
Ruthenacarborane–Phenanthroline Derivatives as Potential Metallodrugs
by
 , and
, and
Martin Kellert
1 ,
,
Imola Sárosi
1,
Rajathees Rajaratnam
2,
Eric Meggers
2,
Peter Lönnecke
1 and
Evamarie Hey-Hawkins
1,* 1
Institute of Inorganic Chemistry, Faculty of Chemistry and Mineralogy, Leipzig University, Johannisallee 29, 04103 Leipzig, Germany
2
Fachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein Straße 4, 35043 Marburg, Germany
*
Author to whom correspondence should be addressed.
Molecules 2020, 25(10), 2322; https://doi.org/10.3390/molecules25102322
Submission received: 10 April 2020
/
Revised: 4 May 2020
/
Accepted: 12 May 2020
/
Published: 15 May 2020
(This article belongs to the Special Issue Boron in Catalysis and Materials Chemistry: A Themed Issue in Honor of Professor Todd B. Marder on the Occasion of His 65th Birthday)
Abstract
:Ruthenium-based complexes have received much interest as potential metallodrugs. In this work, four RuII complexes bearing a dicarbollide moiety, a carbonyl ligand, and a phenanthroline-based ligand were synthesized and characterized, including single crystal diffraction analysis of compounds 2, 4, and 5 and an observed side product SP1. Complexes 2–5 are air and moisture stable under ambient conditions. They show excellent solubility in organic solvents, but low solubility in water.
1. Introduction
Metal-containing compounds are of increasing interest for applications in medicinal chemistry due to their diverse coordination geometries, unusual reactivities, and useful physicochemical properties [1,2,3]. Ruthenium shows a low general toxicity [4] and is an excellent metal for this approach. RuII is usually coordinated in an octahedral or pseudo-octahedral half-sandwich fashion and forms quite stable coordinative or covalent Ru–ligand bonds, which affects cellular metabolism. Furthermore, the reactivity of ruthenium ions is well-known; thus, reactions are predictable and facilitate drug design [5,6,7,8]. To date, predominantly ruthenium half-sandwich complexes have been developed as potential anti-cancer agents, antiproliferative drugs, antibiotics, and immunosuppressants [9,10,11,12,13,14].
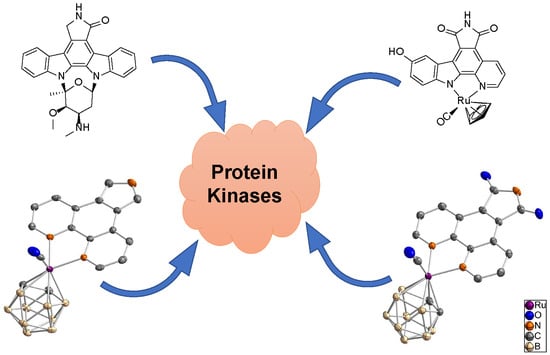
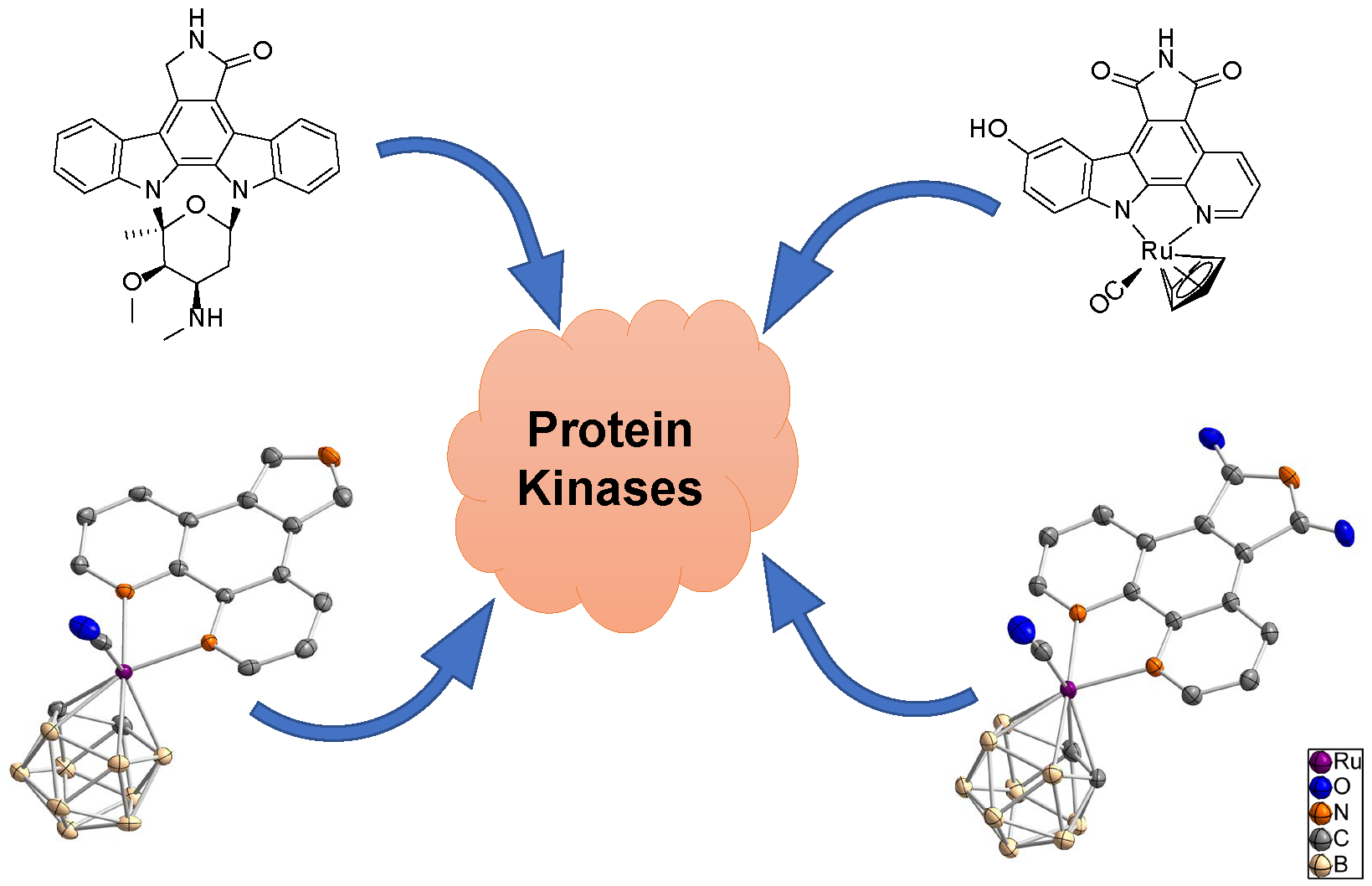
Meggers et al., recently reported an interesting class of organometallic protein kinase inhibitors which were inspired by the alkaloid staurosporine (Figure 1) [4,15,16]. For example, the ruthenium half-sandwich complex DW12 is a potent inhibitor of glycogen synthase kinase 3 (GSK-3), whereas staurosporine constitutes a very unselective inhibitor of a large number of protein kinases [5,16,17,18]. In staurosporine, the important groups for the interaction with protein kinase are the lactam unit, the indolocarbazole heterocycle, and the carbohydrate moiety. In DW12, the ruthenium serves as a purely structural center and enables a geometry, which cannot be easily achieved with purely organic molecules. Thus, the NH group of the maleimide moiety forms a hydrogen bond with the amide carbonyl of Asp133 in the adenosine triphosphate (ATP) binding site of GSK-3. Additionally, one carbonyl group of the maleimide moiety interacts with the NH group of Val135, and the second carbonyl of the same moiety forms a water-mediated hydrogen bond with Asp200. The indole OH group interacts with the carbonyl amide group of Val135. One special feature of DW12 is the presence of the CO ligand, which exhibits a clearly reduced dipolar character due to the interaction with the transition metal ruthenium. Displaying this behavior, the CO ligand is able to undergo an unusual interaction mode with the glycine-rich loop of the ATP binding site. In that way, DW12 achieves a geometry, which seems to be optimal for the ATP binding site of GSK-3, rendering DW12 a more potent inhibitor for GSK-3 than staurosporine.
In order to design novel potential drugs, bioisosteric replacement has become a wide-spread approach [11,14,19,20,21,22,23]. Thus, the development of drugs in which the carborane moiety mimics a phenyl group or is the pharmacophore itself is actively studied [23,24,25,26,27,28,29]. Applications of such carborane-containing drugs are for example cancer therapeutics and enzyme inhibitors [11,14,19,20,22,23,30,31,32,33,34,35,36,37]. Due to its isolobal relationship with the cyclopentadienyl ligand (Cp−) the dicarbollide anion (nido-carborate(2−), C2B9H112−, Cb2−) is a suitable replacement as ligand for transition metals [38,39]. However, distinct activities and reactivities of the respective metal complexes, bearing the Cb2− or the Cp−, are observable and caused by the different charge, size, symmetry, and hybridization of the orbitals of the respective ligands [40]. Furthermore, the dicarbollide moiety is highly hydrophobic and could enhance the transport of the corresponding metallodrug across cellular membranes [19,20,22]. Additionally, these clusters are metabolically stable, which renders them robust compounds in biological media [11,23]. Furthermore, the possible regioselective introduction of specific substituents at either the carbon or the boron vertices of the cluster enables customization of the structure and, therefore, the activity of the metallodrug [19,21].
Former studies have shown the importance of the heteroaromatic bidentate pyridocarbazole and CO ligand in DW12 and related complexes for mimicking staurosporine binding in the ATP binding site [5,17,18,41]. Therefore, the presence of a carbonyl ligand and an aromatic moiety are very important features.
In this work, we report the combination of Cb2− with RuII(CO)–phenanthroline derivatives as mimics for DW12. To our knowledge, this approach to combine the scaffold of an active ruthenium-based protein kinase inhibitor with a dicarbollide moiety has not been pursued before. In DW12, the Cp− ring points away from the ATP binding site towards the aqueous solvent. Thus, there should be sufficient space available in this part of the active site to accommodate larger moieties [42]. Replacing the Cp− ligand with a much bulkier, hydrophobic Cb2− ligand would allow additional van der Waals interactions to be formed with this part of the active site and to profit from the hydrophobic effect which often increases the potency of enzyme inhibitors. Due to the replacement of the cyclopentadienyl ligand with a dicarbollide ligand, the anionic N,N ligand in DW12 must be substituted with a neutral one. As the maleimide moiety in the N,N ligand in DW12 is involved in various hydrogen bond interactions and thus plays an essential role in binding to the ATP binding site of GSK-3, the design of the novel dicarbollide-containing complex should also employ this motif. Therefore, the neutral 5H-pyrrolo[3,4-f][1,10]phenanthroline-5,7(6H)-dione, which is similar to the anionic 9-hydroxy-5H-12λ2-pyrido[2,3-a]pyrrolo[3,4-c]carbazole-5,7(6H)-dione ligand (pyridocarbazole) in DW12, was used as ligand. In combination with the much more hydrophobic and bulkier Cb2− ligand, this should result in increased activity of the respective complex.
2. Results and Discussion
2.1. Ligand and Precursor Syntheses
The precursor 7,8-dicarba-nido-undecaborane(13) (L1) for the dicarbollide moiety was synthesized according to the literature (Scheme 1) [43]. Details about the synthetic procedure are given in the electronic supplementary information.
The phenanthroline derivatives (Figure 2) that were employed in complexation reactions were prepared according to the literature. Even though 5-nitro-1,10-phenanthroline (L2) can be synthesized in excellent yields, it was obtained commercially because of the harsh conditions employed in the synthesis [44]. 1,10-Phenanthroline-5,6-dione (L3) was prepared according to the literature [45,46], and 1,10-phenanthrolinopyrrole (L4) was formed in a Barton-Zard reaction from 5-nitro-1,10-phenanthroline (L2) and ethyl isocyanoacetate under basic conditions followed by hydrolysis of the ester L4’ (Scheme 2) [47,48,49,50,51,52,53,54] The respective synthetic procedure for L4 is given in the electronic supplementary information.
The precursor molecule for further reactions, [3-(CO)3-closo-3,1,2-RuC2B9H11] (1), was prepared in a redox reaction from triruthenium dodecacarbonyl and L1 (Scheme 3) [55,56,57]. The respective synthetic procedure of 1 is given in the electronic supplementary information. Studies showed that the dicarbollide ligand is a much stronger ligand than the Cp ligand [11,39].
2.2. Synthesis of Ruthenium(II) Complexes
The complexes containing phenanthroline derivatives as ligands were prepared via a ligand exchange reaction following a procedure of Jellis and co-workers for other chelating N-donor ligands (Scheme 4) [58]. In this type of reaction, two carbonyl ligands are oxidized to CO2 with stoichiometric amounts of trimethylamine N-oxide [59] to facilitate coordination of one bidentate phenanthroline derivative. For complex 2, both enantiomers R and S were obtained (Ru is the chiral center). No further investigations were carried out to determine the ratio of the two enantiomers, which is assumed to be close to 1:1.
Complexes 2–4 were characterized via NMR, IR, MS, and, in the case of compounds 2 and 4, via single crystal X-ray diffraction. In the 1H-NMR spectrum, compound 3 exhibits three doublets of doublets in the aromatic region as expected for the dione ligand L3. In the infrared spectrum only one absorption band for a C≡O stretching vibration was observed for 2–4 for the single remaining CO ligand. Additionally, in 2, two absorption bands for the symmetric and asymmetric stretching vibration of the NO2 group were observed. In the negative ESI-MS spectrum, compound 3 is observable with one additional bromide ion ([M + Br]−, m/z = 552). No suitable single crystals could be obtained for complex 3. The bifunctional ligand L3 can exhibit several different binding modes: η2-coordination with both nitrogen or both oxygen atoms. In 3, coordination via the nitrogen atoms is assumed, as Ru2+ is a soft Lewis acid [60,61]. It is also possible for L3 to act as a bridging ligand to form complexes with different or additional metal centers. In our case the formation of chain-like oligomers formed by linked complex fragments of 3 are not very likely due to the chosen synthetic procedure.
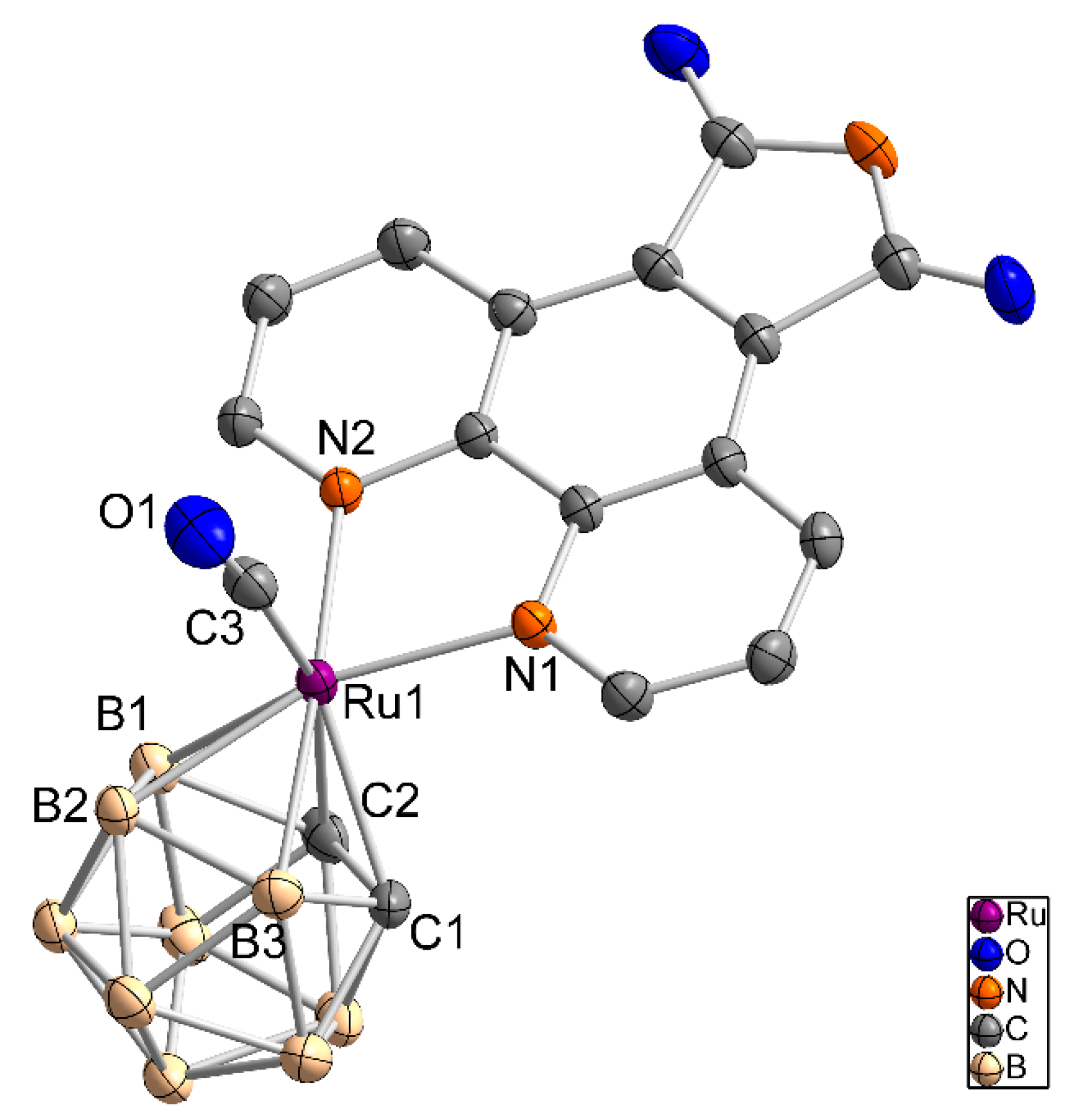
Crystals suitable for single crystal X-ray crystallography were obtained for compounds 2 and 4. Compound 2 crystallizes from a mixture of dichloromethane and n-hexane as orange-red, plate-like crystals in the monoclinic space group P21/n with one additional DCM molecule in the asymmetric unit. The solved structure shows a wR2 value of 9.9% (R1: 17.8%), which is caused by the low quality of the crystals. Nonetheless, the identity of compound 2 is unambiguous (Figure 3, left). Complex 4 crystallizes as yellow plates from a dichloromethane/n-hexane mixture with two independent molecules in the asymmetric unit (Figure 3, right, only one molecule is shown).
Bond lengths and bond angles of 2 and 4 are given in Table 1.
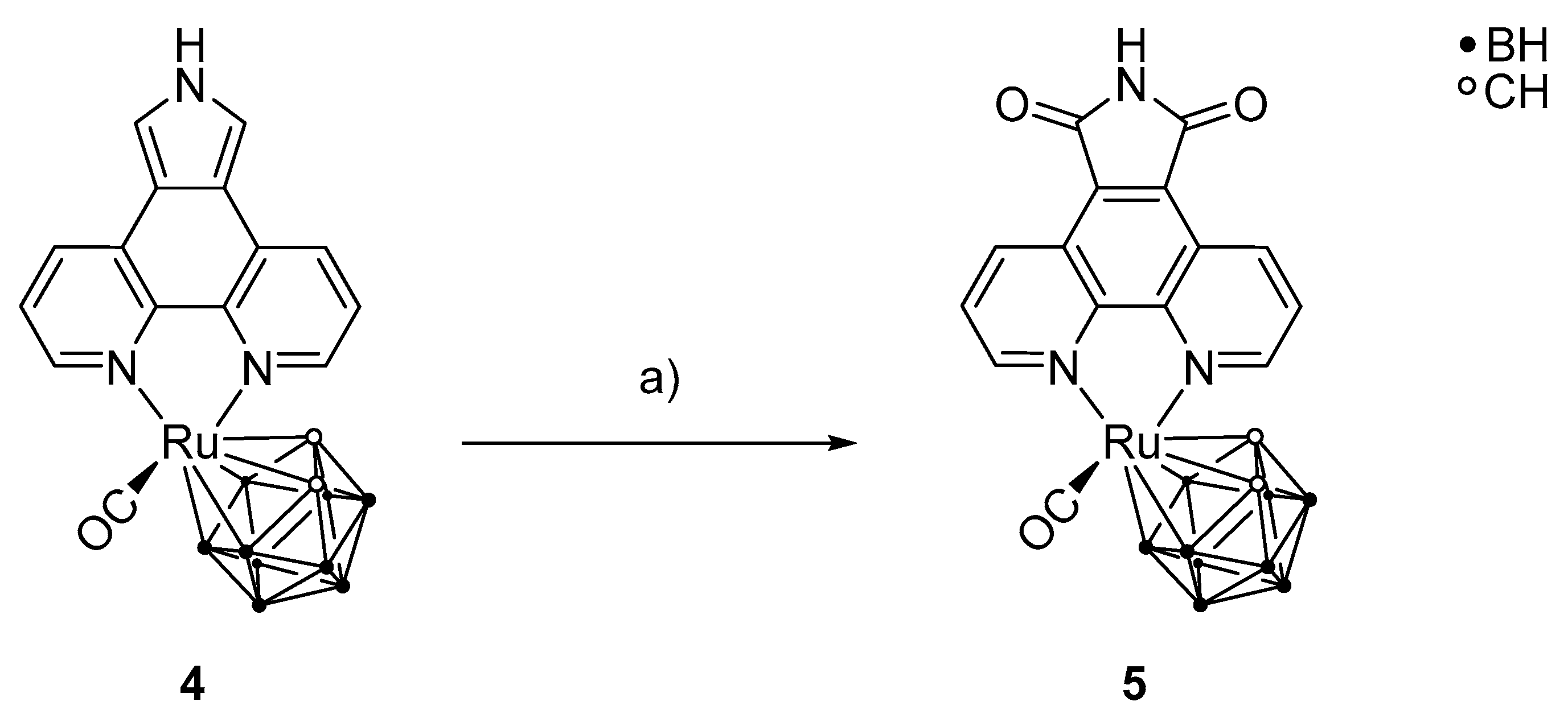
After the successful preparation of 4, a selective oxidation of positions 5 and 7 of the phenanthrolinopyrrole moiety was carried out to prepare [3-(CO)-3,3-{1′,10′-NC5H3(C(CO)(NH)(CO)C)NC5H3-κ2N,N}-closo-3,1,2-RuC2B9H11] (5) (Scheme 5). For this reaction, excess of meta-chloroperoxybenzoic acid (m-CPBA) was used. For stronger oxidizing agents, a polymerization of the pyrrole moiety is observed, and milder oxidizing agents lead to lactam scaffolds only and not to the desired maleimide groups [62,63]. Monitoring the reaction using thin layer chromatography showed that it is necessary to add the oxidizing agent successively in small portions during the reaction period. It was observed that only the ortho positions of the pyrrole moiety were oxidized, whereas the rest of the complex was unaffected.
After column chromatography and recovery of some starting material, the desired complex 5 was isolated in 10% yield as a red, crystalline powder and characterized. In the 1H-NMR spectrum, a low-field shift for the cluster CH and the aromatic CH groups in comparison to 4 was observed. The multiplicity pattern of the aromatic protons indicates an ortho substitution of the pyrrole moiety. Two signals are observed in the negative-mode mass spectrum, namely [M − H+]− and [M − CO − H+]−. The IR spectrum shows the presence of only one CO ligand as well as for the previous complexes. Compound 5 crystallizes from acetonitrile as deep red prisms in the triclinic space group P with three acetonitrile molecules in the asymmetric unit (Figure 4). Bond lengths and bond angles of 5 are given in Table 1.
2.3. Biological Studies
Since the target molecule 5 was designed as a mimic of (R)-DW12, protein kinase Pim1 inhibition studies were carried out but were inconclusive, most likely due to a low solubility of 5 in buffer solutions caused by the high hydrophobicity of the dicarbollide moiety. Additional information about the protein kinase inhibition studies is given in the electronic supplementary information.
As mentioned in the introduction, carborane or metallacarborane derivatives are being studied as drugs or enzyme inhibitors [11,14,19,21,23,27,28,30,35]. Due to the presence of the hydrophobic carborane moiety, lack of solubility in aqueous media is often observed, hampering biological studies. Improved water-solubility was achieved by employing charged species with enhanced solubility [11,14,31,35] or incorporating hydrophilic side chains [23]. A novel approach, developed by Hey-Hawkins et al., overcomes the problematic solubility behavior of metallacarborane complexes by employing bovine serum albumin (BSA) [37]; however, it was also observed that BSA can influence the activity of the solubilized drug in specific enzyme inhibition assays.
3. Materials and Methods
All syntheses were carried out using the Schlenk technique and nitrogen as inert gas. Triruthenium dodecacarbonyl, 1,2-dicarba-closo-dodecaborane(12), 5-nitro-1,10-phenanthroline, ethyl isocyanoacetate, 1,8-diazabicyclo[5.4.0]undec-7-ene, trimethylamine N-oxide and meta-chloroperoxybenzoic acid are commercially available. Trimethylamine N-oxide was dried and purified by sublimation; meta-chloroperoxybenzoic acid was purified by washing with phosphate buffer and drying under reduced pressure. 7,8-Dicarba-nido-undecaborane(13) (L1) [43], 1,10-phenanthrolinopyrrole (L2) [49], [3-(CO)3-closo-3,1,2-RuC2B9H11] (1) [55], and 1,10-phenanthrolino-5,6-dione (L5) [45,46] were synthesized according to the literature. The oxidation-sensitive compound 7,8-dicarba-nido-undecaborane(13) (L1) was stored under nitrogen at −55 °C. Ethyl isocyanoacetate was stored under nitrogen atmosphere and meta-chloroperoxybenzoic acid under normal atmosphere at 4 °C. All other used chemicals were stored under nitrogen atmosphere at ambient temperatures. All solvents, except acetonitrile, benzene, cyclohexane, and methanol, which were dried over calcium hydride, sodium, or calcium oxide, respectively, were taken from the solvent purification system MB SPS-800 (by MBraun). Ethanol was used as a mixture with water. Petrol ether (40–60 °C) for column chromatography was used as provided.
NMR spectra were measured with an ADVANCE DRX 400 spectrometer from Bruker. The spectrometer frequency for 1H and 11B are 400.13 MHz and 128.38 Hz, respectively. As an internal standard, tetramethylsilane was used for 1H-NMR and the Ξ scale was used for 11B-NMR spectroscopy. Data were interpreted with MestReNova [64]. The numbering scheme of all isolated compounds is given in the electronic supplementary information. Positive- or negative-mode low-resolution electrospray ionization mass spectra (ESI-MS) were recorded with an ESI ESQUIRE 3000 PLUS spectrometer with an IonTrap analyzer from Bruker Daltonics. For these measurements, dichloromethane, acetonitrile, methanol, or a mixture of these solvents were used. Infrared spectra were recorded with a Spectrum 2000 IR spectrometer from PerkinElmer in the range of 400 to 4000 cm−1. All samples were prepared as KBr pellets. The determination of the single crystal structures was carried out with a Gemini-S diffractometer from Oxford Diffraction using MoKα radiation (λ = 71.073 pm). The visualization of the structures was carried out with Diamond [65]. Additional crystallographic data are given in the electronic supplementary information. For column chromatography, silica gel 60 Å from the company Acros was used. The particle size was in the range of 0.035 to 0.070 mm. The solvents for semi-inert chromatography were obtained from the solvent purification device MB SPS-800. Thin layer chromatography (TLC) was used to monitor reaction processes using glass plates coated with silica gel 60 F254 from Merck. Carborane-containing spots were stained with a 5% solution of palladium(II) chloride in methanol.
- [3-(CO)-3,3-(L2-κ2N,N)-closo-3,1,2-RuC2B9H11] (2): 0.10 g (0.32 mmol, 1.00 eq.) 1 were placed in a 250 mL round bottom flask and dissolved in 25 mL acetonitrile. With stirring, a solution of 0.05 g (0.64 mmol, 2.00 eq.) trimethylamine N-oxide in 10 mL acetonitrile was added. The mixture was stirred for 10 min. Subsequently, 0.07 g (0.31 mmol, 0.97 eq.) 5-nitro-1,10-phenanthroline (L2), dissolved in 20 mL acetonitrile, were added dropwise to the mixture. The reaction mixture was stirred for 17 h at rt. After the reaction was finished (monitored using TLC), the mixture was filtered and the filtrate was dried under reduced pressure. The product was purified using column chromatography (dichloromethane; Rf = 0.60). 2 (0.02 g, 0.04 mmol, 13%) was obtained as an orange-red crystalline solid. 1H-NMR (400 MHz, CD3CN): δ = 1.00–2.80 (br, 9 H, 9× BH), 3.39 (s, br, 2 H, 2× CH1), 8.05 (m, 2 H, CH3, CH7), 8.88 (d, 3JHH = 8.2 Hz, 1 H, CH6), 9.10 (s, 1 H, CH5), 9.23 (d, 3JHH = 8.6 Hz, 1 H, CH4), 9.49 (d, 3JHH = 5.2 Hz, 1 H, CH8), 9.53 ppm (d, 3JHH = 5.3 Hz, 1 H, CH2); 11B{1H}-NMR (128 MHz, CD3CN): δ = −22.2 (s, 1 B, BH), −21.4 (s, 2 B, BH), −9.8 (s, 2 B, BH), −8.9 (s, 2 B, BH), −6.8 (s, 1 B, BH), −2.0 ppm (s, 1 B, BH); 11B-NMR (128 MHz, CD3CN): δ = −21.8 (m, br, 3 B, BH), −9.3 (m, br, 4 B, BH), −6.8 (d, 1JBH = 139 Hz, 1 B, BH), −2.0 ppm (d, 1JBH = 135 Hz, 1 B, BH); IR (KBr): = 2524 (s, νBH-sp3), 1970 (s, νCO-sp), 1535 (m, νasym.NO2), 1514 (m, νCN-sp2), 1342 cm−1 (m, νsym.NO2); MS (ESI, neg.): found: m/z (%): 549 (100) [M + NO3]−; calcd: m/z: 549 [M + NO3]−.
- [3-(CO)-3,3-(L3-κ2N,N)-closo-3,1,2-RuC2B9H11] (3): 0.10 g (0.32 mmol, 1.00 eq.) 1 were placed in a 250 mL round bottom flask and dissolved in 25 mL acetonitrile. Subsequently, 0.05 g (0.64 mmol, 2.00 eq.) trimethylamine N-oxide, dissolved in 10 mL acetonitrile, were added. The mixture was stirred for 10 min at rt. Then, 0.07 g (0.33 mmol, 1.03 eq.) 1,10-phenanthroline-5,6-dione (L3), dissolved in 15 mL acetonitrile, were added dropwise to the mixture. The reaction mixture was stirred for 84 h at rt. After the reaction was complete (monitored using TLC), the resulting precipitate was filtered off and the filtrate was concentrated under reduced pressure. The product was purified using column chromatography (dichloromethane; Rf = 0.17). Yield of 3: 0.11 g (0.23 mmol, 73%), orange solid. 1H-NMR (400 MHz, CD3CN): δ = 1.00–3.90 (br, 9 H, 9 × BH), 3.37 (s, br, 2 H, 2× CH1), 7.82 (dd, 3JHH = 7.9 Hz, 3JHH = 5.6 Hz, 2 H, 2× CH3), 8.62 (dd, 3JHH = 7.9 Hz, 4JHH = 1.4 Hz, 2 H, 2× CH4), 9.26 ppm (dd, 3JHH = 5.6 Hz, 4JHH = 1.4 Hz, 2 H, 2× CH2); 11B{1H}-NMR (128 MHz, CD3CN): δ = −21.9 (s, br, 3 B, BH), −9.6 (s, br, 4 B, BH), −7.8 (s, 1 B, BH), −1.4 ppm (s, 1 B, BH); 11B-NMR (128 MHz, CD3CN): δ = −21.9 (d, 1JBH = 143 Hz, 3 B, BH), −9.6 (d, 1JBH = 142 Hz, 4 B, BH), −7.8 (d, 1JBH = 144 Hz, 1 B, BH), −1.4 ppm (d, 1JBH = 151 Hz, 1 B, BH); IR (KBr): = 2507 (m, νBH-sp3), 1984 (m, νCO-sp), 1702 (m, νCO-sp2), 1691 (m, νCN-sp2), 802 cm−1 (s, 1,2,3-trisubstituted aromatic ring); MS (ESI, neg.): found: m/z (%): 552 (67) [M + Br]−; calcd: m/z: 552 [M + Br]−.
- [3-(CO)-3,3-(L4-κ2N,N)-closo-3,1,2-RuC2B9H11] (4): 0.20 g (0.63 mmol, 1.00 eq.) 1 were placed in a 250 mL round bottom flask and dissolved in 25 mL acetonitrile. To this solution, 0.10 g (1.26 mmol, 2.00 eq.) trimethylamine N-oxide, dissolved in 10 mL acetonitrile, were added dropwise. The mixture was stirred for 10 min at rt. Then, 0.28 g (1.28 mmol, 2.03 eq.) 1,10-phenanthrolinopyrrole (L4) were added in one portion and the reaction mixture was stirred for 48 h at room temperature. After the reaction was completed (monitored using TLC), the resulting precipitate was filtered off and the filtrate was concentrated under reduced pressure. The product was purified using column chromatography (dichloromethane; Rf = 0.33). Yield of 4: 0.14 g (0.29 mmol, 46%), yellow crystalline solid. 1H-NMR (400 MHz, CD3CN): δ = 0.70–2.80 (br, 9 H, 9× BH), 3.28 (s, br, 2 H, 2× CH1), 7.71 (dd, 3JHH = 8.1 Hz, 3JHH = 5.4 Hz, 2 H, 2× CH3), 7.92 (d, 3JHH = 2.8 Hz, 2 H, 2× CH5), 8.67 (d, 3JHH = 7.7 Hz, 2 H, 2× CH4), 9.03 (d, 3JHH = 5.1 Hz, 2 H, 2× CH2), 10.70 ppm (s, br, 1 H, NH6); 11B{1H}-NMR (128 MHz, CD3CN): δ = −21.7 (s, br, 3 B, BH), −10.1 (s, 2 B, BH), −9.0 (s, 2 B, BH), −7.2 (s, br, 1 B, BH), −2.7 ppm (s, br, 1 B, BH); 11B-NMR (128 MHz, CD3CN): δ = −21.8 (m, 3 B, BH), −8.8 (m, br, 5 B, BH), −2.7 ppm (d, 1JBH = 139 Hz, 1 B, BH); IR (KBr): = 2523 (m, νBH-sp3), 1958(s, νCO-sp), 1639 (w, νCN-sp2), 1600 (w, νCC-sp2), 803 cm−1 (m, 1,2,3-trisubstituted aromatic ring); MS (ESI, neg.): found: m/z (%): 480 (100) [M − H]−; calcd: m/z: 480 [M − H]−.
- [3-(CO)-3,3-{1′,10′-NC5H3(C(CO)(NH)(CO)C)NC5H3-κ2N,N}-closo-3,1,2-RuC2B9H11] (5): 0.31 g (0.65 mmol, 1.00 eq.) 4 were placed in a 250 mL round bottom flask and dissolved in 30 mL acetonitrile. Subsequently, 0.45 g (2.61 mmol, 4.01 eq.) m-CPBA, dissolved in 10 mL acetonitrile, were added under stirring at rt. The reaction mixture was heated under reflux for 84 h. During this time, two additional portions of 0.28 g (1.62 mmol, 2.49 eq.) m-CPBA, dissolved in 10 mL acetonitrile, were added after 24 h and 48 h, respectively. After completion of the reaction (monitored using TLC), the mixture was cooled to rt; the resulting precipitate was filtered off and the filtrate was concentrated under reduced pressure. The product was purified using column chromatography (dichloromethane/acetonitrile, 10:1, (v/v); Rf = 0.52). Yield of 5: 0.03 g (0.06 mmol, 9%, corrected, after recovery of starting material 4: 10%), deep red crystalline powder. In addition, 0.03 g (0.06 mmol) 4 were recovered. 1H-NMR (400 MHz, acetone-d6): δ = 0.82–3.01 (br, 9 H, 9× BH), 3.53 (s, br, 2 H, 2× CH1), 8.31 (dd, 3JHH = 8.3 Hz, 3JHH = 5.2 Hz, 2 H, 2× CH3), 9.59 (dd, 3JHH = 8.3 Hz, 3JHH = 1.4 Hz, 2 H, 2× CH4), 9.73 (dd, 3JHH = 5.3 Hz, 3JHH = 1.4 Hz, 2 H, 2× CH2), 10.74 ppm (s, br, 1 H, NH5); 11B{1H}-NMR (128 MHz, acetone-d6): δ = −21.7 (s, br, 3 B, BH), −9.3 (s, 2 B, BH), −7.5 (s, 3 B, BH), −1.3 ppm (s, 1 B, BH); 11B-NMR (128 MHz, acetone-d6): δ = −21.7 (d, 1JBH = 157 Hz, 3 B, BH), −9.2 (d, 1JBH = 162 Hz, 2 B, BH), −7.5 (d, 1JBH = 150 Hz, 3 B, BH), −1.3 ppm (d, 1JBH = 142 Hz, 1 B, BH); IR (KBr): = 2531 (s, νBH-sp3), 1956 (s, νCO-sp), 1727 (s, νCO-sp2), 1695 cm−1 (m, νCN-sp2); MS (ESI, neg.): found: m/z (%): 510 (100) [M − H]−, 482 (26) [M − CO − H]−; calcd: m/z: 510 [M − H]−, 482 [M − CO − H]−.
4. Conclusions
Four air stable ruthenium(II) half-sandwich complexes, 2‒5, which contain a dicarbollide moiety, a carbonyl ligand, and a phenanthroline derivative, were prepared in moderate to good yields and fully characterized. The complexes were designed to mimic the overall shape and structure of the alkaloid staurosporine and the ruthenium half-sandwich complex DW12. Initial inhibition experiments with the ruthenium(II) complexes 4 and 5 against the protein kinase Pim1 were not conclusive, most likely due to a low solubility of 4 and 5 in buffer solutions caused by the high hydrophobicity of the dicarbollide moiety. Thus, although a high hydrophobicity is beneficial for the inhibition of a biological target molecule, a sufficient solubility in aqueous buffer solution must be warranted, which will need to be addressed in future work.
Supplementary Materials
The electronic supplementary information is available online including the numbering scheme of all isolated compounds, additional synthetic procedures for L1, L4, and 1, the isolation of SP1 and its spectroscopic data, crystallographic information of compounds 2, SP1, 4, and 5, and information about the protein kinase inhibition studies.
Author Contributions
Conceptualization, M.K. and E.H.-H.; data curation, M.K., R.R., and P.L.; formal analysis, M.K. and P.L.; funding acquisition, E.M. and E.H.-H.; investigation, M.K.; methodology, I.S. and R.R.; project administration, M.K. and E.H.-H.; resources, E.M. and E.H.-H.; supervision, I.S., E.M., and E.H.-H.; validation, M.K. and I.S.; visualization, M.K.; writing—original draft, M.K.; writing—review and editing, E.M., P.L., and E.H.-H. All authors have read and agreed to the published version of the manuscript.
Funding
Funding by the “Europäischer Fonds für regionale Entwicklung (EFRE)”, the Free State of Saxony (ESF) and the Graduate School “Leipzig School of Natural Sciences—Building with Molecules and Nano-objects” (BuildMoNa) is gratefully acknowledged.
Acknowledgments
We thank Ramona Oehme, Susann Billig and Claudia Birkemeyer for measuring the mass spectra, Manuela Roßberg and Gunther Wünsche for elemental analysis, and Stefanie Märcker-Recklies for recording the infrared spectra.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Thompson, K.H.; Orvig, C. Metal Complexes in Medicinal Chemistry: New Vistas and Challenges in Drug Design. Dalton Trans. 2006, 761–764. [Google Scholar] [CrossRef]
- Colotti, G.; Ilari, A.; Boffi, A.; Morea, V. Metals and Metal Derivatives in Medicine. Mini Rev. Med. Chem. 2013, 13, 211–221. [Google Scholar] [CrossRef]
- Cohen, S.M. New Approaches for Medicinal Applications of Bioinorganic Chemistry. Curr. Opin. Chem. Biol. 2007, 11, 115–120. [Google Scholar] [CrossRef]
- Meggers, E.; Atilla-Gokcumen, G.E.; Bregman, H.; Maksimoska, J.; Mulcahy, S.P.; Pagano, N.; Williams, D.S. Exploring Chemical Space with Organometallics: Ruthenium Complexes as Protein Kinase Inhibitors. Synlett 2007, 1177–1189. [Google Scholar] [CrossRef]
- Bregman, H.; Williams, D.S.; Atilla, G.E.; Carroll, P.J.; Meggers, E. An Organometallic Inhibitor for Glycogen Synthase Kinase 3. J. Am. Chem. Soc. 2004, 126, 13594–13595. [Google Scholar] [CrossRef] [PubMed]
- Bregman, H.; Williams, D.S.; Meggers, E. Pyrido[2,3-a]pyrrolo[3,4-c]carbazole-5,7(6H)-diones: Synthesis, Cyclometalation, and Protein Kinase Inhibition. Synthesis 2005, 1521–1527. [Google Scholar] [CrossRef]
- Chiara, F.; Rasola, A. GSK-3 and Mitochondria in Cancer Cells. Front. Oncol. 2013, 3, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, P. Protein Kinases—the Major Drug Targets of the Twenty-First Century? Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar] [CrossRef]
- Allardyce, C.S.; Dyson, P.J. Ruthenium in Medicine: Current Clinical Uses and Future Prospects. Platinum Metals Rev. 2001, 45, 62–69. [Google Scholar]
- Dyson, P.J. Systematic Design of a Targeted Organometallic Antitumour Drug in Pre-clinical Development. CHIMIA 2007, 61, 698–703. [Google Scholar] [CrossRef]
- Gozzi, M.; Schwarze, B.; Sárosi, M.-B.; Lönnecke, P.; Drača, D.; Maksimović-Ivanić, D.; Mijatović, S.; Hey-Hawkins, E. Antiproliferative Activity of (η6-Arene)ruthenacarborane Sandwich Complexes Against HCT116 and MCF7 Cell Lines. Dalton Trans. 2017, 46, 12067–12080. [Google Scholar] [CrossRef]
- Süss-Fink, G. Arene Ruthenium Complexes as Anticancer Agents. Dalton Trans. 2010, 39, 1673–1688. [Google Scholar] [CrossRef]
- Gozzi, M.; Murganic, B.; Drača, D.; Popp, J.; Coburger, P.; Maksimović-Ivanić, D.; Mijatović, S.; Hey-Hawkins, E. Quinoline-Conjugated Ruthenacarboranes: Toward Hybrid Drugs with a Dual Mode of Action. Chem. Med. Chem. 2019, 14, 2061–2074. [Google Scholar] [CrossRef] [Green Version]
- Gozzi, M.; Schwarze, B.; Hey-Hawkins, E. Half- and Mixed-sandwich Metallacarboranes for Potential Applications in Medicine. Pure Appl. Chem. 2019, 91, 563–573. [Google Scholar] [CrossRef]
- Bregman, H.; Meggers, E. Ruthenium Half-Sandwich Complexes as Protein Kinase Inhibitors: An N-succinimidyl Ester for Rapid Derivatizations of the Cyclopentadienyl Moiety. Org. Lett. 2006, 8, 5465–5468. [Google Scholar] [CrossRef]
- Atilla-Gokcumen, G.E.; Williams, D.S.; Bregman, H.; Pagano, N.; Meggers, E. Organometallic Compounds with Biological Activity: A Very Selective and Highly Potent Cellular Inhibitor for Glycogen Synthase Kinase 3. ChemBioChem 2006, 7, 1443–1450. [Google Scholar] [CrossRef]
- Atilla-Gokcumen, G.E.; Di Costanzo, L.; Meggers, E. Structure of Anticancer Ruthenium Half-Sandwich Complex Bound to Glycogen Synthase Kinase 3β. J. Biol. Inorg. Chem. 2011, 16, 45–50. [Google Scholar] [CrossRef]
- Rüegg, U.T.; Burgess, G.M. Staurosporine, K-252 and UCN-01: Potent but Nonspecific Inhibitors of Protein Kinases. Trends Pharmacol. Sci. 1989, 10, 218–220. [Google Scholar] [CrossRef]
- Scholz, M.; Hey-Hawkins, E. Carbaboranes as Pharmacophores: Properties, Synthesis, and Application Strategies. Chem. Rev. 2011, 111, 7035–7062. [Google Scholar] [CrossRef]
- Issa, F.; Kassiou, M.; Rendina, L.M. Boron in Drug Discovery: Carboranes as Unique Pharmacophores in Biologically Active Compounds. Chem. Rev. 2011, 111, 5701–5722. [Google Scholar] [CrossRef]
- Grimes, R.N. Carboranes, 3rd ed.; Elsevier, Academic Press: Amsterdam, The Netherland; Boston, MA, USA; Heidelberg, Germany, 2016; ISBN 9780128018941. [Google Scholar]
- Grimes, R.N. Carboranes in the Chemist’s Toolbox. Dalton Trans. 2015, 44, 5939–5956. [Google Scholar] [CrossRef] [PubMed]
- Leśnikowski, Z.J. Challenges and Opportunities for the Application of Boron Clusters in Drug Design. J. Med. Chem. 2016, 59, 7738–7758. [Google Scholar] [CrossRef] [PubMed]
- Marta, G.; Benedikt, S.; Evamarie, H.-H. Half- and Mixed-sandwich Metallacarboranes in Catalysis. In Handbook of Boron Chemistry in Organometallics, Catalysis, Materials and Medicine, 2nd ed.; Imperial College Press/World Scientific Publishing (UK) Ltd.: London, UK, 2018; Volume 2, pp. 27–80. ISBN 9781786344410. [Google Scholar]
- Fujii, S. Expanding the Chemical Space of Hydrophobic Pharmacophores: The Role of Hydrophobic Substructures in the Development of Novel Transcription Modulators. Med. Chem. Commun. 2016, 7, 1082–1092. [Google Scholar] [CrossRef] [Green Version]
- Vincenzi, M.; Bednarska, K.; Leśnikowski, Z.J. Comparative Study of Carborane- and Phenyl-Modified Adenosine Derivatives as Ligands for the A2A and A3 Adenosine Receptors Based on a Rigid in Silico Docking and Radioligand Replacement Assay. Molecules 2018, 23, 1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockmann, P.; Gozzi, M.; Kuhnert, R.; Sárosi, M.-B.; Hey-Hawkins, E. New Keys for Old Locks: Carborane-Containing Drugs as Platforms for Mechanism-based Therapies. Chem. Soc. Rev. 2019, 48, 3497–3512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leśnikowski, Z.J. Recent Developments with Boron as a Platform for Novel Drug Design. Expert Opin. Drug Dis. 2016, 11, 569–578. [Google Scholar] [CrossRef]
- Lesnikowski, Z.J. Boron Units as Pharmacophores-New Applications and Opportunities of Boron Cluster Chemistry. Collect. Czech. Chem. C. 2007, 72, 1646–1658. [Google Scholar] [CrossRef]
- Ahrens, V.M.; Frank, R.; Boehnke, S.; Schütz, C.L.; Hampel, G.; Iffland, D.S.; Bings, N.H.; Hey-Hawkins, E.; Beck-Sickinger, A.G. Receptor-mediated Uptake of Boron-rich Neuropeptide Y Analogues for Boron Neutron Capture Therapy. Chem. Med. Chem. 2015, 10, 164–172. [Google Scholar] [CrossRef]
- Armstrong, A.F.; Valliant, J.F. The Bioinorganic and Medicinal Chemistry of Carboranes: From New Drug Discovery to Molecular Imaging and Therapy. Dalton Trans. 2007, 4240–4251. [Google Scholar] [CrossRef]
- Scholz, M.; Kaluđerović, G.N.; Kommera, H.; Paschke, R.; Will, J.; Sheldrick, W.S.; Hey-Hawkins, E. Carbaboranes as Pharmacophores: Similarities and Differences between Aspirin and Asborin. Eur. J. Med. Chem. 2011, 46, 1131–1139. [Google Scholar] [CrossRef]
- Scholz, M.; Steinhagen, M.; Heiker, J.T.; Beck-Sickinger, A.G.; Hey-Hawkins, E. Asborin Inhibits Aldo/Keto Reductase 1A1. Chem. Med. Chem. 2011, 6, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, B.; Gozzi, M.; Hey-Hawkins, E. Half- and Mixed-sandwich Transition Metal nido-Carboranes(-1) and Metallacarboranes for Medicinal Applications. In Boron-Based Compounds: Potential and Emerging Applications in Medicine; Hey-Hawkins, E., Viñas Teixidor, C., Eds.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2016; pp. 60–108, Online; ISBN 9781119275602. [Google Scholar]
- Neumann, W.; Xu, S.; Sárosi, M.B.; Scholz, M.S.; Crews, B.C.; Ghebreselasie, K.; Banerjee, S.; Marnett, L.J.; Hey-Hawkins, E. nido-Dicarbaborate Induces Potent and Selective Inhibition of Cyclooxygenase-2. Chem. Med. Chem. 2016, 11, 175–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gozzi, M.; Schwarze, B.; Coburger, P.; Hey-Hawkins, E. On the Aqueous Solution Behavior of C-Substituted 3,1,2-Ruthenadicarbadodecaboranes. Inorganics 2019, 7, 91. [Google Scholar] [CrossRef] [Green Version]
- Schwarze, B.; Gozzi, M.; Zilberfain, C.; Rüdiger, J.; Birkemeyer, C.; Estrela-Lopis, I.; Hey-Hawkins, E. Nanoparticle-based Formulation of Metallacarboranes with Bovine Serum Albumin for Application in Cell Cultures. J. Nanopart. Res. 2020, 22, 24. [Google Scholar] [CrossRef]
- Brown, D.A.; Fanning, M.O.; Fitzpatrick, N.J. Molecular Orbital Theory of Organometallic Compounds. 15. A Comparative Study of Ferrocene and π-cyclopentadienyl-(3)-1,2-dicarbollyliron. Inorg. Chem. 1978, 17, 1620–1623. [Google Scholar] [CrossRef]
- Hoffmann, R. Building Bridges Between Inorganic and Organic Chemistry (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 1982, 21, 711–724, Angew. Chem.1982, 94, 725–739. [Google Scholar] [CrossRef]
- Hosmane, N.S. Handbook of Boron Science with Applications in Organometallics, Catalysis, Materials and Medicine; Eagling, R., Ed.; World Scientific Publishing Europe Ltd.: London, UK; Munich, Germany, 2019; ISBN 1786344475. [Google Scholar]
- Debreczeni, J.É.; Bullock, A.N.; Atilla, G.E.; Williams, D.S.; Bregman, H.; Knapp, S.; Meggers, E. Ruthenium Half-sandwich Complexes Bound to Protein Kinase Pim-1. Angew. Chem. Int. Ed. 2006, 45, 1580–1585, Angew. Chem.2006, 118, 1610–1615. [Google Scholar] [CrossRef]
- Feng, L.; Geisselbrecht, Y.; Blanck, S.; Wilbuer, A.; Atilla-Gokcumen, G.E.; Filippakopoulos, P.; Kräling, K.; Celik, M.A.; Harms, K.; Maksimoska, J.; et al. Structurally Sophisticated Octahedral Metal Complexes as Highly Selective Protein Kinase Inhibitors. J. Am. Chem. Soc. 2011, 133, 5976–5986. [Google Scholar] [CrossRef] [Green Version]
- Hlatky, G.G.; Crowther, D.J. Main Group and Transition Metal Cluster Compounds: 38. 7,8-Dicarbaundecaborane(13). Inorg. Syn. 1998, 32, 229–231. [Google Scholar]
- Smith, G.F.; Cagle, F.W., Jr. The Improved Synthesis of 5-Nitro-1,10-phenanthroline. J. Org. Chem. 1947, 12, 781–784. [Google Scholar] [CrossRef]
- Sergeeva, N.N.; Donnier-Marechal, M.; Vaz, G.M.; Davies, A.M.; Senge, M.O. Stability and Spectral Properties of Europium and Zinc Phenanthroline Complexes as Luminescent Probes in High Content Cell-imaging Analysis. J. Inorg. Biochem. 2011, 105, 1589–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Lystrom, L.; Yin, H.; Hetu, M.; Kilina, S.; McFarland, S.A.; Sun, W. Increasing the Triplet Lifetime and Extending the Ground-state Absorption of Biscyclometalated Ir(III) Complexes for Reverse Saturable Absorption and Photodynamic Therapy Applications. Dalton Trans. 2016, 45, 16366–16378. [Google Scholar] [CrossRef] [PubMed]
- Barton, D.H.R.; Kervagoret, J.; Zard, S.Z. An Useful Synthesis of Pyrroles from Nitroolefins. Tetrahedron 1990, 46, 7587–7598. [Google Scholar] [CrossRef]
- Barton, D.H.R.; Zard, S.Z. A New Synthesis of Pyrroles from Nitroalkenes. J. Chem. Soc. Chem. Commun. 1985, 1098–1100. [Google Scholar] [CrossRef]
- Villegas, J.M.; Stoyanov, S.R.; Rillema, D.P. Synthesis and Photochemistry of Ru(II) Complexes Containing Phenanthroline-based Ligands with Fused Pyrrole Rings. Inorg. Chem. 2002, 41, 6688–6694. [Google Scholar] [CrossRef]
- Lash, T.D.; Lin, Y.; Novak, B.H.; Parikh, M.D. Porphyrins with Exocyclic Rings. Part 19: Efficient Syntheses of Phenanthrolinoporphyrins. Tetrahedron 2005, 61, 11601–11614. [Google Scholar] [CrossRef]
- Lash, T.D.; Novak, B.H.; Lin, Y. Synthesis of Phenanthropyrroles and Phenanthrolinopyrroles from Isocyanoacetates: An Extension of the Barton-Zard Pyrrole Condensation. Tetrahedron Lett. 1994, 35, 2493–2494. [Google Scholar] [CrossRef]
- Lin, Y.; Lash, T.D. Porphyrin Synthesis by the "3+1" Methodology: A Superior Approach for the Preparation of Porphyrins with Fused 9,10-Phenanthroline Subunits. Tetrahedron Lett. 1995, 36, 9441–9444. [Google Scholar] [CrossRef]
- Ono, N.; Hironaga, H.; Ono, K.; Kaneko, S.; Murashima, T.; Ueda, T.; Tsukamura, C.; Ogawa, T. A New Synthesis of Pyrroles and Porphyrins Fused with Aromatic Rings. J. Chem. Soc. Perkin Trans. 1 1996, 417–423. [Google Scholar] [CrossRef] [Green Version]
- Ono, N.; Hironaga, H.; Simizu, K.; Ono, K.; Kuwano, K.; Ogawa, T. Synthesis of Pyrroles Annulated with Polycyclic Aromatic Compounds; Precursor Molecules for Low Band Gap Polymers. J. Chem. Soc. Chem. Commun. 1994, 1019–1020. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.; Mullica, D.F.; Sappenfield, E.L.; Stone, F.G.A. Carborane Complexes of Ruthenium: A Convenient Synthesis of [Ru(CO)3(η5-7,8-C2B9H11)] and a Study of Reactions of This Complex. Organometallics 1995, 14, 3516–3526. [Google Scholar] [CrossRef]
- Behnken, P.E.; Hawthorne, M.F. Reactions at the Metal Vertex of a Ruthenacarborane Cluster. Activation of Carbon Monoxide by closo-3,3,3-(CO)3-3,1,2-RuC2B9H11. Inorg. Chem. 1984, 3420–3423. [Google Scholar] [CrossRef]
- Siedle, A.R. Dicarbollide Complexes of Rhodium and Ruthenium. J. Organomet. Chem. 1975, 90, 249–256. [Google Scholar] [CrossRef]
- Jelliss, P.A.; Mason, J.; Nazzoli, J.M.; Orlando, J.H.; Vinson, A.; Rath, N.P.; Shaw, M.J. Synthesis and Characterization of Ruthenacarborane Complexes Incorporating Chelating N-donor Ligands: Unexpected Luminescence from the Complex 3-CO-3,3-κ2-Me2N(CH2)2NMe2-closo-3,1,2-RuC2B9H11. Inorg. Chem. 2006, 45, 370–385. [Google Scholar] [CrossRef] [PubMed]
- Dyson, P.J.; McIndoe, J.S. Transition Metal Carbonyl Cluster Chemistry; Gordon and Breach Science Publishers: Amsterdam, The Netherlands, 2000; ISBN 90-5699-289-9. [Google Scholar]
- Paw, W.; Eisenberg, R. Synthesis, Characterization, and Spectroscopy of Dipyridocatecholate Complexes of Platinum. Inorg. Chem. 1997, 36, 2287–2293. [Google Scholar] [CrossRef]
- Pearsons, R.G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Alamgir, M.; Mitchell, P.S.R.; Bowyer, P.K.; Kumar, N.; Black, D.S. Synthesis of 4,7-Indoloquinones from Indole-7-carbaldehydes by Dakin Oxidation. Tetrahedron 2008, 64, 7136–7142. [Google Scholar] [CrossRef]
- Howard, J.K.; Hyland, C.J.T.; Just, J.; Smith, J.A. Controlled Oxidation of Pyrroles: Synthesis of Highly Functionalized γ-Lactams. Org. Lett. 2013, 15, 1714–1717. [Google Scholar] [CrossRef]
- MestReNova; v12.0.0-20080; Mestrelab Research S.L.: Santiago de Compostela, Spain, 2017.
- Diamond; v4.6.2; Crystal Impact GbR: Bonn, Germany, 1997–2020.
Sample Availability: Samples of the compounds are not available from the authors. |
Figure 1.
Staurosporine, a ruthenium half-sandwich staurosporine mimic (DW12), and the ruthenacarborane complex synthesized and investigated in this study [17].
Figure 1.
Staurosporine, a ruthenium half-sandwich staurosporine mimic (DW12), and the ruthenacarborane complex synthesized and investigated in this study [17].

Scheme 1.
Two-step synthesis of 7,8-dicarba-nido-undecaborane(13) (L1). a) MeOH, KOH, 80 °C, 18 h; b) C6H6, H3PO4, rt, 17 h. Yield over two steps: 70% [43].
Scheme 1.
Two-step synthesis of 7,8-dicarba-nido-undecaborane(13) (L1). a) MeOH, KOH, 80 °C, 18 h; b) C6H6, H3PO4, rt, 17 h. Yield over two steps: 70% [43].

Figure 2.
Phenanthroline-based ligands L2–L4.

Scheme 2.
Preparation of L4 starting from 5-nitro-1,10-phenanthroline (L2) with 1,10-phenanthrolinopyrrole ethyl ester (L4’) as intermediate. a) THF, DBU, ethyl isocyanoacetate, rt, 20 h; b) EtOH, 0.2 M NaOH, 110 °C, 8 h. Yield over two steps: 54%; DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene [49].
Scheme 2.
Preparation of L4 starting from 5-nitro-1,10-phenanthroline (L2) with 1,10-phenanthrolinopyrrole ethyl ester (L4’) as intermediate. a) THF, DBU, ethyl isocyanoacetate, rt, 20 h; b) EtOH, 0.2 M NaOH, 110 °C, 8 h. Yield over two steps: 54%; DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene [49].

Scheme 3.
Preparation of 1 with triruthenium dodecacarbonyl and L1. a) C6H6, 90 °C, 7 h, 60% [55].
Scheme 3.
Preparation of 1 with triruthenium dodecacarbonyl and L1. a) C6H6, 90 °C, 7 h, 60% [55].

Scheme 4.
Preparation of compounds 2–4 from the phenanthroline ligands L2–L4 and the ruthenium(II) complex 1. a) MeCN, trimethylamine N-oxide, rt, 17 h, 13% 2; b) MeCN, trimethylamine N-oxide, rt, 84 h, 73% 3; c) MeCN, trimethylamine N-oxide, rt, 48 h, 46% 4.
Scheme 4.
Preparation of compounds 2–4 from the phenanthroline ligands L2–L4 and the ruthenium(II) complex 1. a) MeCN, trimethylamine N-oxide, rt, 17 h, 13% 2; b) MeCN, trimethylamine N-oxide, rt, 84 h, 73% 3; c) MeCN, trimethylamine N-oxide, rt, 48 h, 46% 4.

Figure 3.
Molecule structure of [3-(CO)-3,3-(L2-κ2N,N)-closo-3,1,2-RuC2B9H11] (2) (left, only the S enantiomer is shown) and [3-(CO)-3,3-(L4-κ2N,N)-closo-3,1,2-RuC2B9H11] (4) (right) as an ellipsoid-stick model with thermal ellipsoids at 50% probability level. Solvent molecules and hydrogen atoms are omitted for clarity.
Figure 3.
Molecule structure of [3-(CO)-3,3-(L2-κ2N,N)-closo-3,1,2-RuC2B9H11] (2) (left, only the S enantiomer is shown) and [3-(CO)-3,3-(L4-κ2N,N)-closo-3,1,2-RuC2B9H11] (4) (right) as an ellipsoid-stick model with thermal ellipsoids at 50% probability level. Solvent molecules and hydrogen atoms are omitted for clarity.

Scheme 5.
Synthesis of [3-(CO)-3,3-{1′,10′-NC5H3(C(CO)(NH)(CO)C)NC5H3-κ2N,N}-closo-3,1,2-RuC2B9H11] (5) from 4 and meta-chloroperoxybenzoic acid (m-CPBA). a) MeCN, m-CPBA, 82 °C, 84 h, 10%.
Scheme 5.
Synthesis of [3-(CO)-3,3-{1′,10′-NC5H3(C(CO)(NH)(CO)C)NC5H3-κ2N,N}-closo-3,1,2-RuC2B9H11] (5) from 4 and meta-chloroperoxybenzoic acid (m-CPBA). a) MeCN, m-CPBA, 82 °C, 84 h, 10%.

Figure 4.
Molecule structure of [3-(CO)-3,3-{1′,10′-NC5H3(C(CO)(NH)(CO)C)NC5H3-κ2N,N}-closo-3,1,2-RuC2B9H11] (5) as an ellipsoid-stick model with thermal ellipsoids at 50% probability level. Solvent molecules and hydrogen atoms are omitted for clarity.
Figure 4.
Molecule structure of [3-(CO)-3,3-{1′,10′-NC5H3(C(CO)(NH)(CO)C)NC5H3-κ2N,N}-closo-3,1,2-RuC2B9H11] (5) as an ellipsoid-stick model with thermal ellipsoids at 50% probability level. Solvent molecules and hydrogen atoms are omitted for clarity.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Selected bond lengths (in pm) and angles (in °) in 2, 4, and 5 in comparison with [3-CO-3,3-(bipy-κ2N,N)-closo-3,1,2-RuC2B9H11] (I) (bipy = 2,2′-bipyridine) [58].
Table 1.
Selected bond lengths (in pm) and angles (in °) in 2, 4, and 5 in comparison with [3-CO-3,3-(bipy-κ2N,N)-closo-3,1,2-RuC2B9H11] (I) (bipy = 2,2′-bipyridine) [58].
| Atom Group | 2 | 4 a | 5 | I |
|---|---|---|---|---|
| Ru(1)–C(1) | 226.5(9) | 222.3(4) [225.0(4)] | 221.3(2) | 217.4(4) |
| Ru(1)–C(2) | 222.7(9) | 217.6(4) [223.4(49] | 222.7(2) | 222.4(4) |
| Ru(1)–B(1) | 220(1) | 222.4(4) [220.1(4)] | 220.6(2) | 227.9(4) |
| Ru(1)–B(2) | 219(1)) | 227.3(4) [220.5(4)] | 224.0(2) | 227.7(4) |
| Ru(1)–B(3) | 221.5(9) | 223.4(4) [223.6(4)] | 224.0(2) | 220.6(5) |
| Ru(1)–C(3) | 185(1) | 185.6(4) [183.5(4)] | 184.6(2) | 186.6(4) |
| Ru(1)–N(1) | 212.9(8) | 210.5(3) [211.6(3)] | 212.2(1) | 209.3(3) |
| Ru(1)–N(2) | 213.6(7) | 212.4(3) [212.2(3)] | 211.9(1) | 213.5(3) |
| N(1)···N(2) | 261(1) | 262.5(5) [262.3(4)] | 262.8(2) | 261.0(5) |
| C(3)–O(1) | 115(1) | 114.7(4) [114.6(4)] | 115.0(2) | 115.4(5) |
| N(1)–Ru(1)–C(3) | 90.9(4) | 91.5(1) [94.9(2)] | 93.9(1) | 90.1(2) |
| N(2)–Ru(1)–C(3) | 91.0(4) | 95.5(1) [94.0(1)] | 92.2(1) | 92.0(1) |
| N(1)–Ru(1)–N(2) | 75.5(3) | 76.7(1) [76.5(1)] | 76.6(1) | 76.3(1) |
| Ru(1)–C(3)–O(1) | 176.4(9) | 173.4(3) [174.1(4)] | 173.8(2) | 175.3(4) |
a values of the second independent molecule of 4 are given in [ ].
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kellert, M.; Sárosi, I.; Rajaratnam, R.; Meggers, E.; Lönnecke, P.; Hey-Hawkins, E. Ruthenacarborane–Phenanthroline Derivatives as Potential Metallodrugs. Molecules 2020, 25, 2322. https://doi.org/10.3390/molecules25102322
AMA Style
Kellert M, Sárosi I, Rajaratnam R, Meggers E, Lönnecke P, Hey-Hawkins E. Ruthenacarborane–Phenanthroline Derivatives as Potential Metallodrugs. Molecules. 2020; 25(10):2322. https://doi.org/10.3390/molecules25102322
Chicago/Turabian StyleKellert, Martin, Imola Sárosi, Rajathees Rajaratnam, Eric Meggers, Peter Lönnecke, and Evamarie Hey-Hawkins. 2020. "Ruthenacarborane–Phenanthroline Derivatives as Potential Metallodrugs" Molecules 25, no. 10: 2322. https://doi.org/10.3390/molecules25102322