In House Validated UHPLC Protocol for the Determination of the Total Hydroxytyrosol and Tyrosol Content in Virgin Olive Oil Fit for the Purpose of the Health Claim Introduced by the EC Regulation 432/2012 for “Olive Oil Polyphenols”

 ,
,  ,
,
Abstract
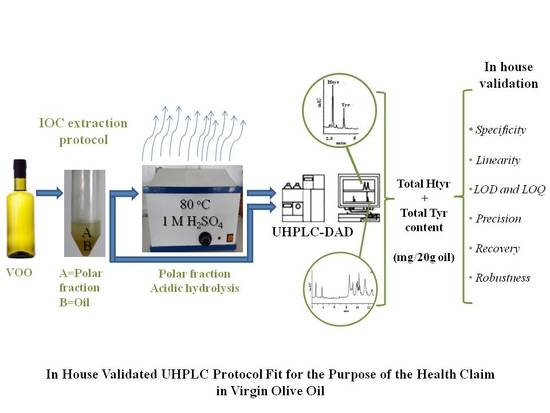
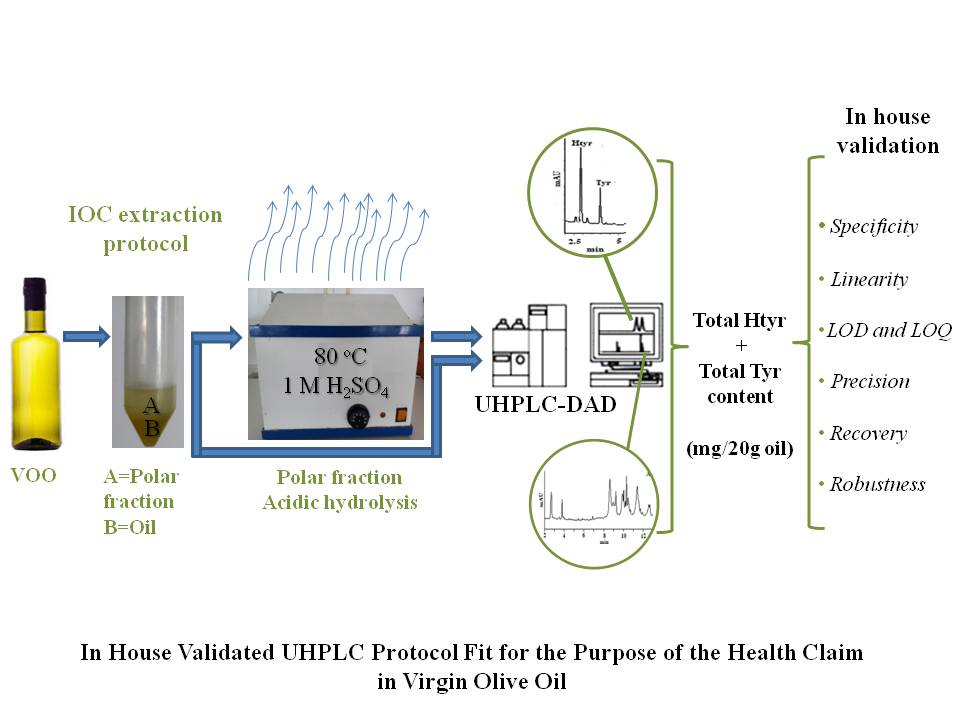
:
1. Introduction
2. Results and Discussion
2.1. Analytical Protocol
2.2. Method Validation
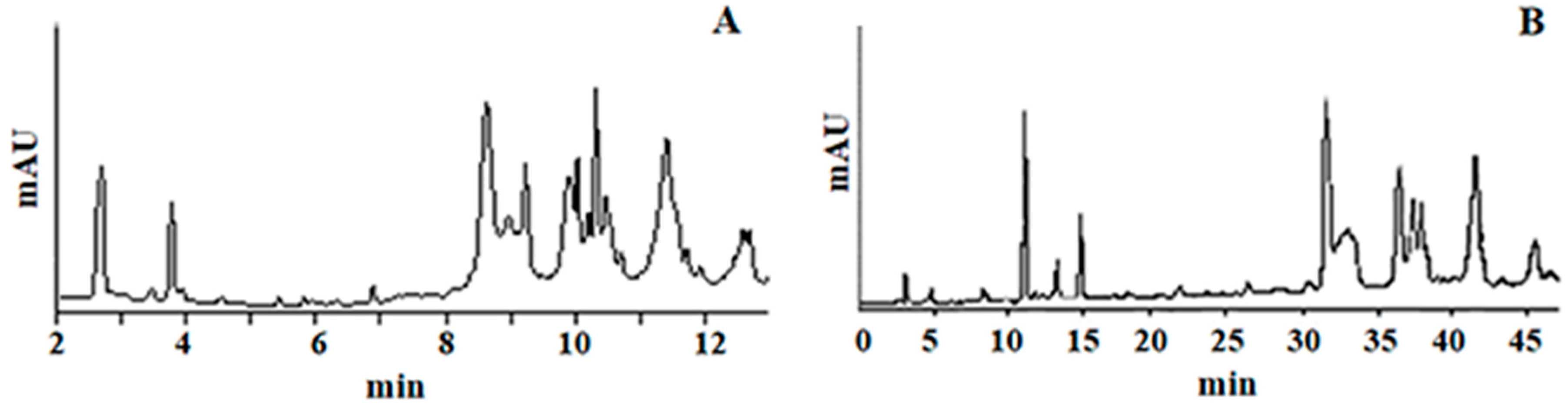
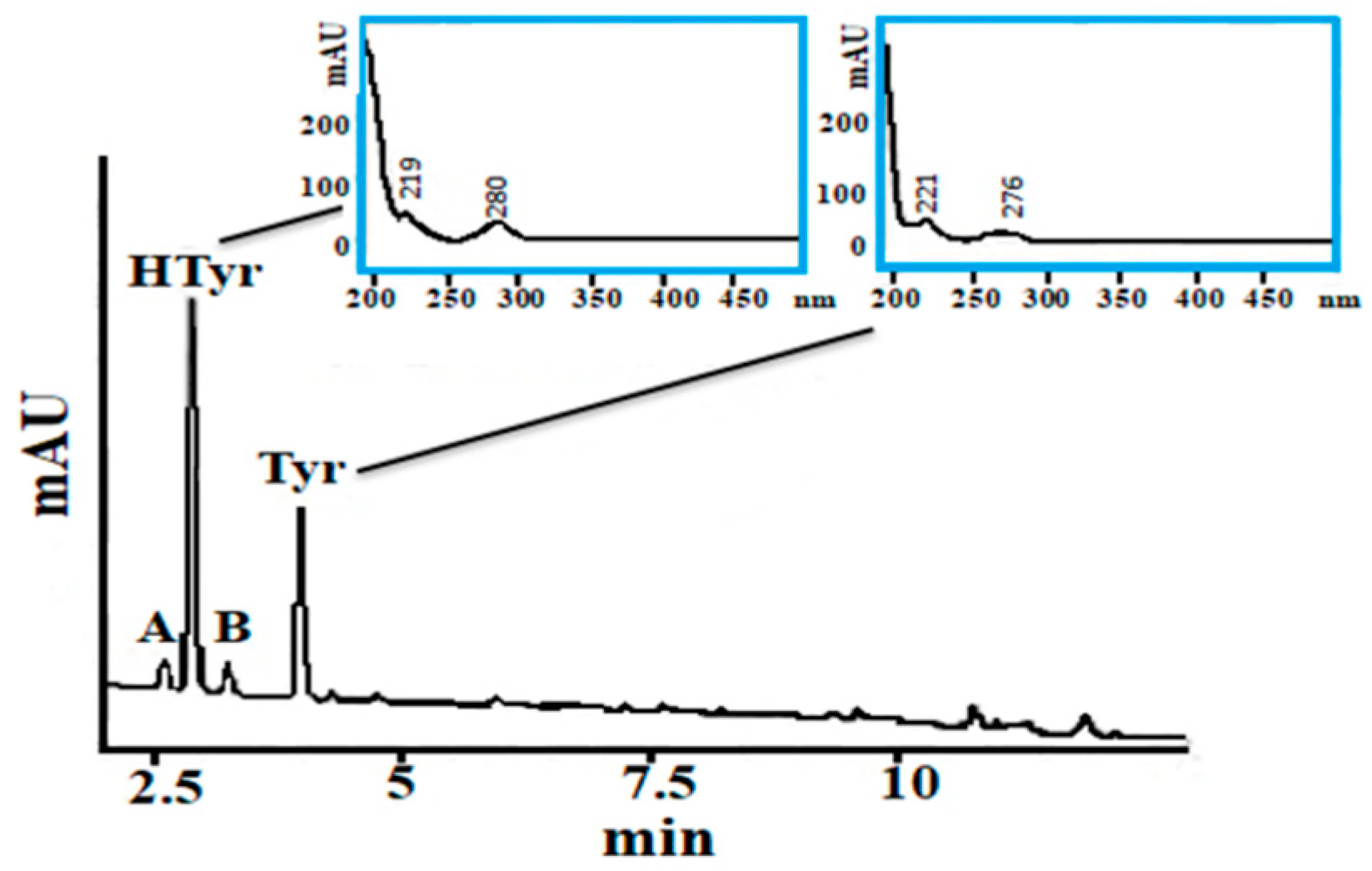
2.2.1. Specificity
2.2.2. Stability of the Analytes
Stability of Standard Solutions
Stability of Spiked Samples
2.2.3. Linearity
2.2.4. Limit of Detection (LOD) and Limit of Quantification (LOQ)
2.2.5. Precision
2.2.6. Recovery Studies
2.2.7. Robustness
2.2.8. Implementation of the In-House Validated UHPLC to VOOs
3. Materials and Methods
3.1. Chemicals
3.2. Samples
3.3. Instrumentation
3.4. Preparation of Standard Solutions
3.5. Sample Preparation
3.5.1. Extraction of the Polar Fraction (PF)
3.5.2. Acidic Hydrolysis
3.6. Folin Ciocalteu Assay
3.7. Chromatographic Conditions
3.8. Method Validation
3.9. Statistical Analyses
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ANOVA | Analysis of variance |
| AOAC | Association of Official Agricultural Chemists |
| CA | caffeic acid |
| DHPA | 3,4-dihydrophenylacetic acid |
| DAD | diode array detector |
| EU | European Union |
| F-C | Folin-Ciocalteu |
| FLD | fluorescence detector |
| HPLC | high performance liquid chromatography |
| HPLC-DAD | high performance liquid chromatography coupled to diode array detector |
| Htyr | Hydroxytyrosol |
| ICH | International Conference on Harmonization |
| IOC | International Olive Council |
| LOD | limit of detection |
| LOQ | limit of quantification |
| MS | mean square |
| PF | polar fraction |
| ROO | refined olive oil |
| RSD | relative standard deviation |
| SD | standard deviation |
| MS/MS | tandem mass spectrometry |
| SS | sum of squares |
| Tyr | Tyrosol |
| UHPLC | ultra-high performance liquid chromatography |
| UHPLC-MS | ultra-high performance liquid chromatography coupled to mass spectrometry |
| UV | ultraviolet |
| USDA | United States Department of Agriculture |
| VOO | virgin olive oil |
References
- European Commission Regulation EC No. 2568/1991 on the characteristics of olive oil and olive-residue oil and on the relevant methods of analysis. Off. J. Eur. Union 1991, L248, 1–128, and amendments M1–M28. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:01991R2568-20151016&from=EN (accessed on 11 February 2019).
- International Olive oil Council. Testing Methods. Available online: http://www.internationaloliveoil.org/estaticos/view/224-testing-methods (accessed on 11 February 2019).
- Alimentarius, C. Codex Standard for Olive Oils and Olive Pomace Oils; Codex Stan. 1981:33-1981. Rev.; FAO: Rome, Italy, 1989. [Google Scholar]
- United States Department of Agriculture. Olive Oil and Olive-Pomace Oil Grades and Standards. Available online: https://www.ams.usda.gov/grades-standards/olive-oil-and-olive-pomace-oil-grades-and-standards (accessed on 11 February 2019).
- European Commission Regulation EC No. 1308/2013 establishing a common organization of the markets in agricultural products and repealing Council Regulations (EEC) No 922/72, (EEC) No 234/79, (EC) No 1037/2001 and (EC) No 1234/2007. Off. J. Eur. Union 2013, L347, 671–854, ANNEX XVI. Available online: https://eur-lex.europa.eu/legal-content/en/TXT/?uri=CELEX:32013R1308 (accessed on 11 February 2019).
- European Commission Regulation EC No. 61/2011 amending Regulation No 2598/91 on the characteristics of olive oil and olive-residue oil and on the relevant methods of analysis. Off. J. Eur. Union 2011, L23, 1–13. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2011:023:0001:0014:EN:PDF (accessed on 11 February 2019).
- European Commission Regulation EC No. 432/2012 establishing a list of permitted health claims made on foods, other than those referring to the reduction of disease risk and to children’s development and health. Off. J. Eur. Union 2012, L136, 1–40. Available online: https://eur-lex.europa.eu/legal-content/GA/TXT/?uri=CELEX:32012R0432 (accessed on 11 February 2019).
- Kalogeropoulos, N.; Tsimidou, M.Z. Antioxidants in Greek virgin olive oils. Antioxidants 2014, 3, 387–413. [Google Scholar] [CrossRef]
- Tsimidou, M.Z.; Boskou, D. The health claim on “olive oil polyphenols” and the need for meaningful terminology and effective analytical protocols. Eur. J. Lipid Sci. Technol. 2015, 117, 1091–1094. [Google Scholar] [CrossRef] [Green Version]
- Tsimidou, M.Z.; Nenadis, N.; Servili, M.; García-González, D.L.; Gallina Toschi, T. Why tyrosol derivatives have to be quantified in the calculation of “olive oil polyphenols” content to support the health claim provisioned in the EC Reg. 432/2012. Eur. J. Lipid Sci. Technol. 2018, 120, 1800098. [Google Scholar] [CrossRef]
- Carrasco-Pancorbo, A.; Cerretani, L.; Bendini, A.; Segura-Carretero, A.; Gallina-Toschi, T.; Fernández-Gutiérrez, A. Analytical determination of polyphenols in olive oils. J. Sep. Sci. 2005, 28, 837–858. [Google Scholar] [CrossRef]
- Bendini, A.; Cerretani, L.; Carrasco-Pancorbo, A.; Gómez-Caravaca, A.; Segura-Carretero, A.; Fernández-Gutiérrez, A.; Lercker, G. Phenolic molecules in virgin olive oils: A survey of their sensory properties, health effects, antioxidant activity and analytical methods. An overview of the last decade. Molecules 2007, 12, 1679–1719. [Google Scholar] [CrossRef]
- Cerretani, L.; Gallina Toschi, T.; Bendini, A. Phenolic fraction of virgin olive oil: An overview on identified compounds and analytical methods for their determination. Funct. Plant Sci. Biotechnol. 2009, 3, 69–80. [Google Scholar]
- Tsimidou, M.Z. Analytical methodologies: Phenolic compounds related to olive oil taste issues. In Handbook of Olive Oil, 2nd ed.; Aparicio-Ruiz, R., Harwod, J., Eds.; Springer: Boston, MA, USA, 2013; pp. 311–333. [Google Scholar]
- Gómez-Caravaca, A.M.; Lozano-Sánchez, J.; Contreras Gámez, M.D.M.; Segura Carretero, A.; Taamalli, A. Bioactive phenolic compounds from Olea europaea: A challenge for analytical chemistry. In Olive and Olive Oil Bioactive Constituents; Boskou, D., Ed.; AOCS Press: Champaign, IL, USA, 2015; pp. 261–298. [Google Scholar]
- International Olive Council (IOC). Determination of Biophenols in Olive Oil by HPLC; COI/T.20/Doc. No. 29; IOC: Madrid, Spain, 2009. [Google Scholar]
- Available online: http://www.internationaloliveoil.org/news/view/666-year-2014-news/483-new-procedure-for-presenting-proposals-for-the-revision-ofioc-standards (accessed on 11 February 2019).
- Romero, C.; Brenes, M. Analysis of total contents of hydroxytyrosol and tyrosol in olive oils. J. Agric. Food Chem. 2012, 60, 9017–9022. [Google Scholar] [CrossRef] [PubMed]
- Mastralexi, A.; Nenadis, N.; Tsimidou, M.Z. Addressing analytical requirements to support health claims on “olive oil polyphenols” (EC regulation 432/2012). J. Agric. Food Chem. 2014, 62, 2459–2461, (Romero, C.; Brenes, M. Comment on addressing analytical requirements to support health claims on “olive oil polyphenols” (EC regulation 432/212). J. Agric. Food Chem. 2014, 62, 10210–10211; Mastralexi, A.; Nenadis, N.; Tsimidou, M.Z. Rebuttal to the comment on addressing analytical requirements to support health claims on “olive oil polyphenols” (EC regulation 432/212). J. Agric. Food Chem. 2014, 62, 10212–10213). [Google Scholar] [CrossRef] [PubMed]
- Purcaro, G.; Codony, R.; Pizzale, L.; Mariani, C.; Conte, L. Evaluation of total hydroxytyrosol and tyrosol in extra virgin olive oils. Eur. J. Lipid Sci. Technol. 2014, 116, 805–811. [Google Scholar] [CrossRef]
- Reboredo-Rodríguez, P.; Valli, E.; Bendini, A.; Di Lecce, G.; Simal-Gándara, J.; Gallina Toschi, T. A widely used spectrophotometric assay to quantify olive oil biophenols according to the health claim (EU Reg. 432/2012). Eur. J. Lipid Sci. Technol. 2016, 118, 1593–1599. [Google Scholar] [CrossRef]
- Ricciutelli, M.; Marconi, S.; Boarelli, M.C.; Caprioli, G.; Sagratini, G.; Ballini, R.; Fiorini, D. Olive oil polyphenols: A quantitative method by high-performance liquid-chromatography-diode-array detection for their determination and the assessment of the related health claim. J. Chromatogr. A 2017, 1481, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Bartella, L.; Mazzotti, F.; Napoli, A.; Sindona, G.; Di Donna, L. A comprehensive evaluation of tyrosol and hydroxytyrosol derivatives in extra virgin olive oil by microwave-assisted hydrolysis and HPLC-MS/MS. Anal. Bioanal. Chem. 2018, 410, 2193–2201. [Google Scholar] [CrossRef] [PubMed]
- Olmo-García, L.; Fernández-Fernández, C.; Hidalgo, A.; Vílchez, P.; Fernández-Gutiérrez, A.; Marchal, R.; Carrasco-Pancorbo, A. Evaluating the reliability of specific and global methods to assess the phenolic content of virgin olive oil: Do they drive to equivalent results? J. Chromatogr. A 2018, 1585, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Mulinacci, N.; Giaccherini, C.; Ieri, F.; Innocenti, M.; Romani, A.; Vincieri, F. Evaluation of lignans and free and linked hydroxy-tyrosol and tyrosol in extra virgin olive oil after hydrolysis processes. J. Sci. Food Agric. 2006, 86, 757–764. [Google Scholar] [CrossRef]
- Brenes, M.; García, A.; García, P.; Garrido, A. Rapid and complete extraction of phenols from olive oil and determination by means of a coulometric electrode array system. J. Agric. Food Chem. 2000, 48, 5178–5183. [Google Scholar] [CrossRef]
- Naushad, M.; Khan, M.R. Ultra Performance Liquid Chromatography Mass Spectrometry: Evaluation and Applications in Food Analysis; CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar]
- Regulation EC No 657/2002 Commission decision of 12 August 2002 Implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off. J. Eur. Union 2002, L221, 8–36. Available online: https://publications.europa.eu/en/publication-detail/-/publication/ed928116-a955-4a84-b10a-cf7a82bad858/language-en (accessed on 11 February 2019).
- Magnusson, B.; Örnemark, U. (Eds.) Eurachem Guide: The Fitness for Purpose of Analytical Methods—A Laboratory Guide to Method Validation and Related Topics, 2nd ed.; Eurachem: Torino, Italy, 2014; ISBN 978-91-87461-59-0. [Google Scholar]
- Nenadis, N.; Mastralexi, A.; Tsimidou, M.Z.; Vichi, S.; Quintanilla-Casas, B.; Donarski, J.; Bailey-Horne, V.; Butinar, B.; Miklavčič, M.; García-González, D.L.; et al. Toward a harmonized and standardized protocol for the determination of total hydroxytyrosol and tyrosol content in virgin olive oil (VOO). Extraction solvent. Eur. J. Lipid Sci. Technol. 2018, 120, 1800099. [Google Scholar] [CrossRef]
- Paiva-Martins, F.; Gordon, M.H. Interactions of ferric ions with olive oil phenolic compounds. J. Agric. Food Chem. 2005, 53, 2704–2709. [Google Scholar] [CrossRef] [PubMed]
- Zafra-Gómez, A.; Luzón-Toro, B.; Capel-Cuevas, S.; Morales, J.C. Stability of hydroxytyrosol in aqueous solutions at different concentration, temperature and with different ionic content: A study using UPLC-MS. Food Nutr. Sci. 2011, 19, 1114–1120. [Google Scholar] [CrossRef]
- Shneider, S. Quality Analysis of Extra Virgin Olive Oils—Part 6 Nutritive Benefits—Phenolic Compounds in Virgin Olive Oil; Application Note Food, Testing and Agriculture. 2016. Available online: https://www.agilent.com/cs/library/applications/5991-3801EN.pdf (accessed on 11 February 2019).
- Available online: https://www.shimadzu.com/an/hplc/column/feature2.html (accessed on 11 February 2019).
- Xu, Q.A. Ultra-High Performance Liquid Chromatography and Its Applications; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Available online: https://www.thermofisher.com/gr/en/home/global/forms/industrial/lc-method-transfer-calculator.html (accessed on 11 February 2019).
- Di Maio, I.; Esposto, S.; Taticchi, A.; Selvaggini, R.; Veneziani, G.; Urbani, S.; Servili, M. HPLC–ESI-MS investigation of tyrosol and hydroxytyrosol oxidation products in virgin olive oil. Food Chem. 2011, 125, 21–28. [Google Scholar] [CrossRef]
- Barwick, V.J. Preparation of Calibration Curves: A Guide to Best Practice; VAM: Teddington, UK, 2003. [Google Scholar]
- Godoy-Caballero, M.P.; Acedo-Valenzuela, M.I.; Galeano-Diaz, T. New reversed phase dispersive liquid–liquid microextraction method for the determination of phenolic compounds in virgin olive oil by rapid resolution liquid chromatography with ultraviolet–visible and mass spectrometry detection. J. Chromatogr. A 2013, 1313, 291–301. [Google Scholar] [CrossRef]
- Celano, R.; Piccinelli, A.L.; Pugliese, A.; Carabetta, S.; Di Sanzo, R.; Rastrelli, L.; Russo, M. Insights into analysis of phenolic secoiridoids in extra virgin olive oil. J. Agric. Food Chem. 2018, 66, 6053–6063. [Google Scholar] [CrossRef]
- Long, G.L.; Winefordner, J.D. Limit of detection. A closer look at the IUPAC definition. Anal. Chem. 1983, 55, 712A–724A. [Google Scholar]
- Lozano-Sánchez, J.; Segura-Carretero, A.; Menendez, J.A.; Oliveras-Ferraros, C.; Cerretani, L.; Fernandez-Gutierrez, A. Prediction of extra virgin olive oil varieties through their phenolic profile. Potential cytotoxic activity against human breast cancer cells. J. Agric. Food Chem. 2010, 58, 9942–9955. [Google Scholar] [CrossRef]
- Becerra-Herrera, M.; Sánchez-Astudillo, M.; Beltrán, R.; Sayago, A. Determination of phenolic compounds in olive oil: New method based on liquid–liquid micro extraction and ultra-high performance liquid chromatography-triple–quadrupole mass spectrometry. LWT-Food Sci. Technol. 2014, 57, 49–57. [Google Scholar] [CrossRef]
- Ballus, C.A.; Quirantes-Piné, R.; Bakhouche, A.; da Silva, L.F.D.O.; de Oliveira, A.F.; Coutinho, E.F.; da Croce, D.M.; Segura-Carretero, A.; Godoy, H.T. Profile of phenolic compounds of Brazilian virgin olive oils by rapid resolution liquid chromatography coupled to electrospray ionisation time-of-flight mass spectrometry (RRLC–ESI-TOF-MS). Food Chem. 2015, 170, 366–377. [Google Scholar] [CrossRef]
- International Conference on Harmonization (ICH) of Technical Requirements for the Registration of Pharmaceuticals for Human Use, Validation of Analytical Procedures: Text and Methodology; ICH-Q2B; ICH: Geneva, Switzerland, 1996.
- Guidelines for standard method performance requirements. In Official Methods of Analysis, 20th ed.; AOAC International: Rockville, MD, USA, 2016; Appendix F; pp. 1–18.
- Alcarón-Flores, M.I.; Romero-González, R.; Frenich, A.G.; Vidal, J.L.M. Analysis of phenolic compounds in olive oil by solid-phase extraction and ultra high performance liquid chromatography–tandem mass spectrometry. Food Chem. 2012, 134, 2465–2472. [Google Scholar] [CrossRef] [PubMed]
- Zar, J.H. Comparing simple linear regression equations. In Biostatistical Analysis: Pearson New International Edition, 5th ed.; Pearson Prentice Hall: Harlow, UK, 2014; pp. 387–404. [Google Scholar]
- De Mendoza, A.E.H.; Campanero, M.A.; Mollinedo, F.; Blanco-Prieto, M.J. Comparative study of A HPLC–MS assay versus an UHPLC–MS/MS for anti-tumoral alkyl lysophospholipid edelfosine determination in both biological samples and in lipid nanoparticulate systems. J. Chromatogr. B 2009, 877, 4035–4041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.N.; Miller, J.C. Statistics and Chemometrics for Analytical Chemistry, 6th ed.; Pearson Education Ltd.: Harlow, UK, 2010. [Google Scholar]
- Shrivastava, A.; Gupta, V. Methods for the determination of limit of detection and limit of quantitation of the analytical methods. Chron. Young Sci. 2011, 2, 21–25. [Google Scholar] [CrossRef]
Sample Availability: Not available by the authors. |



{kind=link}
{kind=link}
{kind=link}
| Samples | Total Phenol Content * | Total Htyr Content | Total Tyr Content |
|---|---|---|---|
| mg/20 g Oil | |||
| VOO-1IOC | 4.74 ± 0.47 b | 3.79 ± 0.32 a | 2.39 ± 0.35 a |
| VOO-1IOCmodified | 5.37 ± 0.32 a | 3.94 ± 0.31 a | 2.25 ± 0.20 a |
| VOO-2IOC | 10.97 ± 0.42 b | 3.80 ± 0.10 a | 5.60 ± 0.18 a |
| VOO-2IOCmodified | 11.62 ± 0.32 a | 3.77 ± 0.03 a | 5.67 ± 0.17 a |
| VOO-3IOC | 5.96 ± 0.20 a | 2.45 ± 0.18 b | 2.99 ± 0.21 b |
| VOO-3IOCmodified | 5.71 ± 0.40 a | 2.81 ± 0.04 a | 3.50 ± 0.02 a |
| Sample | Intra-Day | Inter-Day | ||||||
|---|---|---|---|---|---|---|---|---|
| Total Htyr Content (mg/20 g) | %RSD | Total Tyr Content (mg/20 g) | %RSD | Total Htyr Content (mg/20 g) | %RSD | Total Tyr Content (mg/20 g) | %RSD | |
| VOO-4 | 3.11 ± 0.32 | 10.4 | 5.29 ± 0.31 | 5.9 | 3.03 ± 0.31 | 10.2 | 5.17 ± 0.14 | 2.7 |
| VOO-5 | 1.76 ± 0.11 | 6.0 (n = 5) | 3.37 ± 0.12 | 3.6 (n = 5) | 1.75 ± 0.11 | 6.0 (n = 15) | 3.22 ± 0.19 | 5.8 (n = 15) |
| VOO-6 | 5.55 ± 0.18 | 3.2 | 3.76 ± 0.05 | 1.2 | 5.66 ± 0.12 | 2.1 | 3.66 ± 0.13 | 3.6 |
| ROO-1 * | 0.95 ± 0.06 | 6.4 | 1.73 ± 0.04 | 2.5 | 0.97 ± 0.07 | 7.4 | 1.67 ± 0.12 | 6.9 |
| ROO-2 * | 1.74 ± 0.05 | 2.6 (n = 15) | 3.22 ± 0.10 | 3.0 (n = 15) | 1.79 ± 0.08 | 4.4 (n = 45) | 3.28 ± 0.15 | 4.6 (n = 45) |
| ROO-3 * | 2.67 ± 0.12 | 4.4 | 4.48 ± 0.15 | 3.4 | 2.71 ± 0.11 | 4.2 | 4.58 ± 0.23 | 5.1 |
| Analyte | Working Range | y = a + bx | b ± SD | a ± SD | |
|---|---|---|---|---|---|
| μg/mL | r2 | ||||
| 280 nm | |||||
| 1st column, batch A | |||||
| Htyr | 0.5–100 | 0.996 | y = 11,176.4x + 8415 | 11,176.4 ± 110.6 | 8415 ± 5323 |
| Tyr | 0.5–100 | 0.996 | y = 6109.7x + 4331 | 6109.7 ± 81.2 | 4331 ± 4268 |
| 2nd column, batch B | |||||
| Htyr | 0.5–100 | 0.997 | y = 11,817.4x + 17,025 | 11,817.4 ± 135.5 | 17,025 ± 6520 |
| Tyr | 0.5–100 | 0.994 | y = 7261.9x + 8640 | 7261.9 ± 119.6 | 8640 ± 6288 |
| 3rd column, batch A | |||||
| Htyr | 0.5–100 | 0.999 | y = 10,954.8x − 751 | 10,954.8 ± 62.7 | 751 ± 3019 |
| Tyr | 0.5–100 | 0.998 | y = 6187.8x + 15,359 | 6187.8 ± 62.9 | 15,359 ± 3308 |
| Analyst | Sample | Total Htyr Content | Total Tyr Content |
|---|---|---|---|
| (mg/20 g Oil) | |||
| 1 | VOO-4 | 2.96 ± 0.33 a,* | 5.08 ± 0.26 a,* |
| 2 | 3.11 ± 0.32 a,* | 5.29 ± 0.31 a,* | |
| 1 | VOO-5 | 1.85 ± 0.02 a | 3.45 ± 0.01 a |
| 2 | 1.63 ± 0.03 b | 2.91 ± 0.02 b | |
| 1 | VOO-6 | 5.76 ± 0.01 a | 3.72 ± 0.01 a |
| 2 | 5.68 ± 0.03 a | 3.59 ± 0.03 b | |
| 1 | VOO-7 | 4.52 ± 0.46 a | 5.70 ± 0.02 a |
| 2 | 4.08 ± 0.04 a | 3.86 ± 0.02 b | |
| 1 | VOO-8 | 1.14 ± 0.01 a | 1.54 ± 0.01 a |
| 2 | 1.01 ± 0.02 b | 1.17 ± 0.02 b | |
| Samples | F-C Assay | UHPLC-DAD | HPLC-DAD | ||
|---|---|---|---|---|---|
| Total Phenol Content | Total Htyr Content | Total Tyr Content | Total Htyr Content | Total Tyr Content | |
| (mg CA/20 g Oil) | (mg/20 g Oil) | ||||
| VOO-1 | 4.74 ± 0.47 | 4.33 ± 0.03 a | 2.64 ± 0.10 a | 3.79 ± 0.32 b | 2.39 ± 0.35 a |
| VOO-2 | 10.97 ± 0.42 | 4.72 ± 0.09 a | 7.33 ± 0.23 a | 3.80 ± 0.10 b | 5.60 ± 0.18 b |
| VOO-3 | 5.96 ± 0.42 | 3.41 ± 0.45 a | 4.15 ± 0.41 a | 2.45 ± 0.18 b | 2.99 ± 0.21 b |
| VOO-9 | 2.18 ± 0.14 | 2.38 ± 0.01 a | 3.64 ± 0.06 a | 2.06 ± 0.02 b | 3.27 ± 0.08 b |
| VOO-10 | 3.41 ± 0.24 | 3.28 ± 0.01 a | 4.47 ± 0.03 a | 3.19 ± 0.03 b | 4.34 ± 0.04 b |
| VOO-11 | 3.17 ± 0.04 | 2.34 ± 0.00 b | 3.37 ± 0.01 b | 2.90 ± 0.01 a | 3.91 ± 0.19 a |
| VOO-12 | 2.85 ± 0.20 | 2.69 ± 0.04 b | 5.08 ± 0.01 b | 3.25 ± 0.19 a | 5.80 ± 0.20 a |
| VOO-13 | 3.52 ± 0.07 | 2.65 ± 0.07 a | 2.98 ± 0.02 a | 2.25 ± 0.06 b | 2.84 ± 0.29 a |
| F-C Assay | UHPLC-DAD | HPLC-DAD | |||
|---|---|---|---|---|---|
| Samples | Total Phenol Content | Total Htyr Content | Total Tyr Content | Total Htyr Content | Total Tyr Content |
| (mg CA/20 g Oil) | (mg/20 g Oil) | ||||
| VOO-1 | 4.74 ± 0.47 | 4.84 ± 0.22 a | 2.78 ± 0.18 a | 4.05 ± 0.05 b | 2.39 ± 0.04 a |
| VOO-2 | 10.97 ± 0.42 | 4.48 ± 0.16 a | 7.73 ± 0.40 a | 4.10 ± 0.20 b | 5.95 ± 0.15 b |
| VOO-3 | 5.96 ± 0.42 | 3.64 ± 0.02 a | 4.72 ± 0.10 a | 3.29 ± 0.65 a | 3.84 ± 0.30 b |
| VOO-9 | 2.18 ± 0.14 | 4.42 ± 0.01 a | 5.73 ± 0.03 b | 3.73 ± 0.05 b | 5.95 ± 0.14 a |
| VOO-10 | 3.41 ± 0.24 | 3.11 ± 0.01 b | 4.43 ± 0.05 b | 5.87 ± 0.14 a | 7.71 ± 0.18 a |
| VOO-11 | 3.17 ± 0.04 | 2.90 ± 0.01 b | 4.34 ± 0.03 b | 3.95 ± 0.09 a | 4.77 ± 0.23 a |
| VOO-12 | 2.85 ± 0.20 | 3.68 ± 0.03 a | 5.79 ± 0.03 b | 4.68 ± 0.02 b | 7.87 ± 0.04 a |
| VOO-13 | 3.52 ± 0.07 | 3.61 ± 0.02 a | 3.95 ± 0.03 b | 3.85 ± 0.19 a | 4.32 ± 0.18 a |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsimidou, M.Z.; Sotiroglou, M.; Mastralexi, A.; Nenadis, N.; García-González, D.L.; Gallina Toschi, T. In House Validated UHPLC Protocol for the Determination of the Total Hydroxytyrosol and Tyrosol Content in Virgin Olive Oil Fit for the Purpose of the Health Claim Introduced by the EC Regulation 432/2012 for “Olive Oil Polyphenols”. Molecules 2019, 24, 1044. https://doi.org/10.3390/molecules24061044
Tsimidou MZ, Sotiroglou M, Mastralexi A, Nenadis N, García-González DL, Gallina Toschi T. In House Validated UHPLC Protocol for the Determination of the Total Hydroxytyrosol and Tyrosol Content in Virgin Olive Oil Fit for the Purpose of the Health Claim Introduced by the EC Regulation 432/2012 for “Olive Oil Polyphenols”. Molecules. 2019; 24(6):1044. https://doi.org/10.3390/molecules24061044
Chicago/Turabian StyleTsimidou, Maria Z., Michaela Sotiroglou, Aspasia Mastralexi, Nikolaos Nenadis, Diego L. García-González, and Tullia Gallina Toschi. 2019. "In House Validated UHPLC Protocol for the Determination of the Total Hydroxytyrosol and Tyrosol Content in Virgin Olive Oil Fit for the Purpose of the Health Claim Introduced by the EC Regulation 432/2012 for “Olive Oil Polyphenols”" Molecules 24, no. 6: 1044. https://doi.org/10.3390/molecules24061044