl ether (30 mL), and cooled to 0 °C. Sat. aqueous NaHCO3 (equimolar to formic acid, 323 mmol) was added with vigorous stirring to neutralize the reaction mixture (accompanied by the separation of two layers). The water phase was extracted with diethyl ether (3 × 50 mL) and the combined organic layers were dried and concentrated. The residue was purified by MPLC (gradient AcOEt/hexanes 0/100 to 10/90) to afford 346 mg (66%) of yellowish oil as an inseparable mixture of 17 (l-erythro-17/d-threo-17 63:37) and 20 (l-erythro-20/d-threo-20 58:42, the product of the acetyl group migration of 17 taking place even during MPLC) in ratio 82:18. 1H NMR (600 MHz, CDCl3) δ (l-erythro-17) 0.07 (s, CH3, 3H), 0.11 (s, CH3, 3H), 0.90 (s, tBu, 9H), 0.93 (t, H-6, J = 7.3 Hz, 3H), 1.31–1.36 (m, H-5a, 1H), 1.48–1.55 (m, H-4, H-5b, 3H), 2.12 (s, C(O)CH3, 3H), 2.68 (dd, OH, J = 3.7, 7.7 Hz, 1H), 3.81–3.85 (m, H-1a, 1H), 3.90–3.95 (m, H-1b, H-3, 2H), 4.77 (dt, H-2, J = 3.1, 4.9 Hz, 1H); δ (d-threo-17) 0.08 (s, CH3, 3H), 0.12 (s, CH3, 3H), 0.90 (s, tBu, 9H), 0.91 (t, H-6, J = 7.2 Hz, 3H), 1.24–1.33 (m, H-5a, 1H), 1.35–1.45 (m, H-4a, H-5b, 2H), 1.46–1.54 (m, H-4b, 1H), 2.11 (s, C(O)CH3, 3H), 2.13–2.16 (m, OH, 1H), 3.69–3.73 (m, H-1a, 1H), 3.84–3.90 (m, H-1b, H-3, 2H), 4.87 (ddd, H-2, J = 4.1, 4.8, 6.5 Hz, 1H); δ (l-erythro-20) 0.08 (s, CH3, 3H), 0.09 (s, CH3, 3H), 0.90 (s, tBu, 9H), 0.92 (t, H-6, J = 7.1 Hz, 3H), 1.29–1.34 (m, H-4a, 1H), 1.38–1.44 (m, H-5, 2H), 1.55–1.61 (m, H-4b, 1H), 2.10 (s, C(O)CH3, 3H), 2.32 (d, OH, J = 4.7 Hz, 1H), 3.75-3.78 (m, H-3, 1H), 3.78-3.82 (m, H-2, 1H), 4.06 (dd, H-1a, J = 7.7, 11.6 Hz, 1H), 4.23 (dd, H-1b, J = 3.0, 11.6 Hz, 1H); δ (d-threo-20) 0.08 (s, CH3, 3H), 0.10 (s, CH3, 3H), 0.90 (s, tBu, 9H), 0.92 (t, H-6, J = 7.1 Hz, 3H), 1.31-1.44 (m, H-4a, H-5, 3H), 1.64–1.71 (m, H-4a, 1H), 2.09 (s, C(O)CH3, 3H), 2.40 (d, OH, J = 7.8 Hz, 1H), 3.68-3.71 (m, H-3, 1H), 3.72-3.75 (m, H-2, 1H), 4.05 (dd, H-1a, J = 5.1, 11.3 Hz, 1H), 4.08 (dd, H-1b, J = 7.0, 11.3 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ (l-erythro-17) −4.5, −4.5 (2 × CH3), 14.3 (C-6), 18.2 (tBu), 18.8 (C-5), 21.4 (C(O)CH3), 25.9 (tBu), 36.8 (C-4), 61.9 (C-1), 73.8 (C-3), 76.6 (C-2), 171.2 (C(O)CH3); δ (d-threo-17) = −4.4 (CH3), −4.4 (CH3), 14.4 (C-6), 18.1 (tBu), 18.9 (C-5), 21.3 (C(O)CH3), 25.9 (tBu), 34.8 (C-4), 62.0 (C-1), 71.5 (C-3), 76.7 (C-2), 171.2 (C(O)CH3); δ (l-erythro-20) −4.5 (CH3), −4.3 (CH3), 14.4 (C-6), 18.2 (tBu), 18.4 (C-5), 21.1 (C(O)CH3), 26.0 (tBu), 34.9 (C-4), 65.8 (C-1), 72.6 (C-2), 73.2 (C-3), 171.5 (C(O)CH3); δ (d-threo-20) −4.7 (CH3),-4.1 (CH3), 14.3 (C-6), 18.2 (tBu), 18.6 (C-5), 21.1 (C(O)CH3), 26.0 (tBu), 35.9 (C-4), 66.2 (C-1), 70.8 (C-2), 71.9 (C-3), 171.2 (C(O)CH3); HRMS (ESI) calcd for C14H30O4SiNa+ [M + Na]+: 313.1806, found: 313.1806.
3.8. (2R,3S)-2-O-Acetyl-4,5,6-trideoxy-3-O-tert-butyldimethylsilyl- l-erythro-hexose (l-erythro-10) and (2R,3R)-2-O-acetyl-4,5,6-trideoxy-3-O-tert-butyldimethylsilyl- d-threo-hexose (d-threo-10)
Oxalyl chloride (2.0 M in CH2Cl2, 0.85 mL, 1.70 mmol) was added dropwise to a solution of dimethyl sulfoxide (0.18 mL, 2.56 mmol) in dry CH2Cl2 (3.9 mL) at -78 °C. After 30 min of stirring at −78 °C, a solution of alcohol 17 (330 mg, 82:18 mixture with 20, 0.93 mmol of 17) in dry CH2Cl2 (1.9 mL) was added dropwise. The reaction mixture was stirred at −78 °C for 30 min and Et3N (0.63 mL, 4.54 mmol) was added. After 1 h of stirring at −78 °C, the reaction mixture was allowed to reach room temperature slowly (additional 1 h). Subsequently, the reaction mixture was concentrated, dry diethyl ether was added to the residue, and the mixture was filtered through a short silicagel column. The filtrate was concentrated to give the crude aldehyde 10 which was immediately used in the following step without further purification.
3.9. (3R,4S)-3-O-Acetyl-1-(2-acetoxymethyl-3-methoxyphenyl)-4-O-tert-butyldimethylsilyl-hept-1-ene-3,4- diol (18) and (3R,4R)-3-O-acetyl-1-(2-acetoxymethyl-3-methoxyphenyl)-4-O-tert-butyldimethylsilyl-hept-1-ene-3,4-diol (19)
Sulfone 11 (457 mg, 1.14 mmol) in dry DME (10.5 mL) was slowly added to a solution of the crude aldehyde 10 (theor. 0.93 mmol) in dry DME (10.5 mL) and the mixture was cooled to −60 °C. Subsequently, KHMDS (0.5 M in toluene, 3.97 mL, 1.99 mmol) was added dropwise keeping −60 °C and the reaction mixture was allowed to reach room temperature. The reaction mixture was stirred for 40 min, quenched with sat. aqueous NH4Cl (20 mL), and diluted with AcOEt (20 mL). The aqueous layer was extracted with AcOEt (3 × 20 mL) and the combined organic layers were dried and concentrated. The residue (18/19 70:30) was repeatedly purified by MPLC (gradient AcOEt/hexanes 0/100 to 5/95) and preparative TLC to afford 18 (79 mg, 18%), 19 (22 mg, 5%), and the mixture of 18 and 19 (78 mg, 18%) as colorless oils over two steps (41% overall yield).
1H NMR (600 MHz, CDCl3) δ (18) 0.06 (s, CH3, 3H), 0.10 (s, CH3, 3H), 0.90 (t, H-7, J = 7.0 Hz, 3H), 0.91 (s, tBu, 9H), 1.30-1.36 (m, H-6a, 1H), 1.39–1.48 (m, H-5, H-6b, 3H), 2.06 (s, C(O)CH3), 3H), 2.09 (s, C(O)CH3), 3H), 3.84 (s, OCH3, 3H), 3.85–3.88 (m, H-4, 1H), 5.25 (d, CH2OAc, J = 11.8 Hz, 1H), 5.27 (d, CH2OAc, J = 11.8 Hz, 1H), 5.33 (ddd, H-3, J = 1.0, 2.8, 7.6 Hz, 1H), 6.18 (dd, H-2, J = 7.6, 15.9 Hz, 1H), 6.84 (d, H-4′, J = 8.2 Hz, 1H), 6.85 (d, H-1, J = 15.9 Hz, 1H), 7.11 (d, H-6′, J = 7.8 Hz, 1H), 7.30 (t, H-5′, J = 8.1 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ (18) −4.4 (CH3), −4.2 (CH3), 14.3 (C-7), 18.4 (tBu), 18.9 (C-6), 21.1 (C(O)CH3), 21.5 (C(O)CH3), 26.0 (tBu), 36.3 (C-5), 56.0 (OCH3), 57.7 (CH2OAc), 73.8 (C-4), 77.9 (C-3), 110.3 (C-4′), 118.9 (C-6′), 121.5 (C-2′), 127.5 (C-2), 130.0 (C-5′), 131.2 (C-1), 139.0 (C-1′), 158.6 (C-3′), 170.2 (C(O)CH3), 171.2 (C(O)CH3); HRMS (ESI) calcd for C25H40O6SiNa+ [M + Na]+: 487.2486, found: 487.2486; [α] −48.8 (c 1.131 MeOH).
1H NMR (600 MHz, CDCl3) δ (19) 0.08 (s, CH3, 3H), 0.11 (s, CH3, 3H), 0.89 (t, H-7, J = 7.0 Hz, 3H), 0.90 (s, tBu, 9H), 1.30–1.37 (m, H-6a, 1H), 1.38-1.50 (m, H-5, H-6b, 3H), 2.06 (s, C(O)CH3), 3H), 2.11 (s, C(O)CH3), 3H), 3.80 (ddd, H-4, J = 3.6, 6.0, 7.2 Hz, 1H), 3.84 (s, OCH3, 3H), 5.26 (s, CH2OAc, 2H), 5.38 (ddd, H-3, J = 1.4, 6.1, 6.1 Hz, 1H), 6.11 (dd, H-2, J = 6.2, 15.9 Hz, 1H), 6.83 (d, H-4′, J = 8.3 Hz, 1H), 6.84 (dd, H-1, J = 1.3, 15.9 Hz, 1H), 7.09 (d, H-6′, J = 7.8 Hz, 1H), 7.29 (t, H-5′, J = 8.1 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ (19) −4.4 (CH3), −4.2 (CH3), 14.5 (C-7), 18.2 (tBu), 18.5 (C-6), 21.1 (C(O)CH3), 21.4 (C(O)CH3), 26.0 (tBu), 35.3 (C-5), 56.0 (OCH3), 57.7 (CH2OAc), 73.0 (C-4), 76.8 (C-3), 110.2 (C-4′), 118.8 (C-6′), 121.4 (C-2′), 128.1 (C-2), 129.5 (C-1), 130.0 (C-5′), 139.0 (C-1′), 158.6 (C-3′), 170.2 (C(O)CH3), 171.2 (C(O)CH3); HRMS (ESI) calcd for C25H40O6SiNa+ [M + Na]+: 487.2486, found: 487.2486; [α] +5.5 (c 0.431 MeOH).
3.10. Varioxiranol A (4)
Compound 18 (21.1 mg, 0.045 mmol) was dissolved in methanol (2 mL) and K2CO3 (12.6 mg, 0.091 mol) was added. The reaction mixture was stirred at room temperature for 2.5 h, diluted with AcOEt (4 mL) and with water (4 mL). The water phase was extracted with AcOEt (4 × 2 mL) and combined organic layers were dried and concentrated. The residue was diluted in THF (0.5 mL), the solution was cooled to 0 °C, and TBAF × 3H2O in THF (1.0 M solution, 46 µL, 0.046 mmol) was added. The reaction mixture was stirred at room temperature for 4.5 h and quenched with sat. aqueous NH4Cl (5 mL), the aqueous layer was extracted with CH2Cl2 (3 × 5 mL), and the combined organic layers were dried and concentrated. The residue was purified by FLC (isocratic acetone/CH2Cl2 20/80 then 50/50) to afford varioxiranol A (4, 11.1 mg, 92% over two steps) as colorless crystalline solid that was subsequently recrystallized from AcOEt-hexanes. 1H NMR (600 MHz, CDCl3) δ (4) 0.94 (t, H-7, J = 7.2 Hz, 3H), 1.36-1.43 (m, H-6a, 1H), 1.43–1.48 (m, H-5, 2H), 1.51–1.57 (m, H-6b, 1H), 2.09 (d, OH, J = 4.3 Hz, 1H), 2.20 (t, OH, J = 5.8 Hz, 1H), 2.22 (d, OH, J = 4.6 Hz, 1H), 3.78–3.82 (m, H-4, 1H), 3.87 (s, OCH3, 3H), 4.26–4.30 (m, H-3, 1H), 4.76–4.83 (m, CH2OH, 2H), 6.19 (dd, H-2, J = 6.9, 15.8 Hz, 1H), 6.84 (d, H-4′, J = 8.2 Hz, 1H), 7.02 (d, H-1, J = 15.8 Hz, 1H), 7.10 (d, H-6′, J = 7.9 Hz, 1H), 7.25 (t, H-5′, J = 8.2 Hz, 1H); 13C NMR (150 MHz, acetone-d6) δ (4) 14.5 (C-7), 19.8 (C-6), 35.7 (C-5), 55.5 (CH2OH), 56.1 (OCH3), 75.0 (C-4), 76.7 (C-3), 110.4 (C-4′), 119.4 (C-6′), 127.9 (C-2′), 129.2 (C-1), 129.3 (C-5′), 133.7 (C-2), 139.6 (C-1′), 158.9 (C-3′); HRMS (ESI) calcd for C15H22O4Na+ [M+Na]+: 289.1410, found: 289.1410; [α] +2.5 (c 0.207 MeOH); mp 120–121 °C.
3.11. 4-epi-Varioxiranol A (9)
Compound 19 (20.8 mg, 0.045 mmol) was dissolved in methanol (2 mL) and K2CO3 (12.4 mg, 0.090 mmol) was added. The reaction mixture was stirred at room temperature for 2.5 h and diluted with AcOEt (4 mL) and with water (4 mL). The water phase was extracted with AcOEt (4 × 2 mL) and combined organic layers were dried and concentrated. The residue was diluted in THF (0.5 mL), the solution was cooled to 0 °C, and TBAF × 3H2O in THF (1.0 M solution, 45 µL, 0.045 mmol) was added. The reaction mixture was stirred at room temperature for 4.5 h and quenched with sat. aqueous NH4Cl (5 mL). The aqueous layer was extracted with CH2Cl2 (3 × 5 mL) and the combined organic layers were dried and concentrated. The residue was purified by FLC (isocratic acetone/CH2Cl2 30/70) to afford 4-epi-varioxiranol A (9, 6.8 mg, 57% over two steps) that was subsequently recrystallized from CH2Cl2-hexanes yielding colorless crystalline solid. 1H NMR (600 MHz, CDCl3) δ (9) 0.94 (t, H-7, J = 7.1 Hz, 3H), 1.39–1.45 (m, H-6a, 1H), 1.46–1.56 (m, H-5, H-6b, 3H), 2.20 (t, OH, J = 6.3 Hz, 1H), 2.28 (d, OH, J = 4.3 Hz, 1H), 2.40 (d, OH, J = 4.3 Hz, 1H), 3.56–3.61 (m, H-4, 1H), 3.87 (s, OCH3, 3H), 4.10–4.14 (m, H-3, 1H), 4.78 (dd, CH2OH, J = 6.3, 12.1 Hz, 1H), 4.81 (dd, CH2OH, J = 6.3, 12.1 Hz, 1H), 6.12 (dd, H-2, J = 6.8, 15.8 Hz, 1H), 6.84 (d, H-4′, J = 8.2 Hz, 1H), 7.04 (d, H-1, J = 15.8 Hz, 1H), 7.08 (d, H-6′, J = 7.8 Hz, 1H), 7.25 (t, H-5′, J = 8.0 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ (9) 14.2 (C-7), 19.0 (C-6), 35.4 (C-5), 55.8 (OCH3), 56.6 (CH2OH), 74.5 (C-4), 76.3 (C-3), 110.0 (C-4′), 119.4 (C-6′), 126.4 (C-2′), 129.1 (C-5′), 129.8 (C-1), 132.6 (C-2), 137.7 (C-1′), 158.2 (C-3′); HRMS (ESI) calcd for C15H22O4Na+ [M + Na]+: 289.1410, found: 289.1410; [α] +45.0 (c 0.094 MeOH); mp 112–113 °C.
Supplementary Materials
The following are available online, S1: Copies of 1H NMR and 13C NMR spectra for all new compounds.
Author Contributions
Conceptualization, A.L. and T.G.; synthesis, M.Š. and M.P.; data curation and NMR analysis, J.D.; X-ray analysis, J.M.; writing—original draft preparation, A.L. and T.G.
Funding
This research received no external funding.
Acknowledgments
This work was supported by Slovak Grant Agencies (VEGA No. 1/0552/18, APVV-14-0147 and ASFU, Bratislava, ITMS projects No. 26240120025 and 26240220084).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zadar, P.; Frisvad, J.C.; Gunde-Cimerman, N.; Varga, J.; Samson, R.A. Four new species of Emericella from the Mediterranean region of Europe. Mycologia 2008, 100, 779–795. [Google Scholar] [CrossRef]
- Dunn, A.W.; Johnstone, R.A.W. Fungal metabolites. Part 7. Structures of C25 compounds from Aspergillus variecolor. J. Chem. Soc. Perkin Trans. 1 1979, 2113–2117. [Google Scholar] [CrossRef]
- Dunn, A.W.; Johnstone, R.A.W. Fungal metabolites. Part 8. Isolation of 2-methoxy-6-(3,4-dihydroxyhepta-1,5-dienyl)benzyl alcohol. J. Chem. Soc. Perkin Trans. 1 1979, 2122–2123. [Google Scholar] [CrossRef]
- Markovič, M.; Lopatka, P.; Koóš, P.; Gracza, T. First total synthesis of natural andytriol and a biomimetic approach to varioxiranes. Tetrahedron 2015, 71, 8407–8415. [Google Scholar] [CrossRef]
- Malmstrøm, J.; Christophersen, C.; Barrero, A.F.; Oltra, J.E.; Justicia, J.; Rosales, A. Bioactive Metabolites from a Marine-Derived Strain of the Fungus Emericella variecolor. J. Nat. Prod. 2002, 65, 364–367. [Google Scholar] [CrossRef]
- Wu, Q.; Wu, C.; Long, H.; Chen, R.; Liu, D.; Proksch, P.; Guo, P.; Lin, W. Varioxiranols A–G and 19-O-Methyl-22-methoxypre-shamixanthone, PKS and Hybrid PKS-Derived Metabolites from a Sponge-Associated Emericella variecolor Fungus. J. Nat. Prod. 2015, 78, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Liangsakul, J.; Pornpakakul, S.; Sangvichien, E.; Muangsin, N.; Sihanonth, P. Emervaridione and varioxiranediol, two new metabolites from the endophytic fungus, Emericella variecolor. Tetrahedron Lett. 2011, 52, 6427–6430. [Google Scholar] [CrossRef]
- He, Y.; Hu, Z.; Li, Q.; Huang, J.; Li, X.-N.; Zhu, H.; Liu, J.; Wang, J.; Xue, Y.; Zhang, Y. Bioassay-Guided Isolation of Antibacterial Metabolites from Emericella sp. TJ29. J. Nat. Prod. 2017, 80, 2399–2405. [Google Scholar] [CrossRef] [PubMed]
- Palík, M.; Karlubíková, O.; Lásiková, A.; Kožíšek, J.; Gracza, T. Total Synthesis of (+)-Varitriol. Eur. J. Org. Chem. 2009, 709–715. [Google Scholar] [CrossRef]
- Palík, M.; Karlubíková, O.; Lackovičová, D.; Lásiková, A.; Gracza, T. Formal synthesis of (+)-varitriol. Application of Pd(II)/Cu(II)-catalysed bicyclisation of unsaturated polyols. Tetrahedron 2010, 66, 5244–5249. [Google Scholar] [CrossRef]
- Karlubíková, O.; Palík, M.; Lásiková, A.; Gracza, T. An Efficient Total Synthesis of (+)-Varitriol from D-Ribonolactone. Synthesis 2010, 3449–3452. [Google Scholar] [CrossRef]
- Caletková, O.; Lásiková, A.; Hajdúch, M.; Džubák, P.; Gracza, T. Synthesis and antitumour activity of varitriol and its analogues. ARKIVOC 2012, 365–383. [Google Scholar] [CrossRef]
- Antošová, A.; Šípošová, K.; Bednáriková, Z.; Lásiková, A.; Dohánošová, J.; Gracza, T.; Gažová, Z. Natural tetrahydrofuran derivatives reduce insulin amyloid aggregation. Eur. Biophys. J. 2013, 42, S62. [Google Scholar] [CrossRef]
- Schmid, C.R.; Bryant, J.D. d-(R)-Glyceraldehyde acetonide. Org. Synth. 1995, 72, 6–8. [Google Scholar] [CrossRef]
- de Napoli, L.; Messere, A.; Palomba, D.; Piccialli, V.; Evidente, A.; Gennaro Piccialli, G. Studies toward the Synthesis of Pinolidoxin, a Phytotoxic Nonenolide from the Fungus Ascochyta pinodes. Determination of the Configuration at the C-7, C-8, and C-9 Chiral Centers and Stereoselective Synthesis of the C6–C18 Fragment. J. Org. Chem. 2000, 65, 3432–3442. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.N.; Micalizio, G.C.; Roush, W.R. Enantio- and Diastereoselective Synthesis of Cyclic β-Hydroxy Allylsilanes via Sequential Aldehyde γ-Silylallylboration and Ring-Closing Metathesis Reactions. Org. Lett. 2003, 5, 1693–1696. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Betteridge, P.W.; Carruthers, J.R.; Copper, R.I.; Prout, K.; Watkin, D.J. CRYSTALS version 12: Software for guided crystal structure analysis. J. Appl. Crystallogr. 2003, 36, 1487. [Google Scholar] [CrossRef]
- Dolomanov, O.; Bourhis, L.J.; Gildea, R.I.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Parsons, S.; Flack, H.D.; Wagner, T. Use of intensity quotients and differences in absolute structure refinement. Acta Crystallogr. 2013, B69, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Hooft, R.W.W.; Straver, L.H.; Spek, A.L. Determination of absolute structure using Bayesian statistics on Bijvoet differences. J. Appl. Crystallogr. 2008, 41, 96–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the compounds are available from the authors. |
Figure 1.
Natural compounds isolated from a strain of Emericella variecolor 1–8.

Scheme 1.
Retrosynthetic analysis of 4 and 9.

Scheme 2.
Synthesis of natural varioxiranol A 4 and 4-epi-varioxiranol A 9. Reagents and conditions: (a) prop-1-ylmagnesium chloride, THF, Et2O, r.t., 1 h; (b) TBSCl, imidazole, CH2Cl2, 0 °C to r.t., 23 h; (c) TFA (50%), CH2Cl2, r.t., 1 h; (d) TrCl, Et3N, DMAP, CH2Cl2, 0 °C to r.t., 15 h; (e) Ac2O, DMAP, CH2Cl2, r.t., 30 min; (f) HCOOH/Et2O (1/1), r.t., 50 min; (g) DMSO, (COCl)2, Et3N, CH2Cl2, −78 °C to r.t., 2.5 h; (h) KHMDS, dimethoxyethane, sulfone 11, CH2Cl2, −60 °C to r.t., 40 min; (i) K2CO3, MeOH, r.t. 2.5 h; (j) TBAF × 3H2O, THF, 0 °C to r.t., 4.5 h.
Scheme 2.
Synthesis of natural varioxiranol A 4 and 4-epi-varioxiranol A 9. Reagents and conditions: (a) prop-1-ylmagnesium chloride, THF, Et2O, r.t., 1 h; (b) TBSCl, imidazole, CH2Cl2, 0 °C to r.t., 23 h; (c) TFA (50%), CH2Cl2, r.t., 1 h; (d) TrCl, Et3N, DMAP, CH2Cl2, 0 °C to r.t., 15 h; (e) Ac2O, DMAP, CH2Cl2, r.t., 30 min; (f) HCOOH/Et2O (1/1), r.t., 50 min; (g) DMSO, (COCl)2, Et3N, CH2Cl2, −78 °C to r.t., 2.5 h; (h) KHMDS, dimethoxyethane, sulfone 11, CH2Cl2, −60 °C to r.t., 40 min; (i) K2CO3, MeOH, r.t. 2.5 h; (j) TBAF × 3H2O, THF, 0 °C to r.t., 4.5 h.

Figure 2.
A ball-and-stick view of crystal structures 4 and 9.


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lásiková, A.; Doháňošová, J.; Štiblariková, M.; Parák, M.; Moncol, J.; Gracza, T. First Total Synthesis of Varioxiranol A. Molecules 2019, 24, 862. https://doi.org/10.3390/molecules24050862
AMA Style
Lásiková A, Doháňošová J, Štiblariková M, Parák M, Moncol J, Gracza T. First Total Synthesis of Varioxiranol A. Molecules. 2019; 24(5):862. https://doi.org/10.3390/molecules24050862
Chicago/Turabian StyleLásiková, Angelika, Jana Doháňošová, Mária Štiblariková, Martin Parák, Ján Moncol, and Tibor Gracza. 2019. "First Total Synthesis of Varioxiranol A" Molecules 24, no. 5: 862. https://doi.org/10.3390/molecules24050862