
Activity and Affinity of Pin1 Variants


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
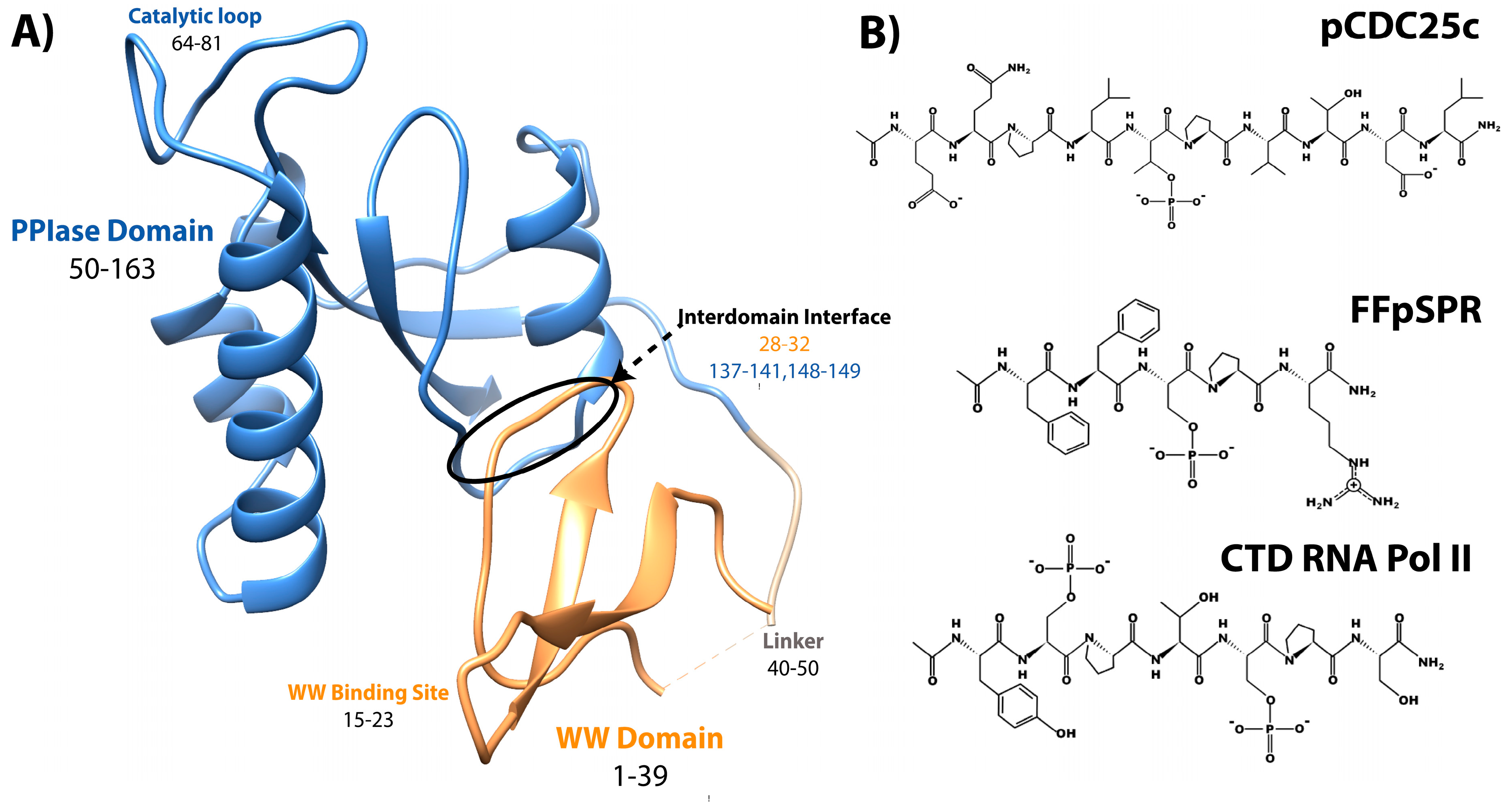
2. Mutation Impact on Affinity to Various Ligands
2.1. Methods for Determination of Affinity and Activity
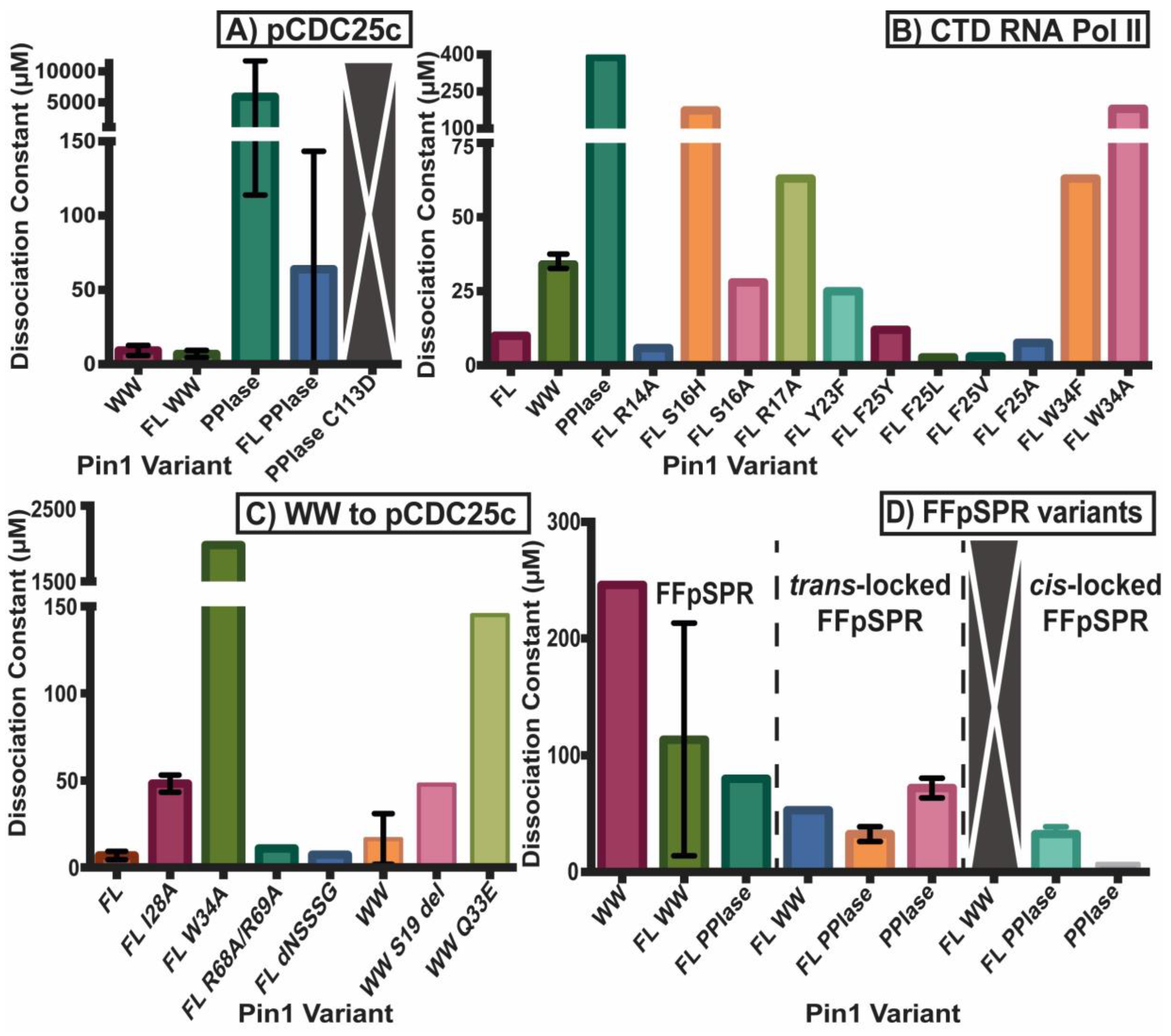
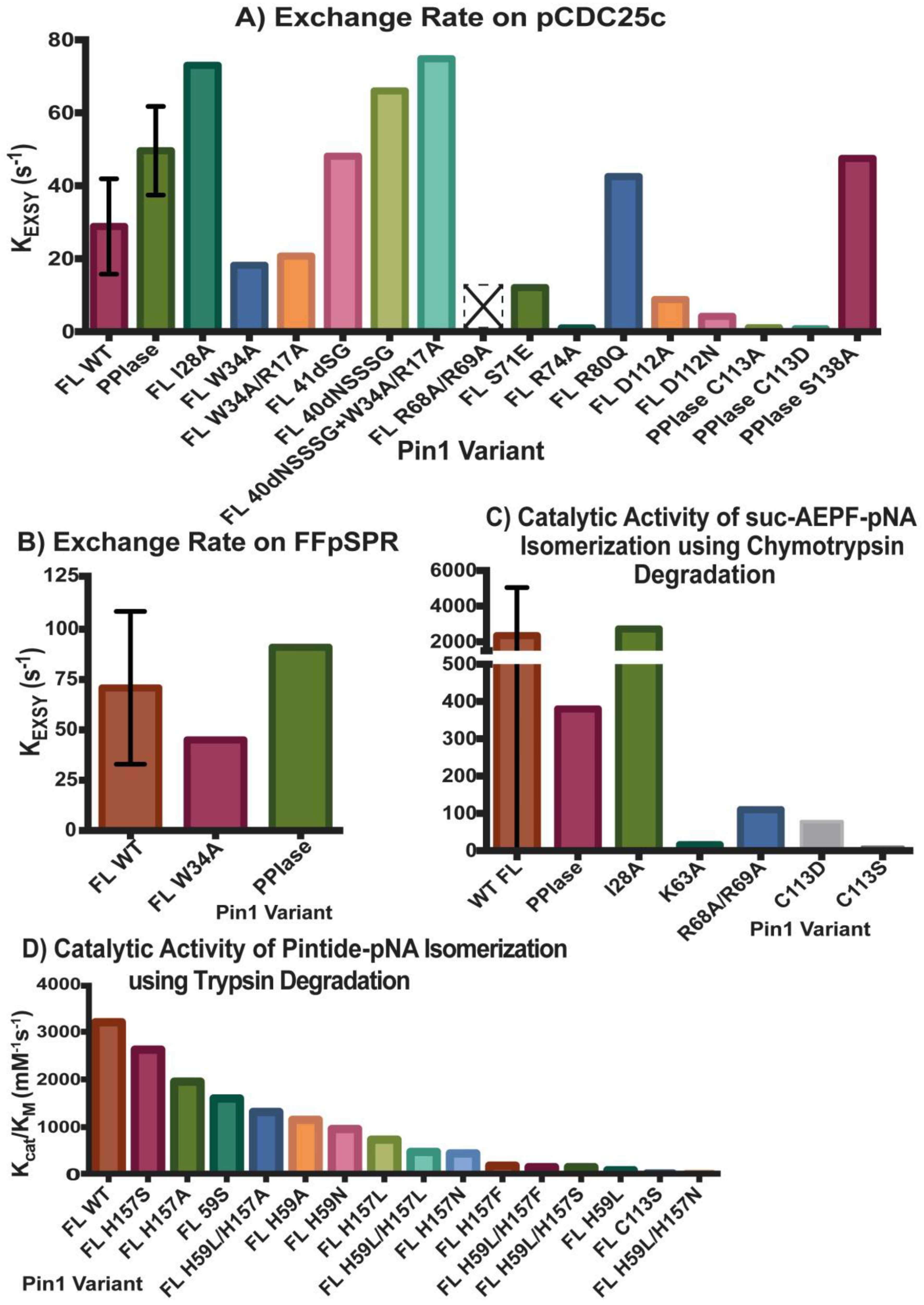
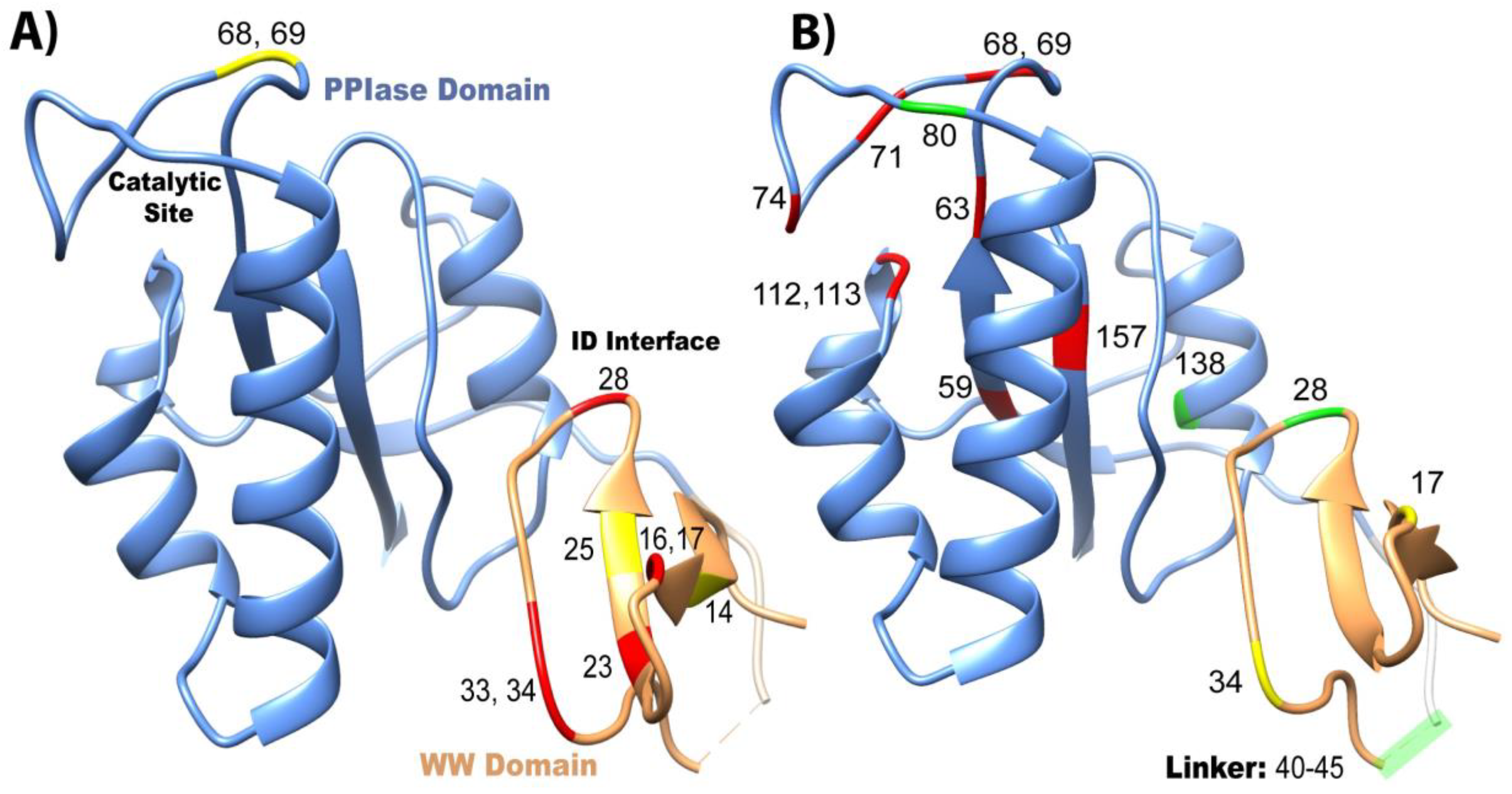
2.2. Experimental Mutant-Dependent Affinities and Activities
2.2.1. Mutations in the WW Domain
2.2.2. Mutations in the PPIase Domain
2.2.3. Mutations in the Interdomain Interface and Linker
2.2.4. Impact on cis- and trans-Locked Inhibitors on Pin1
2.2.5. Differences in Allosteric Responses Due to pCDC25c Versus FFpSPR Binding
3. Conclusions
Supplementary Materials
Funding
Conflicts of Interest
References
- Ping Lu, K.; Hanes, S.D.; Hunter, T. A human peptidyl–prolyl isomerase essential for regulation of mitosis. Nature 1996, 380, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.Z.; Kops, O.; Werner, A.; Lu, P.J.; Shen, M.; Stoller, G.; Küllertz, G.; Stark, M.; Fischer, G.; Lu, K.P. Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and tau proteins. Mol. Cell 2000, 6, 873–883. [Google Scholar] [CrossRef]
- Yaffe, M.B.; Schutkowski, M.; Shen, M.; Zhou, X.Z.; Stukenberg, P.T.; Rahfeld, J.U.; Xu, J.; Kuang, J.; Kirschner, M.W.; Fischer, G.; et al. Sequence-specific and phosphorylation dependent proline isomerization: A potential mitotic regulatory mechanism. Science 1997, 278, 1957–1960. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, R.; Lu, K.P.; Hunter, T.; Noel, J.P. Structural and Functional Analysis of the Mitotic Rotamase Pin1 suggest substrate recognition is phosphorylation dependent. Cell 1997, 89, 875–886. [Google Scholar] [CrossRef]
- Wilson, K.A.; Bouchard, J.J.; Peng, J.W. Interdomain interactions support interdomain communication in human pin1. Biochemistry 2013, 52, 6968–6981. [Google Scholar] [CrossRef]
- Jacobs, D.M.; Saxena, K.; Vogtherr, M.; Bernadó, P.; Pons, M.; Fiebig, K.M. Peptide binding induces large scale changes in inter-domain mobility in human Pin1. J. Biol. Chem. 2003, 278, 26174–26182. [Google Scholar] [CrossRef]
- Bayer, E.; Goettsch, S.; Mueller, J.W.; Griewel, B.; Guiberman, E.; Mayr, L.M.; Bayer, P. Structural analysis of the mitotic regulator hPin1 in solution: Insights into domain architecture and substrate binding. J. Biol. Chem. 2003, 278, 26183–26193. [Google Scholar] [CrossRef]
- Bouchard, J.J.; Xia, J.; Case, D.A.; Peng, J.W. Enhanced Sampling of Interdomain Motion Using Map-Restrained Langevin Dynamics and NMR: Application to Pin1. J. Mol. Biol. 2018, 430, 2164–2180. [Google Scholar] [CrossRef]
- Zhu, W.; Li, Y.; Liu, M.; Zhu, J.; Yang, Y. Uncorrelated Effect of Interdomain Contact on Pin1 Isomerase Activity Reveals Positive Catalytic Cooperativity. J. Phys. Chem. Lett. 2019, 10, 1272–1278. [Google Scholar] [CrossRef]
- Peng, J.W. Investigating dynamic interdomain allostery in Pin1. Biophys. Rev. 2015, 7, 239–249. [Google Scholar] [CrossRef]
- Liou, Y.-C.; Zhou, X.Z.; Lu, K.P. Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem. Sci. 2011, 36, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Kimzey, A.; Sauter, G.; Sowadski, J.M.; Lu, K.P.; Wang, D.-G. Prevalent Overexpression of Prolyl Isomerase Pin1 in Human Cancers. Am. J. Pathol. 2004, 164, 1727–1737. [Google Scholar] [CrossRef]
- Wulf, G.M.; Ryo, A.; Wulf, G.G.; Lee, S.W.; Niu, T.; Petkova, V.; Lu, K.P. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. EMBO J. 2001, 20, 3459–3472. [Google Scholar] [CrossRef] [PubMed]
- Ryo, A.; Nakamura, M.; Wulf, G.; Liou, Y.-C.; Lu, K.P. Pin1 regulates turnover and subcellular localization of β-catenin by inhibiting its interaction with APC. Nat. Cell Biol. 2001, 3, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Ryo, A.; Suizu, F.; Yoshida, Y.; Perrem, K.; Liou, Y.-C.; Wulf, G.; Rottapel, R.; Yamaoka, S.; Lu, K.P. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol. Cell 2003, 12, 1413–1426. [Google Scholar] [CrossRef]
- Lam, P.B.; Burga, L.N.; Wu, B.P.; Hofstatter, E.W.; Lu, K.; Wulf, G.M. Prolyl isomerase Pin1 is highly expressed in Her2-positive breast cancer and regulates erbB2 protein stability. Mol. Cancer 2008, 7, 91. [Google Scholar] [CrossRef]
- Rajbhandari, P.; Finn, G.; Solodin, N.M.; Singarapu, K.K.; Sahu, S.C.; Markley, J.L.; Kadunc, K.J.; Ellison-Zelski, S.J.; Kariagina, A.; Haslam, S.Z.; et al. Regulation of Estrogen Receptor N-Terminus Conformation and Function by Peptidyl Prolyl Isomerase Pin1. Mol. Cell. Biol. 2012, 32, 445–457. [Google Scholar] [CrossRef]
- Rustighi, A.; Zannini, A.; Tiberi, L.; Sommaggio, R.; Piazza, S.; Sorrentino, G.; Nuzzo, S.; Tuscano, A.; Eterno, V.; Benvenuti, F.; et al. Prolyl-isomerase Pin1 controls normal and cancer stem cells of the breast. EMBO Mol. Med. 2014, 6, 99–119. [Google Scholar] [CrossRef]
- Lu, P.-J.; Wulf, G.; Zhou, X.Z.; Davies, P.; Lu, K.P. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature 1999, 399, 784–788. [Google Scholar] [CrossRef]
- Kondo, A.; Albayram, O.; Zhou, X.Z.; Lu, K.P. Pin1 Knockout Mice: A Model for the Study of Tau Pathology in Alzheimer’s Disease; Humana Press: New York, NY, USA, 2017; pp. 415–425. [Google Scholar]
- Butterfield, D.A.; Abdul, H.M.; Opii, W.; Newman, S.F.; Joshi, G.; Ansari, M.A.; Sultana, R. REVIEW: Pin1 in Alzheimer’s disease. J. Neurochem. 2006, 98, 1697–1706. [Google Scholar] [CrossRef]
- Pastorino, L.; Sun, A.; Lu, P.-J.; Zhou, X.Z.; Balastik, M.; Finn, G.; Wulf, G.; Lim, J.; Li, S.-H.; Li, X.; et al. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-β production. Nature 2006, 440, 528–534. [Google Scholar] [CrossRef] [PubMed]
- El Boustani, M.; De Stefano, L.; Caligiuri, I.; Mouawad, N.; Granchi, C.; Canzonieri, V.; Tuccinardi, T.; Giordano, A.; Rizzolio, F. A Guide to PIN1 Function and Mutations Across Cancers. Front. Pharmacol. 2019, 9, 1477. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Boyd-Kimball, D.; Poon, H.F.; Cai, J.; Pierce, W.M.; Klein, J.B.; Markesbery, W.R.; Zhou, X.Z.; Lu, K.P.; Butterfield, D.A. Oxidative modification and down-regulation of Pin1 in Alzheimer’s disease hippocampus: A redox proteomics analysis. Neurobiol. Aging 2006, 27, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Driver, J.A.; Zhou, X.Z.; Lu, K.P. Pin1 dysregulation helps to explain the inverse association between cancer and Alzheimer’s disease. Biochim. Biophys. Acta 2015, 1850, 2069–2076. [Google Scholar] [CrossRef]
- Zhou, X.Z.; Lu, K.P. The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target. Nat. Rev. Cancer 2016, 16, 463–478. [Google Scholar] [CrossRef]
- Min, S.-H.; Zhou, X.Z.; Lu, K.P. The role of Pin1 in the development and treatment of cancer. Arch. Pharm. Res. 2016, 39, 1609–1620. [Google Scholar] [CrossRef]
- Bouchard, J.J. Ensemble Interpretation of Domain Mobility in Modular Protein Pin1 by NMR and Molecular Dynamics; University of Notre Dame: South Bend, IN, USA, 2014. [Google Scholar]
- Shaw, P.E. Peptidyl-prolyl isomerases: A new twist to transcription. EMBO Rep. 2002, 3, 521–526. [Google Scholar] [CrossRef]
- Lu, K.P.; Zhou, X.Z. The prolyl isomerase PIN1: A pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 904–916. [Google Scholar] [CrossRef]
- Yeh, E.S.; Means, A.R. PIN1, the cell cycle and cancer. Nat. Rev. Cancer 2007, 7, 381–388. [Google Scholar] [CrossRef]
- Lin, C.-H.; Li, H.-Y.; Lee, Y.-C.; Calkins, M.J.; Lee, K.-H.; Yang, C.-N.; Lu, P.-J. Landscape of Pin1 in the cell cycle. Exp. Biol. Med. 2015, 240, 403–408. [Google Scholar] [CrossRef]
- Matena, A.; Rehic, E.; Hönig, D.; Kamba, B.; Bayer, P. Structure and function of the human parvulins Pin1 and Par14/17. Biol. Chem. 2018, 399, 101–125. [Google Scholar] [CrossRef]
- Jeener, J.; Meier, B.H.; Bachmann, P.; Ernst, R.R. Investigation of exchange processes by two-dimensional NMR spectroscopy. J. Chem. Phys. 1979, 71, 4546–4553. [Google Scholar] [CrossRef]
- Kofron, J.L.; Kuzmic, P.; Kishore, V.; Colón-Bonilla, E.; Rich, D.H. Determination of kinetic constants for peptidyl prolyl cis-trans isomerases by an improved spectrophotometric assay. Biochemistry 1991, 30, 6127–6134. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Tochio, N.; Wang, J.; Tamari, Y.; Uewaki, J.I.; Utsunomiya-Tate, N.; Igarashi, K.; Shiraki, T.; Kobayashi, N.; Tate, S.I. The C113D mutation in human Pin1 causes allosteric structural changes in the phosphate binding pocket of the ppiase domain through the tug of war in the dual-histidine motif. Biochemistry 2014, 53, 5568–5578. [Google Scholar] [CrossRef] [PubMed]
- Innes, B.T.; Bailey, M.L.; Brandl, C.J.; Shilton, B.H.; Litchfield, D.W. Non-catalytic participation of the pin1 peptidyl-prolyl isomerase domain in target binding. Front. Physiol. 2013, 4. [Google Scholar] [CrossRef]
- Jäger, M.; Dendle, M.; Kelly, J.W. Sequence determinants of thermodynamic stability in a WW domain—An all-β-sheet protein. Protein Sci. 2009, 18, 1806–1813. [Google Scholar] [CrossRef]
- Behrsin, C.D.; Bailey, M.L.; Bateman, K.S.; Hamilton, K.S.; Wahl, L.M.; Brandl, C.J.; Shilton, B.H.; Litchfield, D.W. Functionally Important Residues in the Peptidyl-prolyl Isomerase Pin1 Revealed by Unigenic Evolution. J. Mol. Biol. 2007, 365, 1143–1162. [Google Scholar] [CrossRef]
- Verdecia, M.A.; Bowman, M.E.; Lu, K.P.; Hunter, T.; Noel, J.P. Structural basis for phosphoserine-proline recognition by group IV WW domains. Nat. Struct. Biol. 2000, 7, 639–643. [Google Scholar] [CrossRef]
- Lu, P.J.; Zhou, X.Z.; Shen, M.; Lu, K.P. Function of WW domains as phosphoserine- or phosphothreonine-binding modules. Science 1999, 283, 1325–1328. [Google Scholar] [CrossRef]
- Peng, T.; Zintsmaster, J.S.; Namanja, A.T.; Peng, J.W. Sequence-specific dynamics modulate recognition specificity in WW domains. Nat. Struct. Mol. Biol. 2007, 14, 325–331. [Google Scholar] [CrossRef]
- Jäger, M.; Zhang, Y.; Bieschke, J.; Nguyen, H.; Dendle, M.; Bowman, M.E.; Noel, J.P.; Gruebele, M.; Kelly, J.W. Structure-function-folding relationship in a WW domain. Proc. Natl. Acad. Sci. USA 2006, 103, 10648–10653. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Mahoney, B.J.; Zhang, M.; Zintsmaster, J.S.; Peng, J.W. Negative Regulation of Peptidyl-Prolyl Isomerase Activity by Interdomain Contact in Human Pin1. Structure 2015, 23, 2224–2233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Case, D.A.; Peng, J.W. Propagated Perturbations from a Peripheral Mutation Show Interactions Supporting WW Domain Thermostability. Structure 2018, 26, 1474–1485.e5. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kawasaki, R.; Uewaki, J.I.; Rashid, A.U.R.; Tochio, N.; Tate, S.I. Dynamic allostery modulates catalytic activity by modifying the hydrogen bonding network in the catalytic site of human Pin1. Molecules 2017, 22, 992. [Google Scholar] [CrossRef]
- Bailey, M.L.; Shilton, B.H.; Brandl, C.J.; Litchfield, D.W. The dual histidine motif in the active site of Pin1 has a structural rather than catalytic role. Biochemistry 2008, 47, 11481–11489. [Google Scholar] [CrossRef]
- Lee, T.H.; Chen, C.H.; Suizu, F.; Huang, P.; Schiene-Fischer, C.; Daum, S.; Zhang, Y.J.; Goate, A.; Chen, R.H.; Zhou, X.Z.; et al. Death-Associated Protein Kinase 1 Phosphorylates Pin1 and Inhibits Its Prolyl Isomerase Activity and Cellular Function. Mol. Cell 2011, 42, 147–159. [Google Scholar] [CrossRef]
- Mahoney, B.J.; Zhang, M.; Zintsmaster, J.S.; Peng, J.W. Extended Impact of Pin1 Catalytic Loop Phosphorylation Revealed by S71E Phosphomimetic. J. Mol. Biol. 2018, 430, 710–721. [Google Scholar] [CrossRef]
- Namanja, A.T.; Wang, X.J.; Xu, B.; Mercedes-Camacho, A.Y.; Wilson, K.A.; Etzkorn, F.A.; Peng, J.W. Stereospecific gating of functional motions in Pin1. Proc. Natl. Acad. Sci. USA 2011, 108, 12289–12294. [Google Scholar] [CrossRef]
- Rangasamy, V.; Mishra, R.; Sondarva, G.; Das, S.; Lee, T.H.; Bakowska, J.C.; Tzivion, G.; Malter, J.S.; Rana, B.; Lu, K.P.; et al. Mixed-lineage kinase 3 phosphorylates prolyl-isomerase Pin1 to regulate its nuclear translocation and cellular function. Proc. Natl. Acad. Sci. USA 2012, 109, 8149–8154. [Google Scholar] [CrossRef]
- Namanja, A.T.; Wang, X.J.; Xu, B.; Mercedes-Camacho, A.Y.; Wilson, B.D.; Wilson, K.A.; Etzkorn, F.A.; Peng, J.W. Toward flexibility-activity relationships by NMR spectroscopy: Dynamics of Pin1 ligands. J. Am. Chem. Soc. 2010, 132, 5607–5609. [Google Scholar] [CrossRef]
- Namanja, A.T.; Peng, T.; Zintsmaster, J.S.; Elson, A.C.; Shakour, M.G.; Peng, J.W. Substrate Recognition Reduces Side-Chain Flexibility for Conserved Hydrophobic Residues in Human Pin1. Structure 2007, 15, 313–327. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Born, A.; Henen, M.A.; Vögeli, B. Activity and Affinity of Pin1 Variants. Molecules 2020, 25, 36. https://doi.org/10.3390/molecules25010036
Born A, Henen MA, Vögeli B. Activity and Affinity of Pin1 Variants. Molecules. 2020; 25(1):36. https://doi.org/10.3390/molecules25010036
Chicago/Turabian StyleBorn, Alexandra, Morkos A. Henen, and Beat Vögeli. 2020. "Activity and Affinity of Pin1 Variants" Molecules 25, no. 1: 36. https://doi.org/10.3390/molecules25010036
APA StyleBorn, A., Henen, M. A., & Vögeli, B. (2020). Activity and Affinity of Pin1 Variants. Molecules, 25(1), 36. https://doi.org/10.3390/molecules25010036