High Consistency of Structure-Based Design and X-Ray Crystallography: Design, Synthesis, Kinetic Evaluation and Crystallographic Binding Mode Determination of Biphenyl-N-acyl-β-d-Glucopyranosylamines as Glycogen Phosphorylase Inhibitors

 ,
,  and
and 
Abstract
:
1. Introduction
2. Results
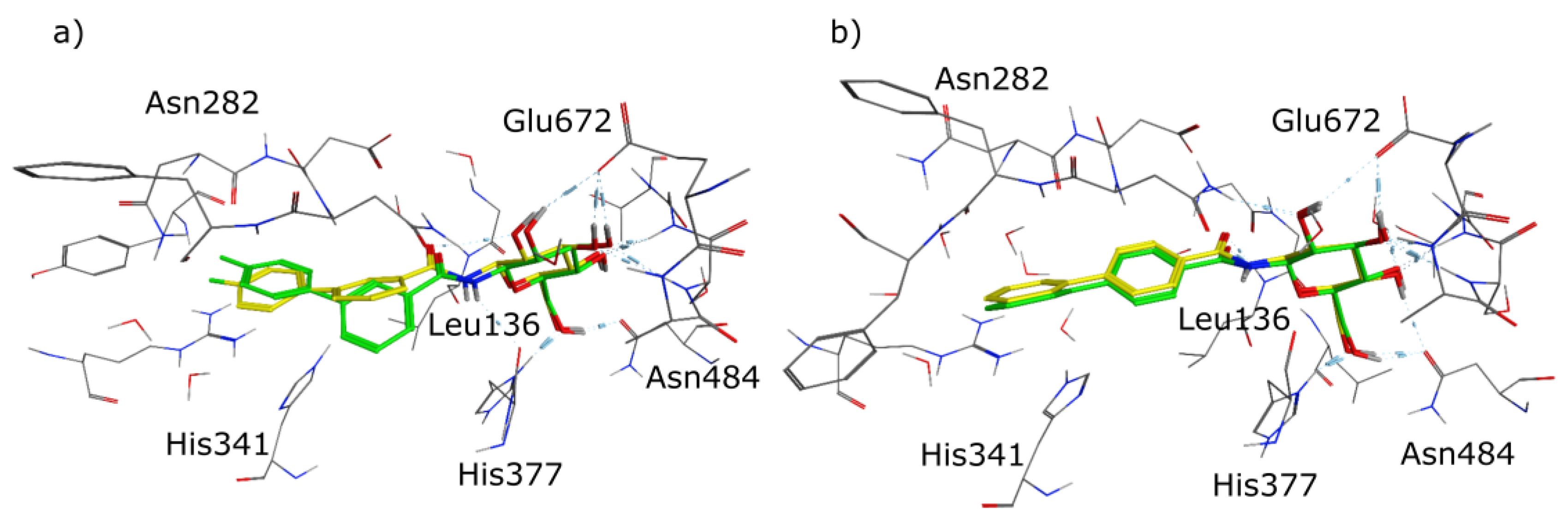
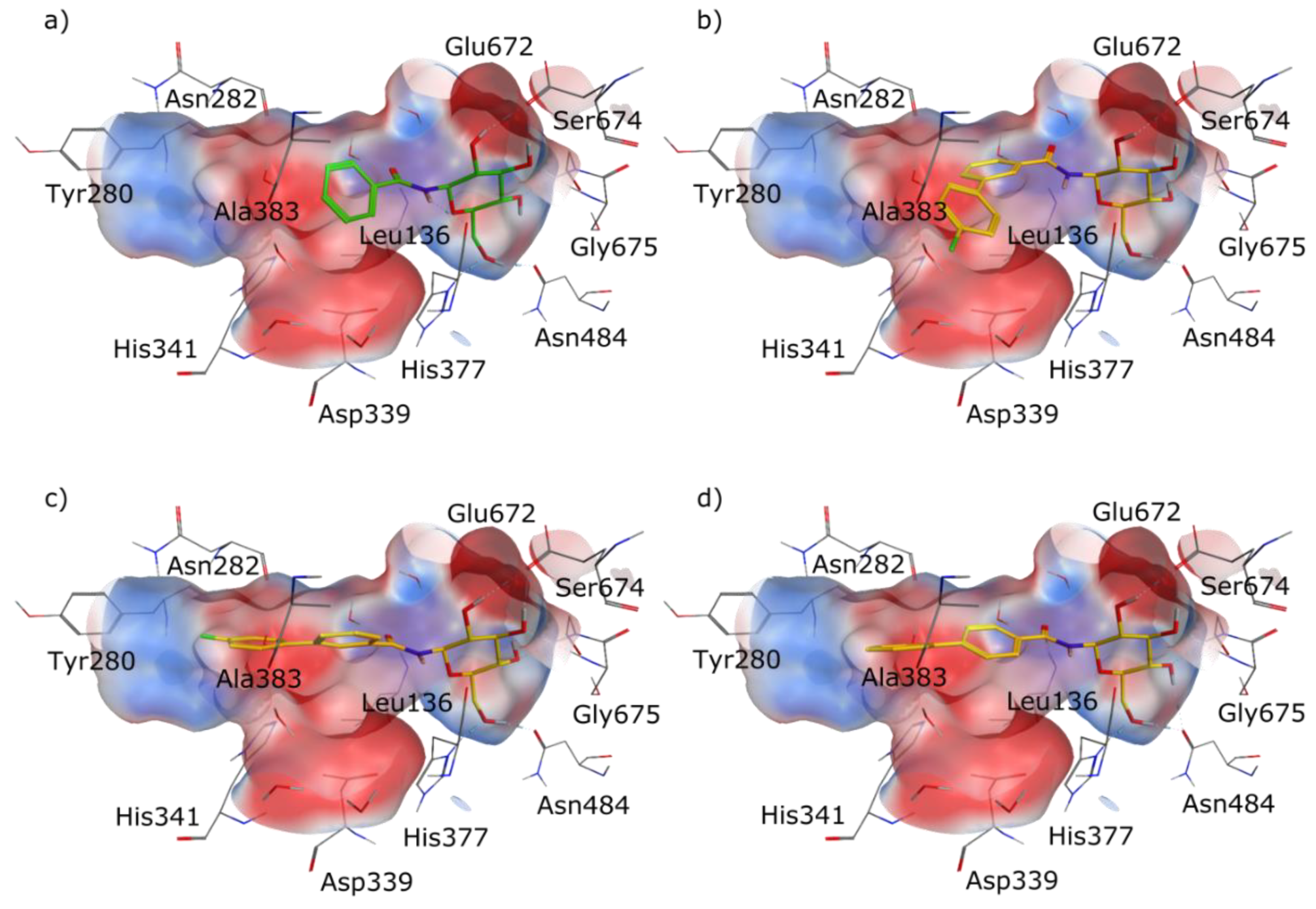
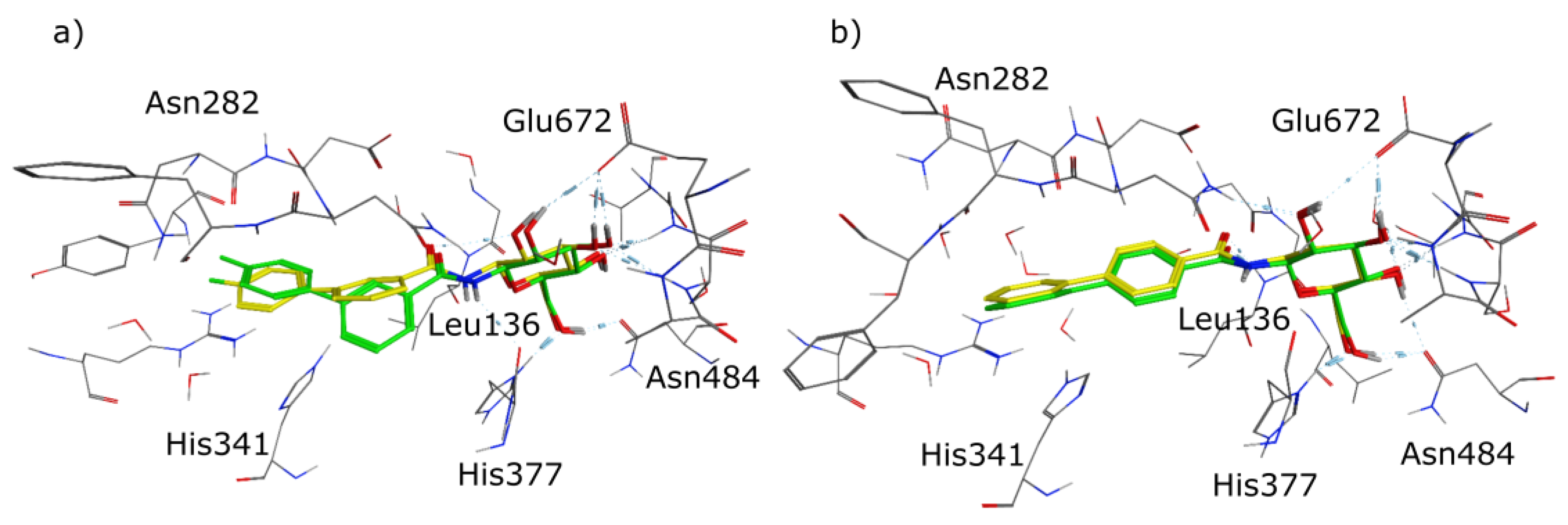
2.1. Molecular Modelling
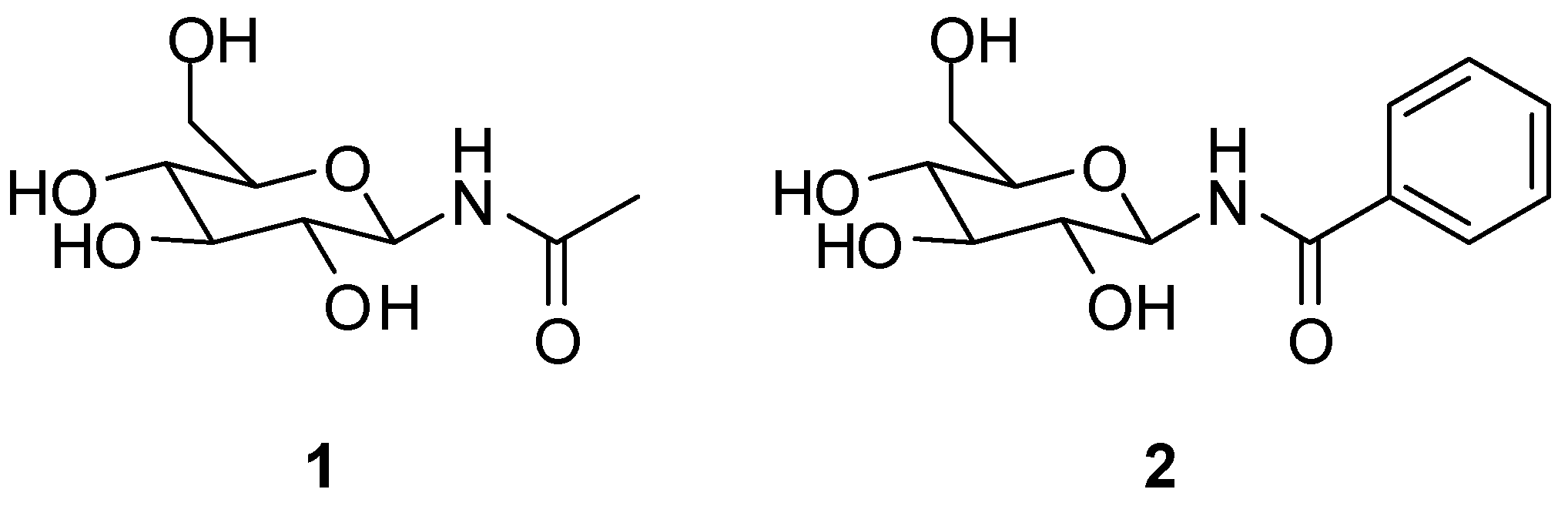
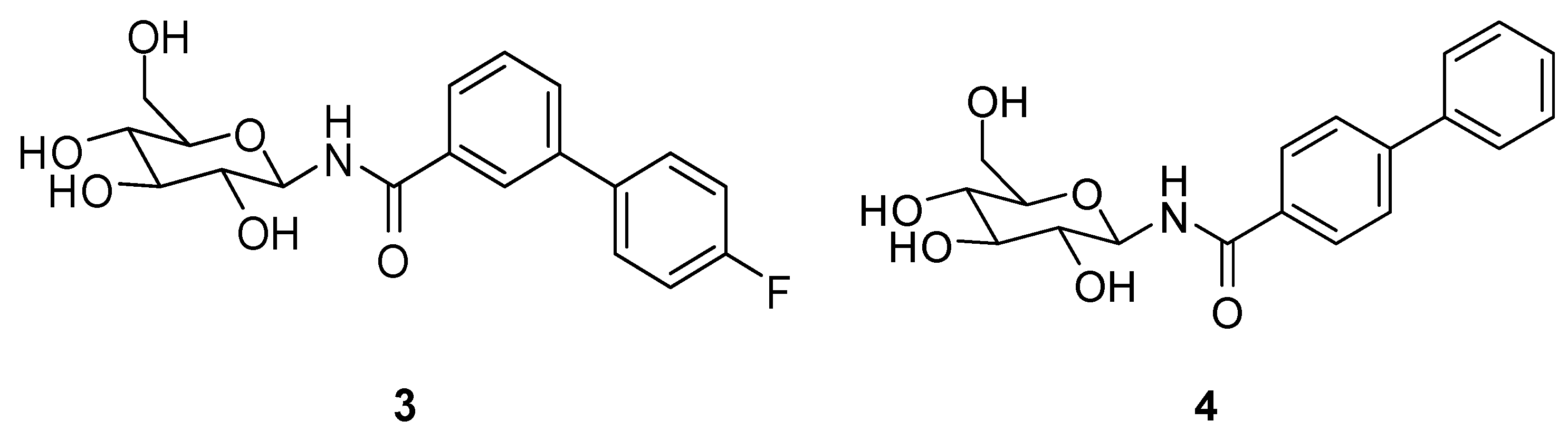
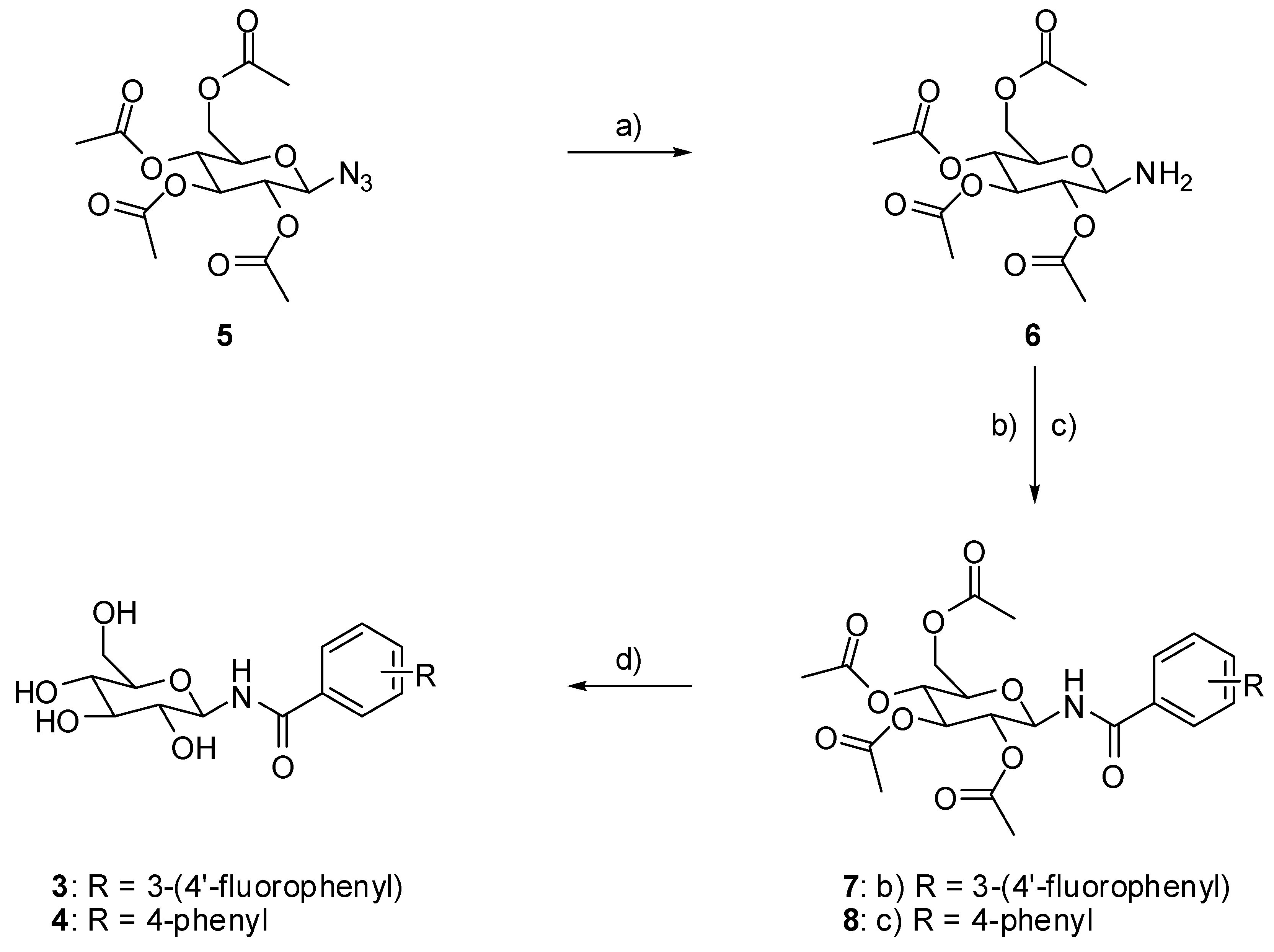
2.2. Chemistry
2.3. Biological Evaluation
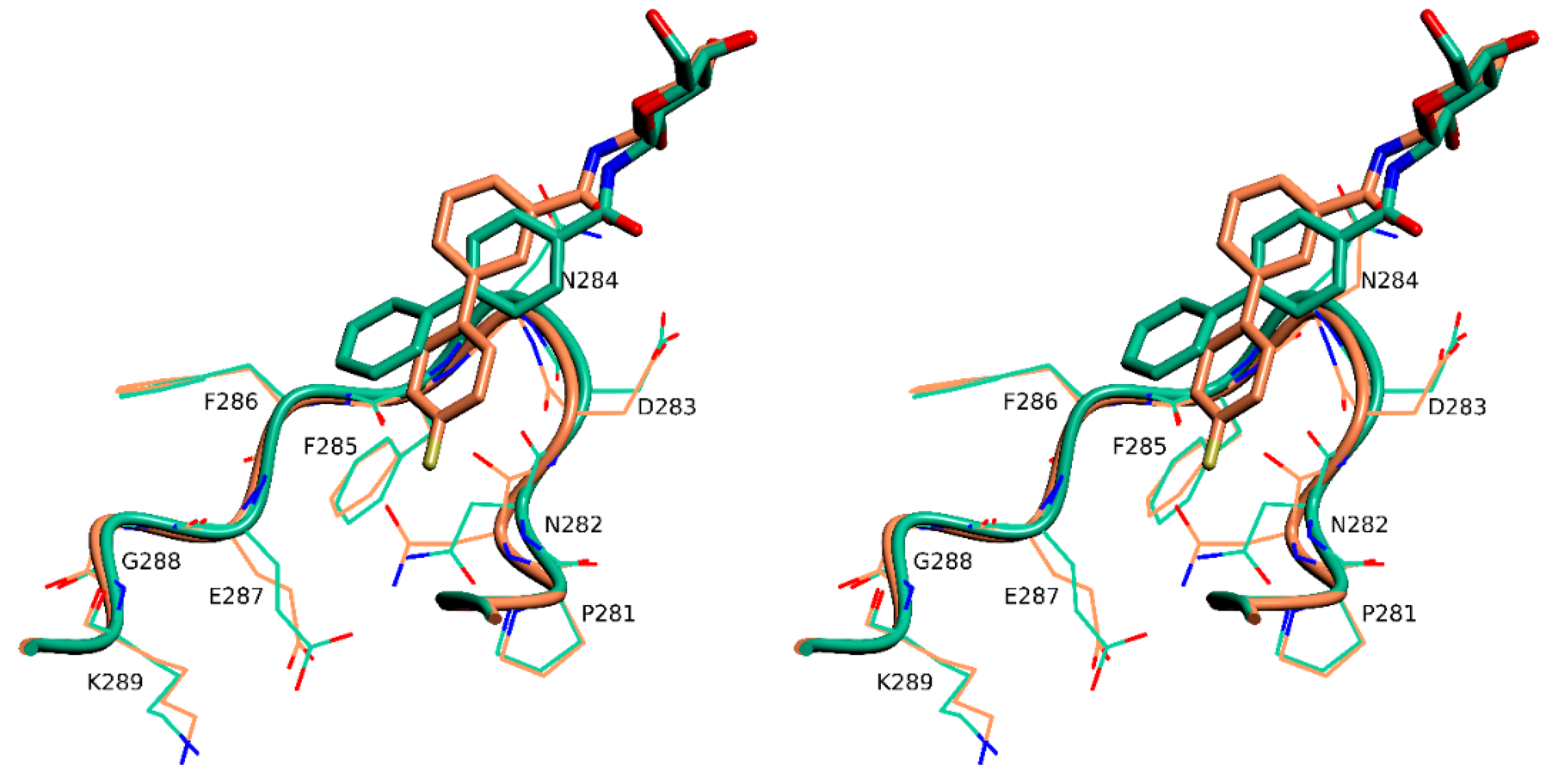
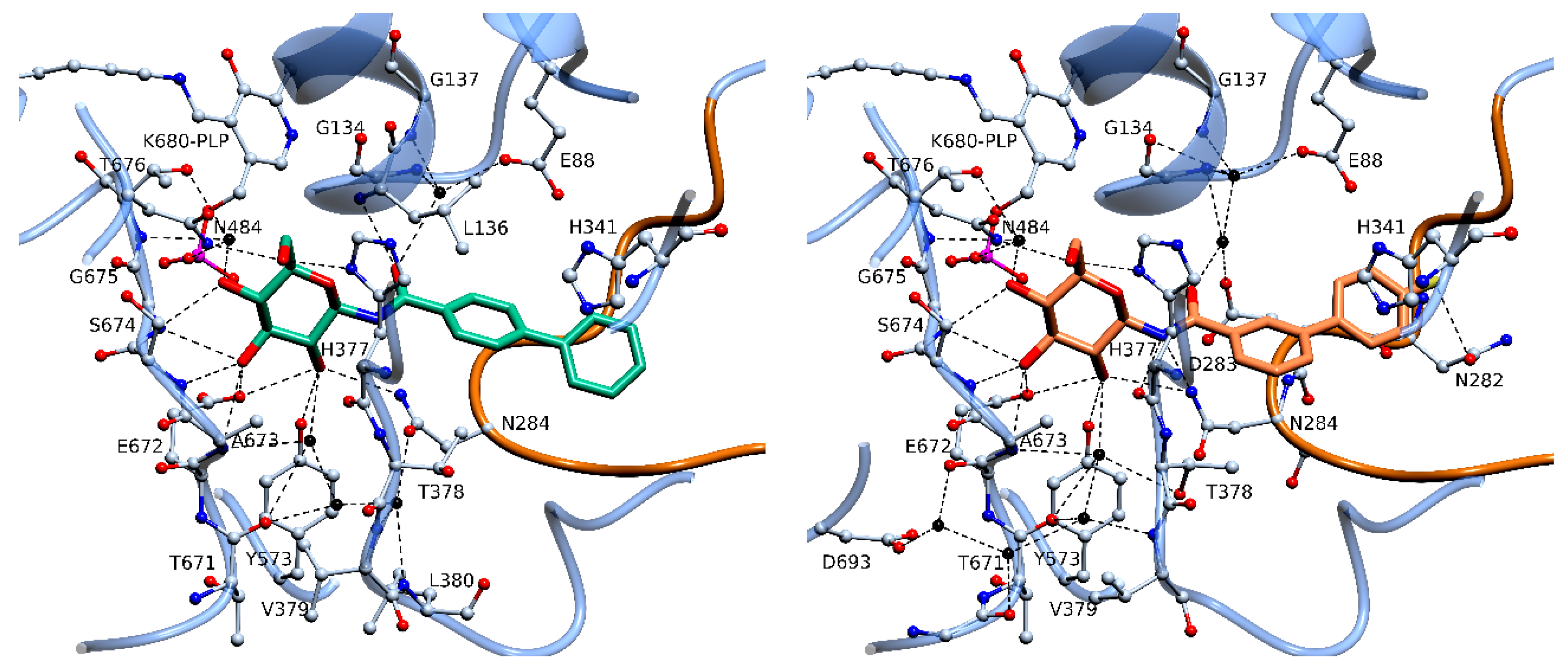
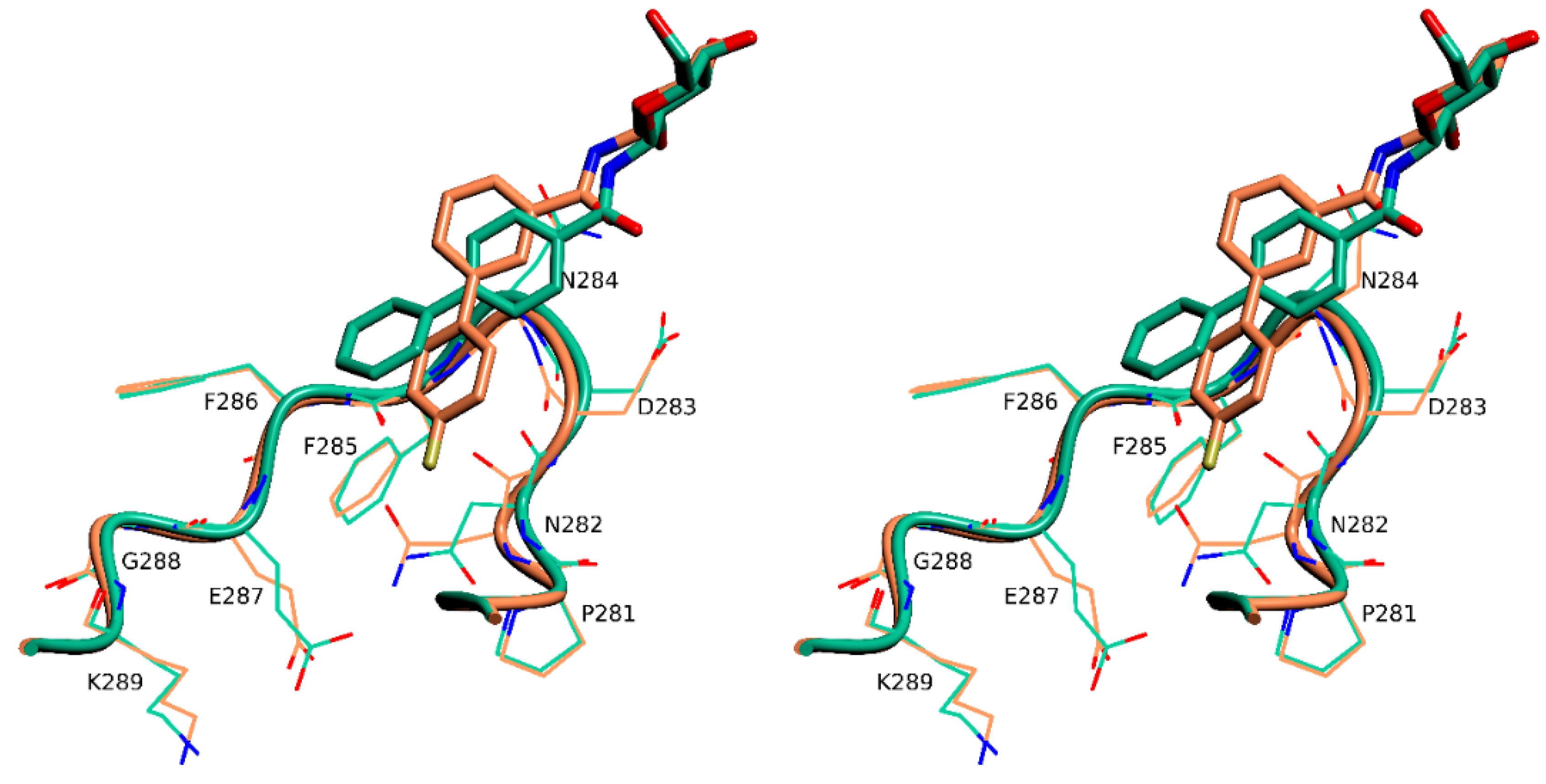
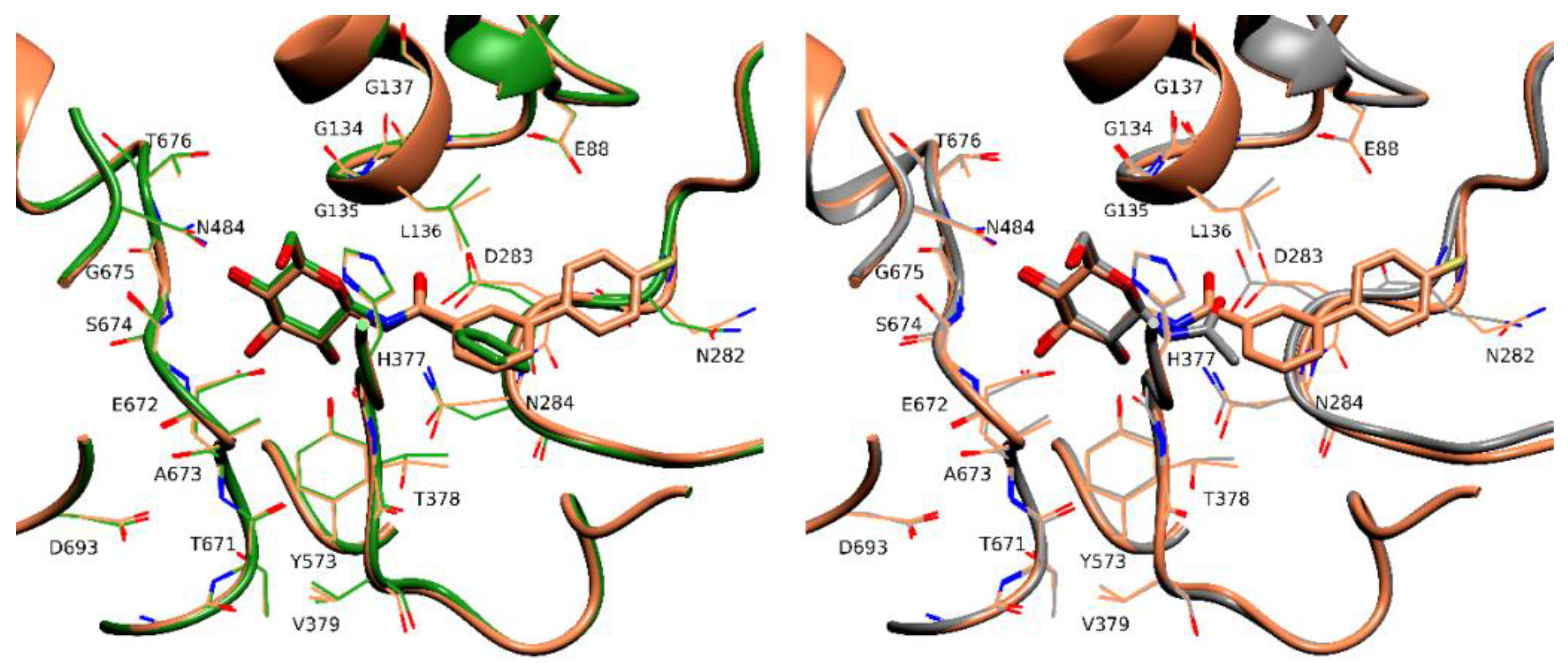
2.4. X-Ray Crystallography
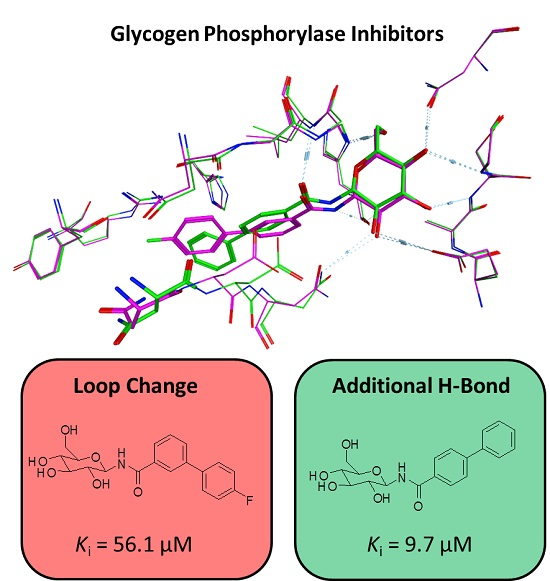
3. Discussion
4. Materials and Methods
4.1. General
4.2. Chemistry
4.3. In Silico Studies
4.4. Kinetics
4.5. X-Ray Crystallography
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Donnier-Maréchal, M.; Vidal, S. Glycogen phosphorylase inhibitors: A patent review (2013–2015). Expert Opin. Ther. Pat. 2016, 26, 199–212. [Google Scholar]
- Hayes, J.M.; Kantsadi, A.L.; Leonidas, D.D. Natural products and their derivatives as inhibitors of glycogen phosphorylase: Potential treatment for type 2 diabetes. Phytochem. Rev. 2014, 13, 471–498. [Google Scholar]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [PubMed]
- Oikonomakos, N.G. Glycogen phosphorylase as a molecular target for type 2 diabetes therapy. Curr. Protein Pept. Sci. 2002, 3, 561–586. [Google Scholar] [CrossRef]
- Leonidas, D.D.; Oikonomakos, N.G.; Papageorgiou, A.C.; Xenakis, A.; Cazianis, C.T.; Bem, F. The ammonium sulfate activation of phosphorylase b. FEBS Lett. 1990, 261, 23–27. [Google Scholar]
- Somsák, L. Glucose derived inhibitors of glycogen phosphorylase. Comptes Rendus Chim. 2011, 14, 211–223. [Google Scholar] [CrossRef]
- Watson, K.A.; Mitchell, E.P.; Johnson, L.N.; Cruciani, G.; Son, J.C.; Bichard, C.J.; Fleet, G.W.; Oikonomakos, N.G.; Kontou, M.; Zographos, S.E. Glucose analogue inhibitors of glycogen phosphorylase: From crystallographic analysis to drug prediction using GRID force-field and GOLPE variable selection. Acta Crystallogr. D Biol. Crystallogr. 1995, 51, 458–472. [Google Scholar]
- Watson, K.A.; Mitchell, E.P.; Johnson, L.N.; Bichard, C.J.F.; Orchard, M.G.; Fleet, G.W.J.; Oikonomakos, N.G.; Leonidas, D.D.; Son, J.C. Design of Inhibitors of Glycogen Phosphorylase: A Study of .alpha.- and .beta.-C-Glucosides and 1-Thio-.beta.-D-glucose Compounds. Biochemistry 1994, 33, 5745–5758. [Google Scholar]
- Chrysina, E.D.; Bokor, É.; Alexacou, K.M.; Charavgi, M.D.; Oikonomakos, G.N.; Zographos, S.E.; Leonidas, D.D.; Oikonomakos, N.G.; Somsák, L. Amide-1,2,3-triazole bioisosterism: The glycogen phosphorylase case. Tetrahedron Asymmetry 2009, 20, 733–740. [Google Scholar]
- Kerekes, A.D.; Esposite, S.J.; Doll, R.J.; Tagat, J.R.; Yu, T.; Xiao, Y.; Zhang, Y.; Prelusky, D.B.; Tevar, S.; Gray, K.; et al. Aurora Kinase Inhibitors Based on the Imidazo[1,2-a]pyrazine Core: Fluorine and Deuterium Incorporation Improve Oral Absorption and Exposure. J. Med. Chem. 2011, 54, 201–210. [Google Scholar] [PubMed]
- Yamazaki, S.; Shen, Z.; Jiang, Y.; Smith, B.J.; Vicini, P. Application of Target-Mediated Drug Disposition Model to Small Molecule Heat Shock Protein 90 Inhibitors. Drug Metab. Dispos. 2013, 41, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.A.K.; Hoy, V.J.; O’Hagan, D.; Smith, G.T. How good is fluorine as a hydrogen bond acceptor? Tetrahedron 1996, 52, 12613–12622. [Google Scholar]
- Auffinger, P.; Hays, F.A.; Westhof, E.; Ho, P.S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 16789–16794. [Google Scholar] [PubMed]
- Fischer, T.; Riedl, R. Targeted Fluoro Positioning for the Discovery of a Potent and Highly Selective Matrix Metalloproteinase Inhibitor. ChemistryOpen 2017, 6, 192–195. [Google Scholar]
- Kyriakis, E.; Solovou, T.G.A.; Kun, S.; Czifrák, K.; Szőcs, B.; Juhász, L.; Bokor, É.; Stravodimos, G.A.; Kantsadi, A.L.; Chatzileontiadou, D.S.M.; et al. Probing the β-pocket of the active site of human liver glycogen phosphorylase with 3-(C-β-d-glucopyranosyl)-5-(4-substituted-phenyl)-1, 2, 4-triazole inhibitors. Bioorganic Chem. 2018, 77, 485–493. [Google Scholar]
- Oikonomakos, N.G.; Kontou, M.; Zographos, S.E.; Watson, K.A.; Johnson, L.N.; Bichard, C.J.F.; Fleet, G.W.J.; Acharya, K.R. N-acetyl-β-D-glucopyranosylamine: A potent T-state inhibitor of glycogen phosphorylase. A comparison with α-D-glucose. Protein Sci. 1995, 4, 2469–2477. [Google Scholar]
- Newgard, C.B.; Nakano, K.; Hwang, P.K.; Fletterick, R.J. Sequence analysis of the cDNA encoding human liver glycogen phosphorylase reveals tissue-specific codon usage. Proc. Natl. Acad. Sci. USA 1986, 83, 8132–8136. [Google Scholar]
- Rath, V.L.; Ammirati, M.; LeMotte, P.K.; Fennell, K.F.; Mansour, M.N.; Danley, D.E.; Hynes, T.R.; Schulte, G.K.; Wasilko, D.J.; Pandit, J. Activation of Human Liver Glycogen Phosphorylase by Alteration of the Secondary Structure and Packing of the Catalytic Core. Mol. Cell 2000, 6, 139–148. [Google Scholar] [CrossRef]
- Kyriakis, E.; Stravodimos, G.A.; Kantsadi, A.L.; Chatzileontiadou, D.S.; Skamnaki, V.T.; Leonidas, D.D. Natural flavonoids as antidiabetic agents. The binding of gallic and ellagic acids to glycogen phosphorylase b. FEBS Lett. 2015, 589, 1787–1794. [Google Scholar] [PubMed]
- Kantsadi, A.L.; Bokor, É.; Kun, S.; Stravodimos, G.A.; Chatzileontiadou, D.S.M.; Leonidas, D.D.; Juhász-Tóth, É.; Szakács, A.; Batta, G.; Docsa, T.; et al. Synthetic, enzyme kinetic, and protein crystallographic studies of C-β-d-glucopyranosyl pyrroles and imidazoles reveal and explain low nanomolar inhibition of human liver glycogen phosphorylase. Eur. J. Med. Chem. 2016, 123, 737–745. [Google Scholar]
- Somsák, L.; Czifrák, K.; Tóth, M.; Bokor, E.; Chrysina, E.D.; Alexacou, K.-M.; Hayes, J.M.; Tiraidis, C.; Lazoura, E.; Leonidas, D.D.; et al. New inhibitors of glycogen phosphorylase as potential antidiabetic agents. Curr. Med. Chem. 2008, 15, 2933–2983. [Google Scholar] [PubMed]
- Project, C.C. Number 4 The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994, 50, 760–763. [Google Scholar]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [PubMed]
- Gerber, P.R.; Müller, K. MAB, a generally applicable molecular force field for structure modelling in medicinal chemistry. J. Comput. Aided Mol. Des. 1995, 9, 251–268. [Google Scholar]
- Fischer, E.H.; Krebs, E.G. [49a] Muscle phosphorylase b: x Glucose-1-phosphate+ Gn ⇄ Gn+x + x inorganic phosphate(where Gn designates glycogen containing n glucose residues). In Methods in Enzymology; Academic Press: Boston, MA, USA, 1962; Volume 5, pp. 369–373. [Google Scholar]
- Hayes, J.M.; Skamnaki, V.T.; Archontis, G.; Lamprakis, C.; Sarrou, J.; Bischler, N.; Skaltsounis, A.-L.; Zographos, S.E.; Oikonomakos, N.G. Kinetics, in silico docking, molecular dynamics, and MM-GBSA binding studies on prototype indirubins, KT5720, and staurosporine as phosphorylase kinase ATP-binding site inhibitors: The role of water molecules examined. Proteins Struct. Funct. Bioinforma. 2011, 79, 703–719. [Google Scholar]
- Saheki, S.; Takeda, A.; Shimazu, T. Assay of inorganic phosphate in the mild pH range, suitable for measurement of glycogen phosphorylase activity. Anal. Biochem. 1985, 148, 277–281. [Google Scholar]
- Leatherbarrow, R.J. GraFit Version 6.0; Erithacus Software Limited: Staines, UK, 2007. [Google Scholar]
- Agilent Technologies UK Ltd. CrysAlisPro Software System, 1.171.35.11; Agilent Technologies UK Ltd.: Oxford, UK, 2011. [Google Scholar]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar]
- Vagin, A.A.; Steiner, R.A.; Lebedev, A.A.; Potterton, L.; McNicholas, S.; Long, F.; Murshudov, G.N. REFMAC5 dictionary: Organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2184–2195. [Google Scholar] [PubMed]
- Long, F.; Nicholls, R.A.; Emsley, P.; Gražulis, S.; Merkys, A.; Vaitkus, A.; Murshudov, G.N. AceDRG: A stereochemical description generator for ligands. Acta Crystallogr. Sect. Struct. Biol. 2017, 73, 112–122. [Google Scholar]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [PubMed]
- Joosten, R.P.; Long, F.; Murshudov, G.N.; Perrakis, A. The PDB_REDO server for macromolecular structure model optimization. IUCrJ 2014, 1, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [PubMed]
- McNicholas, S.; Potterton, E.; Wilson, K.S.; Noble, M.E.M. Presenting your structures: The CCP4mg molecular-graphics software. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 386–394. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds 3 and 4 are available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fischer, T.; Koulas, S.M.; Tsagkarakou, A.S.; Kyriakis, E.; Stravodimos, G.A.; Skamnaki, V.T.; Liggri, P.G.V.; Zographos, S.E.; Riedl, R.; Leonidas, D.D. High Consistency of Structure-Based Design and X-Ray Crystallography: Design, Synthesis, Kinetic Evaluation and Crystallographic Binding Mode Determination of Biphenyl-N-acyl-β-d-Glucopyranosylamines as Glycogen Phosphorylase Inhibitors. Molecules 2019, 24, 1322. https://doi.org/10.3390/molecules24071322
Fischer T, Koulas SM, Tsagkarakou AS, Kyriakis E, Stravodimos GA, Skamnaki VT, Liggri PGV, Zographos SE, Riedl R, Leonidas DD. High Consistency of Structure-Based Design and X-Ray Crystallography: Design, Synthesis, Kinetic Evaluation and Crystallographic Binding Mode Determination of Biphenyl-N-acyl-β-d-Glucopyranosylamines as Glycogen Phosphorylase Inhibitors. Molecules. 2019; 24(7):1322. https://doi.org/10.3390/molecules24071322
Chicago/Turabian StyleFischer, Thomas, Symeon M. Koulas, Anastasia S. Tsagkarakou, Efthimios Kyriakis, George A. Stravodimos, Vassiliki T. Skamnaki, Panagiota G.V. Liggri, Spyros E. Zographos, Rainer Riedl, and Demetres D. Leonidas. 2019. "High Consistency of Structure-Based Design and X-Ray Crystallography: Design, Synthesis, Kinetic Evaluation and Crystallographic Binding Mode Determination of Biphenyl-N-acyl-β-d-Glucopyranosylamines as Glycogen Phosphorylase Inhibitors" Molecules 24, no. 7: 1322. https://doi.org/10.3390/molecules24071322
APA StyleFischer, T., Koulas, S. M., Tsagkarakou, A. S., Kyriakis, E., Stravodimos, G. A., Skamnaki, V. T., Liggri, P. G. V., Zographos, S. E., Riedl, R., & Leonidas, D. D. (2019). High Consistency of Structure-Based Design and X-Ray Crystallography: Design, Synthesis, Kinetic Evaluation and Crystallographic Binding Mode Determination of Biphenyl-N-acyl-β-d-Glucopyranosylamines as Glycogen Phosphorylase Inhibitors. Molecules, 24(7), 1322. https://doi.org/10.3390/molecules24071322