
A Dual Ligand Sol–Gel Organic-Silica Hybrid Monolithic Capillary for In-Tube SPME-MS/MS to Determine Amino Acids in Plasma Samples


Abstract
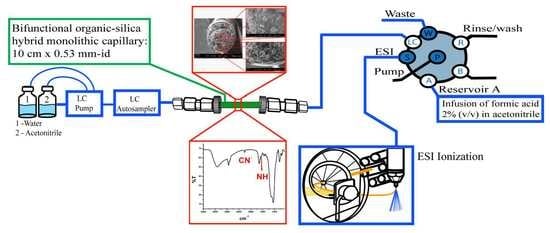
:
1. Introduction
2. Results
2.1. Hybrid Monolithic Capillary Synthesis
2.2. Characterization of Hybrid Silica Monoliths
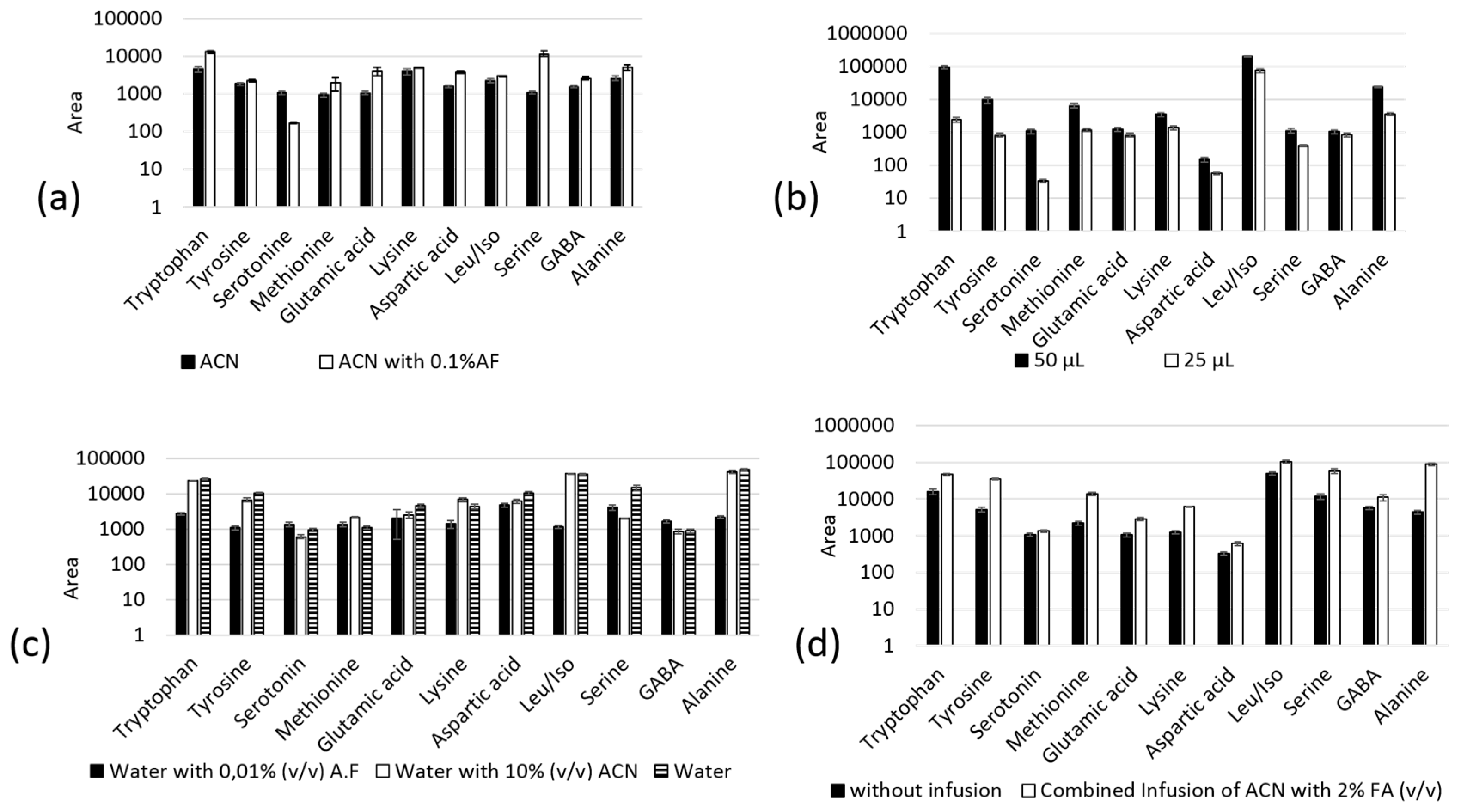
2.3. In-Tube SPME-MS/MS Optimization
2.4. Adsorption Experiments
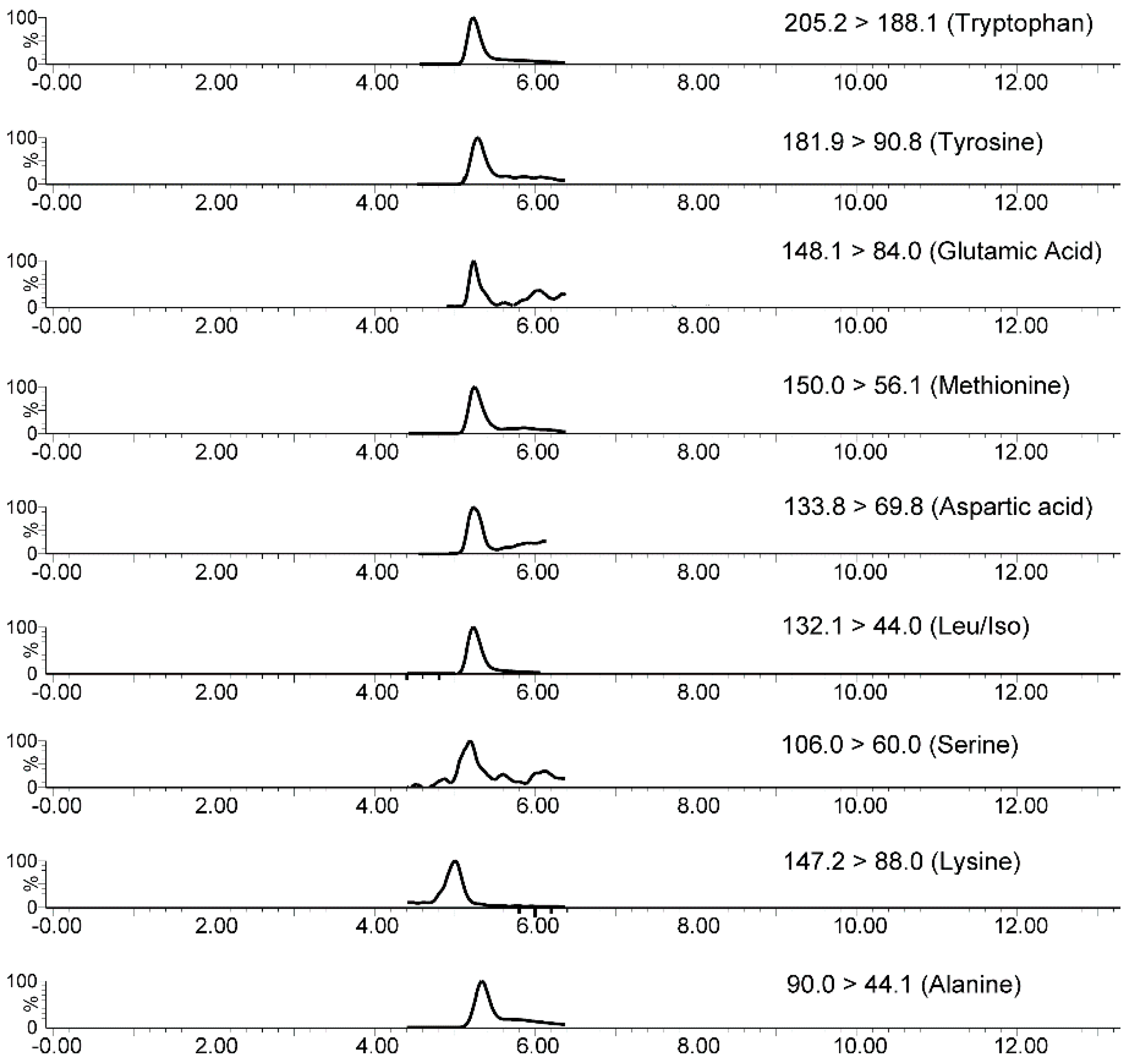
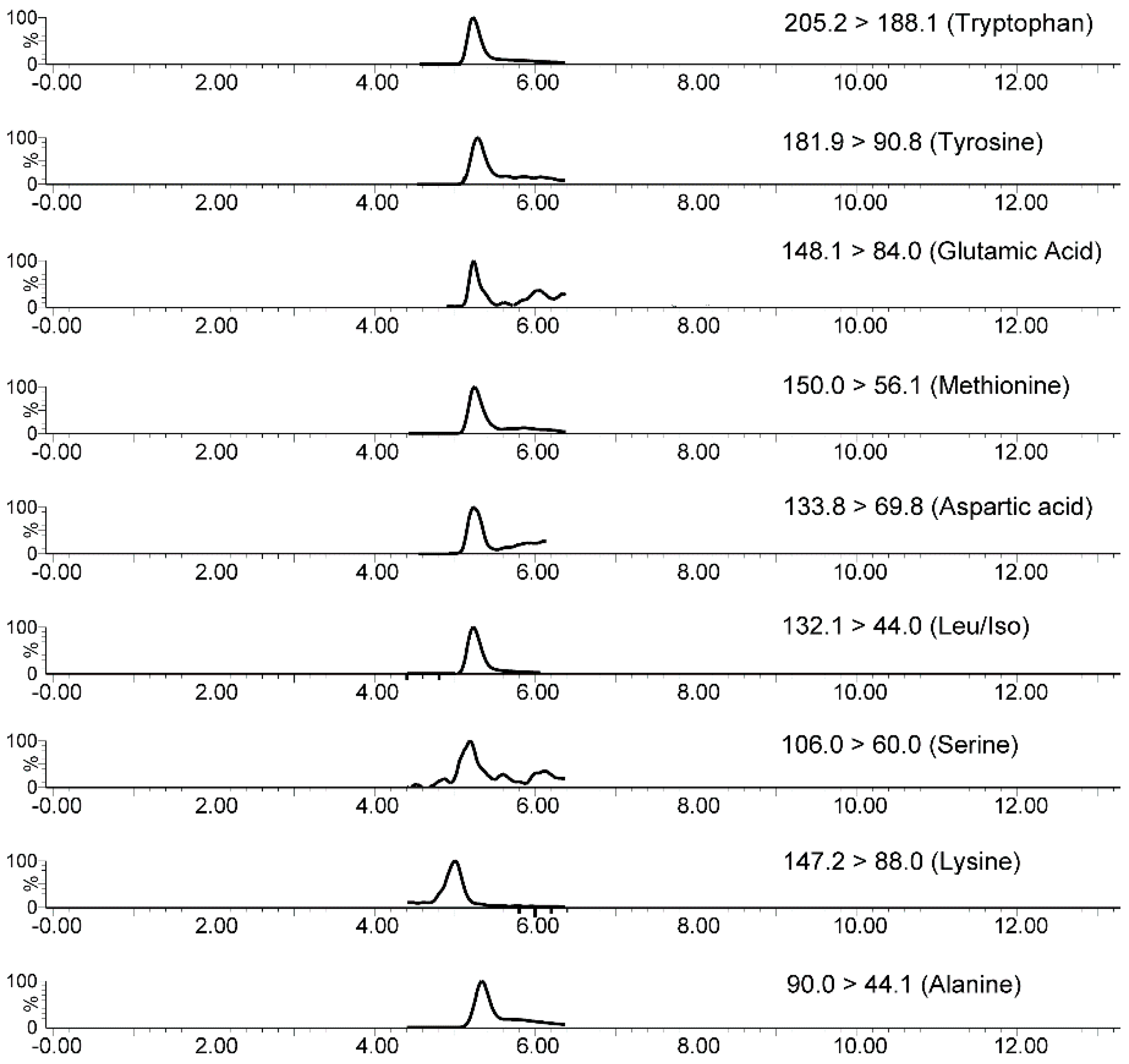
2.5. In-Tube SPME-MS/MS Analytical Validation
2.6. Comparison of the Proposed Method with Literature Methods
3. Materials and Methods
3.1. Standards and Reagents
3.2. Synthesis of Hybrid Silica-Based Monolithic Capillaries Bearing Amino and Cyano Groups
3.3. Hybrid Silica Monolithic Capillary Characterization
3.4. Plasma Samples
3.5. MS/MS Conditions
3.6. In-Tube SPME Procedure
3.7. Adsorption Capacity
3.8. Analytical Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- McGrath, J.; Saha, S.; Welham, J.; El Saadi, O.; MacCauley, C.; Chant, D. A systematic review of the incidence of schizophrenia: The distribution of rates and the influence of sex, urbanicity, migrant status and methodology. BMC Med. 2004, 2, 13. [Google Scholar] [CrossRef]
- De Luca, V.; Viggiano, E.; Messina, G.; Viggiano, A.; Borlido, C.; Viggiano, A.; Monda, M. Peripheral amino Acid levels in schizophrenia and antipsychotic treatment. Psychiatry Investig. 2008, 5, 203–208. [Google Scholar] [CrossRef]
- Javitt, D.C. Excitatory Amino Acids in Schizophrenia: Both What You Have, and What You Do With Them. Biol. Psychiatry. 2018, 83, 470–472. [Google Scholar] [CrossRef]
- Saleem, S.; Shaukat, F.; Gul, A.; Arooj, M.; Malik, A. Potential role of amino acids in pathogenesis of schizophrenia. Int. J. Health Sci. 2017, 11, 63. [Google Scholar]
- Balu, D.T.; Coyle, J.T. The NMDA receptor ‘glycine modulatory site’ in schizophrenia: D-serine, glycine, and beyond. Curr. Opin. Pharmacol. 2015, 20, 109–115. [Google Scholar] [CrossRef]
- Cao, B.; Wang, D.; Brietzke, E.; McIntyre, R.S.; Pan, Z.; Cha, D.; Rosenblat, J.D.; Zuckerman, H.; Liu, Y.; Xie, Q.; Wang, J. Characterizing amino-acid biosignatures amongst individuals with schizophrenia: A case–control study. Amino Acids 2018, 50, 1013–1023. [Google Scholar] [CrossRef]
- Panizzutti, R.; Fisher, M.; Garrett, C.; Man, W.H.; Sena, W.; Madeira, C.; Vinogradov, S. Association between increased serum d-serine and cognitive gains induced by intensive cognitive training in schizophrenia. Schizophr. Res. 2018, in press. [Google Scholar] [CrossRef]
- MacKay, M.-A.B.; Kravtsenyuk, M.; Thomas, R.; Mitchell, N.D.; Dursun, S.M.; Baker, G.B. D-Serine: Potential Therapeutic Agent and/or Biomarker in Schizophrenia and Depression? Front. Psychiatry 2019, 10, 25. [Google Scholar] [CrossRef]
- Domingues, D.S.; Crevelin, E.J.; Moraes, L.A.B.; Hallak, J.C.E.; Crippa, J.A.S.; Queiroz, M.E.C. Simultaneous determination of amino acids and neurotransmitters in plasma samples from schizophrenic patients by hydrophilic interaction liquid chromatography with tandem mass spectrometry. J. Sep. Sci. 2015, 38, 780–787. [Google Scholar] [CrossRef]
- Altamura, C.A.; Mauri, M.C.; Ferrara, A.; Moro, A.R.; D’andrea, G.; Zamberlan, F.; Meltzer, H.Y. Plasma and platelet excitatory amino acids in psychiatric disorders. Am. J. Psychiatry 1993, 150, 1731–1733. [Google Scholar]
- Cai, H.-L.; Zhu, R.-H. Determination of dansylated monoamine and amino acid neurotransmitters and their metabolites in human plasma by liquid chromatography–electrospray ionization tandem mass spectrometry. Anal. Biochem. 2010, 396, 103–111. [Google Scholar] [CrossRef]
- Queiroz, M.E.C.; Melo, L.P. Selective capillary coating materials for in-tube solid-phase microextraction coupled to liquid chromatography to determine drugs and biomarkers in biological samples: A review. Anal. Chim. Acta 2014, 826, 1–11. [Google Scholar] [CrossRef]
- Saito, A.; Hamano, M.; Kataoka, H. Simultaneous analysis of multiple urinary biomarkers for the evaluation of oxidative stress by automated online in-tube solid-phase microextraction coupled with negative/positive ion-switching mode liquid chromatography–tandem mass spectrometry. J. Sep. Sci. 2018, 41, 2743–2749. [Google Scholar] [CrossRef]
- Lashgari, M.; Yamini, Y. Fiber-in-tube solid-phase microextraction of caffeine as a molecular tracer in wastewater by electrochemically deposited layered double hydroxide. J. Sep. Sci. 2018, 41, 2393–2400. [Google Scholar] [CrossRef]
- Inukai, T.; Kaji, S.; Kataoka, H. Analysis of nicotine and cotinine in hair by on-line in-tube solid-phase microextraction coupled with liquid chromatography-tandem mass spectrometry as biomarkers of exposure to tobacco smoke. J. Pharm. Biomed. Anal. 2018, 156, 272–277. [Google Scholar] [CrossRef]
- Serra-Mora, P.; Jornet-Martinez, N.; Moliner-Martinez, Y.; Campíns-Falcó, P. In tube-solid phase microextraction-nano liquid chromatography: Application to the determination of intact and degraded polar triazines in waters and recovered struvite. J. Chromatogr. A 2017, 1513, 51–58. [Google Scholar] [CrossRef]
- Luo, X.; Li, G.; Hu, Y. In-tube solid-phase microextraction based on NH 2-MIL-53 (Al)-polymer monolithic column for online coupling with high-performance liquid chromatography for directly sensitive analysis of estrogens in human urine. Talanta 2017, 165, 377–383. [Google Scholar] [CrossRef]
- Andrade, M.A.; Lanças, F.M. Determination of Ochratoxin A in wine by packed in-tube solid phase microextraction followed by high performance liquid chromatography coupled to tandem mass spectrometry. J. Chromatogr. A 2017, 1493, 41–48. [Google Scholar] [CrossRef]
- Bu, Y.; Feng, J.; Tian, Y.; Wang, X.; Sun, M.; Luo, C. An organically modified silica aerogel for online in-tube solid-phase microextraction. J. Chromatogr. A 2017, 1517, 203–208. [Google Scholar] [CrossRef]
- Wu, F.; Wang, J.; Zhao, Q.; Jiang, N.; Lin, X.; Xie, Z.; Li, J.; Zhang, Q. Detection of trans-fatty acids by high performance liquid chromatography coupled with in-tube solid-phase microextraction using hydrophobic polymeric monolith. J. Chromatogr. B 2017, 1040, 214–221. [Google Scholar] [CrossRef]
- Feng, J.; Wang, X.; Tian, Y.; Bu, Y.; Luo, C.; Sun, M. Electrophoretic deposition of graphene oxide onto carbon fibers for in-tube solid-phase microextraction. J. Chromatogr. A 2017, 1517, 209–214. [Google Scholar] [CrossRef]
- Bu, Y.; Feng, J.; Sun, M.; Zhou, C.; Luo, C. Facile and efficient poly (ethylene terephthalate) fibers-in-tube for online solid-phase microextraction towards polycyclic aromatic hydrocarbons. Anal. Bioanal. Chem. 2016, 408, 4871–4882. [Google Scholar] [CrossRef]
- Bu, Y.; Feng, J.; Wang, X.; Tian, Y.; Sun, M.; Luo, C. In situ hydrothermal growth of polyaniline coating for in-tube solid-phase microextraction towards ultraviolet filters in environmental water samples. J. Chromatogr. A 2017, 1483, 48–55. [Google Scholar] [CrossRef]
- Sun, M.; Feng, J.; Bu, Y.; Luo, C. Ionic liquid coated copper wires and tubes for fiber-in-tube solid-phase microextraction. J. Chromatogr. A 2016, 1458, 1–8. [Google Scholar] [CrossRef]
- Xiang, X.; Shang, B.; Wang, X.; Chen, Q. PEEK tube-based online solid-phase microextraction–high-performance liquid chromatography for the determination of yohimbine in rat plasma and its application in pharmacokinetics study. Biomed. Chromatogr. 2017, 31, 3866. [Google Scholar] [CrossRef]
- Ou, J.; Liu, Z.; Wang, H.; Lin, H.; Dong, J.; Zou, H. Recent development of hybrid organic-silica monolithic columns in CEC and capillary LC. Electrophoresis 2015, 36, 62–75. [Google Scholar] [CrossRef]
- Svec, F. Stellan Hjertén’s contribution to the development of monolithic stationary phases. Electrophoresis 2008, 29, 1593–1603. [Google Scholar] [CrossRef] [Green Version]
- Siouffi, A.-M. Silica gel-based monoliths prepared by the sol–gel method: Facts and figures. J. Chromatogr. A 2003, 1000, 801–818. [Google Scholar] [CrossRef]
- Li, Z.; Rodriguez, E.; Azaria, S.; Pekarek, A.; Hage, D.S. Affinity monolith chromatography: A review of general principles and applications. Electrophoresis 2017, 38, 2837–2850. [Google Scholar] [CrossRef]
- Zajickova, Z. Advances in the development and applications of organic–silica hybrid monoliths. J. Sep. Sci. 2017, 40, 25–48. [Google Scholar] [CrossRef]
- Zheng, M.-M.; Wang, S.-T.; Hu, W.-K.; Feng, Y.-Q. In-tube solid-phase microextraction based on hybrid silica monolith coupled to liquid chromatography–mass spectrometry for automated analysis of ten antidepressants in human urine and plasma. J. Chromatogr. A 2010, 1217, 7493–7501. [Google Scholar] [CrossRef]
- de Souza, I.D.; Domingues, D.S.; Queiroz, M.E. Hybrid silica monolith for microextraction by packed sorbent to determine drugs from plasma samples by liquid chromatography–tandem mass spectrometry. Talanta 2015, 140, 166–175. [Google Scholar] [CrossRef]
- Domingues, D.S.; de Souza, I.D.; Queiroz, M.E.C. Analysis of drugs in plasma samples from schizophrenic patients by column-switching liquid chromatography-tandem mass spectrometry with organic–inorganic hybrid cyanopropyl monolithic column. J. Chromatogr. B 2015, 993, 26–35. [Google Scholar] [CrossRef]
- Brothier, F.; Pichon, V. Immobilized antibody on a hybrid organic–inorganic monolith: Capillary immunoextraction coupled on-line to nanoLC-UV for the analysis of microcystin-LR. Anal. Chim. Acta 2013, 792, 52–58. [Google Scholar] [CrossRef]
- Gómez-Ríos, G.A.; Reyes-Garcés, N.; Bojko, B.; Pawliszyn, J. Biocompatible solid-phase microextraction nanoelectrospray ionization: An unexploited tool in bioanalysis. Anal. Chem. 2015, 88, 1259–1265. [Google Scholar] [CrossRef]
- Santos, M.G.; Tavares, I.M.C.; Barbosa, A.F.; Bettini, J.; Figueiredo, E.C. Analysis of tricyclic antidepressants in human plasma using online-restricted access molecularly imprinted solid phase extraction followed by direct mass spectrometry identification/quantification. Talanta 2017, 163, 8–16. [Google Scholar] [CrossRef]
- Li, W.; Fries, D.P.; Malik, A. Sol–gel stationary phases for capillary electrochromatography. J. Chromatogr. A 2004, 1044, 23–52. [Google Scholar] [CrossRef]
- Hench, L.L.; West, J.K. The sol-gel process. Chem. Rev. 1990, 90, 33–72. [Google Scholar] [CrossRef]
- Yan, L.; Zhang, Q.; Zhang, J.; Zhang, L.; Li, T.; Feng, Y.; Zhang, L.; Zhang, W.; Zhang, Y. Hybrid organic–inorganic monolithic stationary phase for acidic compounds separation by capillary electrochromatography. J. Chromatogr. A 2004, 1046, 255–261. [Google Scholar] [CrossRef]
- Rodríguez, R.; Flores, M.; Gómez, J.; Castaño, V.M. Master behaviour for gelation in fluoride-catalyzed gels. Mater. Lett. 1992, 15, 242–247. [Google Scholar] [CrossRef]
- Moreira, J.E.; Cesar, M.L.; Aegerter, M.A. Light scattering of silica particles in solution. J. Non-Cryst. Solids 1990, 121, 394–396. [Google Scholar] [CrossRef]
- Iler, R.K. Chemistry of Silica: Solubility, Polymerization, Colloid and Surface Properties, and Biochemistry; John Wiley & Sons: New York, NY, USA, 1979. [Google Scholar]
- Kulkarni, S.; Fang, L.; Alhooshani, K.; Malik, A. Sol–gel immobilized cyano-polydimethylsiloxane coating for capillary microextraction of aqueous trace analytes ranging from polycyclic aromatic hydrocarbons to free fatty acids. J. Chromatogr. A 2006, 1124, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Li, F. Removal of Cu(II) from aqueous solution by adsorption onto functionalized SBA-16 mesoporous silica. Microporous Mesoporous Mater. 2008, 116, 116–122. [Google Scholar] [CrossRef]
- Al-Oweini, R.; El-Rassy, H. Synthesis and characterization by FTIR spectroscopy of silica aerogels prepared using several Si(OR)4 and R′′Si(OR′)3 precursors. J. Mol. Struct. 2009, 919, 140–145. [Google Scholar] [CrossRef]
- Innocenzi, P. Infrared spectroscopy of sol–gel derived silica-based films: A spectra-microstructure overview. J. Non-Cryst. Solids 2003, 316, 309–319. [Google Scholar] [CrossRef]
- Wahab, M.A.; Kim, II; Ha, C.-S. Hybrid periodic mesoporous organosilica materials prepared from 1,2-bis(triethoxysilyl)ethane and (3-cyanopropyl)triethoxysilane. Microporous Mesoporous Mater. 2004, 69, 19–27. [Google Scholar] [CrossRef]
- Pasternack, R.M.; Rivillon Amy, S.; Chabal, Y.J. Attachment of 3-(Aminopropyl)triethoxysilane on Silicon Oxide Surfaces: Dependence on Solution Temperature. Langmuir 2008, 24, 12963–12971. [Google Scholar] [CrossRef]
- Peña-Alonso, R.; Rubio, F.; Rubio, J.; Oteo, J.L. Study of the hydrolysis and condensation of γ-Aminopropyltriethoxysilane by FT-IR spectroscopy. J. Mater. Sci. 2007, 42, 595–603. [Google Scholar] [CrossRef]
- Chiang, C.-H.; Ishida, H.; Koenig, J.L. The structure of γ-aminopropyltriethoxysilane on glass surfaces. J. Colloid Interface Sci. 1980, 74, 396–404. [Google Scholar] [CrossRef]
- Qiu, H.; Lv, L.; Pan, B.-C.; Zhang, Q.-J.; Zhang, W.-M.; Zhang, Q.-X. Critical review in adsorption kinetic models. J. Zhejiang Univ. Sci. A 2009, 10, 716–724. [Google Scholar] [CrossRef]
- Largitte, L.; Pasquier, R. A review of the kinetics adsorption models and their application to the adsorption of lead by an activated carbon. Chem. Eng. Res. Des. 2016, 109, 495–504. [Google Scholar] [CrossRef]
- Lagergren, S.K. About the theory of so-called adsorption of soluble substances. Sven. Vetenskapsakad. Handingarl 1898, 24, 1–39. [Google Scholar]
- FDA. Guidance for Industry: Bioanalytical Method Validation. Available online: https://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf (accessed on 17 April 2019).
- EMA. Guideline on Bioanalytical Method Validation. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (accessed on 17 April 2019).
- Kollipara, S.; Bende, G.; Agarwal, N.; Varshney, B.; Paliwal, J. International Guidelines for Bioanalytical Method Validation: A Comparison and Discussion on Current Scenario. Chromatographia 2011, 73, 201–217. [Google Scholar] [CrossRef]
- Schaefer, A.; Piquard, F.; Haberey, P. Plasma amino-acids analysis: Effects of delayed samples preparation and of storage. Clin. Chim. Acta 1987, 164, 163–169. [Google Scholar] [CrossRef]
- González, O.; Blanco, M.E.; Iriarte, G.; Bartolomé, L.; Maguregui, M.I.; Alonso, R.M. Bioanalytical chromatographic method validation according to current regulations, with a special focus on the non-well defined parameters limit of quantification, robustness and matrix effect. J. Chromatogr. A 2014, 1353, 10–27. [Google Scholar] [CrossRef]
- Zhang, L.; Gionfriddo, E.; Acquaro, V.; Pawliszyn, J. Direct Immersion Solid-Phase Microextraction Analysis of Multi-class Contaminants in Edible Seaweeds by Gas Chromatography-Mass Spectrometry. Anal. Chim. Acta 2018, 1031, 83–97. [Google Scholar] [CrossRef]
- Otto, M. Chemometrics: Statistics and Computer Application in Analytical Chemistry; John Wiley & Sons: New York, NY, USA, 2016. [Google Scholar]
- Li, Q.Z.; Huang, Q.X.; Li, S.C.; Yang, M.Z.; Rao, B. Simultaneous Determination of Glutamate, Glycine, and Alanine in Human Plasma Using Precolumn Derivatization with 6-Aminoquinolyl-N-hydroxysuccinimidyl Carbamate and High-Performance Liquid Chromatography. Korean J Physiol. Pharmacol. 2012, 16, 355–360. [Google Scholar] [CrossRef]
- Han, M.; Xie, M.; Han, J.; Yuan, D.; Yang, T.; Xie, Y. Development and validation of a rapid, selective, and sensitive LC-MS/MS method for simultaneous determination of D-and L-amino acids in human serum: Application to the study of hepatocellular carcinoma. Anal. Bioanal. Chem. 2018, 410, 2517–2531. [Google Scholar] [CrossRef]
- Le, A.; Ng, A.; Kwan, T.; Cusmano-Ozog, K.; Cowan, T.M. (LC-MS/MS). J. Chromatogr. B 2014, 944, 166–174. [Google Scholar] [CrossRef]
- Harder, U.; Koletzko, B.; Peissner, W. Quantification of 22 plasma amino acids combining derivatization and ion-pair LC-MS/MS. J. Chromatogr. B 2011, 879, 495–504. [Google Scholar] [CrossRef]
- De Gisi, S.; Lofrano, G.; Grassi, M.; Notarnicola, M. Characteristics and adsorption capacities of low-cost sorbents for wastewater treatment: A review. Sustain. Mater. Technol. 2016, 9, 10–40. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, D.S.; Eisenhofer, G.; Kopin, I.J. Sources and Significance of Plasma Levels of Catechols and Their Metabolites in Humans. J. Pharmacol. Exp. Ther. 2003, 305, 800–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Duan, J.-a.; Qian, D.; Tang, Y.; Qian, Y.; Wu, D.; Su, S.; Shang, E. Rapid determination of amino acids in fruits of Ziziphus jujuba by hydrophilic interaction ultra-high-performance liquid chromatography coupled with triple-quadrupole mass spectrometry. J. Agric. Food Chem. 2013, 61, 2709–2719. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Procedure | TEOS (µL) | APTES (µL) | CN-TEOS (µL) | H2O/EtOH (µL) | TEOS/APTES/CN-TEOS (µL) | Aging Temperature (°C) |
|---|---|---|---|---|---|---|
| 1 | 56 | 28 | 28 | 100:20 | 2:1:1 | 60 |
| 2 | 38 | 38 | 38 | 100:20 | 1:1:1 | 60 |
| 3 | 56 | 56 | 0 | 100:20 | 1:1:0 | 60 |
| 4 | 56 | 0 | 56 | 100:20 | 1:0:1 | 60 |
| 5 | 56 | 28 | 28 | 50:50 | 2:1:1 | 60 |
| 6 | 56 | 28 | 28 | 50:50 | 2:1:1 | 22 |
| Analyte | Precursor Ion | Product Ion (Quantification) | DP (V) | CE (V) | Product Ion (Identification) |
|---|---|---|---|---|---|
| Tryptophan | 205.2 | 146.0 | 20 | 12 | 188.1 |
| Methionine | 150.0 | 56.0 | 20 | 15 | 104.0 |
| Methionine d3 | 153.1 | 56.0 | 20 | 15 | 107.1 |
| Tyrosine | 182.1 | 136.1 | 25 | 15 | 90.8 |
| Leucine/Isoleucine | 132.1 | 86.0 | 20 | 10 | 44.0 |
| GABA | 104.1 | 87.0 | 30 | 15 | 45.0 |
| Serotonin | 177.1 | 115.1 | 20 | 36 | 104.9 |
| Glutamic acid | 148.1 | 84.0 | 25 | 15 | 102.1 |
| Lysine | 147.2 | 88.0 | 25 | 15 | 107.0 |
| Aspartic acid | 134.1 | 74.0 | 20 | 12 | 88.0 |
| Serine | 106.0 | 60.0 | 20 | 10 | 88.0 |
| Alanine | 90.0 | 44.0 | 20 | 10 | 62.0 |
| Alanine 13C315N | 94.2 | 47.1 | 20 | 10 | 64.8 |
| MOBILE PHASE A: Water B: Acetonitrile | ||||
|---|---|---|---|---|
| Time (min) | % A | % B | Valve Position | Comments |
| 0.0 | 0 | 100 | 1 | pre-concentration of analytes and exclusion of plasma macromolecules |
| 2.0 | 100 | 0 | 2 | Elution of analytes from monolithic capillary to mass spectrometer |
| 4.0 | 100 | 0 | 2 | Post capillary infusion of acetonitrile with 0.1% formic acid |
| 7.0 | 100 | 0 | 1 | Final elution step and start of gradient elution to clean up the capillary column |
| Validation Parameters | Ala | Leu/Iso | Met | Ty | Try | Ser | Glu | Lys | Asp |
|---|---|---|---|---|---|---|---|---|---|
| Linearity (R2) | 0.995 | 0.993 | 0.998 | 0.995 | 0.997 | 0.990 | 0.996 | 0.991 | 0.993 |
| Slope Intercept | 0.0013 0.4277 | 0.0110 2.4736 | 0.0704 0.6808 | 0.0057 0.1844 | 0.0251 0.6792 | 0.0024 0.3963 | 0.0022 0.0746 | 0.0046 0.0859 | 0.0057 0.0092 |
| LOF (p-value) | 0.997 | 0.803 | 0.489 | 0.251 | 0.876 | 0.892 | 0.972 | 0.251 | 0.875 |
| Linear range (nmol mL−1) | 45–360 | 15–300 | 12–102 | 10–102 | 9–96 | 12–210 | 12–90 | 12–102 | 6–36 |
| Student’s t-test (p-value) | 0.520 | 0.087 | 0.125 | 0.907 | 0.079 | 0.077 | 0.219 | 0.280 | 0.244 |
| Plasma Concentration (nmol mL−1) | Ala | Leu/Iso | Met | Ty | Try | Ser | Glu | Lys | Asp |
|---|---|---|---|---|---|---|---|---|---|
| Average values | 270.8 ± 60.1 | 246.1 ± 28.0 | 18.3 ± 5.1 | 40.5 ± 10.4 | 37.1 ± 9.7 | 143.0 ± 48.6 | 31.8 ± 11.2 | 20.6 ± 4.0 | 11.2 ± 7.5 |
| Analytes | Sample Preparation | Sample Volume (µL) | Analytical Method | Elution of Analytes (min) | validation Parameters (Intra and Inter Assays) | Ref. |
|---|---|---|---|---|---|---|
| 10 amino acids | Protein precipitation | Plasma 50 | UHPLC-MS/MS-Ascentis® Express HILIC column (4.6 × 100 mm, 2.7 µm). MP: A = Ammonium acetate solution 10 mM; B = acetonitrile with 0.1% FA | 3.2 | LLOQ: 9.7–13.3 nmol mL−1 Precision: 2–10% (CV) Accuracy: −2.1–9.9% (RSE) | [9] |
| 33 Amino acids | Protein precipitation | Plasma 100 | Two columns: 1 - PGC column (Thermo Fisher Scientific, 3 µm Hypercarb, 4.6 mm i.d. × 50 mm), and 2 - fused-core column (Advanced Materials Technology, 2.7 µm Halo C18, 2.1 mm i.d. × 100 mm) | 9.4 | LLOQ: 0.1–10.0 nmol mL−1 Precision: 1.2–9.2% (CV) Accuracy: N.A | [63] |
| 20 amino acids | Protein precipitation | Serum 100 | UHPLC-MS/MS CROWNPAK CR-I(+) column (3.0-mm i.d. × 150 mm, 5 μm) | 10.1 | LLOQ: 0.1–10.0 nmol mL−1 Precision: 2.6–10.1% (CV) Accuracy: −12.8–12.4% (RSE) | [62] |
| 22 amino acids | Protein precipitation | Plasma 10 | HPLC-MS/MS Two Agilent Zorbax SB-C18 columns, (2.1 mm × 50 mm, 1.8 µm) | 35.0 | LLOQ: 0.01–0.07 nmol mL−1 Precision: 1.0–15.0% (CV) Accuracy: −12.8–12.4% | [64] |
| 10 amino acids | In-tube SPME | Plasma 200 | In-tube SPME-MS/MS with post capillary infusion | 5.2 | LLOQ: 6–45 nmol mL−1 Precision: 1.1–19.0% (CV) Accuracy: −14.4–19.6% (RSE) | This work |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miranda, L.F.C.; Gonçalves, R.R.; C. Queiroz, M.E. A Dual Ligand Sol–Gel Organic-Silica Hybrid Monolithic Capillary for In-Tube SPME-MS/MS to Determine Amino Acids in Plasma Samples. Molecules 2019, 24, 1658. https://doi.org/10.3390/molecules24091658
Miranda LFC, Gonçalves RR, C. Queiroz ME. A Dual Ligand Sol–Gel Organic-Silica Hybrid Monolithic Capillary for In-Tube SPME-MS/MS to Determine Amino Acids in Plasma Samples. Molecules. 2019; 24(9):1658. https://doi.org/10.3390/molecules24091658
Chicago/Turabian StyleMiranda, Luis F. C., Rogéria R. Gonçalves, and Maria E. C. Queiroz. 2019. "A Dual Ligand Sol–Gel Organic-Silica Hybrid Monolithic Capillary for In-Tube SPME-MS/MS to Determine Amino Acids in Plasma Samples" Molecules 24, no. 9: 1658. https://doi.org/10.3390/molecules24091658