3.2. Chemistry
1,8-Dihydroxy-3-(2-hydroxyethoxy)-6-methylanthracene-9,10-dione (2) To a mixture of emodin (10.0 g, 37.0 mmol) in dry DMF (150 mL) were added Cs2CO3 (13.2 g, 40.5 mmol) and 2-iodoethanol (19.1 g, 111 mmol) at room temperature. After stirring for 36 h at 60 °C, the resulting mixture was evaporated under reduced pressure and then mixed with water (500 mL). The pH value of aqueous phase was adjusted to around 5 with 10% hydrochloric acid solution. The yellow precipitate was collected and washed with water to give the crude product, which was in further purification by triturating twice with ethyl acetate (100 mL) and filtered to afford compound 2 (7.6 g, 65%) as a brown solid; 1H-NMR δ 12.17 (s, 1H), 11.99 (s, 1H), 7.54 (d, J = 1.6 Hz, 1H), 7.21 (d, J = 2.6 Hz, 2H), 6.89 (d, J = 2.5 Hz, 1H), 4.98 (t, J = 5.5 Hz, 1H), 4.26–4.15 (m, 2H), 3.76 (q, J = 5.2 Hz, 2H), 2.44 (s, 3H); 13C-NMR δ 189.75, 180.93, 165.58, 164.30, 161.41, 148.42, 134.61, 132.66, 124.11, 120.48, 113.26, 109.60, 107.95, 106.89, 70.76, 59.20, 21.49; ESI, m/z: 315.05 [M + H]+.
General procedure A for preparation of compounds 3a–3z, 4a–4b, and 7a–7l
To a mixture of compound 2 (1.00 mmol) in dry dichloromethane (20 mL) were added various N-Boc amino acids (1.20 mmol), dicyclohexyl carbodiimide (DCC) (4.50 mmol) and 4-(N,N-dimethlyamino)pyridine (DMAP) (1.00 mmol) at 0 °C. After stirring about 30 min to 5 h at 0 °C, TLC analysis showed the complete consumption of compound 2, and then the resulting mixture was added dropwise TFA (5 mL) at the same temperature and kept stirring for anther about 2 h. The insoluble side product was filtered out and the filtrate was evaporated to give the residue, which was purified by reverse phase flash chromatography with the following conditions: Column: Spherical C18, 20–40 μm, 330 g; Mobile Phase A: Water (plus 5 mM TFA); Mobile Phase B: ACN; Flow rate: 80 mL/min; Gradient: 5% B gradient in 10 min, 25% B–45% B gradient in 25 min; Detector: 254 nm. The fractions containing the desired product were collected at around 40% B and concentrated under reduced pressure to afford compounds 3a–3z in 20% to 50% yields.
2-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-2-oxoethanaminium 2,2,2-trifluoroacetate (3a). According to the general procedure A, compound 2 was treated with N-Boc-glycine and then purified by reverse phase flash chromatography to give compound 3a: Yellow solid; yield, 45%; 1H-NMR δ 9.76 (brs, 3H), 7.50 (s, 1H), 7.19 (s, 1H), 7.16 (s, 1H), 6.90 (s, 1H), 4.56 (d, J = 4.9 Hz, 2H), 4.44 (d, J = 5.1 Hz, 2H), 3.90 (s, 2H), 2.42 (s, 3H); 13C-NMR δ 189.89, 181.04, 167.77, 164.74, 164.25, 161.46, 148.58, 134.82, 132.73, 124.23, 120.57, 113.38, 110.06, 107.80, 107.08, 66.71, 63.44, 21.50; 19F-NMR δ − 73.56; ESI, m/z: 372.1 [M + H − CF3COOH]+.
(R)-1-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-1-oxopropan-2-aminium 2,2,2-trifluoroacetate (3b). According to the general procedure A, compound 2 was treated with N-Boc-d-alanine and then purified by reverse phase flash chromatography to give compound 3b: Yellow solid; yield, 50%; 1H-NMR δ 9.50 (brs, 3H), 7.44 (d, J = 1.7 Hz, 1H), 7.14 (s, 1H), 7.11 (d, J = 2.5 Hz, 1H), 6.86 (d, J = 2.5 Hz, 1H), 4.65–4.50 (m, 2H), 4.43 (t, J = 4.5 Hz, 2H), 4.18 (q, J = 7.2 Hz, 1H), 2.40 (s, 3H), 1.44 (d, J = 7.2 Hz, 3H); 13C-NMR δ 189.78, 180.88, 170.03, 164.76, 164.24, 161.44, 148.53, 134.71, 132.62, 124.17, 120.52, 113.26, 109.96, 107.76, 107.04, 66.66, 63.58, 47.87, 21.49, 15.72; 19F-NMR δ − 73.53; ESI, m/z: 386.15 [M + H − CF3COOH]+.
(S)-1-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-1-oxopropan-2-aminium 2,2,2-trifluoroacetate (3c). According to the general procedure A, compound 2 was treated with N-Boc-l-alanine and then purified by reverse phase flash chromatography to give compound 3c: Yellow solid; yield, 48%; 1H-NMR δ 9.85 (brs, 3H), 7.47 (d, J = 1.7 Hz, 1H), 7.16 (s, 1H), 7.14 (d, J = 2.5 Hz, 1H), 6.88 (d, J = 2.5 Hz, 1H), 4.64–4.53 (m, 2H), 4.45 (d, J = 4.3 Hz, 2H), 4.17 (q, J = 7.2 Hz, 1H), 2.41 (s, 3H), 1.43 (d, J = 7.1 Hz, 3H); 13C-NMR δ 189.82, 180.95, 170.13, 164.77, 164.25, 161.45, 148.54, 134.76, 132.67, 124.19, 120.54, 113.31, 110.00, 107.77, 107.07, 66.67, 63.56, 47.88, 21.49, 15.80; 19F-NMR δ − 73.54; ESI, m/z: 386.15 [M + H − CF3COOH]+.
(S)-1-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-3-hydroxy-1-oxopropan-2-aminium 2,2,2-trifluoroacetate (3d). According to the general procedure A, compound 2 was treated with N-Boc-O-tert-butyl-l-serine and then purified by reverse phase flash chromatography to give compound 3d: Yellow solid; yield, 20%; 1H-NMR δ 7.51 (s, 1H), 7.18 (s, 1H), 7.17 (d, J = 2.5 Hz, 1H), 6.90 (d, J = 2.5 Hz, 1H), 5.53 (s, 1H), 4.52 (d, J = 3.6 Hz, 2H), 4.42 (d, J = 3.3 Hz, 2H), 4.16 (s, 1H), 3.83–3.73 (m, 2H), 2.40 (s, 3H); 13C-NMR δ 189.93, 181.12, 168.16, 164.81, 164.25, 161.46, 148.57, 134.85, 132.77, 124.23, 120.57, 113.41, 110.07, 107.85, 107.11, 66.69, 63.63, 59.53, 54.25, 21.49; 19F-NMR δ − 73.48; ESI, m/z: 402.05 [M + H − CF3COOH]+.
(R)-1-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-3-methyl-1-oxobutan-2-aminium 2,2,2-trifluoroacetate (3e). According to the general procedure A, compound 2 was treated with N-Boc-d-valine and then purified by reverse phase flash chromatography to give compound 3e: Yellow solid; yield, 46%; 1H-NMR δ 8.90 (brs, 2H), 7.49 (s, 1H), 7.18 (s, 1H), 7.15 (d, J = 2.5 Hz, 1H), 6.90 (d, J = 2.5 Hz, 1H), 4.67 (d, J = 12.7 Hz, 1H), 4.60–4.39 (m, 3H), 4.00 (d, J = 4.5 Hz, 1H), 2.42 (s, 3H), 2.18 (dd, J = 12.4, 6.5 Hz, 1H), 0.99 (dd, J = 14.8, 6.9 Hz, 6H); 13C-NMR δ 189.89, 181.04, 168.97, 164.68, 164.26, 161.46, 148.57, 134.85, 132.73, 124.23, 120.57, 113.37, 110.09, 107.74, 107.05, 66.65, 63.45, 57.24, 29.44, 21.50, 18.13, 17.37; 19F-NMR δ − 73.55; ESI, m/z: 414.15 [M + H − CF3COOH]+.
(S)-1-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-3-methyl-1-oxobutan-2-aminium 2,2,2-trifluoroacetate (3f). According to the general procedure A, compound 2 was treated with N-Boc-l-valine and then purified by reverse phase flash chromatography to give compound 3f: Yellow solid; yield, 48%; 1H-NMR δ 8.84 (brs, 2H), 7.48 (d, J = 1.6 Hz, 1H), 7.17 (s, 1H), 7.14 (d, J = 2.5 Hz, 1H), 6.89 (d, J = 2.6 Hz, 1H), 4.74–4.62 (m, 1H), 4.57–4.39 (m, 3H), 4.00 (d, J = 4.5 Hz, 1H), 2.42 (s, 3H), 2.22–2.14 (m, 1H), 0.99 (dd, J = 15.0, 6.9 Hz, 6H); 13C-NMR δ 189.87, 181.00, 168.95, 164.67, 164.26, 161.46, 148.56, 134.82, 132.71, 124.22, 120.56, 113.35, 110.07, 107.74, 107.04, 66.65, 63.45, 57.23, 29.43, 21.50, 18.13, 17.37; 19F-NMR δ − 73.54; ESI, m/z: 414.15 [M + H − CF3COOH]+.
(S)-1-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-4-methyl-1-oxopentan-2-aminium 2,2,2-trifluoroacetate (3g). According to the general procedure A, compound 2 was treated with N-Boc-l-leucine and then purified by reverse phase flash chromatography to give compound 3g: Yellow solid; yield, 42%; 1H-NMR δ 9.43 (brs, 3H), 7.48 (d, J = 1.7 Hz, 1H), 7.17 (s, 1H), 7.14 (d, J = 2.5 Hz, 1H), 6.89 (d, J = 2.5 Hz, 1H), 4.68–4.58 (m, 1H), 4.56–4.42 (m, 3H), 4.05 (t, J = 7.1 Hz, 1H), 2.42 (s, 3H), 1.81–1.54 (m, 3H), 0.88 (d, J = 2.2 Hz, 3H), 0.87 (d, J = 2.3 Hz, 3H); 13C-NMR δ 189.84, 180.97, 170.02, 164.77, 164.27, 161.46, 148.57, 134.78, 132.68, 124.22, 120.57, 113.33, 110.02, 107.82, 107.03, 66.59, 63.59, 50.56, 23.68, 22.06, 21.85, 21.50; 19F-NMR δ − 73.51; ESI, m/z: 428.1 [M + H − CF3COOH]+.
(2S)-1-(2-(4,5-dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-3-methyl-1-oxo-pentan-2-aminium 2,2,2-trifluoroacetate (3h). According to the general procedure A, compound 2 was treated with N-Boc-l-isoleucine and then purified by reverse phase flash chromatography to give compound 3h: Yellow solid; yield, 42%; 1H-NMR δ 7.52 (s, 1H), 7.21 (s, 1H), 7.17 (d, J = 2.5 Hz, 1H), 6.92 (d, J = 2.5 Hz, 1H), 4.73–4.63 (m, 1H), 4.56–4.41 (m, 3H), 4.06 (d, J = 3.9 Hz, 1H), 2.44 (s, 3H), 1.89 (s, 1H), 1.47 (m, 1H), 1.35–1.24 (m, 1H), 0.93 (d, J = 6.9 Hz, 3H), 0.87 (t, J = 7.3 Hz, 3H); 13C-NMR δ 189.93, 181.10, 168.86, 164.69, 164.28, 161.47, 148.59, 134.88, 132.77, 124.26, 120.59, 113.42, 110.11, 107.79, 107.05, 66.62, 63.43, 56.13, 36.05, 25.09, 21.50, 14.06, 11.44; 19F-NMR δ − 73.50; ESI, m/z: 428.1 [M + H − CF3COOH]+.
(S)-1-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-4-(methylthio)-1-oxobutan-2-aminium 2,2,2-trifluoroacetate (3i). According to the general procedure A, compound 2 was treated with N-Boc-l-methionine and then purified by reverse phase flash chromatography to give compound 3i: Yellow solid; yield, 38%; 1H-NMR δ 7.51 (d, J = 1.7 Hz, 1H), 7.20 (s, 1H), 7.18 (d, J = 2.5 Hz, 1H), 6.91 (d, J = 2.6 Hz, 1H), 4.63 (d, J = 12.5 Hz, 1H), 4.58–4.37 (m, 3H), 4.18 (t, J = 6.2 Hz, 1H), 2.67–2.52 (m, 2H), 2.42 (s, 3H), 2.05 (dd, J = 8.2, 5.9 Hz, 2H), 2.00 (s, 3H); 13C-NMR δ 189.95, 181.10, 169.41, 164.76, 164.28, 161.48, 148.62, 134.89, 132.78, 124.26, 120.61, 113.42, 110.13, 107.79, 107.11, 66.63, 63.71, 50.99, 29.61, 28.21, 21.53, 14.20; 19F-NMR δ − 73.47; ESI, m/z: 446.1 [M + H − CF3COOH]+.
1-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-2-methyl-1-oxopropan-2-aminium 2,2,2-trifluoroacetate (3j). According to the general procedure A, compound 2 was treated with N-Boc-Aib-OH and then purified by reverse phase flash chromatography to give compound 3j: Yellow solid; yield, 38%; 1H-NMR δ 11.96 (br, 2H), 8.51 (br, 3H), 7.54 (s, 1H), 7.34–7.14 (m, 2H), 6.94 (d, J = 2.5 Hz, 1H), 4.65–4.54 (m, 2H), 4.47 (t, J = 4.3 Hz, 2H), 2.44 (s, 3H), 1.47 (s, 6H); 13C-NMR δ 189.33, 180.18, 171.67, 164.71, 164.21, 161.37, 148.37, 134.23, 132.13, 123.96, 120.35, 112.78, 109.52, 107.66, 106.79, 66.61, 63.79, 55.98, 23.37, 21.47; 19F-NMR δ − 73.54; ESI, m/z: 400.2 [M + H − CF3COOH]+.
1-((2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)carbonyl)cyclopropan-aminium 2,2,2-trifluoroacetate (3k). According to the general procedure A, compound 2 was treated with 1-(Boc-amino)cyclopropanecarboxylic acid and then purified by reverse phase flash chromatography to give compound 3k: Yellow solid; yield, 28%; 1H-NMR δ 11.96 (brs, 1H), 8.87 (brs, 2H), 7.52 (d, J = 1.7 Hz, 1H), 7.24–7.10 (m, 2H), 6.92 (d, J = 2.6 Hz, 1H), 4.53 (dd, J = 5.9, 2.7 Hz, 2H), 4.43 (dd, J = 5.8, 2.8 Hz, 2H), 2.43 (s, 3H), 1.50–1.41 (m, 2H), 1.38–1.30 (m, 2H); 13C-NMR δ 189.28, 180.10, 169.83, 164.69, 164.22, 161.39, 148.39, 134.15, 132.07, 123.98, 120.37, 112.73, 109.54, 107.67, 106.74, 66.57, 63.78, 33.87, 21.51, 13.69; 19F-NMR δ − 73.55; ESI, m/z: 398.2 [M + H − CF3COOH]+.
1-((2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)carbonyl)cyclobutan-aminium 2,2,2-trifluoroacetate (3l). According to the general procedure A, compound 2 was treated with 1-(Boc-amino)cyclobutanecarboxylic acid and then purified by reverse phase flash chromatography to give compound 3l: Yellow solid; yield, 28%; 1H-NMR δ 11.95 (brs, 1H), 8.73 (brs, 2H), 7.52 (d, J = 1.6 Hz, 1H), 7.30–7.09 (m, 2H), 6.95 (d, J = 2.6 Hz, 1H), 4.74–4.58 (m, 2H), 4.51 (t, J = 4.3 Hz, 2H), 2.54 (m, 2H), 2.43 (s, 3H), 2.37 (m, , 2H), 2.03 (m, 2H); 13C-NMR δ 189.10, 179.79, 170.89, 164.71, 164.22, 161.36, 148.28, 134.00, 131.88, 123.85, 120.24, 112.54, 109.30, 107.63, 106.62, 66.66, 63.68, 56.86, 29.63, 21.42, 14.41; 19F-NMR δ − 73.51; ESI, m/z: 412.2 [M + H − CF3COOH]+.
3-((2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)carbonyl)oxetan-3-aminium 2,2,2-trifluoroacetate (3m). According to the general procedure A, compound 2 was treated with 3-Boc-amino-3-oxetanecarboxylic acid and then purified by reverse phase flash chromatography to give compound 3m: Yellow solid; yield, 37%; 1H-NMR δ 11.97 (brs, 2H), 8.76 (brs, 3H), 7.55 (d, J = 1.7 Hz, 1H), 7.23 (d, J = 2.3 Hz, 2H), 6.97 (d, J = 2.5 Hz, 1H), 4.83 (d, J = 7.6 Hz, 2H), 4.65 (dd, J = 9.7, 6.2 Hz, 4H), 4.53 (dd, J = 5.3, 2.9 Hz, 2H), 2.44 (s, 3H); 13C-NMR δ 189.35, 180.21, 168.11, 164.71, 164.22, 161.38, 148.43, 134.24, 132.14, 124.01, 120.40, 112.80, 109.54, 107.69, 106.82, 75.75, 66.58, 64.24, 57.15, 21.48; 19F-NMR δ − 73.51; ESI, m/z: 414.1 [M + H − CF3COOH]+.
1-((2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)carbonyl)cyclopentan-aminium 2,2,2-trifluoroacetate (3n). According to the general procedure A, compound 2 was treated with N-Boc-aminocyclopentanecarboxylic acid and then purified by reverse phase flash chromatography to give compound 3n: Yellow solid; yield, 31%; 1H-NMR δ 11.98 (brs, 2H), 8.47 (brs, 3H), 7.55 (d, J = 1.6 Hz, 1H), 7.30–7.12 (m, 2H), 6.96 (d, J = 2.5 Hz, 1H), 4.58 (dd, J = 6.0, 2.8 Hz, 2H), 4.48 (t, J = 4.2 Hz, 2H), 2.45 (s, 3H), 2.18 (dd, J = 14.0, 6.0 Hz, 2H), 1.89–1.69 (m, 6H); 13C-NMR δ 189.38, 180.26, 172.16, 164.74, 164.23, 161.39, 148.39, 134.31, 132.19, 123.99, 120.36, 112.84, 109.58, 107.69, 106.81, 66.63, 64.33, 63.73, 36.31, 25.09, 21.46; 19F-NMR δ − 73.49; ESI, m/z: 425.1 [M + H − CF3COOH]+.
(R)-2-((2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)carbonyl)-pyrrolidinium 2,2,2-trifluoroacetate (3o). According to the general procedure A, compound 2 was treated with N-Boc-d-proline and then purified by reverse phase flash chromatography to give compound 3o: Yellow solid; yield, 44%; 1H-NMR δ 7.52 (d, J = 1.7 Hz, 1H), 7.20 (s, 1H), 7.18 (d, J = 2.6 Hz, 1H), 6.92 (d, J = 2.6 Hz, 1H), 4.64–4.53 (m, 2H), 4.51–4.42 (m, 3H), 3.27–3.16 (m, 2H), 2.43 (s, 3H), 2.36–2.19 (m, 1H), 2.07–1.86 (m, 3H); 13C-NMR δ 189.96, 181.16, 168.92, 164.80, 164.28, 161.49, 148.62, 134.91, 132.80, 124.30, 120.62, 113.44, 110.14, 107.78, 107.17, 66.67, 63.87, 58.63, 45.59, 27.79, 23.00, 21.52; 19F-NMR δ − 73.51; ESI, m/z: 412.1 [M + H − CF3COOH]+.
(S)-2-((2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)carbonyl)-pyrrolidinium 2,2,2-trifluoroacetate (3p). According to the general procedure A, compound 2 was treated with N-Boc-l-proline and then purified by reverse phase flash chromatography to give compound 3p: Yellow solid; yield, 37%; 1H-NMR δ 7.50 (d, J = 1.7 Hz, 1H), 7.19 (t, J = 1.3 Hz, 1H), 7.17 (d, J = 2.6 Hz, 1H), 6.91 (d, J = 2.5 Hz, 1H), 4.67–4.54 (m, 2H), 4.52–4.38 (m, 3H), 3.30–3.16 (m, 2H), 2.42 (s, 3H), 2.32–2.25 (m, 1H), 2.09–1.82 (m, 3H); 13C-NMR δ 189.93, 181.09, 168.91, 164.79, 164.27, 161.48, 148.61, 134.87, 132.77, 124.26, 120.60, 113.41, 110.11, 107.77, 107.15, 66.67, 63.86, 58.62, 45.58, 27.80, 23.00, 21.52; 19F-NMR δ − 73.50; ESI, m/z: 412.1 [M + H − CF3COOH]+.
(R)-2-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-2-oxo-1-phenyl-ethanaminium 2,2,2-trifluoroacetate (3q). According to the general procedure A, compound 2 was treated with N-Boc-d-phenylglycine and then purified by reverse phase flash chromatography to give compound 3q: Yellow solid; yield, 39%; 1H-NMR δ 11.97 (brs, 2H), 8.89 (brs, 3H), 7.56 (d, J = 1.7 Hz, 1H), 7.49 (dd, J = 6.8, 3.0 Hz, 2H), 7.41 (dd, J = 5.1, 2.1 Hz, 3H), 7.23 (s, 1H), 7.08 (d, J = 2.5 Hz, 1H), 6.82 (d, J = 2.5 Hz, 1H), 5.38 (s, 1H), 4.58 (q, J = 5.0 Hz, 2H), 4.38–4.31 (m, 2H), 2.44 (s, 3H); 13C-NMR δ 189.06, 179.81, 168.64, 164.44, 164.18, 161.33, 148.26, 133.91, 132.65, 131.90, 129.70, 129.09, 128.48, 123.79, 120.33, 112.54, 109.26, 107.69, 106.46, 66.40, 63.82, 55.81, 21.41; 19F-NMR δ − 73.46; ESI, m/z: 448.1 [M + H − CF3COOH]+.
(S)-2-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-2-oxo-1-phenyl-ethanaminium 2,2,2-trifluoroacetate (3r). According to the general procedure A, compound 2 was treated with N-Boc-l-phenylglycine and then purified by reverse phase flash chromatography to give compound 3r: Yellow solid; yield, 45%; 1H-NMR δ 11.95 (brs, 1H), 8.84 (br, 2H), 7.56 (d, J = 1.7 Hz, 1H), 7.49 (dd, J = 6.8, 3.0 Hz, 2H), 7.41 (dd, J = 5.1, 2.1 Hz, 3H), 7.23 (s, 1H), 7.08 (d, J = 2.5 Hz, 1H), 6.82 (d, J = 2.5 Hz, 1H), 5.37 (s, 1H), 4.58 (q, J = 5.0 Hz, 2H), 4.38–4.31 (m, 2H), 2.44 (s, 3H); 13C-NMR δ 189.90, 181.06, 168.50, 164.62, 164.23, 161.47, 148.57, 134.73, 132.76, 132.41, 129.51, 128.89, 128.15, 124.22, 120.58, 113.39, 110.01, 107.93, 106.94, 66.50, 63.82, 55.41, 21.50; 19F-NMR δ − 73.45; ESI, m/z: 448.1 [M + H − CF3COOH]+.
(R)-2-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-1-(4-hydroxyphenyl)-2-oxoethanaminium 2,2,2-trifluoroacetate (3s). According to the general procedure A, compound 2 was treated with N-Boc-d-4-hydroxyphenylglycine and then purified by reverse phase flash chromatography to give compound 3s: Yellow solid; yield, 42%; 1H-NMR δ 11.99 (brs, 2H), 9.73 (s, 1H), 8.66 (brs, 3H), 7.57 (s, 1H), 7.26 (d, J = 7.8 Hz, 3H), 7.13 (s, 1H), 6.85 (s, 1H), 6.72 (d, J = 8.1 Hz, 2H), 5.22 (s, 1H), 4.57 (s, 2H), 4.35 (s, 2H), 2.46 (s, 3H); 13C-NMR δ 189.65, 180.61, 168.86, 164.62, 164.25, 161.45, 158.62, 148.47, 134.46, 132.48, 129.67, 124.09, 122.44, 120.50, 115.64, 113.12, 109.76, 107.81, 106.79, 66.57, 63.71, 55.11, 21.51; 19F-NMR δ − 73.43; ESI, m/z: 464.2 [M + H − CF3COOH]+.
(S)-2-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-1-(4-hydroxyphenyl)-2-oxoethanaminium 2,2,2-trifluoroacetate (3t). According to the general procedure A, compound 2 was treated with N-Boc-l-4-hydroxyphenylglycine and then purified by reverse phase flash chromatography to give compound 3t: Yellow solid; yield, 40%; 1H-NMR δ 11.96 (brs, 1H), 9.73 (s, 1H), 8.73 (brs, 2H), 7.57 (s, 1H), 7.25 (d, J = 9.1 Hz, 3H), 7.15–7.11 (m, 1H), 6.84 (s, 1H), 6.72 (d, J = 8.2 Hz, 2H), 5.21 (s, 1H), 4.63–4.49 (m, 2H), 4.35 (s, 2H), 2.45 (s, 3H); 13C-NMR δ 189.65, 180.61, 168.90, 164.63, 164.27, 161.46, 158.63, 148.49, 134.46, 132.49, 129.69, 124.11, 122.47, 120.51, 115.65, 113.13, 109.76, 107.83, 106.79, 66.59, 63.72, 55.12, 21.53; 19F-NMR δ − 73.44; ESI, m/z: 464.1 [M + H − CF3COOH]+.
(R)-2-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-1-(4-fluorophenyl)-2-oxoethanaminium 2,2,2-trifluoroacetate (3u). According to the general procedure A, compound 2 was treated with (R)-N-Boc-4-fluorophenylglycine and then purified by reverse phase flash chromatography to give compound 3u: Yellow solid; yield, 45%; 1H-NMR δ 11.97 (brs, 2H), 8.87 (brs, 3H), 7.57–7.49 (m, 3H), 7.27–7.19 (m, 3H), 7.06 (d, J = 2.6 Hz, 1H), 6.81 (d, J = 2.5 Hz, 1H), 5.41 (s, 1H), 4.72–4.45 (m, 2H), 4.35 (t, J = 4.3 Hz, 2H), 2.44 (s, 3H); 13C-NMR δ 189.41, 180.29, 168.43, 164.53, 164.20, 161.44, 148.39, 134.23, 132.24, 130.77, 130.68, 128.86, 128.83, 123.99, 120.39, 115.98, 115.77, 112.87, 109.54, 107.74, 106.64, 66.44, 63.88, 54.79, 21.44; 19F-NMR δ − 73.44, − 111.56; ESI, m/z: 466.1 [M + H − CF3COOH]+.
(S)-2-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-1-(4-fluorophenyl)-2-oxoethanaminium 2,2,2-trifluoroacetate (3v). According to the general procedure A, compound 2 was treated with (S)-N-Boc-4-fluorophenylglycine and then purified by reverse phase flash chromatography to give compound 3v: Yellow solid; yield, 43%; 1H-NMR (400 MHz, DMSO-d6) δ 11.95 (brs, 2H), 8.89 (brs, 3H), 7.57–7.49 (m, 3H), 7.27–7.19 (m, 3H), 7.05 (d, J = 2.6 Hz, 1H), 6.80 (d, J = 2.5 Hz, 1H), 5.41 (s, 1H), 4.74–4.47 (m, 2H), 4.34 (t, J = 4.3 Hz, 2H), 2.44 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 189.87, 180.99, 168.35, 164.62, 164.23, 161.46, 148.57, 134.69, 132.73, 130.60, 130.52, 128.72, 128.69, 124.20, 120.57, 115.92, 115.70, 113.38, 109.96, 107.85, 106.88, 66.48, 63.90, 54.63, 21.49; 19F-NMR (376 MHz, DMSO-d6) δ − 73.48, − 111.55; ESI, m/z: 466.1 [M + H − CF3COOH]+.
(R)-1-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-1-oxo-3-phenyl-propan-2-aminium 2,2,2-trifluoroacetate (3w). According to the general procedure A, compound 2 was treated with N-Boc-d-phenylalanine and then purified by reverse phase flash chromatography to give compound 3w: Yellow solid; yield, 48%; 1H-NMR δ 11.90 brs, 1H), 8.60 (brs, 2H), 7.47 (s, 1H), 7.29–7.21 (m, 5H), 7.16 (s, 1H), 7.13–7.09 (m, 1H), 6.83 (d, J = 2.5 Hz, 1H), 4.58–4.22 (m, 5H), 3.23–3.00 (m, 2H), 2.40 (s, 3H); 13C-NMR δ 189.88, 181.02, 169.06, 164.69, 164.27, 161.48, 148.57, 134.76, 134.47, 132.71, 129.41, 128.54, 127.28, 124.22, 120.59, 113.37, 110.02, 107.87, 107.01, 66.52, 63.60, 53.14, 36.01, 21.52; 19F-NMR δ − 73.47; ESI, m/z: 462.1 [M + H − CF3COOH]+.
(S)-1-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-1-oxo-3-phenyl-propan-2-aminium 2,2,2-trifluoroacetate (3x). According to the general procedure A, compound 2 was treated with N-Boc-l-phenylalanine and then purified by reverse phase flash chromatography to give compound 3x: Yellow solid; yield, 45%; 1H-NMR δ 11.90 (brs, 1H), 8.59 (brs, 2H), 7.47 (d, J = 1.7 Hz, 1H), 7.31–7.21 (m, 5H), 7.16 (d, J = 1.6 Hz, 1H), 7.12 (d, J = 2.5 Hz, 1H), 6.84 (d, J = 2.5 Hz, 1H), 4.67–4.13 (m, 5H), 3.26–2.96 (m, 2H), 2.40 (s, 3H); 13C-NMR δ 189.88, 181.03, 169.05, 164.69, 164.27, 161.48, 148.57, 134.76, 134.46, 132.72, 129.41, 128.54, 127.28, 124.22, 120.59, 113.37, 110.02, 107.87, 107.01, 66.53, 63.60, 53.14, 36.00, 21.52; 19F-NMR δ − 73.51; ESI, m/z: 462.1 [M + H − CF3COOH]+.
(R)-1-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-3-(4-hydroxyphenyl)-1-oxopropan-2-aminium 2,2,2-trifluoroacetate (3y). According to the general procedure A, compound 2 was treated with N-Boc-O-tert-butyl-l-tyrosine and then purified by reverse phase flash chromatography to give compound 3y: Yellow solid; yield, 26%; 1H-NMR δ 9.33 (s, 1H), 7.54 (d, J = 1.6 Hz, 1H), 7.22 (s, 1H), 7.20 (d, J = 2.5 Hz, 1H), 7.01 (d, J = 2.0 Hz, 1H), 6.99 (d, J = 2.1 Hz, 1H), 6.91 (d, J = 2.5 Hz, 1H), 6.64 (d, J = 2.0 Hz, 1H), 6.62 (d, J = 1.9 Hz, 1H), 4.59–4.31 (m, 4H), 4.27 (t, J = 6.2 Hz, 1H), 3.06–2.91 (m, 2H), 2.43 (s, 3H); 13C-NMR δ 190.03, 181.19, 169.29, 164.76, 164.30, 161.50, 156.67, 148.62, 134.95, 132.86, 130.44, 124.27, 124.18, 120.64, 115.32, 113.49, 110.17, 107.82, 107.09, 66.63, 63.60, 53.39, 35.30, 21.54; 19F-NMR δ − 73.49; ESI, m/z: 478.1 [M + H − CF3COOH]+.
(S)-1-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-3-(4-hydroxyphenyl)-1-oxopropan-2-aminium 2,2,2-trifluoroacetate (3z). According to the general procedure A, compound 2 was treated with N-Boc-l-tyrosine and then purified by reverse phase flash chromatography to give compound 3z: Yellow solid; yield, 23%; 1H-NMR δ 11.04 (s, 1H), 7.50 (d, J = 5.7 Hz, 2H), 7.34 (d, J = 8.0 Hz, 1H), 7.26–7.11 (m, 3H), 7.06 (t, J = 7.5 Hz, 1H), 6.96 (t, J = 7.5 Hz, 1H), 6.85 (d, J = 2.5 Hz, 1H), 4.56–4.18 (m, 5H), 3.27 (d, J = 6.5 Hz, 2H), 2.41 (s, 3H); 13C-NMR δ 189.93, 181.08, 169.46, 164.76, 164.25, 161.48, 148.59, 136.18, 134.83, 132.77, 126.88, 124.89, 124.23, 121.17, 120.60, 118.60, 117.87, 113.39, 111.56, 110.09, 107.80, 107.09, 106.24, 66.49, 63.71, 52.70, 26.20, 21.52; 19F-NMR δ − 73.47; ESI, m/z: 501.15 [M + H − CF3COOH]+.
(R)-1-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-N-methyl-1-oxo-propan-2-aminium 2,2,2-trifluoroacetate (4a). According to the general procedure A, compound 2 was treated with Boc-N-methyl-d-alanine and then purified by reverse phase flash chromatography to give compound 4a: Yellow solid; yield, 48%; 1H-NMR δ 11.93 (brs, 2H), 9.18 (brs, 2H), 7.50 (d, J = 1.6 Hz, 1H), 7.29–7.12 (m, 2H), 6.91 (d, J = 2.5 Hz, 1H), 4.65–4.53 (m, 2H), 4.47 (d, J = 4.3 Hz, 2H), 4.20 (q, J = 7.1 Hz, 1H), 2.61 (s, 3H), 2.42 (s, 3H), 1.45 (d, J = 7.1 Hz, 3H); 13C-NMR δ 189.11, 179.90, 169.48, 164.62, 164.18, 161.36, 148.27, 134.02, 131.90, 123.98, 120.23, 112.58, 109.48, 107.57, 106.63, 66.56, 63.68, 55.23, 30.60, 21.38, 14.04; 19F-NMR δ − 73.51; ESI, m/z: 400.2 [M + H − CF3COOH]+.
(S)-1-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-N-methyl-1-oxo-propan-2-aminium 2,2,2-trifluoroacetate (4b). According to the general procedure A, compound 2 was treated with Boc-N-methyl-l-alanine and then purified by reverse phase flash chromatography to give compound 4b: Yellow solid; yield, 45%; 1H-NMR δ 12.15 (brs, 1H), 11.96 (brs, 1H), 9.11 (s, 2H), 7.53 (d, J = 1.6 Hz, 1H), 7.24–7.17 (m, 2H), 6.93 (d, J = 2.6 Hz, 1H), 4.68–4.52 (m, 2H), 4.47 (t, J = 4.6 Hz, 2H), 4.19 (q, J = 7.2 Hz, 1H), 2.61 (s, 3H), 2.44 (s, 3H), 1.44 (d, J = 7.2 Hz, 3H); 13C-NMR δ 189.12, 179.88, 169.52, 164.55, 164.14, 161.31, 148.29, 134.03, 131.92, 123.87, 120.24, 112.58, 109.36, 107.49, 106.66, 66.54, 63.67, 55.29, 30.62, 21.40, 14.07; 19F-NMR δ − 73.48; ESI, m/z: 400.2 [M + H − CF3COOH]+.
General procedure B for preparation of compounds 5a and 5b
To a solution of compound 4a or 4b (100 mg, 0.25 mmol) and paraformaldehyde (37% aqueous solution, 62 mg, 0.76 mmol) in MeOH (15 mL) was added NaBH3CN (24 mg, 0.38 mmol) at 0 °C. After stirring 2 h at room temperature, the reaction was quenched by TFA (0.1 mL). The resulting reaction solution was used directly in purification by reverse phase flash chromatography with the following conditions: Column: Spherical C18, 20–40 μm, 330 g; Mobile Phase A: Water (plus 5 mM TFA); Mobile Phase B: ACN; Flow rate: 80 mL/min; Gradient: 5% B gradient in 10 min, 25% B–45% B gradient in 25 min; Detector: 254 nm. The fractions containing the desired product were collected at around 40% B and concentrated under reduced pressure to afford compounds 5a or 5b in 85% or 89% yield, respectively.
(R)-1-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-N,N-dimethyl-1-oxo-propan-2-aminium 2,2,2-trifluoroacetate (5a). Yellow solid; yield, 85%; 1H-NMR δ 12.17 (s, 1H), 11.95 (s, 1H), 10.23 (s, 1H), 7.53 (s, 1H), 7.25–7.14 (m, 2H), 6.93 (d, J = 2.5 Hz, 1H), 4.63–4.56 (m, 2H), 4.52–4.45 (m, 2H), 4.37 (q, J = 7.2 Hz, 1H), 2.80 (s, 6H), 2.43 (s, 3H), 1.48 (d, J = 7.2 Hz, 3H); 13C-NMR δ 189.60, 180.57, 168.53, 164.69, 164.23, 161.43, 148.51, 134.54, 132.41, 124.14, 120.49, 113.05, 109.81, 107.65, 107.00, 66.56, 63.87, 61.48, 21.52, 11.71; 19F-NMR δ − 73.59; ESI, m/z: 414.2 [M + H − CF3COOH]+.
(S)-1-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)-N,N-dimethyl-1-oxo-propan-2-aminium 2,2,2-trifluoroacetate (5b). Yellow solid; yield, 89%; 1H-NMR δ 12.15 (s, 1H), 11.93 (s, 1H), 10.35 (s, 1H), 7.50 (s, 1H), 7.27–7.08 (m, 2H), 6.91 (d, J = 2.6 Hz, 1H), 4.59 (d, J = 4.5 Hz, 2H), 4.47 (t, J = 4.3 Hz, 2H), 4.39 (q, J = 7.1 Hz, 1H), 2.82 (s, 6H), 2.42 (s, 3H), 1.49 (d, J = 7.2 Hz, 3H); 13C-NMR δ 189.12, 179.87, 168.50, 164.53, 164.15, 161.32, 148.31, 134.04, 131.91, 123.87, 120.26, 112.57, 109.37, 107.48, 106.68, 66.47, 63.85, 61.55, 21.41, 11.82; 19F-NMR δ − 73.75; ESI, m/z: 414.2 [M + H − CF3COOH]+.
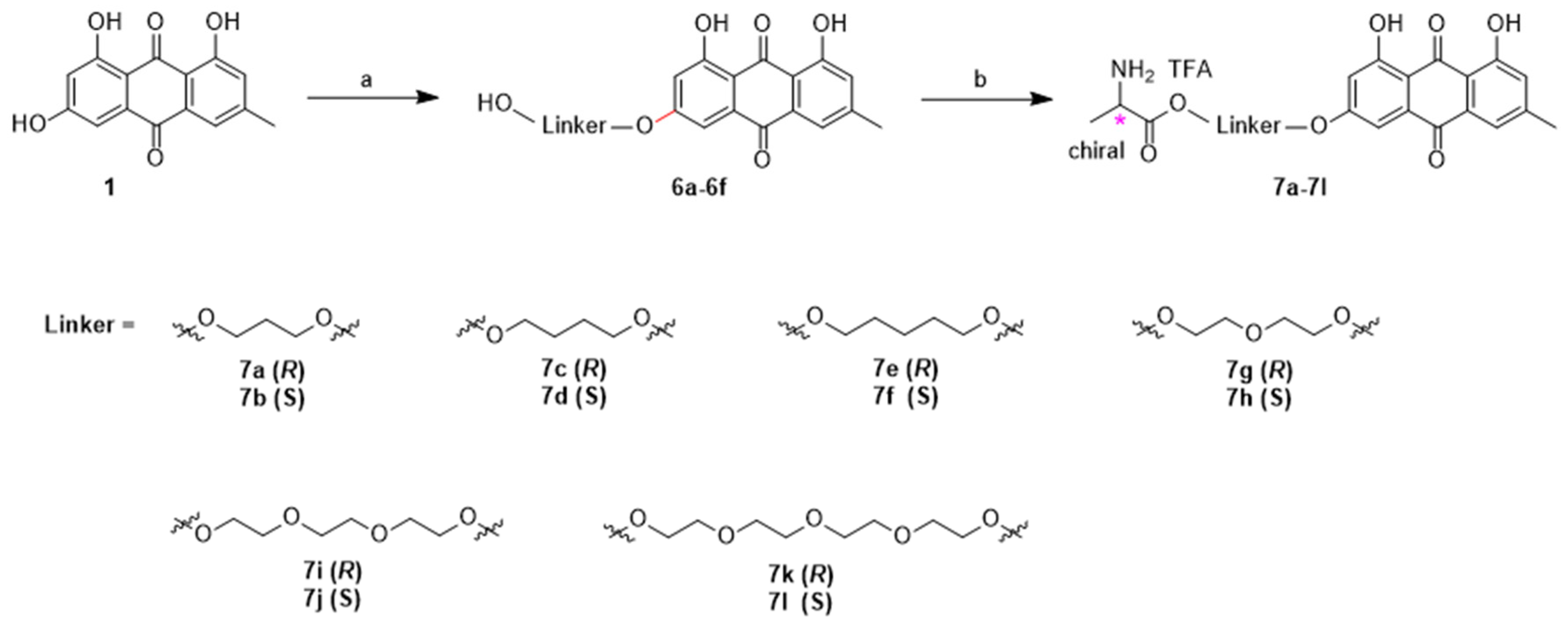
General procedure C for preparation of compounds 6a–6f
To a mixture of emodin (10 mmol) in dry DMF (50 mL) were added Cs2CO3 (12 mmol) and hydroxybromides or iodides (30 mmol) at room temperature. After stirring for 36 h at 60 °C, the resulting mixture was evaporated under reduced pressure and then mixed with water (100 mL). The pH value of aqueous phase was adjusted to around 5 with 10% hydrochloric acid solution, extracted with dichloromethane (2 × 100 mL). The combined organic layer was washed with brine (200 mL), dried over anhydrous sodium sulfate and evaporated to dryness. The crude product was purified by silica gel column chromatography with 1%–10% ethyl acetate in petroleum to afford compounds 6a–6f.
1,8-Dihydroxy-3-(3-hydroxypropsoxy)-6-methylanthracene-9,10-dione (6a). According to the general procedure C, emodin was treated with 3-iodopropan-1-ol and then purified by silica gel column chromatography to give compound 6a: Brown solid; yield, 55%; 1H-NMR δ 12.14 (s, 1H), 11.96 (s, 1H), 7.51 (s, 1H), 7.21–7.13 (m, 2H), 6.85 (d, J = 2.5 Hz, 1H), 4.62 (t, J = 5.2 Hz, 1H), 4.23 (t, J = 6.3 Hz, 2H), 3.58 (q, J = 5.9 Hz, 2H), 2.43 (s, 3H), 1.95–1.87 (m, 2H); 13C-NMR δ 189.78, 180.98, 165.53, 164.32, 161.42, 148.44, 134.66, 132.69, 124.13, 120.51, 113.30, 109.60, 107.84, 106.83, 65.99, 57.00, 31.71, 21.49; ESI, m/z: 329.1 [M + H]+.
1,8-Dihydroxy-3-(4-hydroxybutoxy)-6-methylanthracene-9,10-dione (6b). According to the general procedure C, emodin was treated with 4-bromobutan-1-ol and then purified by silica gel column chromatography to give compound 6b: Brown solid; yield, 47%; 1H-NMR δ 12.16 (s, 1H), 11.98 (s, 1H), 7.53 (s, 1H), 7.23–7.14 (m, 2H), 6.86 (d, J = 2.5 Hz, 1H), 4.49 (d, J = 5.5 Hz, 1H), 4.18 (t, J = 6.5 Hz, 2H), 3.47 (q, J = 6.0 Hz, 2H), 2.43 (s, 3H), 1.80 (t, J = 7.5 Hz, 2H), 1.58 (p, J = 6.7 Hz, 2H); 13C-NMR δ 189.83, 181.06, 165.53, 164.36, 161.44, 148.47, 134.71, 132.74, 124.16, 120.54, 113.35, 109.62, 107.92, 106.84, 68.81, 60.29, 28.76, 25.14, 21.52; ESI, m/z: 343.1 [M + H]+.
1,8-Dihydroxy-3-(5-hydroxypentyloxy)-6-methylanthracene-9,10-dione (6c). According to the general procedure C, emodin was treated with 5-bromopentan-1-ol and then purified by silica gel column chromatography to give compound 6c: Brown solid; yield, 52%; 1H-NMR δ 12.15 (s, 1H), 11.96 (s, 1H), 7.51 (s, 1H), 7.17 (d, J = 16.9 Hz, 2H), 6.84 (s, 1H), 4.40 (s, 1H), 4.15 (t, J = 6.5 Hz, 2H), 3.45 (t, J = 6.5 Hz, 2H), 2.43 (s, 3H), 1.77 (d, J = 10.1 Hz, 2H), 1.48 (s, 4H); 13C-NMR δ 189.50, 180.55, 165.39, 164.26, 161.35, 148.28, 134.34, 132.41, 123.97, 120.36, 113.02, 109.30, 107.75, 106.58, 68.79, 60.57, 32.12, 28.20, 21.94, 21.44; ESI, m/z: 357.1 [M + H]+.
1,8-Dihydroxy-3-(2-(2-hydroxyethoxy)ethoxy)-6-methylanthracene-9,10-dione (6d). According to the general procedure C, emodin was treated with 2-(2-bromoethoxy)ethanol and then purified by silica gel column chromatography to give compound 6d: Brown solid; yield, 38%; 1H-NMR δ 12.14 (s, 1H), 11.95 (s, 1H), 7.50 (d, J = 1.6 Hz, 1H), 7.19 (d, J = 1.6 Hz, 1H), 7.17 (d, J = 2.5 Hz, 1H), 6.88 (d, J = 2.5 Hz, 1H), 4.65 (s, 1H), 4.35–4.25 (m, 2H), 3.86–3.76 (m, 2H), 3.53 (d, J = 13.1 Hz, 4H), 2.42 (s, 3H); 13C-NMR δ 189.80, 180.93, 165.31, 164.29, 161.42, 148.47, 134.66, 132.67, 124.14, 120.52, 113.28, 109.71, 107.85, 106.95, 72.48, 68.49, 68.47, 60.22, 21.51; ESI, m/z: 359.2 [M + H]+.
1,8-Dihydroxy-3-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)-6-methylanthracene-9,10-dione (6e). According to the general procedure C, emodin was treated with 2-(2-(2-bromoethoxy)ethoxy)ethanol and then purified by silica gel column chromatography to give compound 6e: Brown solid; yield, 42%; 1H-NMR δ 12.01 (s, 1H), 11.82 (s, 1H), 7.34 (d, J = 1.6 Hz, 1H), 7.06 (d, J = 1.6 Hz, 1H), 7.00 (d, J = 2.6 Hz, 1H), 6.75 (d, J = 2.5 Hz, 1H), 4.57 (br, 1H), 4.33–4.14 (m, 2H), 3.86–3.73 (m, 2H), 3.70–3.55 (m, 4H), 3.53–3.41 (m, 4H), 2.36 (s, 3H); 13C-NMR δ 189.52, 180.51, 165.18, 164.21, 161.35, 148.36, 134.35, 132.37, 124.00, 120.39, 112.99, 109.44, 107.74, 106.79, 72.39, 70.01, 69.78, 68.50, 68.37, 60.22, 21.46; ESI, m/z: 403.2 [M + H]+.
1,8-Dihydroxy-3-(2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethoxy)-6-methylanthracene-9,10-dione (6f). According to the general procedure C, emodin was treated with 2-(2-(2-(2-bromoethoxy)ethoxy)ethoxy)ethanol and then purified by silica gel column chromatography to give compound 6f: Brown solid; yield, 30%; 1H-NMR δ 12.13 (s, 1H), 11.94 (s, 1H), 7.49 (d, J = 1.6 Hz, 1H), 7.18 (t, J = 1.3 Hz, 1H), 7.15 (d, J = 2.5 Hz, 1H), 6.87 (d, J = 2.5 Hz, 1H), 4.57 (t, J = 5.5 Hz, 1H), 4.34–4.26 (m, 2H), 3.83–3.75 (m, 2H), 3.62 (dd, J = 6.1, 3.5 Hz, 2H), 3.58–3.45 (m, 8H), 3.42 (d, J = 5.1 Hz, 2H), 2.42 (s, 3H); 13C-NMR δ 189.74, 180.83, 165.29, 164.27, 161.41, 148.45, 134.59, 132.60, 124.11, 120.50, 113.21, 109.65, 107.85, 106.95, 72.34, 69.97, 69.84, 69.80, 69.76, 68.51, 68.41, 60.19, 21.50; ESI, m/z: 447.2 [M + H]+.
(R)-1-(3-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)propoxy)-1-oxopropan-2-aminium 2,2,2-trifluoroacetate (7a). According to the general procedure A, compound 6a was treated with N-Boc-d-alanine and then purified by reverse phase flash chromatography to give compound 7a: Yellow solid; yield, 70%; 1H-NMR δ 7.52 (d, J = 1.7 Hz, 1H), 7.23–7.16 (m, 2H), 6.88 (d, J = 2.5 Hz, 1H), 4.40–4.33 (m, 2H), 4.28 (t, J = 6.2 Hz, 2H), 4.15 (q, J = 7.1 Hz, 1H), 2.43 (s, 3H), 2.14 (p, J = 6.3 Hz, 2H), 1.42 (d, J = 7.1 Hz, 3H); 13C-NMR δ 189.85, 181.06, 169.89, 165.17, 164.26, 161.45, 148.52, 134.77, 132.73, 124.19, 120.55, 113.35, 109.86, 107.75, 107.00, 65.30, 62.31, 47.92, 27.59, 21.49, 15.68; 19F-NMR δ − 73.50; ESI, m/z: 400.2 [M + H − CF3COOH]+.
(S)-1-(3-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)propoxy)-1-oxopropan-2-aminium 2,2,2-trifluoroacetate (7b). According to the general procedure A, compound 6a was treated with N-Boc-l-alanine and then purified by reverse phase flash chromatography to give compound 7b: Yellow solid; yield, 68%; 1H-NMR δ 7.54 (s, 1H), 7.22–7.20 (m, 2H), 6.89 (s, 1H), 4.38–4.33 (m, 2H), 4.31 (t, J = 6.2 Hz, 2H), 4.15 (q, J = 7.1 Hz, 1H), 2.44 (s, 3H), 2.14 (p, J = 6.3 Hz, 2H), 1.41 (d, J = 7.1 Hz, 3H); 13C-NMR δ 189.19, 180.04, 170.06, 164.99, 164.19, 161.34, 148.26, 134.04, 132.04, 123.88, 120.28, 112.69, 109.21, 107.56, 106.56, 65.34, 62.38, 48.11, 27.71, 21.45, 15.78; 19F-NMR δ − 73.47; ESI, m/z: 400.2 [M + H − CF3COOH]+.
(R)-1-(4-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)butoxy)-1-oxopropan-2-aminium 2,2,2-trifluoroacetate (7c). According to the general procedure A, compound 6b was treated with N-Boc-d-alanine and then purified by reverse phase flash chromatography to give compound 7c: Yellow solid; yield, 65%; 1H-NMR δ 8.31 (brs, 2H), 7.56 (s, 1H), 7.21 (dd, J = 13.9, 1.9 Hz, 2H), 6.90(d, J = 2.5 Hz, 1H), 4.30–4.22 (m, 4H), 4.14 (q, J = 7.2 Hz, 1H), 2.45 (s, 3H), 1.92–1.76 (m, 4H), 1.41 (d, J = 7.2 Hz, 3H); 13C-NMR δ 189.85, 181.11, 170.03, 165.36, 164.33, 161.45, 148.50, 134.77, 132.76, 124.20, 120.54, 113.38, 109.76, 107.86, 106.92, 68.32, 65.24, 47.90, 24.73, 24.62, 21.49, 15.74; 19F-NMR δ − 73.44; ESI, m/z: 414.2 [M + H − CF3COOH]+.
(S)-1-(4-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)butoxy)-1-oxopropan-2-aminium 2,2,2-trifluoroacetate (7d). According to the general procedure A, compound 6b was treated with N-Boc-l-alanine and then purified by reverse phase flash chromatography to give compound 7d: Yellow solid; yield, 68%; 1H-NMR δ 7.53 (d, J = 1.6 Hz, 1H), 7.20 (dd, J = 13.9, 1.9 Hz, 2H), 6.88 (d, J = 2.5 Hz, 1H), 4.31–4.20 (m, 4H), 4.14 (q, J = 7.2 Hz, 1H), 2.44 (s, 3H), 1.92–1.76 (m, 4H), 1.41 (d, J = 7.2 Hz, 3H); 13C-NMR δ 189.24, 180.16, 170.12, 165.17, 164.23, 161.33, 148.23, 134.08, 132.13, 123.89, 120.27, 112.77, 109.16, 107.63, 106.48, 68.28, 65.24, 48.01, 24.84, 24.70, 21.45, 15.81; 19F-NMR δ − 73.47; ESI, m/z: 414.1 [M + H − CF3COOH]+.
(R)-1-(5-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)pentyloxy)-1-oxopropan-2-aminium 2,2,2-trifluoroacetate (7e). According to the general procedure A, compound 6c was treated with N-Boc-d-alanine and then purified by reverse phase flash chromatography to give compound 7e: Yellow solid; yield, 70%; 1H-NMR δ 12.00 (brs, 2H), 8.32 (brs, 3H), 7.54 (d, J = 1.6 Hz, 1H), 7.22 (s, 1H), 7.18 (d, J = 2.5 Hz, 1H), 6.88 (d, J = 2.5 Hz, 1H), 4.27–4.10 (m, 5H), 2.44 (s, 3H), 1.83–1.76 (m, 2H), 1.75–1.68 (m, 2H), 1.56–1.46 (m, 2H), 1.40 (d, J = 7.1 Hz, 3H); 13C-NMR δ 189.15, 180.02, 170.15, 165.20, 164.22, 161.32, 148.17, 133.96, 132.05, 123.84, 120.22, 112.69, 109.02, 107.59, 106.36, 68.55, 65.45, 48.03, 27.95, 27.74, 21.76, 21.44, 15.81; 19F-NMR δ − 73.46; ESI, m/z: 428.2 [M + H − CF3COOH]+.
(S)-1-(5-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)pentyloxy)-1-oxopropan-2-aminium 2,2,2-trifluoroacetate (7f). According to the general procedure A, compound 6c was treated with N-Boc-l-alanine and then purified by reverse phase flash chromatography to give compound 7f: Yellow solid; yield, 66%; 1H-NMR δ 11.97 (brs, 2H), 8.38 (brs, 3H), 7.52 (s, 1H), 7.21 (s, 1H), 7.17 (d, J = 2.5 Hz, 1H), 6.87 (d, J = 2.5 Hz, 1H), 4.27–4.10 (m, 5H), 2.43 (s, 3H), 1.83–1.76 (m, 2H), 1.75–1.68 (m, 2H), 1.56–1.46 (m, 2H), 1.40 (d, J = 7.1 Hz, 3H); 13C-NMR δ 189.72, 180.94, 170.02, 165.41, 164.31, 161.42, 148.42, 134.63, 132.65, 124.12, 120.48, 113.27, 109.60, 107.82, 106.79, 68.59, 65.42, 47.86, 27.81, 27.57, 21.67, 21.47, 15.72; 19F-NMR δ − 73.49; ESI, m/z: 428.2 [M + H − CF3COOH]+.
(R)-1-(2-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)ethoxy)-1-oxo-propan-2-aminium 2,2,2-trifluoroacetate (7g). According to the general procedure A, compound 6d was treated with N-Boc-d-alanine and then purified by reverse phase flash chromatography to give compound 7g: Yellow solid; yield, 65%; 1H-NMR δ 9.46 (brs, 3H), 7.53 (s, 1H), 7.22 (s, 1H), 7.19 (s, 1H), 6.90 (s, 1H), 4.39–4.27 (m, 4H), 4.11 (q, J = 7.1 Hz, 1H), 3.82 (d, J = 4.6 Hz, 2H), 3.76 (d, J = 4.6 Hz, 2H), 2.44 (s, 3H), 1.38 (d, J = 7.1 Hz, 3H); 13C-NMR δ 189.83, 181.01, 170.27, 165.24, 164.29, 161.46, 148.52, 134.73, 132.71, 124.21, 120.54, 113.33, 109.81, 107.84, 107.01, 68.48, 68.36, 68.12, 64.78, 47.88, 21.52, 15.85; 19F-NMR δ − 73.46; ESI, m/z: 430.2 [M + H − CF3COOH]+.
(S)-1-(2-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)ethoxy)-1-oxo-propan-2-aminium 2,2,2-trifluoroacetate (7h). According to the general procedure A, compound 6d was treated with N-Boc-l-alanine and then purified by reverse phase flash chromatography to give compound 7h: Yellow solid; yield, 67%; 1H-NMR δ 7.54 (d, J = 1.7 Hz, 1H), 7.22 (s, 1H), 7.20 (d, J = 2.6 Hz, 1H), 6.91 (d, J = 2.5 Hz, 1H), 4.40–4.26 (m, 4H), 4.12 (q, J = 7.2 Hz, 1H), 3.82 (t, J = 4.4 Hz, 2H), 3.75 (t, J = 4.6 Hz, 2H), 2.44 (s, 3H), 1.38 (d, J = 7.2 Hz, 3H); 13C-NMR δ 189.86, 181.05, 170.26, 165.25, 164.30, 161.46, 148.53, 134.76, 132.74, 124.21, 120.55, 113.36, 109.84, 107.85, 107.03, 68.48, 68.36, 68.11, 64.79, 47.88, 21.52, 15.85; 19F-NMR δ − 73.45; ESI, m/z: 430.1 [M + H − CF3COOH]+.
(R)-1-(2-(2-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)ethoxy)ethoxy)-1-oxopropan-2-aminium 2,2,2-trifluoroacetate (7i). According to the general procedure A, compound 6e was treated with N-Boc-d-alanine and then purified by reverse phase flash chromatography to give compound 7i: Yellow solid; yield, 66%; 1H-NMR δ 9.64 (s, 3H), 7.53 (s, 1H), 7.22 (s, 1H), 7.19 (d, J = 2.5 Hz, 1H), 6.90 (d, J = 2.6 Hz, 1H), 4.29 (d, J = 15.1 Hz, 4H), 4.18–4.05 (m, 1H), 3.80 (d, J = 4.5 Hz, 2H), 3.67 (t, J = 4.5 Hz, 6H), 2.44 (s, 3H), 1.39 (d, J = 7.1 Hz, 3H); 13C-NMR δ 189.70, 180.82, 170.23, 165.25, 164.27, 161.42, 148.46, 134.59, 132.58, 124.14, 120.48, 113.20, 109.67, 107.80, 106.94, 69.93, 69.78, 68.53, 68.41, 68.04, 64.85, 47.87, 21.50, 15.82; 19F-NMR δ − 73.46; ESI, m/z: 474.2 [M + H − CF3COOH]+.
(S)-1-(2-(2-(2-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)ethoxy)ethoxy)ethoxy)-1-oxopropan-2-aminium 2,2,2-trifluoroacetate (7j). According to the general procedure A, compound 6e was treated with N-Boc-l-alanine and then purified by reverse phase flash chromatography to give compound 7j: Yellow solid; yield, 65%; 1H-NMR δ 9.53 (brs, 3H), 7.51 (d, J = 1.6 Hz, 1H), 7.20 (d, J = 1.6 Hz, 1H), 7.17 (d, J = 2.5 Hz, 1H), 6.88 (d, J = 2.5 Hz, 1H), 4.37–4.21 (m, 4H), 4.12 (q, J = 7.1 Hz, 1H), 3.84–3.76 (m, 2H), 3.67 (t, J = 4.6 Hz, 2H), 3.65–3.56 (m, 4H), 2.43 (s, 3H), 1.39 (d, J = 7.2 Hz, 3H); 13C-NMR δ 189.87, 181.06, 170.21, 165.31, 164.30, 161.46, 148.53, 134.76, 132.75, 124.21, 120.56, 113.37, 109.82, 107.86, 107.04, 69.92, 69.76, 68.53, 68.44, 68.02, 64.88, 47.86, 21.52, 15.80; 19F-NMR δ − 73.49; ESI, m/z: 474.2 [M + H − CF3COOH]+.
(R)-1-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)-13-oxo-3,6,9,12-tetraoxapenta-decan-14-aminium 2,2,2-trifluoroacetate (7k). According to the general procedure A, compound 6f was treated with N-Boc-d-alanine and then purified by reverse phase flash chromatography to give compound 7k: Yellow solid; yield, 66%; 1H-NMR δ 9.76 (brs, 3H), 7.46 (d, J = 1.6 Hz, 1H), 7.16 (s, 1H), 7.13 (d, J = 2.6 Hz, 1H), 6.85 (d, J = 2.5 Hz, 1H), 4.37–4.21 (m, 4H), 4.11 (q, J = 7.2 Hz, 1H), 3.85–3.76 (m, 2H), 3.68–3.60 (m, 4H), 3.59–3.52 (m, 6H), 2.41 (s, 3H), 1.40 (d, J = 7.1 Hz, 3H); 13C-NMR δ 189.81, 181.00, 170.17, 165.30, 164.28, 161.44, 148.49, 134.71, 132.71, 124.17, 120.52, 113.32, 109.73, 107.86, 107.02, 69.96, 69.75, 68.51, 68.42, 67.97, 64.84, 47.85, 21.50, 15.78; 19F-NMR δ − 73.54; ESI, m/z: 518.2 [M + H − CF3COOH]+.
(S)-1-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)-13-oxo-3,6,9,12-tetraoxa-pentadecan-14-aminium 2,2,2-trifluoroacetate (7l). According to the general procedure A, compound 6f was treated with N-Boc-l-alanine and then purified by reverse phase flash chromatography to give compound 7l: Yellow solid; yield, 65%; 1H-NMR δ 9.73 (brs, 3H), 7.53 (d, J = 1.7 Hz, 1H), 7.21 (s, 1H), 7.19 (d, J = 2.5 Hz, 1H), 6.89 (d, J = 2.6 Hz, 1H), 4.31 (q, J = 5.6, 4.2 Hz, 3H), 4.28–4.24 (m, 1H), 4.11 (q, J = 7.1 Hz, 1H), 3.80 (dd, J = 5.5, 3.2 Hz, 2H), 3.67–3.59 (m, 4H), 3.58–3.53 (m, 6H), 2.44 (s, 3H), 1.39 (d, J = 7.1 Hz, 3H); 13C-NMR δ 189.74, 180.89, 170.20, 165.28, 164.29, 161.44, 148.48, 134.63, 132.63, 124.16, 120.50, 113.24, 109.70, 107.84, 106.97, 69.98, 69.77, 68.52, 68.42, 67.99, 64.86, 47.86, 21.51, 15.80; 19F-NMR δ − 73.47; ESI, m/z: 518.2 [M + H − CF3COOH]+.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}