Tri(boryl)alkanes and Tri(boryl)alkenes: The Versatile Reagents


Department Química Física i Inorgànica, University Rovira i Virgili, 43007 Tarragona, Spain
*
Author to whom correspondence should be addressed.
Molecules 2020, 25(7), 1758; https://doi.org/10.3390/molecules25071758
Submission received: 26 March 2020
/
Revised: 7 April 2020
/
Accepted: 8 April 2020
/
Published: 10 April 2020
(This article belongs to the Special Issue Boron in Catalysis and Materials Chemistry: A Themed Issue in Honor of Professor Todd B. Marder on the Occasion of His 65th Birthday)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The interest of organoboron chemistry in organic synthesis is growing, together with the development of new and versatile polyborated reagents. Here, the preparation of 1,1,1-tri(boryl)alkanes, 1,2,3-tri(boryl)alkanes, 1,1,2-tri(boryl)alkanes, as well as 1,1,2-tri(boryl)alkenes as suitable and accessible polyborated systems is demonstrated as being easily applied in the construction of new carbon-carbon and carbon-heteroatom bonds. Synthetic procedures and limitations have been collected to demonstrate the powerful strategies to construct selective molecules, taking advantages of the easy transformation of carbon-boron bond in multiple functionalities, under the total control of chemo- and stereoselectivity.
1. Introduction
The possibility of preparing a compound that contains three C-B bonds within its formula enhances the power towards polyfunctionalization strategies in a sequential manner. The fact that the three boryl units can be bonded to the same C or different carbons of the same single compound opens a large number of possibilities to construct a new functional product, especially from the perspective that the presence of more than one boryl group contributes to the stabilization of the intermediate carbanions formed, facilitating the transformation of the C-B bond, even under room temperature conditions. Both tri(boryl)alkanes and tri(boryl)alkenes preserve the stereoselectivity along the transformations and the current approaches to functionalize chemoselectively one boryl unit versus the other two, which nowadays open an unlimited opportunity to stimulate the creativity of synthetic chemists towards accessible protocols for useful applications.
2. 1,1,1-Tri(boryl)alkanes
2.1. Synthetic Approaches
Triborylmethane compounds have originally been prepared by Matteson and co-workers, developing a reliable synthetic procedure by mixing dimethoxyboron chloride, Cl-B(OMe)2, with CHCl3 and Li (Scheme 1) [1,2]. By using the appropriate chlorinated reagents, related triboryl compounds can be easily prepared, giving access to a new generation of organopolyborated species.
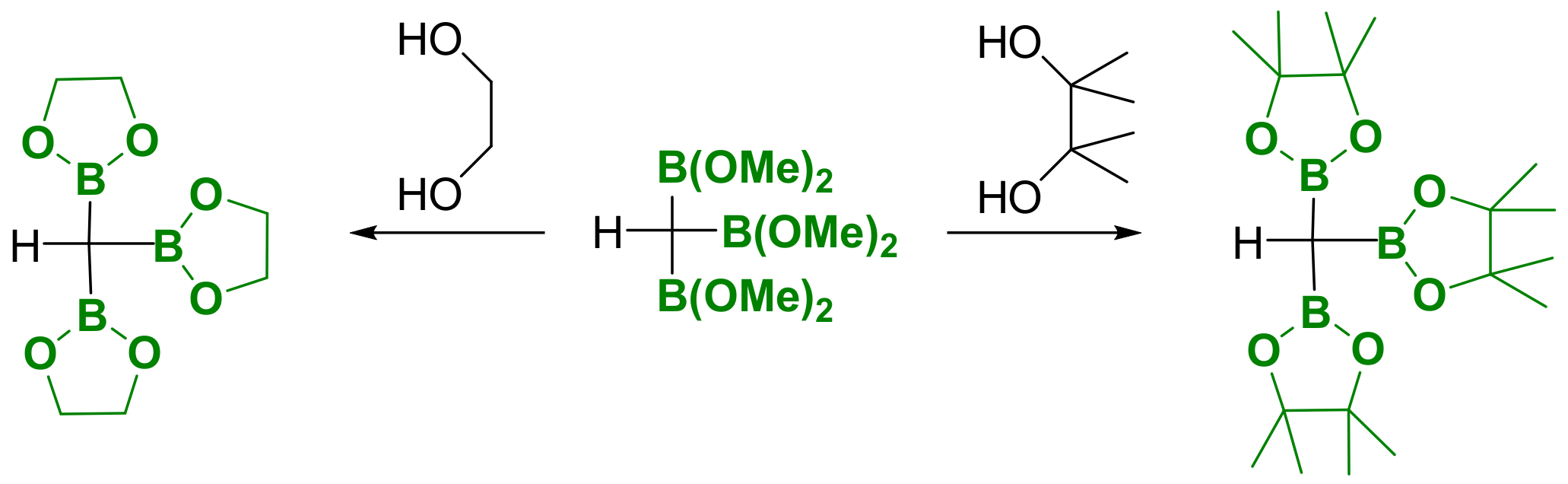
The lability of dimethoxyboryl moieties contributes to several difficulties in isolating the corresponding organotriboronates, and consequently, cyclic boronic esters have been prepared, as they are generally more stable towards hydrolysis. The transesterification of tris(dimethoxyboryl)methane with ethylenglycol or pinacol provides a direct access to the cyclic esters ethylenglycolboryl and pinacolboryl [3], with the expected degree of stability (Scheme 2) [4].
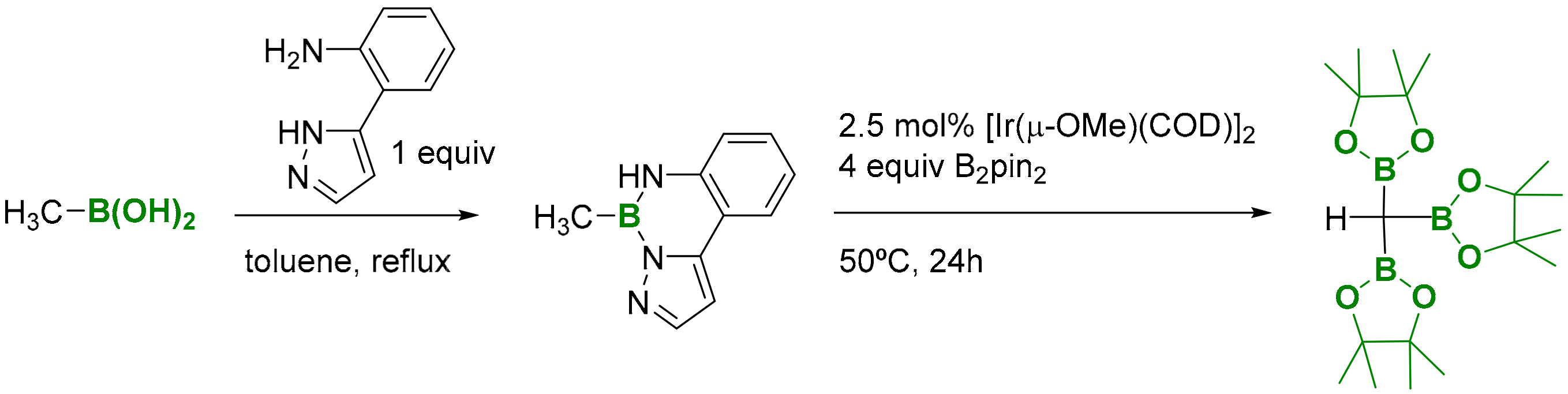
In particular, the synthesis of tris(pinacolboryl)methane has been recently redesigned by Suginome and co-workers, via the α,α-diborylation of pyrazolylaniline-modified methylboronic acid with B2pin2 in the presence of [Ir(μ-OMe)(COD)]2 as catalyst (Scheme 3) [5].
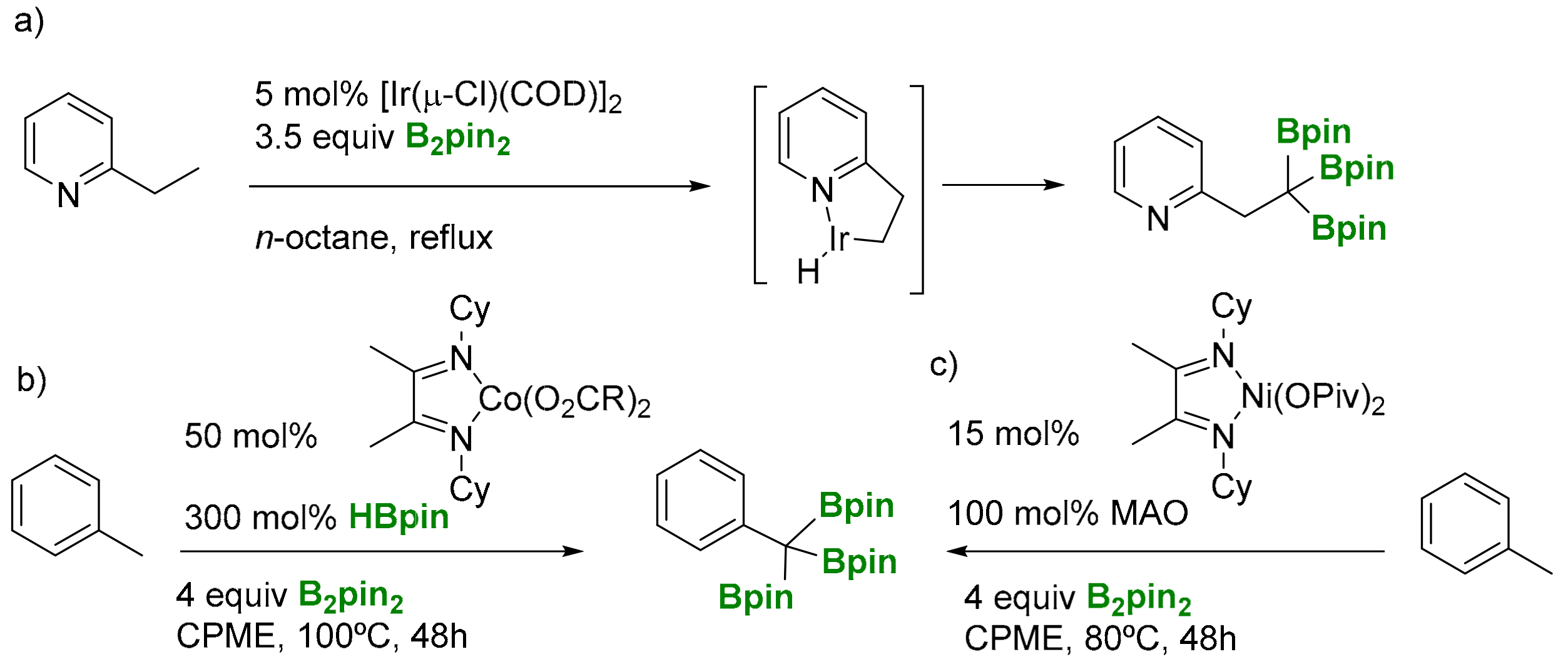
Triborylalkane derivatives have been prepared via the triple borylation of terminal C(sp3)-H bonds with transition-metal complexes. Sato, Mita and co-workers determined the first catalytic C(sp3)–H triborylation at a single carbon with the assistance of a nitrogen directing group [6]. Model substrate 2-ethylpyridine forms a five-membered metallacycle intermediate, via C(sp3)–H activation with Ir from [Ir(μ-Cl)(COD)]2 catalyst, that subsequently reacts with 4 equiv of B2pin2 towards site-selective C(sp3)–H borylation leading to the desired triborylated product (Scheme 4a). Triborylation of terminal C(sp3)-H bonds in toluene was also observed by Chirik and co-workers by using 50 mol% of α-diimine cobalt bis(carboxylate) precatalysts and 4 equiv of B2pin2 (Scheme 4b) [7]. Similarly, α-diimine nickel bis(carboxylate) complex enables the selective preparation of benzyltriboronates via the triborylation of benzylic C−H bonds (Scheme 4c) [8].
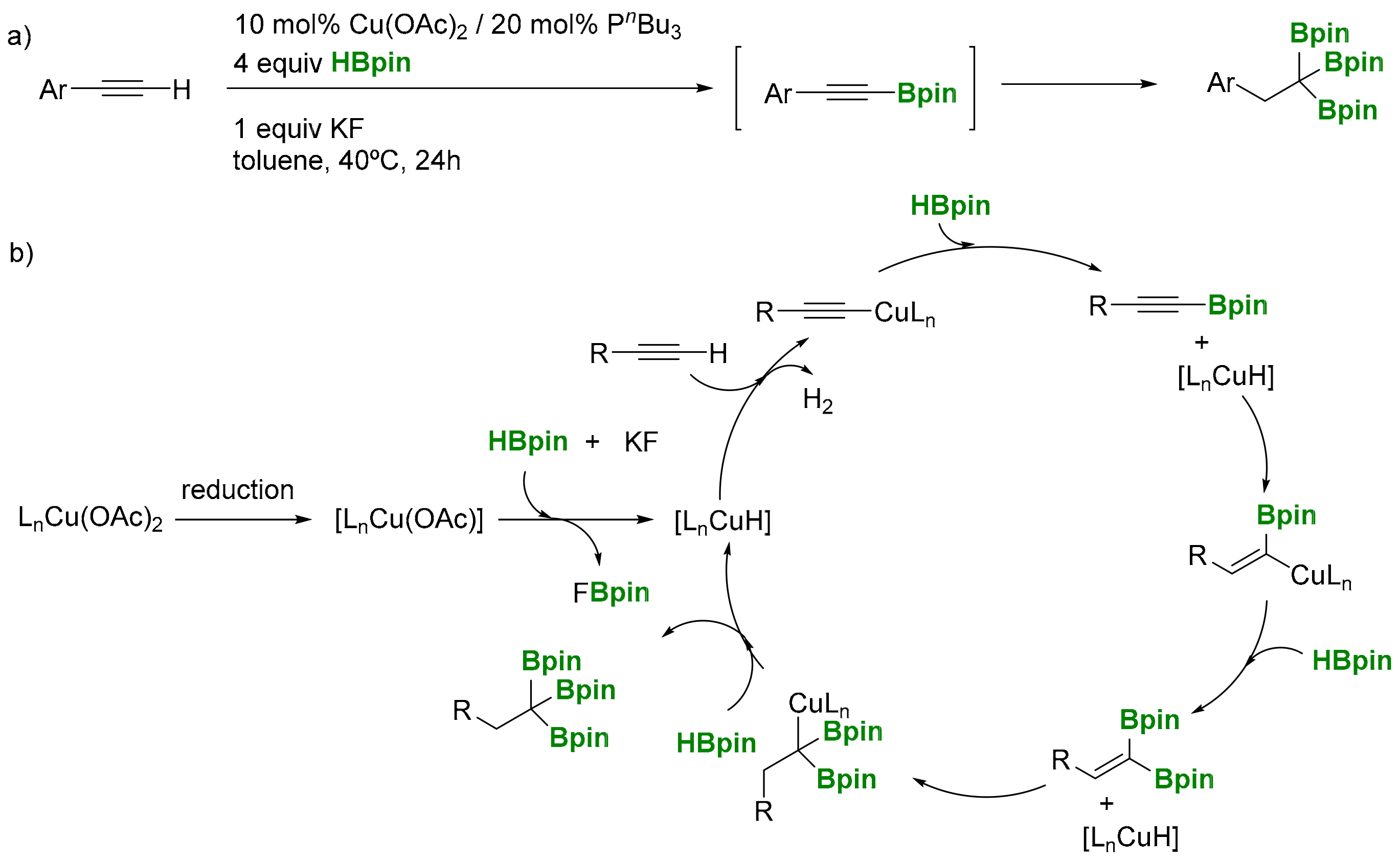
The sequential dehydrogenative borylation/hydroboration of terminal alkynes with Cu complexes contributes to the expansion of the triborylalkane derivatives synthesis. Marder and co-workers have developed a one-step synthesis via the dehydrogenative borylation of alkynes followed by the double hydroboration of 1-alkynylboronate intermediate with HBpin (HBpin = pinacolborane) catalyzed by Cu(OAc)2 (Scheme 5a) [9]. Interestingly, other copper sources, such as CuCl2, CuCl, and Cu(OTf)2, resulted in being inactive in this transformation. The base KF was essential in a stoichiometric manner, presumably by promoting the Cu-H formation in an early step of the catalytic cycle (Scheme 5b). This strategy complements a pioneer attempt by Marder and co-workers to perform the Rh-catalyzed reaction of ethynylarenes with HBcat (HBcat = catecholborane), to obtain the corresponding tris(catecholboryl)alkane, via vinylboronate intermediate [10]. Catechol-substituted monoborylacetylene could be regioselectively hydroborted via the transition-metal free syn-addition of catecholborane favoring the formation of the 1,1-diborylalkene, that can be further hydroborated selectively with HBCl2 to provide the corresponding 1,1,1-triborylethane. The subsequent substitution of Cl atoms by cathecol leads to the desired tris(catecholboryl)ethane [11]. Further treatment of tris(catecholboryl)ethane with tBuLi results in the formation of 1,1,1-tris[di(tert-butyl)boryl]ethane (Scheme 6).
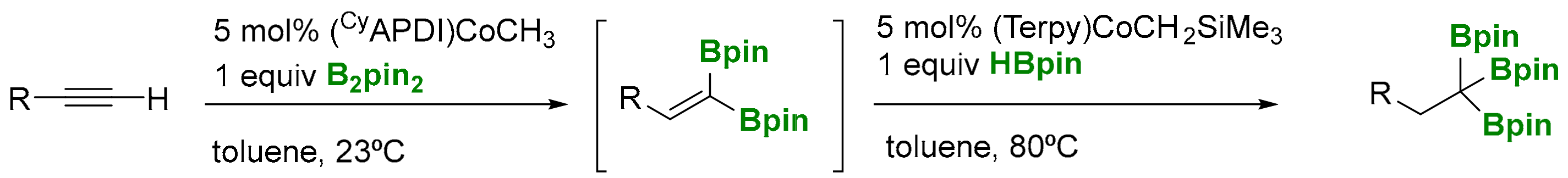
Alternatively, Chirik and co-workers have demonstrated that a cyclohexylsubstituted pyridine(diimine) cobalt methyl complex allows the dehydrogenative 1,1-diboration of terminal alkynes with B2pin2 at room temperature. This 1,1-diborylalkene intermediate can accomplish a consecutive cobalt-catalyzed hydroboration with HBpin to generate the desired of 1,1,1-triborylalkane (Scheme 7) [12]. The methodology is described as a one pot-sequential 1,1-diboration of terminal alkynes with B2pin2, followed by hydroboration with HBpin, although two different types of cobalt complexes are used for each step.
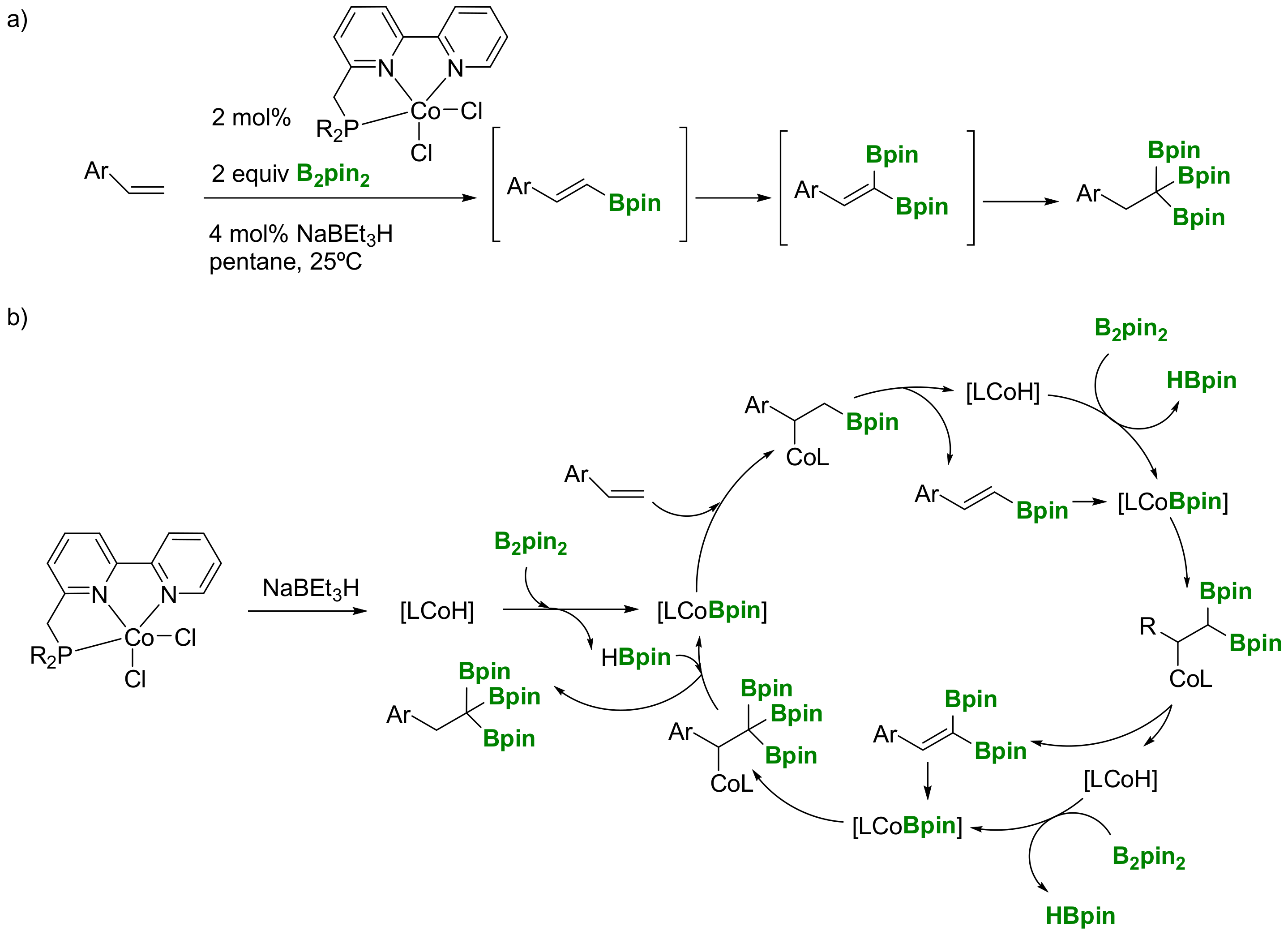
Huang and co-workers have developed a Co(I)-catalyzed double dehydrogenative borylation of vinylarenes with B2pin2 to generate a 1,1-diborylalkene intermediate, which undergoes hydroboration with pinacolborane to give 1,1,1-tris(boronates) in high yield (Scheme 8a) [13]. The dehydrogenative borylation of alkenes leading to vinylic boron compounds was first reported in 1992 [14], and it is understood as being the result of a catalytic hydroboration of an alkene with a monohydrideborane, followed by the elimination of hydrogen. However, Huang and co-workers have postulated a mechanism where the dehydrogenative borylation occurs by the insertion of vinylarenes into the Co−B bond to generate a β-boryl-substituted Co(I) species, which undergoes β-hydride elimination to give monoborylalkenes and the Cu-H intermediate that regenerates the Co-B catalytic species via σ-bond methatesis with B2pin2, maintaining the same type of reactivity along the catalytic cycle (Scheme 8b). As a limitation, this protocol cannot be applied for 1,2- and 1,1-disubstituted vinylarenes as well as vinylalkanes, since 1,1,1-tris(boronates) were not formed.
2.2. Synthetic Applications
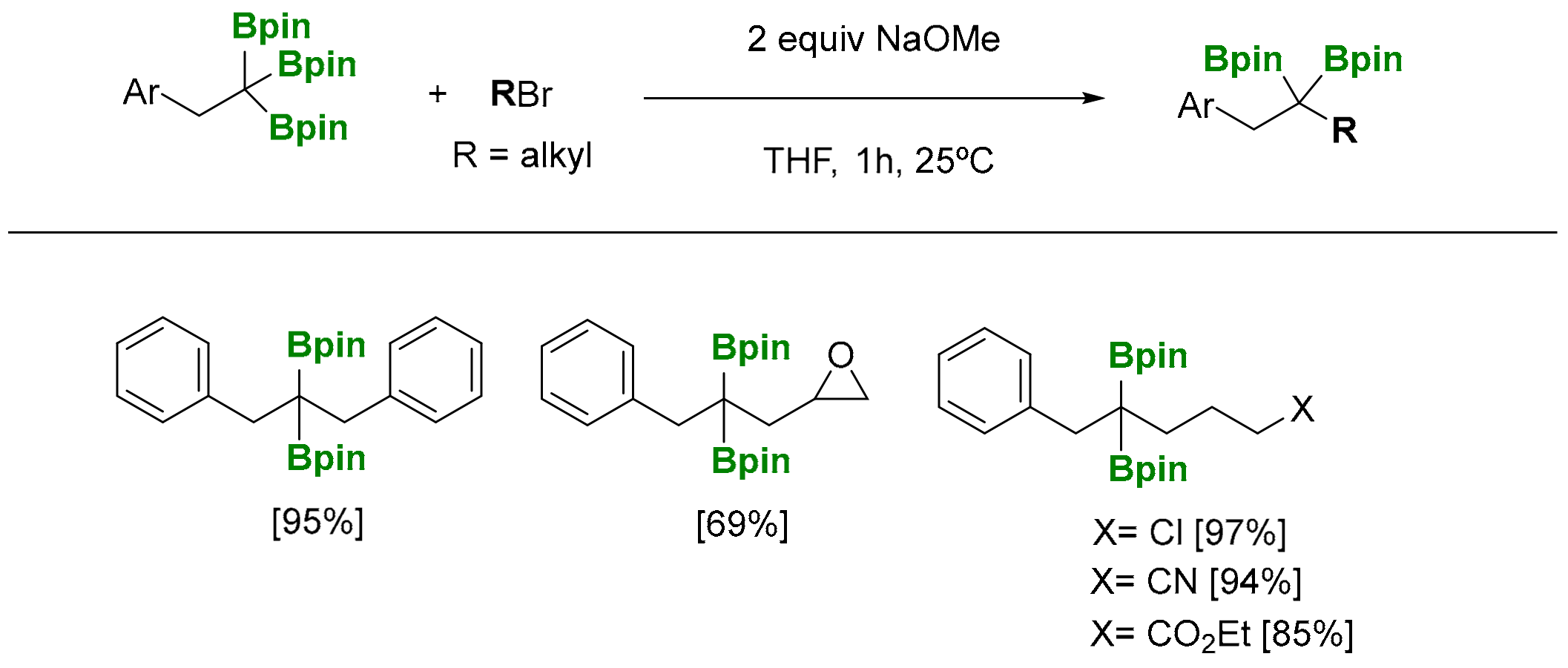
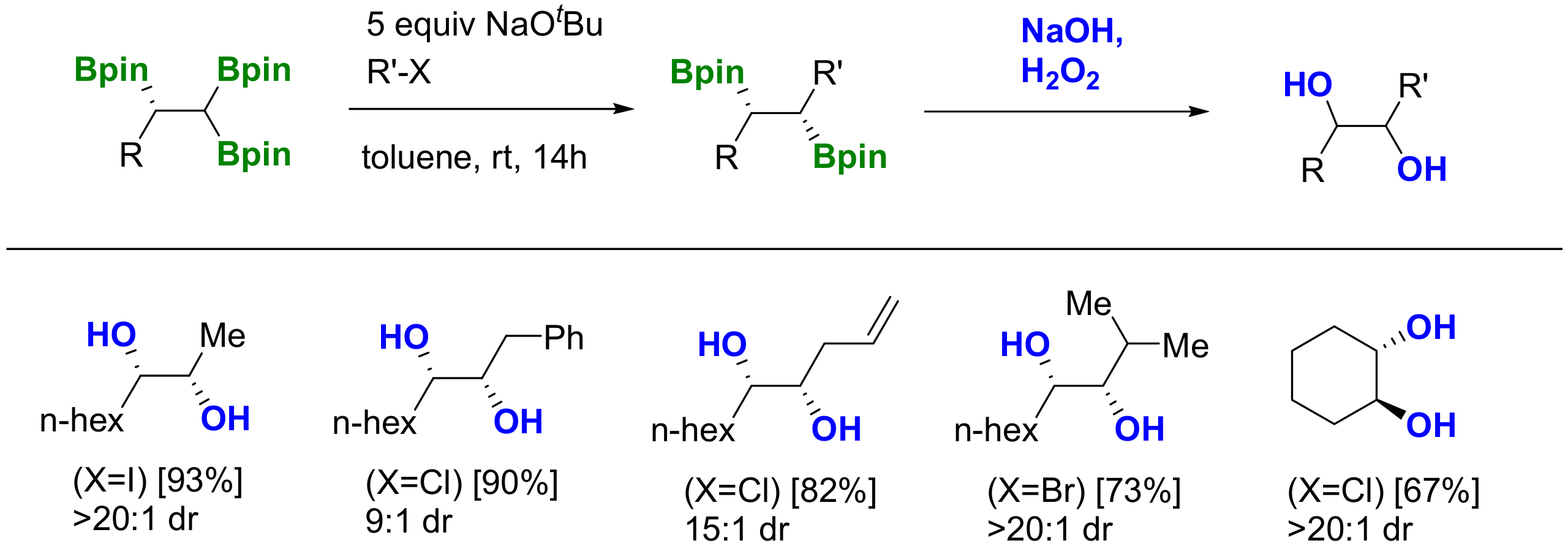
The reactivity of 1,1,1-tris(pinacolboryl)alkanes has been launched by alkoxide-assisted deborylative pathways, via α-bis(boryl) carbanion formation, that can be eventually trapped by different electrophiles [4]. The deborylative alkylation of 1,1,1-tris(boronate) products has been applied to the synthesis of internal geminal bis(boronates), by reaction with alkyl bromides in the presence of NaOMe (1.5 equiv), at 25 °C (Scheme 9) [13]. Interestingly, 1,1,1-tris(boronates) seems to be more reactive for deborylative alkylations than the analogue 1,1-bis(boronates), as the reactions with the latter require the use of an excess of NaOtBu (3 equiv) [15]. The reaction is compatible with diverse functionalities in the electrophiles, including cyanide, propylene epoxide, ester, halides and amines.
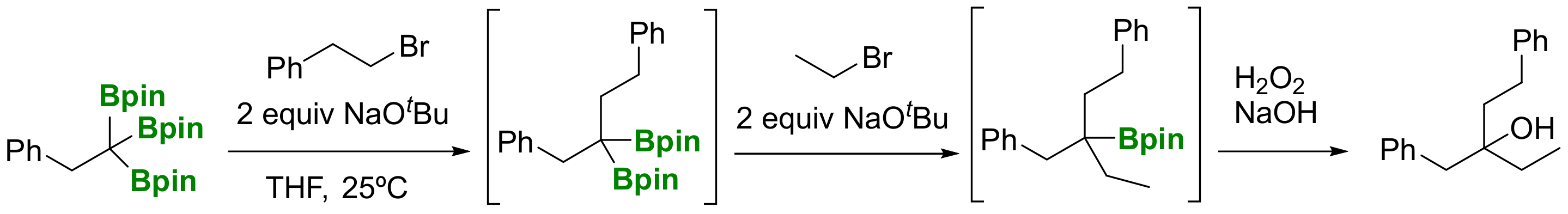
Double deborylative alkylation delivers tertiary boronic esters, as well as carbocyclic derivatives. Using 1,n-dihalides as electrophiles and NaOtBu (4 equiv), the base-assisted deborylation acts twice on the triborylated reagent, to facilitate the formation of α-vinylboronates (when n = 0) and cyclic organoboronates (when n = 1–5) at room temperature within 6h (Scheme 10) [9]. Following the same synthetic strategy, different alkyl groups can be introduced in a stepwise manner through sequential deborylative alkylations, providing access to tertiary boronic esters, which, after in situ oxidation with H2O2/NaOH, furnished tertiary alcohols containing three different alkyl groups in moderate yield (Scheme 11) [9].
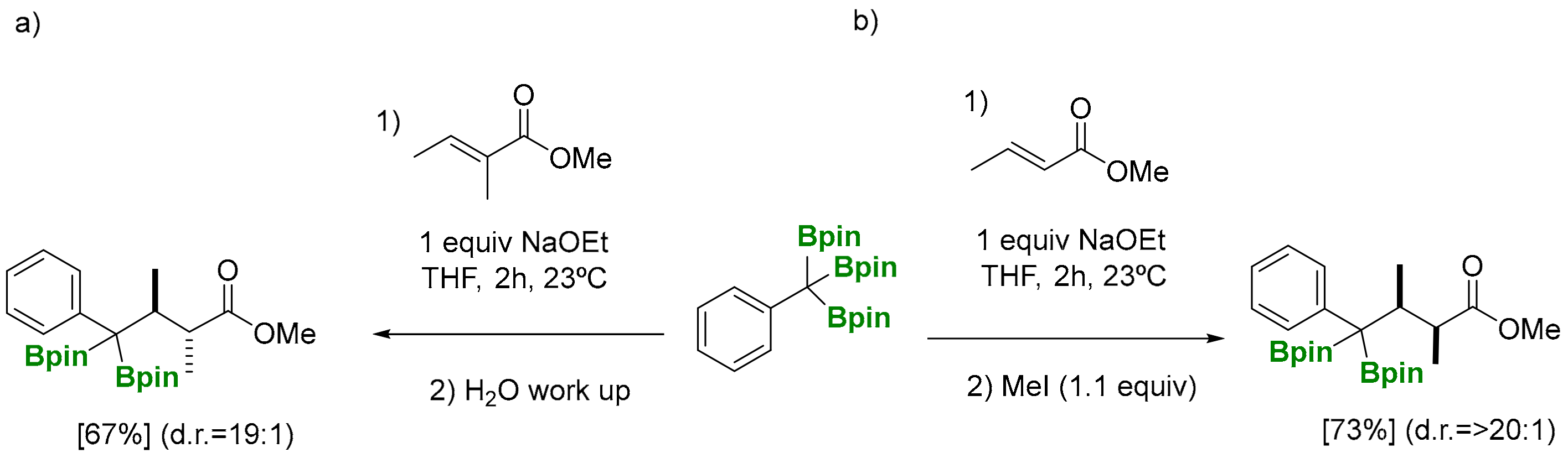
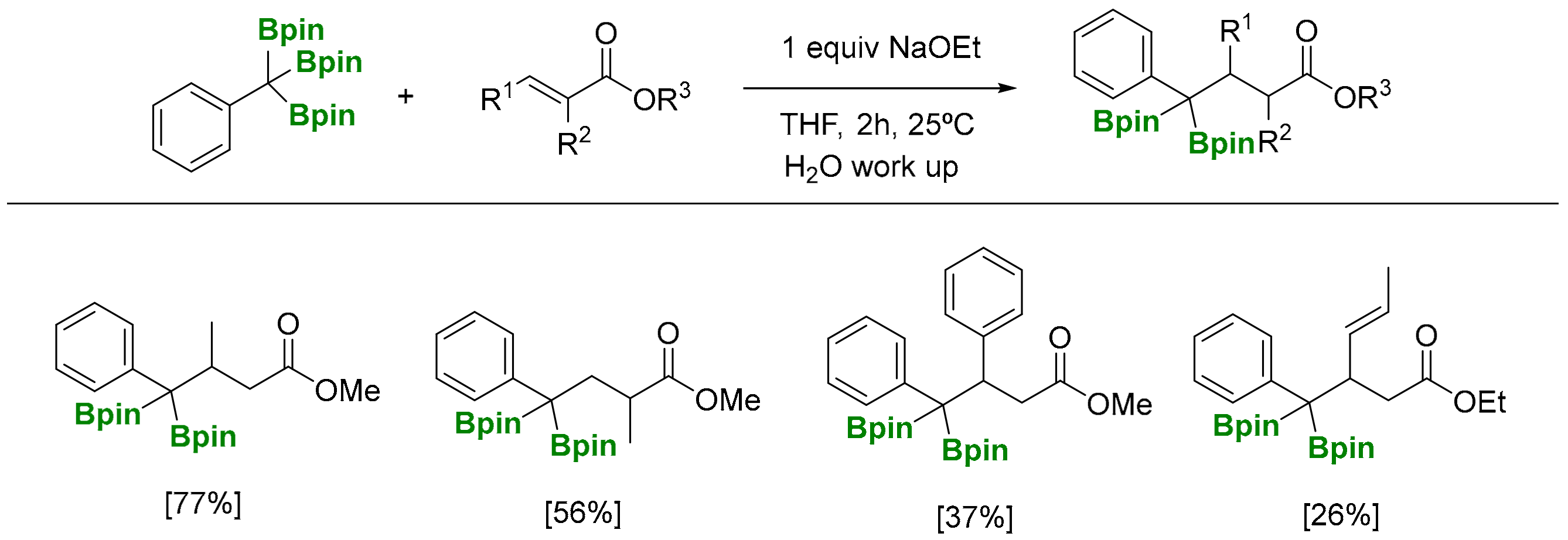
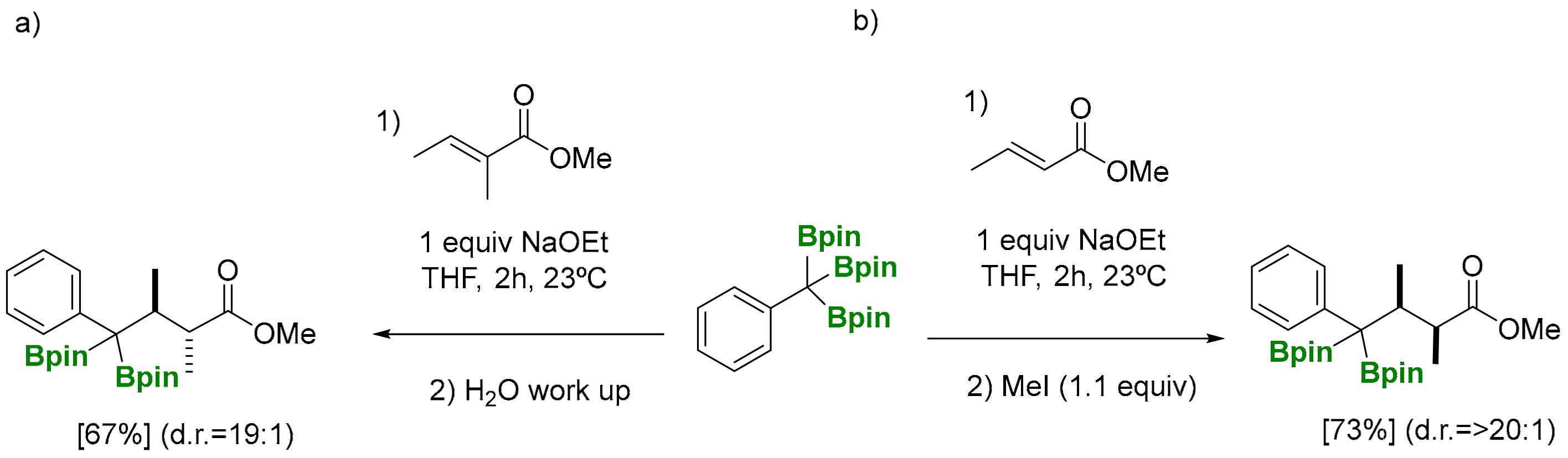
The deborylative conjugate addition of 1,1,1-tris(pinacolboryl)alkanes has been pursued through a NaOEt (1 equiv) assisted reaction to generate benzylic α-diboryl anions that can act as soft nucleophiles to be added to (E)-methylcrotonate, at room temperature, followed by aqueous work up (Scheme 12) [8]. Products formed from the deborylative conjugate addition to phenyl-substituted (E)-methyl cinnamate, as well as α,β,γ,δ-unsaturated ethyl sorbate, suffered from a competitive 1,2-addition to the more electron deficient enoates (Scheme 12) [8]. The reaction can be diastereoselective for α-subtituted substrates, such as methyl (E)-2-methylbut-2-enoate, in favor of the anti diastereomer. The stereocontrol at the α-position of the ester group has been suggested to occur during the protonation when the reaction is quenched (Scheme 13a) [8]. Remarkably, when the deborylative conjugate addition of the benzylic α-diboryl anion to (E)-methylcrotonate was quenched with iodomethane, the resulting product was isolated in >20:1 as the syn diastereomer (Scheme 13b) [8], highlighting the exclusive ability of boronate groups to act as masking groups for soft nucleophilic anions and also as elements for substrate-controlled diastereoselectivity.
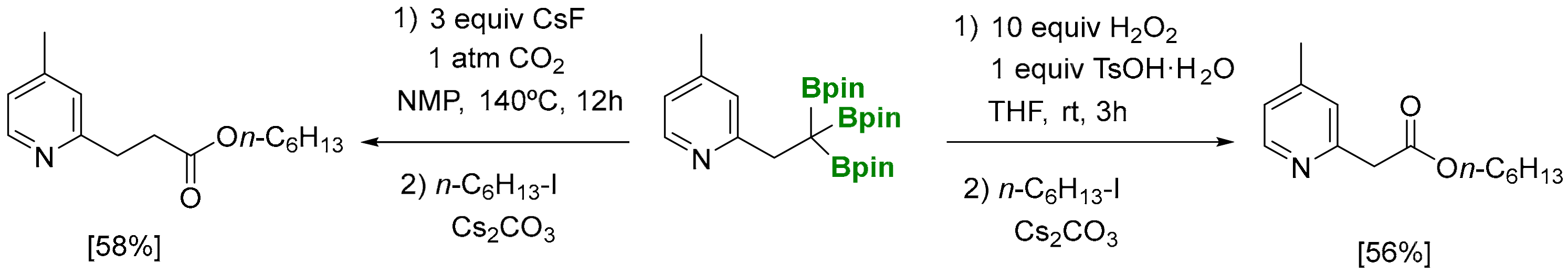
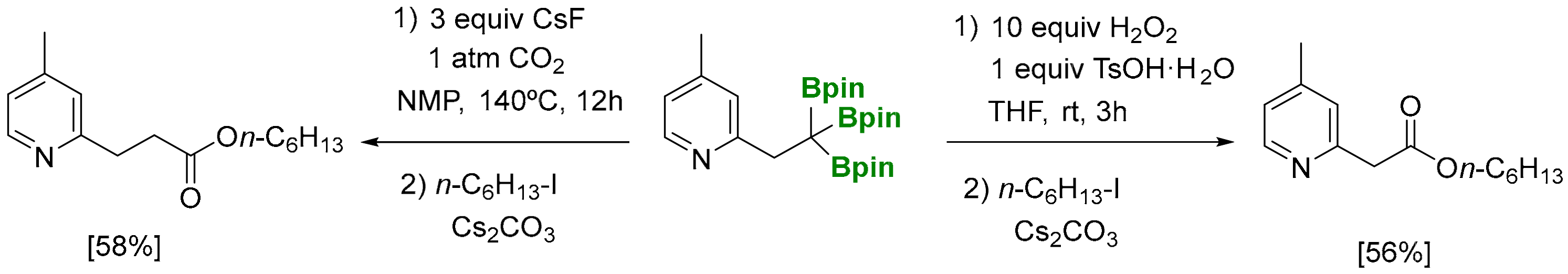
The carboxylation of 1,1,1-tris(pinacolboryl)alkanes, using CsF as a mild activator for deborylation [16], has been explored under 1 atm of CO2, providing the corresponding carboxylated products in moderate yields, after esterification with RI (Scheme 14) [6]. In addition, the sequence based on the trioxidation of 1,1,1-tris(pinacolboryl)alkanes using H2O2 in the presence of an acidic additive such as TsOH·H2O prevents protodeborylation pathways, and n-C6H13-I allows the isolation of hexyl 2-(4-methylpyridin-2-yl)acetate in moderate yield. Both methods’ results are useful, since carboxylic acids with complementary chain lengths can be prepared by selecting the appropriate reagents to interact with the tris(boryl)alkane.
Other interesting reactivity in C-C bond formation from tris(boryl)alkane involves deborylative Boron–Wittig olefination, via the B-O elimination pathway. Originally, the proof of concept was established by the addition of methyllithium to tris(ethylenedioxyboryl)methane, to generate in situ the corresponding bis(ethylenedioxyboryl)methide salt, that interacts with aldehydes or ketones to give the corresponding alkene monoboronic esters (Scheme 15) [17,18].
The analogue 1,1,1-tris(boryl)methide salt has exclusively been prepared via base-assisted deborylation of the corresponding tetra(boryl)methane (C(BO2C2Me4)4) [19], promoting the condensation with aldehydes and ketones to obtain 1,1-gem-diborylalkenes (Scheme 16) [20].
The reactivity developed from 1,1,1-tris(boryl)alkanes offers a significant number of applications, since their activation in the presence of bases generates the corresponding carbanions, which are strongly stabilized by delocalization, with vacant p-orbitals of the remaining two boron atoms. The possibility to perform further functionalization of the remaining C-B bonds eventually leads to new strategic synthetic protocols, emphasizing the straightforward methodology, as well as the smooth reaction conditions.
3. 1,2,3-Tri(boryl)alkanes
3.1. Synthetic Approaches
The introduction of three vicinal boryl moieties in a one-pot protocol represents a challenging reaction. The triboration of alkynes towards 1,2,3-triborated compounds has been accomplished by Yoshida and co-workers [21] via the Cu–PCy3 catalysed borylcupration of alkyl-substituted propargyl ethers (Scheme 17a). This approach demonstrates the easy access to multifunctinalized organoboron compounds that contain two alkenyl boryl and one allylboryl moiety. Mechanistically, it has been suggested that alkyl-substituted propargyl ethers insert regioselectively into Cu-B bonds generating the alkenyl copper where Cu is located next to OMe group. Subsequently, σ-bond methatesis with B2pin2 facilitates the formation of π-allyl species that after a second σ-bond methatesis with the diboron reagent provides the triborated product (Scheme 17b). However, phenyl-substituted propargyl ethers react towards the diborated product, presumably as a consequence of the regioselective alkenyl copper species formation, with Cu next to Ph group, and eventual σ-bond metathesis with B2pin2 (Scheme 17b).
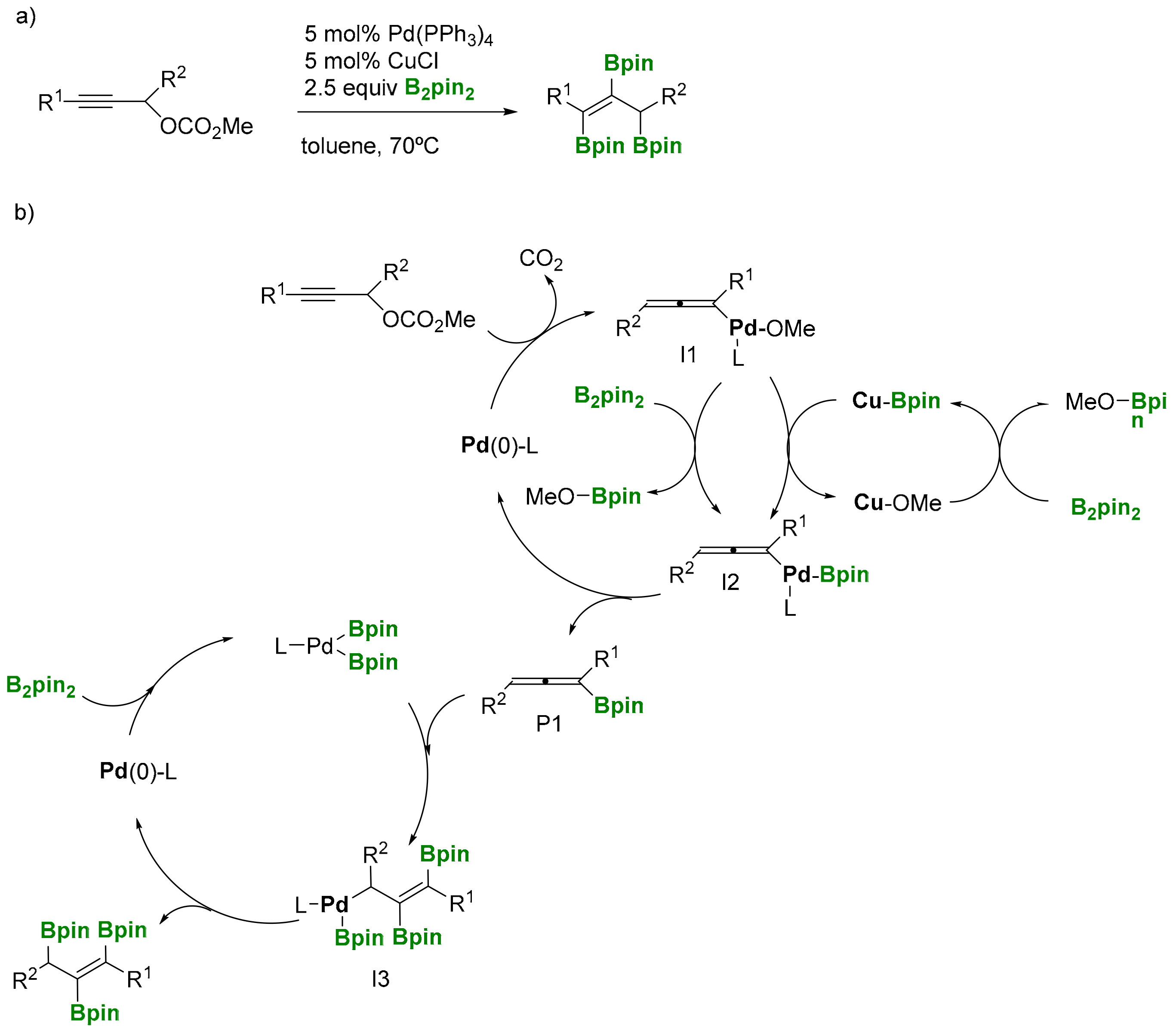
Alternatively, Ma and co-workers [22] developed a highly selective 1,2,3-triboration of propargylic carbonates towards (E)-propen-1,2,3-triboronates, by the catalytic systems Pd(PPh3)4 and CuCl as co-catalyst (Scheme 18a). Mechanistically, two catalytic cycles have been suggested. The first one is based on the Pd(0) catalyzed SN2′-type oxidative addition of propargylic carbonates to give 1,2-allenyl palladium intermediate I1 (Scheme 18b), with the concomitant release of CO2. In the following step, I1 undergoes transmetallation with Cu-Bpin species, previously formed from Cu-OMe and B2pin2, in order to generate the intermediate I2. Subsequent reductive elimination allows the formation of 1,2-allenyl boronate P1, that releases from the catalytic cycle 1. Interestingly, it has been suggested that P1 might insert in a Pd-B bond in catalytic cycle 2, producing η1-allylic intermediate I3, which undergoes reductive elimination to afford the final triborated product with exclusive (E)- stereoselectivity (Scheme 18b).
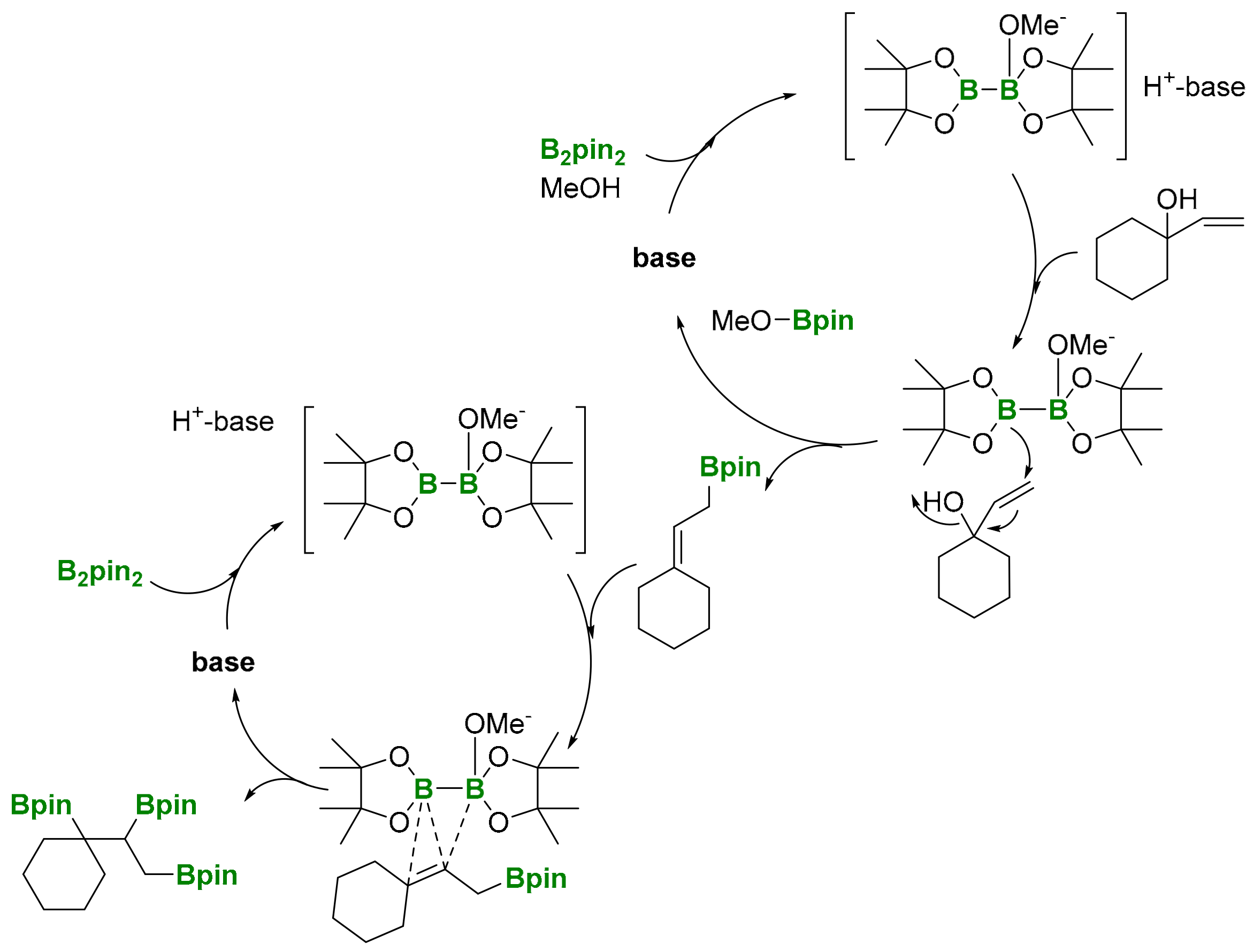
All the previous efforts devoted to introducing three vicinal C–B bonds into a one-pot sequential reaction required transition metal complexes to activate the borane reagent and facilitate the addition to the unsaturated substrates. However, Fernández, Szabó and co-workers [23] were first to observe the possibility of conducting the 1,2,3-triboration reaction in a transition-metal free context, using allylic alcohols as substrates and conducting a one-pot allylic borylation/diboration sequence (Scheme 19). The reaction takes place in the presence of catalytic amount of base Cs2CO3, that contributes to the generation of MeO- from MeOH. The methoxy group activates the B2pin2, through intermediate [Hbase][MeOB2pin2], and promotes the nucleophilic attack of the Bpin moiety to the terminal position of the tertiary allylic alcohol (Scheme 20).
The initial mechanism of this reaction can be described as an SN2′-type process. In the absence of MeOH or base, the reaction does not take place. After the transition-metal free allylic borylation, the allylic boronate product can interact with the [Hbase][MeOB2pin2] adduct and complete the diboration reaction [24], to generate the 1,2,3-triborated product (Scheme 20). This unprecedented tandem allylic borylation/diboration reaction in the absence of transition-metal catalysts had not been observed before and served as the basis of a new methodology towards polyborated compounds.
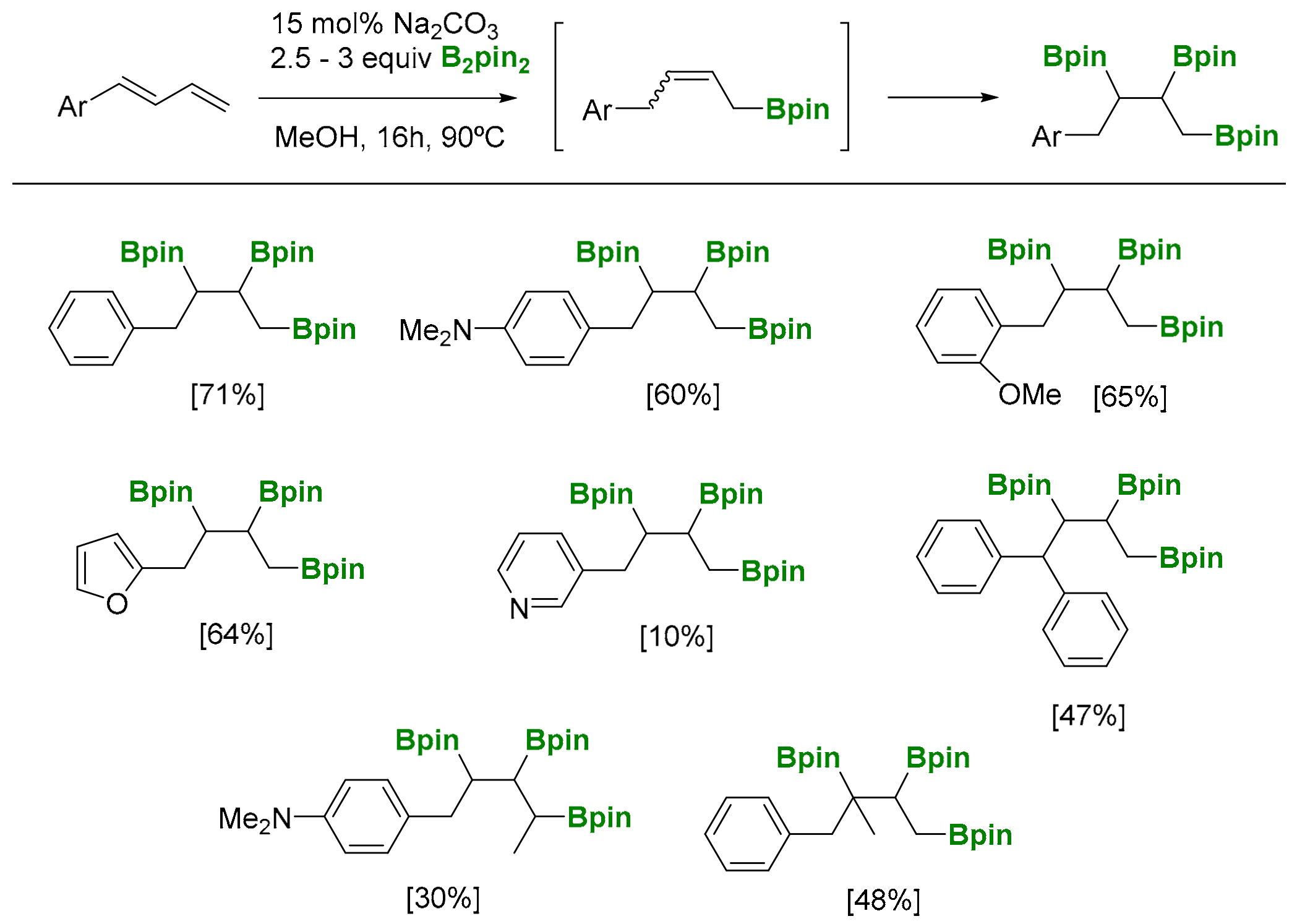
Fernández and co-workers also explored the viability of performing a direct 1,2,3-triboration reaction in a transition-metal free scenario via the 1,4-hydroboration of 1,3-dienes [25], followed by the in situ diboration reaction of the resultant allylic boronate (Scheme 21) [26]. This work represents an unprecedented tandem 1,4-hydroboration/diboration reaction, that is generalized for terminal and internal 1,3-dienes.
The nucleophilic attack of the Bpin moiety from [Hbase][MeOB2pin2] takes place to the less hindered position of the conjugated dienes throughout a SN2′-borylative process. The resulting allylic boronate product conducts a transition-metal free diboration reaction of the internal C=C bond, giving access to the 1,2,3-triborated product (Scheme 22) [26].
3.2. Synthetic Applications
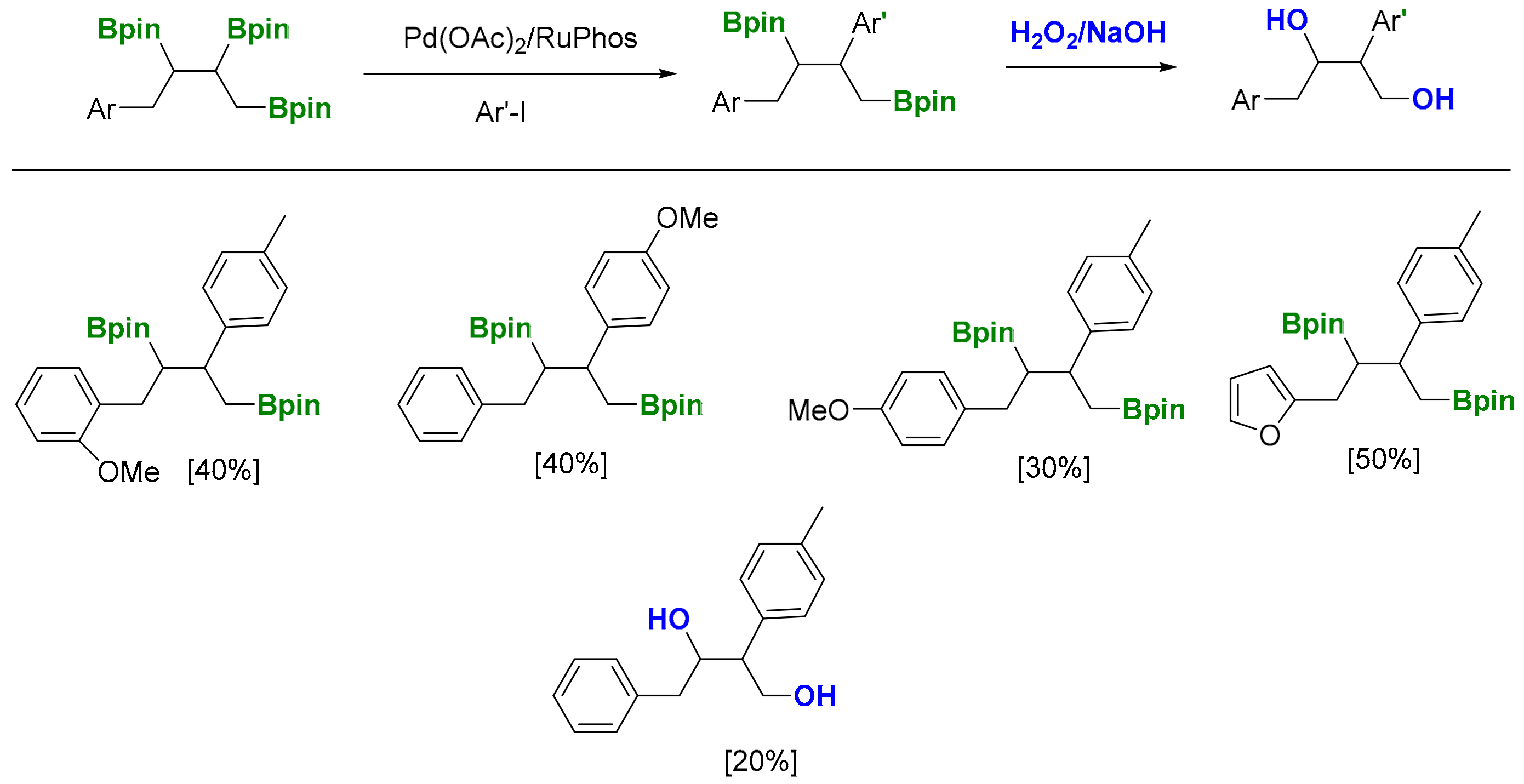
The polyfunctionalization of 1,2,3-triborated products can be accomplished throughout the cross-coupling reaction. Interestingly, in the presence of the 1 equiv of aryliodide and Pd complex, it was possible to exclusively functionalize the internal C-B bond of the triborated product (Scheme 23) [26]. The reaction was general for para- and ortho-substituted aryl systems, as well as for furyl groups. The in situ oxidation of the resulting 1,3-diborated products, in the presence of H2O2/NaOH, allowed access to 1,3-butanediol 2,3-diaryl systems in a straightforward manner (Scheme 23) [26].
Aggarwal and co-workers have reported an alternative functionalization of 1,2,3-triborated compounds following a photoredoxcatalyzed mono-deboronation. Initially the reaction proceeds through the generation of primary β-boryl radicals that undergo a rapid 1,2-boron shift to form thermodinamically favored secondary followed by a subsequent 1,2-boron shift towards tertiary radicals (Scheme 24). This protocol allows for the selective transformation of the more hindered boronic ester from the 1,2,3-triborated substrate [23], conducting its radical addition to tert-butyl-acrylate [27]. The product is obtained with high selectivity, highlighting the thermodynamic control that favors the most stabilized radical center as the site of the reaction.
4. 1,1,2-Tri(boryl)alkanes
4.1. Synthetic Approaches
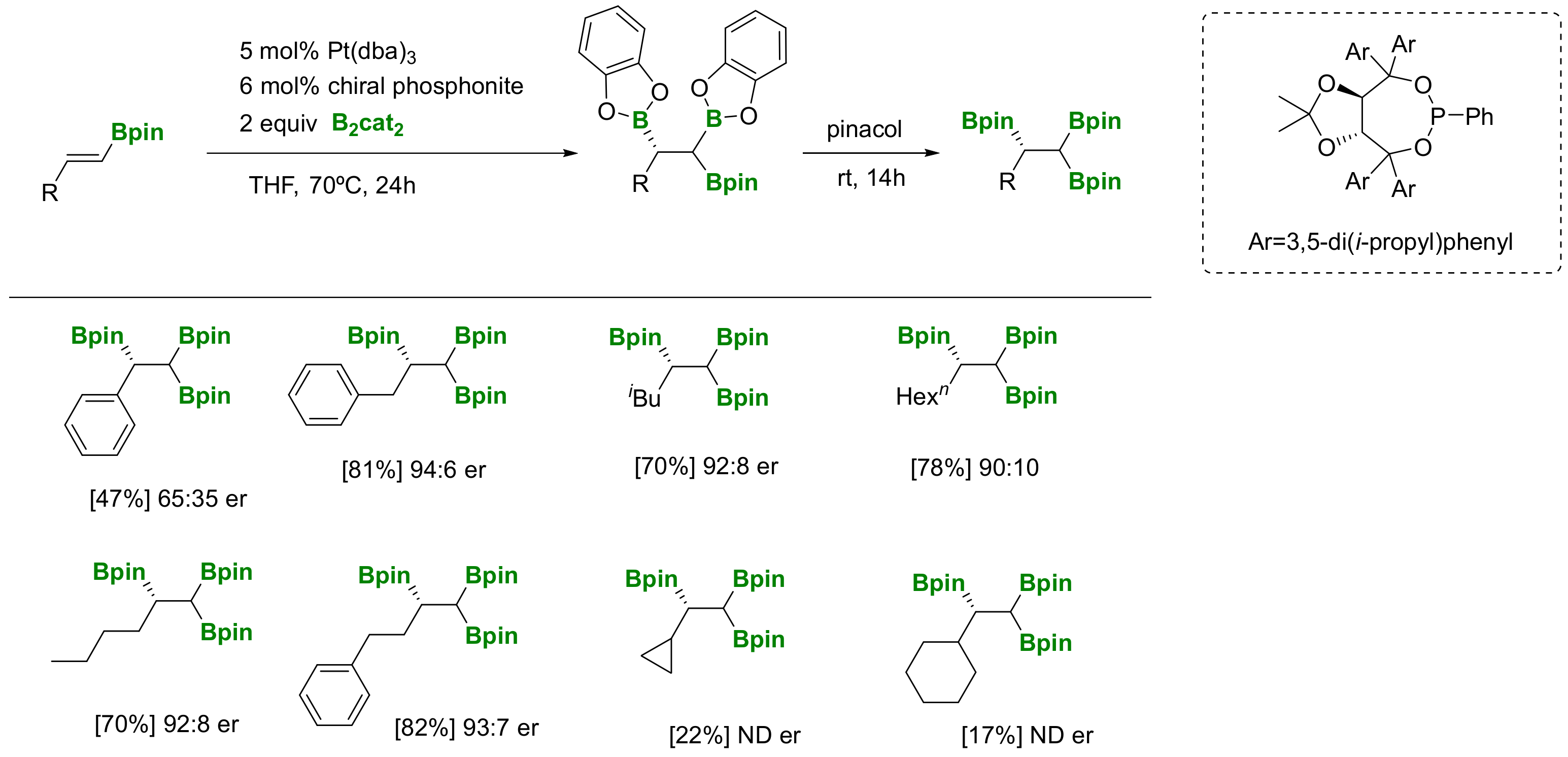
The preparation of versatile 1,1,2-tri(boryl)alkanes has been conducted via the Pt-catalyzed diboration of vinylboronates, (Scheme 25) [28]. Morken and co-workers employed chiral phosphonite to induce enantioselectivity along the internal C-B bond formation in the presence of Pt(dba)3 and bis(catecholato)diboron. Further treatment of the triborated product with pinacol, allowed the purification of the desired stable products with moderate to high enantioselectivity. The reaction has been considered of general application, although limitations are related to α-substituted substrates that suffered from lower reactivity and selectivity [28].
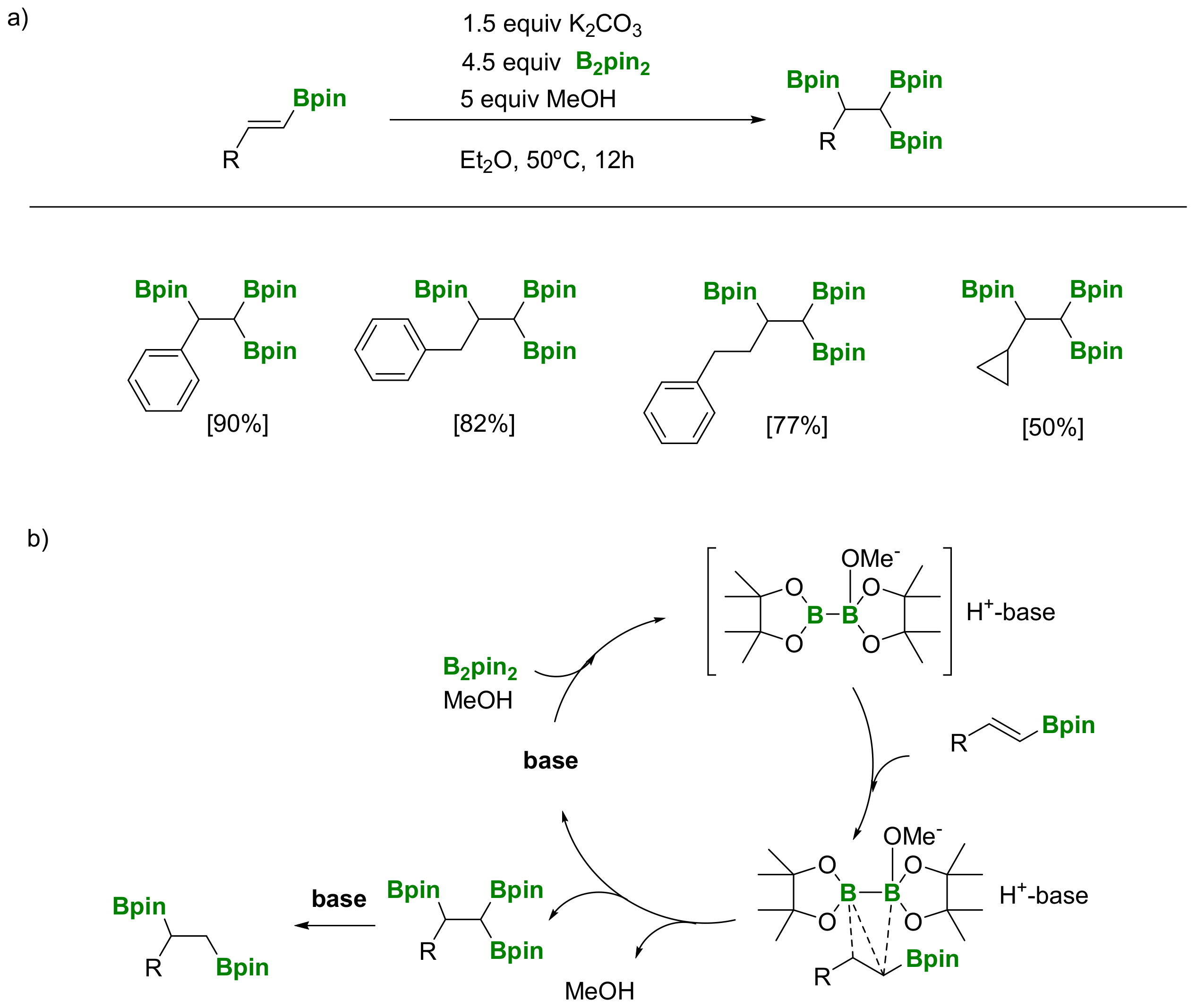
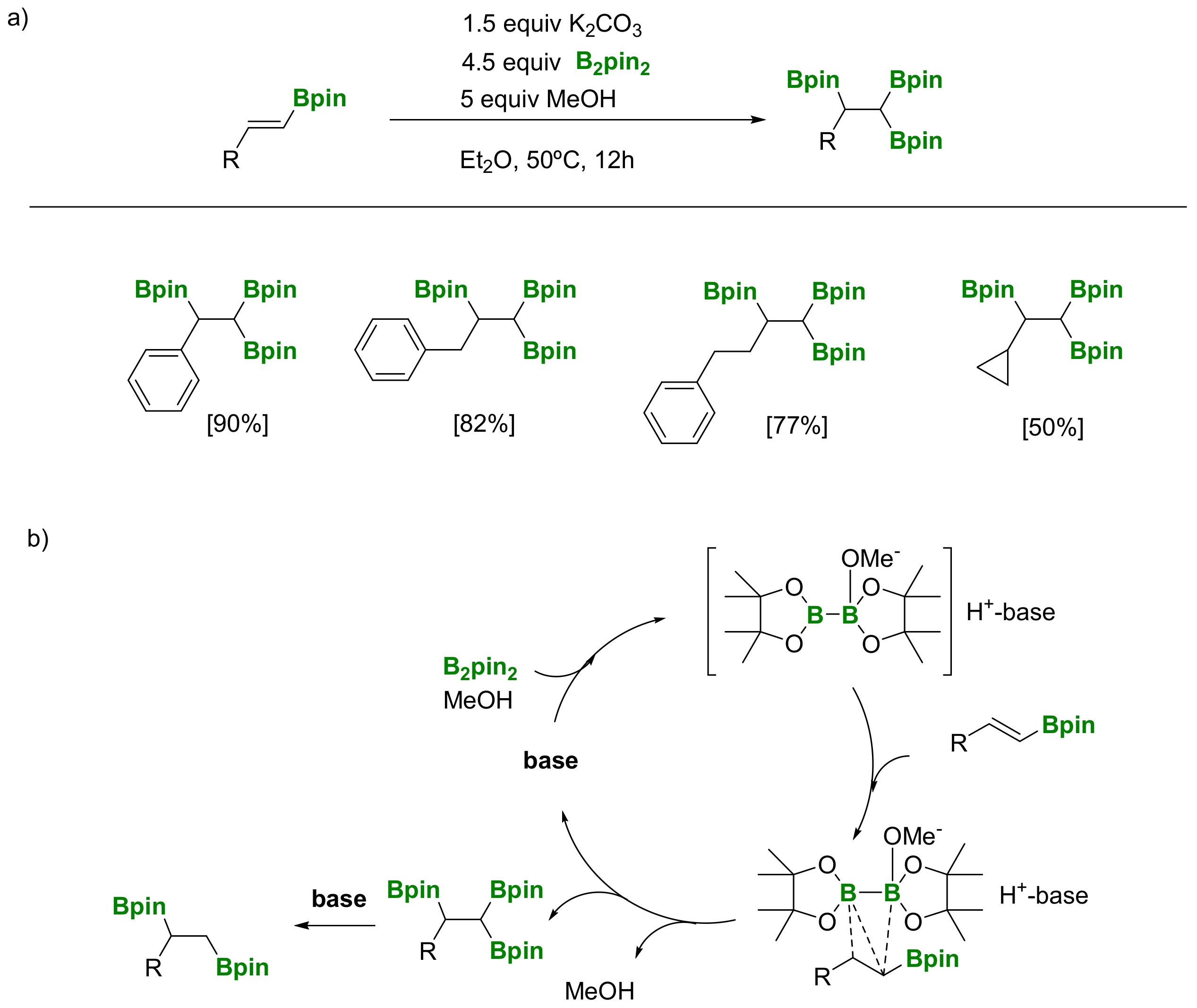
Another approach, launched by Song and co-workers [29], involves transition-metal free base-controlled 1,1,2-tris(boryl)alkane synthesis via diboration of alkenylboronates with B2pin2 (Scheme 26a). The reaction conditions for the synthesis of 1,1,2-tris(boryl)alkane were strictly optimized, since the corresponding 1,2-di(boryl)alkanes can also be formed as byproducts, depending on the base used. K2CO3 (1.5 equivalents) resulted in the optimized one, although 4.5 equivalents of B2pin2 were required. The substrate scope is broad and several functionalized terminal alkynes, such as propargyl amides and propargyl sulfonamides, are compatible. From a mechanistic point of view, a nucleophilic attack from the adduct [Hbase][MeOB2pin2], (generated in situ from B2pin2/base/MeOH) to the alkenylboronate substrate (Scheme 26b) has been suggested, as it was described on the transition-metal free diboration of alkenes developed by Fernández and co-workers in 2011 [24].
4.2. Synthetic Applications
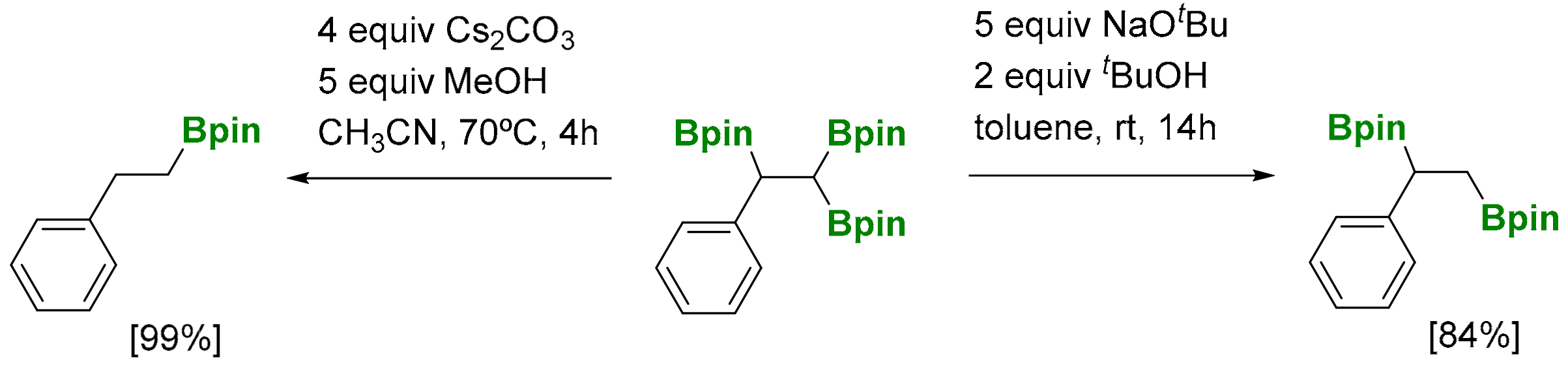
The versatility of 1,1,2-tri(boryl)alkanes is based on the construction of new C-C and C-heteroatom bonds, since the three boronate moieties can be distinguished in a stepwise manner. Song and co-workers proved the selective deprotonation on the geminal diboryl moiety, when NaOtBu is used as base (5 equiv) producing the 1,2-bis(boronate) product (Scheme 27) [29]. However, in the presence of Cs2CO3 (4 equiv) the 1,1,2-tri(boryl)alkane is converted into the linear alkyl monoboronate product (Scheme 27) [29].
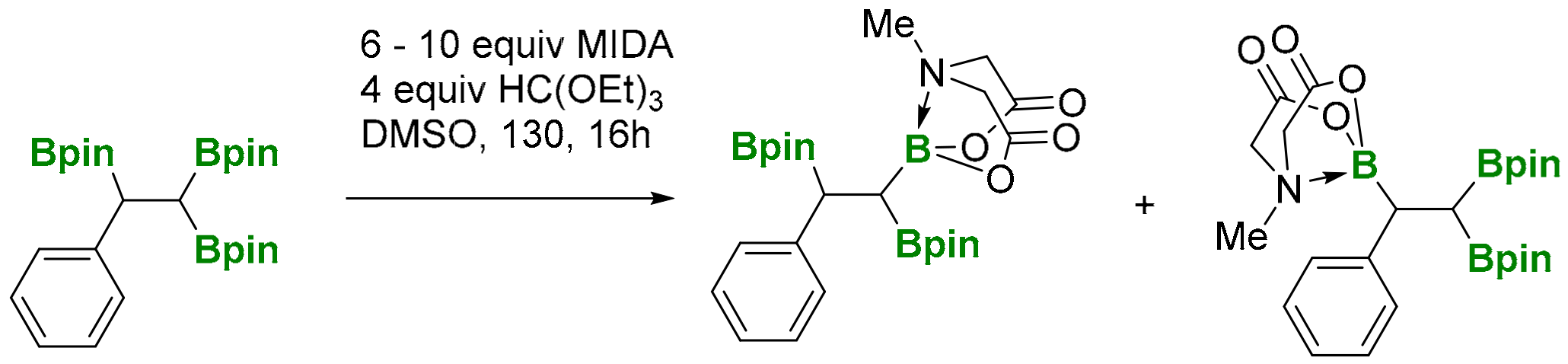
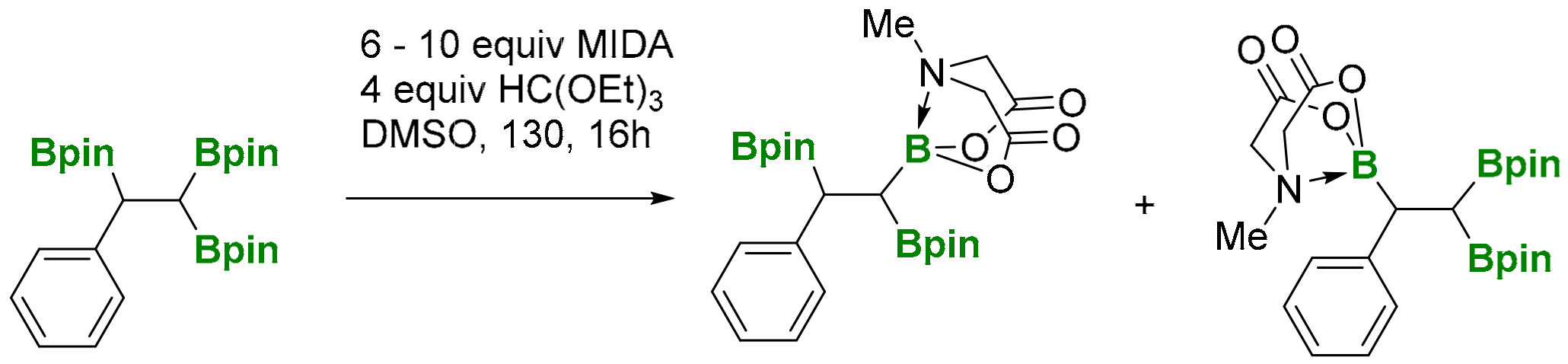
A simple transformation from 1,1,2-tri(boryl)alkane into mixed Bpin and B-MIDA triboronate compounds (MIDA=N-(methyliminodiacetic acid) has been developed by Sharma and co-workers [30]. However, the experiment showed that the treatment of 1,1,2-tri(pinacolboryl)alkane with MIDA provided a mixture (1:1 ratio by NMR) of the two mono-B(MIDA)-diBpin species, possessing the B(MIDA) group vicinal to Bpin or geminal to the Bpin moiety (Scheme 28). Unfortunately, the mixture resulted in being inseparable, using silica gel chromatography.
Deborylative alkylation reactions with chiral 1,1,2-tris-(boronates) were performed in the group of Morken [28], and they found that five equivalents of NaOtBu in toluene were efficient to form the new C-C bond from one of the geminal boryl moieties. The syn diastereoisomer was the predominant product, obtained as 1,2-diol upon oxidative workup. The reaction was applied for primary and seconary electrophiles (Scheme 29). The deborylative alkylation with alkenyl electrophiles represents a suitable method for the synthesis of internal vicinal bis(boronates), in the presence of terminal alkenes. When the substrate contains alkyl halide groups, the vinyl boronate diboration enables the construction of 1,1,2-tri(boryl)alkanes that may undergo intramolecular alkylation to form cyclic 1,2-bis(boronates). This method allows the construction of five, six and seven membered rings, which, after in situ oxidation, provides anti-1,2-diol (Scheme 29).
5. 1,1,2-Tri(boryl)alkenes
5.1. Synthetic Approaches
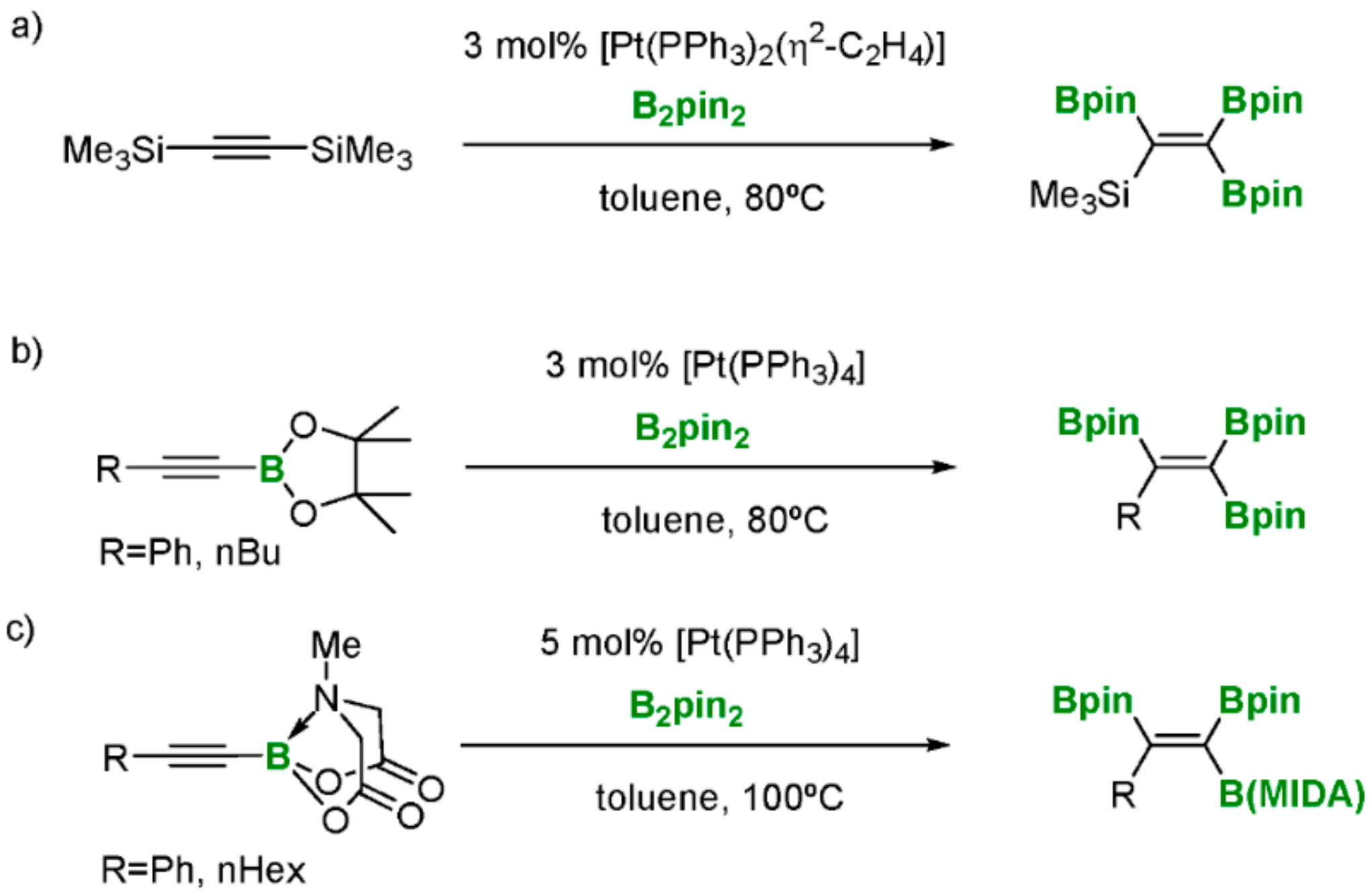
The preparation of polyborated alkenes has originally been accomplished by Marder and co-workers, via the tandem desilylative borylation of bis(trimethylsilyl)acetylene and the subsequent diboration with B2pin2 in the presence of [Pt(PPh3)2(η2-C2H4)] as catalyst (Scheme 30a) [31]. Similarly, Srebnik and co-workers established the convenience of using [Pt(PPh3)4] as catalyst to diborate 1-alkynylboronates to easily obtain the expected 1,1,2-triborylalkenes (Scheme 30b) [32]. More extensively, Nishihara and co-workers launched a more general platinum catalyzed diboration of phenylethynyl MIDA boronate with B2pin2 that proceeds to yield 1,1,2-triboryl-2-phenylethene, with two different classes of the boron functionalities (Scheme 30c) [33]. This protocol uses the MIDA boronic group in the substrate, which is considered a masked group [34], towards strategic further selective C-B functionalization.
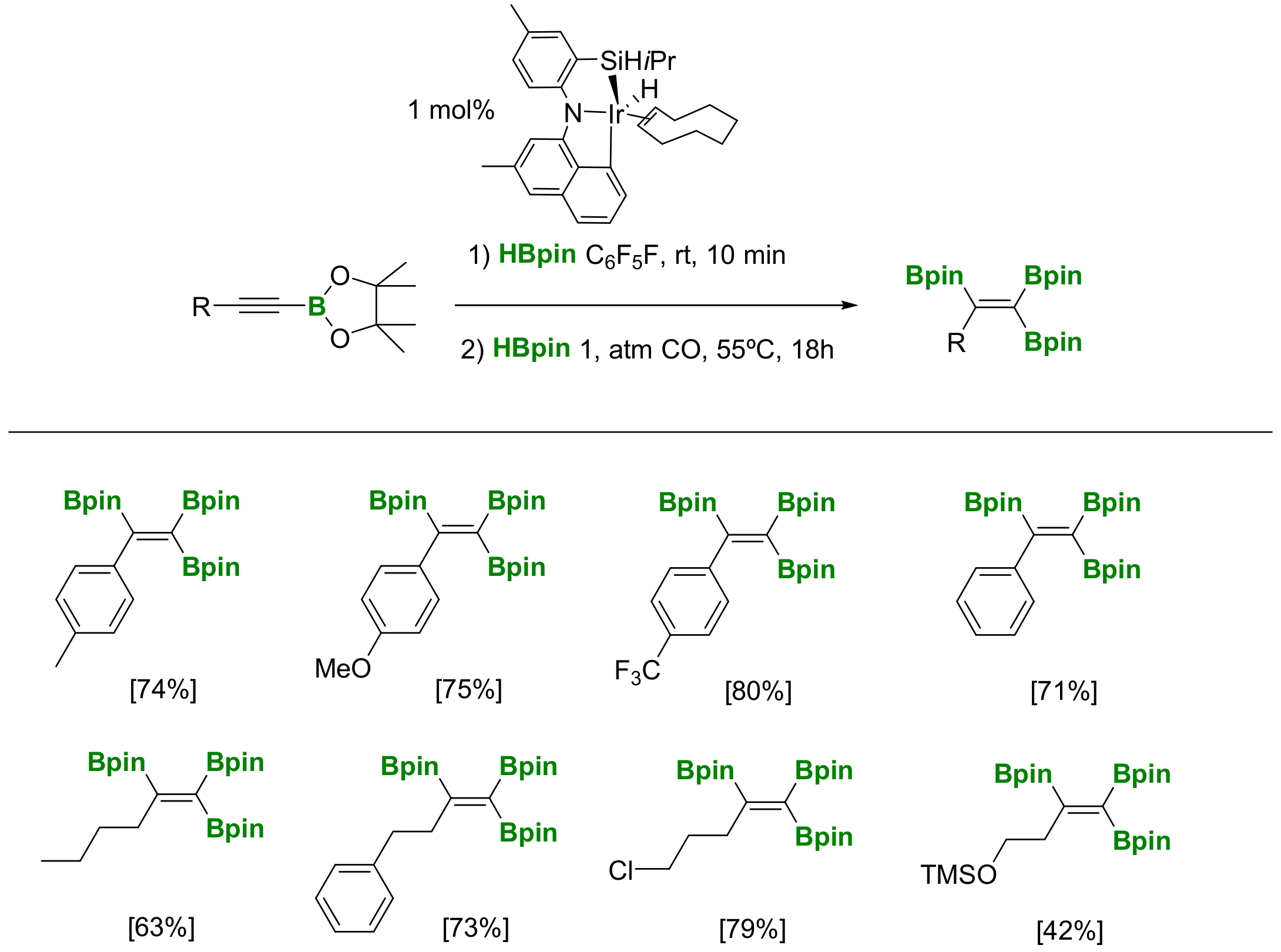
Moving from diboration to the hyroboration of alkynylboronates, Ozerov and co-workers discovered that pinacolborane (HBpin) can be efficiently added to terminal alkynes in the presence of iridium SiNC pincer complex, to efficiently prepare alkynylboronates. The following pathway involved the dehydrogenative diboration with HBpin and the same iridum complex under an atmosphere of CO (Scheme 31) [35]. The formation of 1,1,2-tri(boryl)alkanes through this protocol can be extended to a large number of substrates. However, the authors noted that the mechanism remains unclear, discarding the intermediacy of a hydroborated product or diboration with B2pin2.
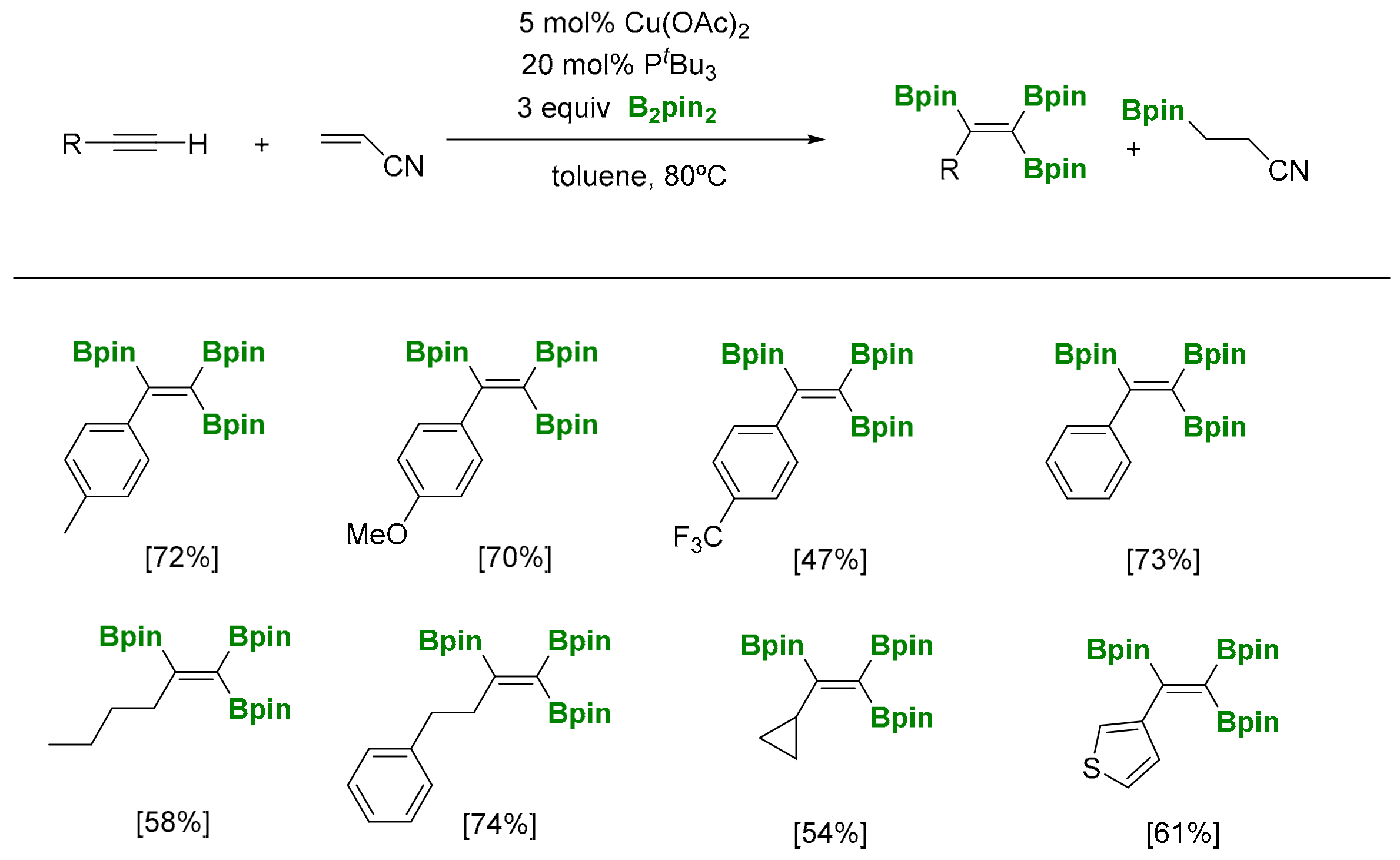
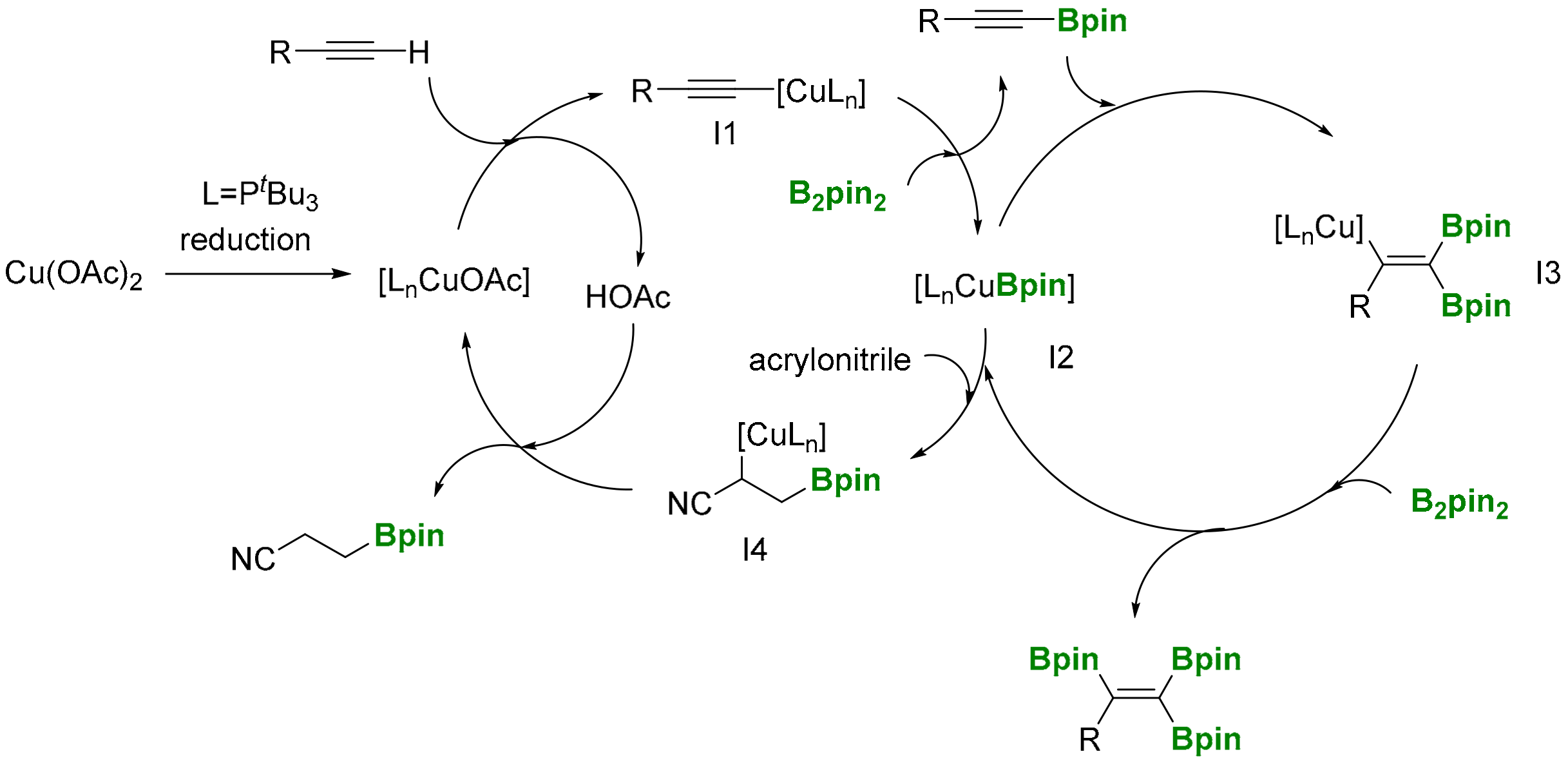
Marder and co-workers have developed a copper catalyzed triboration protocol for the synthesis of 1,1,2-triborylalkenes, using Cu(OAc)2 and PnBu3 as suitable ligands, and with B2pin2 as a boron source (Scheme 32) [36]. The use of stoichiometric amounts of acrylonitrile as additive seems to be required as hydrogen (B-H) acceptor, in order to avoid by-products reactions. The reaction features mild reaction conditions, broad substrate scope and good functional group tolerance.
The mechanism has been suggested on the basis of a coordination of terminal alkynes to [LnCuOAc], which is formed from Cu(OAc)2, the ligand and a reductive pathway (Scheme 33). Cu(OTf)2 or CuCl2 resulted inactive catalytic systems. Alternatively, the addition of KOAc to CuCl2 and CuCl reactivated the system, indicating that the −OAc plays an important role in the reaction. The reactivity of [LnCuOAc] with the terminal alkyne provides the intermediate I1, which interacts with B2pin2 through σ-bond methatesis to afford the alkynylboronate and the Cu-Bpin species I2. Consequently, insertion of the alkynylboronate into Cu-Bpin generates the alkenylcopper I3 that eventually reacts with B2pin2 through σ-bond methatesis, to generate the desired 1,1,2-tri(boryl)alkene (Scheme 33). Acrylonirile is added, because its hydroboration is faster than that of alkynes, avoiding the alkyne hydroboration side reaction [36].
5.2. Synthetic Applications
The transformation of 1,1,2-tri(boryl)alkenes can be conducted by palladium-catalyzed arylation reaction with aryl iodides. Pd(dba)2 was found to be the best catalyst to give the arylated product and the new C-C is selectively formed in trans to the B(MIDA) moiety (Scheme 34). HPtBu3BF4 was selected as the best ligand to modify the palladium complex in the presence of three equivalents of K3PO4 as the base [33]. The limitation of this protocol concerns the aryl halide used, since aryl iodide results in being very efficient for the cross-coupling reaction, and the aryl bromide and aryl chloride show a low or null reactivity (Scheme 34). Additionally, other electrophiles such as ethyl iodide or (E)-octenyl iodide were subjected to the Suzuki–Miyaura coupling reactions, but no desired products were formed. Despite the fact that some limitations were observed, this method represents the first example of obtaining gem-diborylated olefins with two different boryl groups via a chemoselective arylation. The chemoselectivity can be explained simply by a steric effect, because even the addition of two equivalents of aryl iodides did not provide the diarylated products [33].
The versatility of 1,1,2-tri(pinacolboryl)alkenes was also demonstrated, due to complementarily stereoselective Suzuki-Miyaura coupling reactions with aryl halides catalyzed by Pd(PPh3)4 in the presence of three equivalents of K3PO4 as the base (Scheme 35) [35]. The (E)-stereochemistry of the diborated product becomes one of the few examples described in the literature, and becomes a potential synthetic strategy to obtain trans-diaryldiborylalkenes in a simple way.
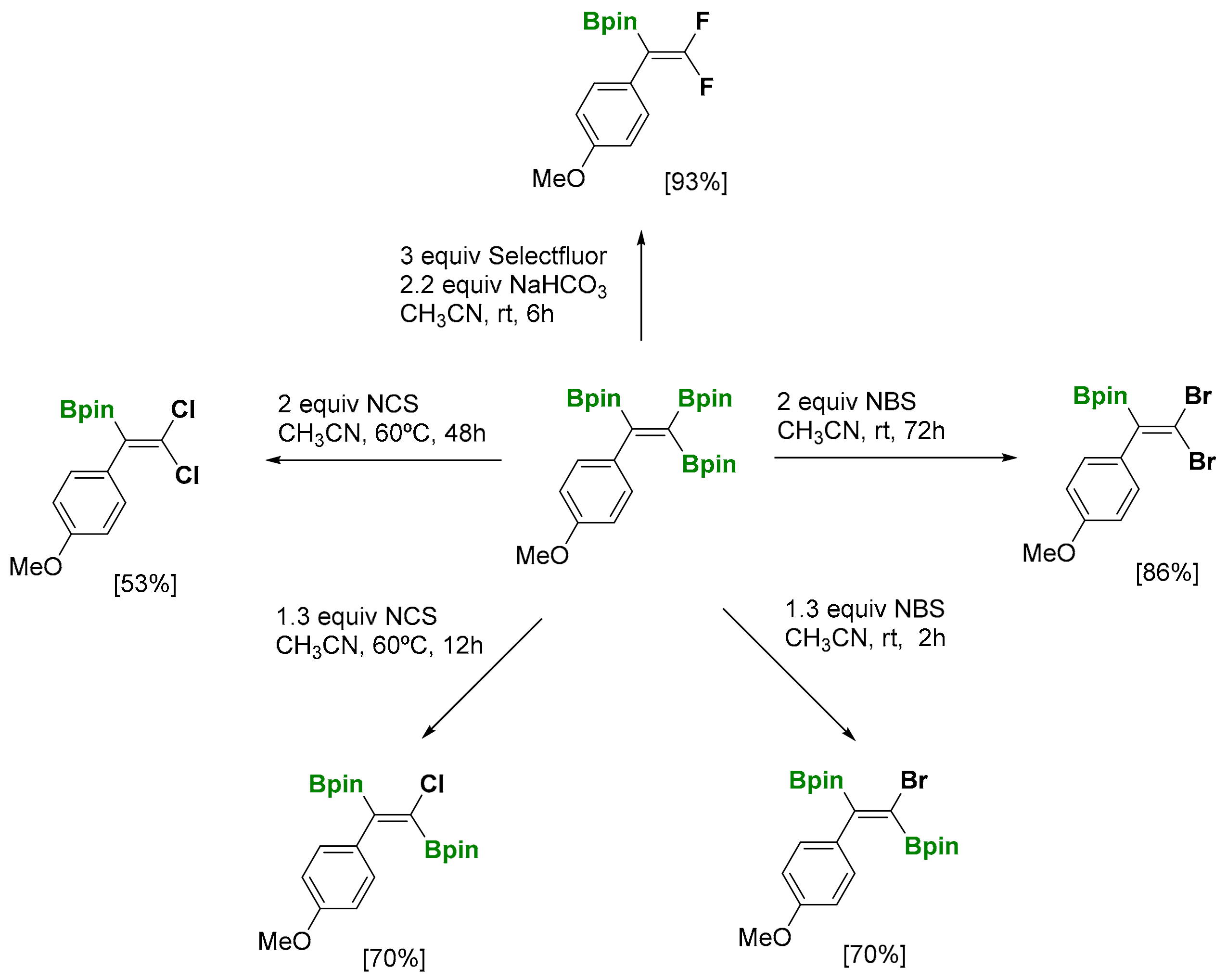
The utility of the 1,1,2-tri(pinacolboryl)alkenes has also explored different ways to be functionalized through difluorination (with Selectfluor) to afford gem-difluoroborylakenes (Scheme 36) [36]. Similarly, dibromination (with NBS) and dichlorination (with NCS) of 1,1,2-tri(pinacolboryl)alkenes facilitated the formation of gem-dihaloborylakenes (Scheme 36). The use of a lower amount of NBS or NCS, as well as lower reaction times, generated trans-stereoselectively towards the monohalo-diborylated alkene (Scheme 36).
6. Conclusions
The preparation of 1,1,1-tri(boryl)alkanes, 1,2,3-tri(boryl)alkanes, 1,1,2-tri(boryl)alkanes, as well as 1,1,2-tri(boryl)alkenes, appears to be a useful tool to synthesize organic compounds through selective C-B functionalization. The assistance of vicinal or geminal boryl units to the selective functionalization of C(sp3)-B or C(sp2-B) fragments becomes crucial. In part, this advantage is due to the enhanced α-borylcarbanions stability as intermediates in synthesis. Within the last decade, intensive efforts are devoted to prepare tri(boryl)alkanes and tri(boryl)alkenes, and significant applications have illustrated the powerful tool to construct molecules in a selective way. We can only expect that their interest will keep growing, attending to their real utility.
Funding
This research was supported by MINECO through project CTQ2016-80328-P. O. Salvadó thanks Martí Franques grant.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Castle, R.B.; Matteson, D.S. A Methanetetraboronic Ester. J. Am. Chem. Soc. 1968, 90, 2194. [Google Scholar] [CrossRef]
- Castle, R.B.; Matteson, D.S. Methanetetraboronic and methanetriboronic esters. J. Organomet. Chem. 1969, 20, 19–28. [Google Scholar] [CrossRef]
- Matteson, D.S.; Thomas, J.R. C-alkylation of methanetetraboronic and methanetriboronic esters. J. Organomet. Chem. 1970, 24, 263–271. [Google Scholar] [CrossRef]
- Matteson, D.S. Methanetetraboronic and methanetriboronic esters as synthetic intermediates. Synthesis 1975, 1975, 147–158. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Ishibashi, A.; Suginome, M. Boryl-Directed, Ir-Catalyzed C(sp3)–H Borylation of Alkylboronic Acids Leading to Site-Selective Synthesis of Polyborylalkanes. Org. Lett. 2019, 21, 6235–6240. [Google Scholar] [CrossRef] [PubMed]
- Mita, T.; Ikeda, Y.; Michigami, K.; Sato, Y. Iridium-catalyzed triple C(sp3)–H borylations: Construction of triborylated sp3-carbon centers. Chem. Commun. 2013, 49, 5601–5603. [Google Scholar] [CrossRef] [PubMed]
- Palmer, W.N.; Obligacion, J.V.; Pappas, I.; Chirik, P.J. Cobalt-Catalyzed Benzylic Borylation: Enabling Polyborylation and Functionalization of Remote, Unactivated C(sp3)−H Bonds. J. Am. Chem. Soc. 2016, 138, 766–769. [Google Scholar] [CrossRef]
- Palmer, W.N.; Zarate, C.; Chirik, P.J. Benzyltriboronates: Building Blocks for Diastereoselective Carbon−Carbon Bond Formation. J. Am. Chem. Soc. 2017, 139, 2589–2592. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ming, W.; Zhang, Y.; Friedrich, A.; Marder, T.B. Copper-Catalyzed Triboration: Straightforward, Atom-Economical Synthesis of 1,1,1-Triborylalkanes from Terminal Alkynes and HBpin. Angew. Chem. Int. Ed. 2019, 58, 18923–18927. [Google Scholar] [CrossRef] [Green Version]
- Baker, R.T.; Nguyen, P.; Marder, T.B.; Westcott, S.A. Transition Metal Catalyzed Diboration of Vinylarenes. Angew. Chem. Int. Ed. Engl. 1995, 34, 1336–1338. [Google Scholar] [CrossRef]
- Gu, Y.; Pritzkow, H.; Siebert, W. Synthesis and Reactivity of Monoborylacetylene Derivatives. Eur. J. Inorg. Chem. 2001, 200, 373–379. [Google Scholar] [CrossRef]
- Krautwald, S.; Bezdek, M.J.; Chirik, P.J. Cobalt-Catalyzed 1,1-Diboration of Terminal Alkynes: Scope, Mechanism, and Synthetic Applications. J. Am. Chem. Soc. 2017, 139, 3868–3875. [PubMed] [Green Version]
- Zhang, L.; Huang, Z. Synthesis of 1,1,1-Tris(boronates) from Vinylarenes by Co-Catalyzed Dehydrogenative Borylations−Hydroboration. J. Am. Chem. Soc. 2015, 137, 15600–15603. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Lloyd-Jones, G.C. Vinylborane Formation in Rhodium-catalysed Hydroborations; Ligand-free Homogeneous Catalysis. J. Chem. Soc. Chem. Commun. 1992, 710–712. [Google Scholar] [CrossRef]
- Hong, K.; Liu, X.; Morken, J.P. Simple Access to Elusive α-Boryl Carbanions and Their Alkylation: An Umpolung Construction for Organic Synthesis. J. Am. Chem. Soc. 2014, 136, 10581–10584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nave, S.; Sonawane, R.P.; Elford, T.G.; Aggarwal, V.K. Protodeboronation of Tertiary Boronic Esters: Asymmetric Synthesis of Tertiary Alkyl Stereogenic Centers. J. Am. Chem. Soc. 2010, 132, 17096–17098. [Google Scholar] [CrossRef]
- Matteson, D.S.; Moody, R.J.; Jesthi, P.K. Reaction of aldehydes and ketones with a boron-substituted carbanion, bis(ethylenedioxyboryl)methide. Simple aldehyde homologation. J. Am. Chem. Soc. 1975, 97, 5608–5609. [Google Scholar] [CrossRef]
- Matteson, D.S.; Moody, R.J. Homologation of carbonyl compounds to aldehydes with lithium bis(ethylenedioxyboryl)methide. J. Org. Chem. 1980, 45, 1091–1095. [Google Scholar] [CrossRef]
- Matteson, D.S.; Davies, R.A.; Hagelee, L.A. A bromomethanetriboronic ester. J. Organomet. Chem. 1974, 69, 45–52. [Google Scholar] [CrossRef]
- Matteson, D.S.; Tripathy, P.B. Alkene-1,1-diboronic esters from the condensation of triborylmethide anions with ketones or aldehydes. J. Organomet. Chem. 1974, 69, 53–62. [Google Scholar]
- Yoshida, H.; Kawashima, S.; Takemoto, Y.; Okada, K.; Ohshita, J.; Takaki, K. Copper-Catalyzed Borylation Reactions of Alkynes and Arynes. Angew. Chem. Int. Ed. 2012, 51, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Cao, T.; Han, Y.; Lin, W.; Liu, Q.; Tang, Y.; Zhai, Y.; Jia, M.; Zhang, W.; Zhu, T.; et al. Regio- and (E)-Stereoselective Triborylation of propargylic Carbonates. Chin. J. Chem. 2017, 35, 1251–1255. [Google Scholar] [CrossRef]
- Miralles, N.; Alam, R.; Szabó, K.; Fernández, E. Transition-Metal-Free Borylation of Allylic and Propargylic Alcohols. Angew. Chem. Int. Ed. 2016, 55, 4303–4307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonet, A.; Pubill-Ulldemolions, C.; Bo, C.; Gulyás, H.; Fernández, E. Transition-metal-free diboration reaction by activation of diboron compounds with simple Lewis bases. Angew. Chem. Int. Ed. 2011, 50, 7158. [Google Scholar] [CrossRef]
- Maza, R.J.; Davenport, E.; Miralles, N.; Carbó, J.; Fernández, E. Transition-Metal-Free Allylic Borylation of 1,3-Dienes. Org. Lett. 2019, 21, 2251–2254. [Google Scholar] [CrossRef]
- Davenport, E.; Fernández, E. Transition-metal-free synthesis of vicinal triborated compounds and selective functionalisation of the internal C-B bond. Chem. Commun. 2018, 54, 10104–10107. [Google Scholar]
- Kaiser, D.; Noble, A.; Fasano, V.; Aggarwal, V.K. 1,2-Boron Shifts of β-Boryl Radicals Generated from Bis-boronic Esters Using Photoredox Catalysis. J. Am. Chem. Soc. 2019, 141, 14104–14109. [Google Scholar] [CrossRef]
- Coombs, J.R.; Zhang, L.; Morken, J.P. Enantiomerically Enriched Tris(boronates): Readily Accessible Conjunctive Reagents for Asymmetric Synthesis. J. Am. Chem. Soc. 2014, 136, 16140–16143. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Yan, J.; Yang, K.; Chen, F.; Song, Q. Base-controlled highly selective synthesis of alkyl 1,2-bis(boronates) or 1,1,2-tris(boronates) from terminal alkynes. Green Chem. 2017, 19, 3997–4001. [Google Scholar] [CrossRef]
- Lin, S.; Wang, L.; Aminoleslami, N.; Lao, Y.; Yagel, C.; Sharma, A. A modular and concise approach to MIDA acylboronates via chemoselective oxidation of unsymmetrical geminal diborylalkane: Unlocking access to a novel class of acylborons. Chem. Sci. 2019, 10, 4684–4687. [Google Scholar] [CrossRef] [Green Version]
- Lesley, G.; Nguyen, P.; Taylor, N.J.; Marder, T.B.; Scott, A.J.; Clegg, W.; Norman, N.C. Synthesis and Characterization of Platinum(II)-Bis(boryl) Catalyst Precursors for Diboration of Alkynes and Diynes: Molecular Structures of cis-[(PPh3)2Pt(B-4-Butcat)2], cis-[(PPh3)2Pt(Bcat)2], cis-[(dppe)Pt(Bcat)2], cis-[(dppb)Pt(Bcat)2], (E)-(4-MeOC6H4)C(Bcat)CH(Bcat), (Z)-(C6H5)C(Bcat)C(C6H5)(Bcat), and (Z,Z)-(4-MeOC6H4)C(Bcat)C(Bcat)C(Bcat)C(4-MeOC6H4)(Bcat) (cat = 1,2-O2C6H4; dppe ) Ph2PCH2CH2PPh2; dppb ) Ph2P(CH2)4PPh2. Organometallics 1996, 15, 5137–5154. [Google Scholar]
- Ali, H.A.; Quntar, A.E.A.A.; Goldberg, I.; Srebnik, M. Platinum(0)-Catalyzed Diboration of Alkynylboronates and Alkynylphosphonates with Bis(pinacolato)diborane(4): Molecular Structures of [((Me4C2O2)B)(C6H5)C=C(P(O)(OC2H5)2)(B(O2C2Me4))] and [((Me4C2O2)B)(C4H9)C=C(B(O2C2Me4))2]. Organometallics 2002, 21, 4533–4539. [Google Scholar] [CrossRef]
- Hyodo, K.; Suetsugu, M.; Nishihara, Y. Diborylation of Alkynyl MIDA Boronates and Sequential Chemoselective Suzuki−Miyaura Couplings: A Formal Carboborylation of Alkynes. Org. Lett. 2014, 16, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Mancilla, T.; Contreras, R.; Wrackmeyer, B.J. New bicyclic organylboronicesters derived from iminodiacetic acids. J. Organomet. Chem. 1986, 307, 1–6. [Google Scholar] [CrossRef]
- Lee, C.-I.; Shih, E.-C.; Zhou, J.; Reibenspies, J.H.; Ozerov, O.V. Synthesis of Triborylalkenes from Terminal Alkynes by Iridium-Catalyzed Tandem C-H Borylation and Diboration. Angew. Chem. Int. Ed. 2015, 54, 14003–14007. [Google Scholar] [CrossRef]
- Liu, X.; Ming, W.; Friedrich, A.; Kerner, F.; Marder, T.B. Copper-Catalyzed Triboration of Terminal Alkynes using B2pin2: Efficient synthesis of 1,1,2-triborylakenes. Angew. Chem. Int. Ed. 2020, 59, 304–309. [Google Scholar] [CrossRef] [Green Version]
Scheme 1.
Synthesis of H(B(OMe)2)3.

Scheme 2.
Synthesis of H(Bpin)3 from H(B(OMe)2)3.
Scheme 3.
Synthesis of H(Bpin)3 from Me-B(OH)2.
Scheme 4.
Transition metal catalytized C(sp3)–H triborylation at a single carbon.

Scheme 5.
(a) sequential dehydrogenative borylation/hydroboration of terminal alkynes; (b) mechanistic proposal.
Scheme 5.
(a) sequential dehydrogenative borylation/hydroboration of terminal alkynes; (b) mechanistic proposal.
Scheme 6.
Synthesis of 1,1,1-tris(catecholboryl)alkane and 1,1,1-tris[di(tert-butyl)boryl]ethane.

Scheme 7.
Cobalt-catalyzed dehydrogenative 1,1-diboration of terminal alkynes.
Scheme 8.
(a) Co(I)-catalyzed double dehydrogenative borylation of vinylarenes; (b) mechanistic proposal.
Scheme 8.
(a) Co(I)-catalyzed double dehydrogenative borylation of vinylarenes; (b) mechanistic proposal.
Scheme 9.
Reactivity of 1,1,1-tris(pinacolboryl)alkanes with RBr.
Scheme 10.
Double deborylative alkylation of 1,1,1-tris(pinacolboryl)alkanes.

Scheme 11.
Sequential deborylative alkylations, followed by in situ oxidation with H2O2/NaOH, towards tertiary alcohols.
Scheme 11.
Sequential deborylative alkylations, followed by in situ oxidation with H2O2/NaOH, towards tertiary alcohols.
Scheme 12.
Deborylative conjugate addition of 1,1,1-tris(pinacolboryl)alkanes.
Scheme 13.
(a) diastereoselective deborylative conjugate addition of 1,1,1-tris(pinacolboryl)alkanes; (b) diastereoselective sequential deborylative conjugate addition / electrophilic methylation.
Scheme 13.
(a) diastereoselective deborylative conjugate addition of 1,1,1-tris(pinacolboryl)alkanes; (b) diastereoselective sequential deborylative conjugate addition / electrophilic methylation.
Scheme 14.
Carboxylation of 1,1,1-tris(pinacolboryl)alkanes.
Scheme 15.
Deborylative Boron–Wittig olefination from tris(boryl)alkanes.
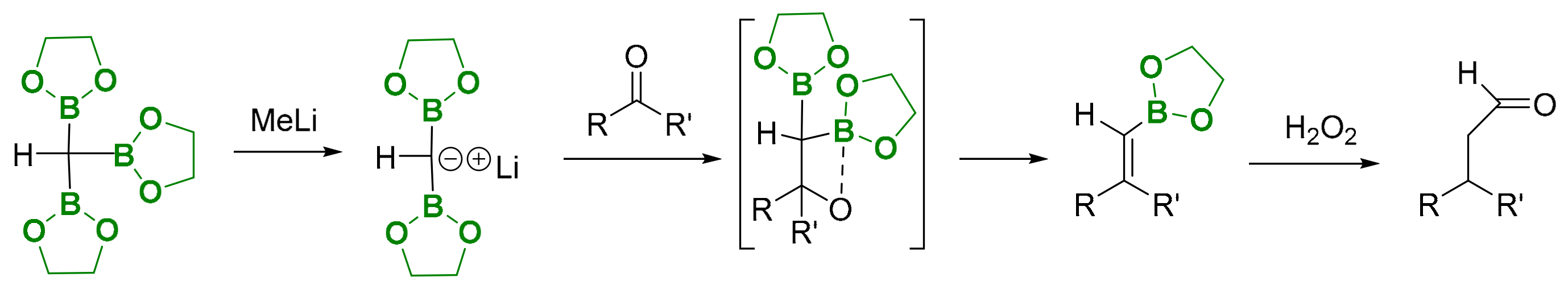
Scheme 16.
Deborylation of tetra(boryl)methane and condensation with aldehydes and ketones.

Scheme 17.
(a) Borylcupration of alkyl-substituted propargyl ethers; (b) mechanistic proposal.

Scheme 18.
(a) 1,2,3-Triboration of propargylic carbonates; (b) mechanistic proposal.
Scheme 19.
Transition metal-free one-pot allylic borylation/diboration sequence.

Scheme 20.
Mechanistic proposal of transition metal-free 1,2,3-triboration of allylic alcohols.
Scheme 21.
Transition metal-free 1,2,3-triboration of conjugated dienes.

Scheme 22.
Mechanistic proposal of transition metal-free 1,2,3-triboration of conjugated dienes.

Scheme 23.
Selective polyfunctionalization of 1,2,3-triborated products.
Scheme 24.
Photoredoxcatalyzed mono-deboronation of 1,2,3-triborated products.

Scheme 25.
Synthesis of 1,1,2-tri(boryl)alkanes via bis(catecholato)diboron.
Scheme 26.
(a) transition-metal free 1,1,2-tris(boryl)alkane synthesis; (b) mechanistic proposal.
Scheme 27.
Selective deprotonation on the geminal diboryl moiety in 1,1,2-tri(boryl)alkanes.
Scheme 28.
Transformation from 1,1,2-tri(boryl)alkane into mixed Bpin and B-MIDA triboronate compounds.
Scheme 28.
Transformation from 1,1,2-tri(boryl)alkane into mixed Bpin and B-MIDA triboronate compounds.
Scheme 29.
Deborylative alkylation with chiral 1,1,2-tris-(boronates).
Scheme 30.
(a) Tandem desilylative borylation of bis(trimethylsilyl)acetylene and the subsequent Pt-catalyzed diboration; (b) Pt-catalyzed diboration of 1-alkynylboronates; (c) platinum-catalyzed diboration of phenylethynyl MIDA boronate.
Scheme 30.
(a) Tandem desilylative borylation of bis(trimethylsilyl)acetylene and the subsequent Pt-catalyzed diboration; (b) Pt-catalyzed diboration of 1-alkynylboronates; (c) platinum-catalyzed diboration of phenylethynyl MIDA boronate.
Scheme 31.
Dehydrogenative diboration with HBpin of alkynylboronates.
Scheme 32.
Copper catalyzed triboration of alkynes.
Scheme 33.
Mechanistic proposal of copper catalyzed triboration of alkynes.
Scheme 34.
Selective palladium catalyzed arylation of 1,1,2-tri(boryl)alkenes.

Scheme 35.
Selective palladium catalyzed arylation of 1,1,2-tri(boryl)alkenes.

Scheme 36.
Selective functionalization of 1,1,2-tri(boryl)alkenes.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Salvadó, O.; Fernández, E. Tri(boryl)alkanes and Tri(boryl)alkenes: The Versatile Reagents. Molecules 2020, 25, 1758. https://doi.org/10.3390/molecules25071758
AMA Style
Salvadó O, Fernández E. Tri(boryl)alkanes and Tri(boryl)alkenes: The Versatile Reagents. Molecules. 2020; 25(7):1758. https://doi.org/10.3390/molecules25071758
Chicago/Turabian StyleSalvadó, Oriol, and Elena Fernández. 2020. "Tri(boryl)alkanes and Tri(boryl)alkenes: The Versatile Reagents" Molecules 25, no. 7: 1758. https://doi.org/10.3390/molecules25071758