Computational and Crystallographic Examination of Naphthoquinone Based Diarylethene Photochromes

 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
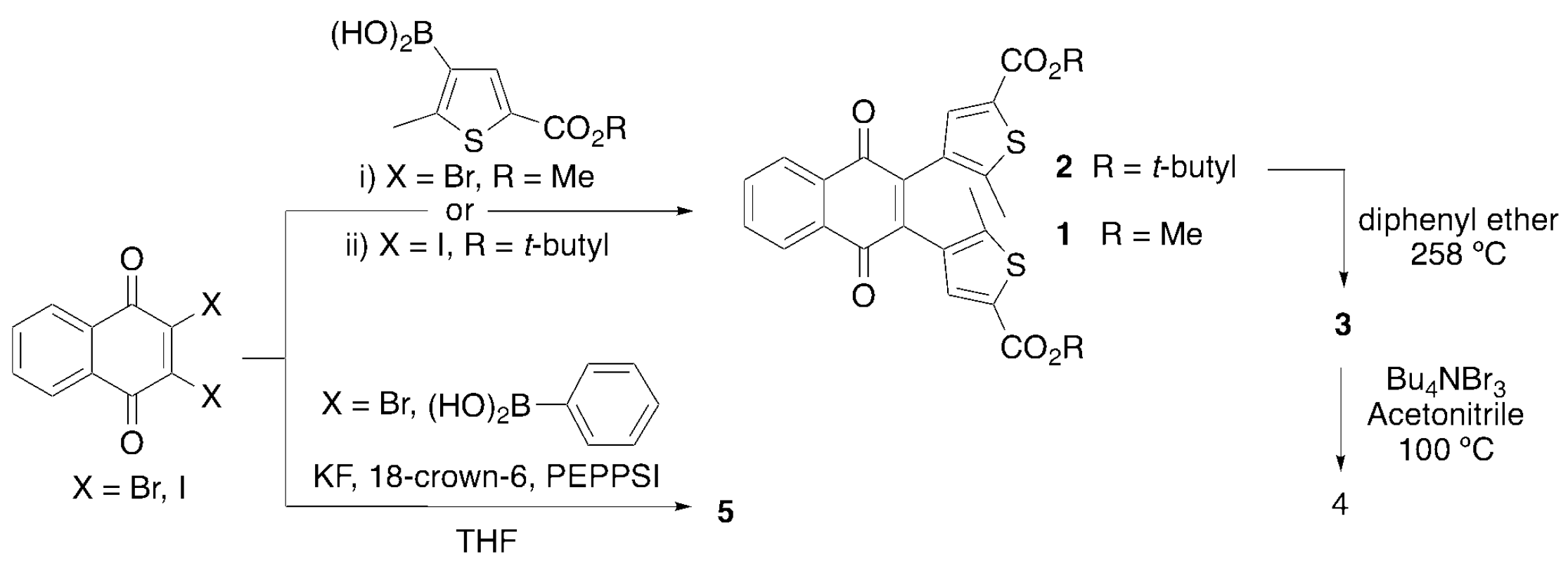
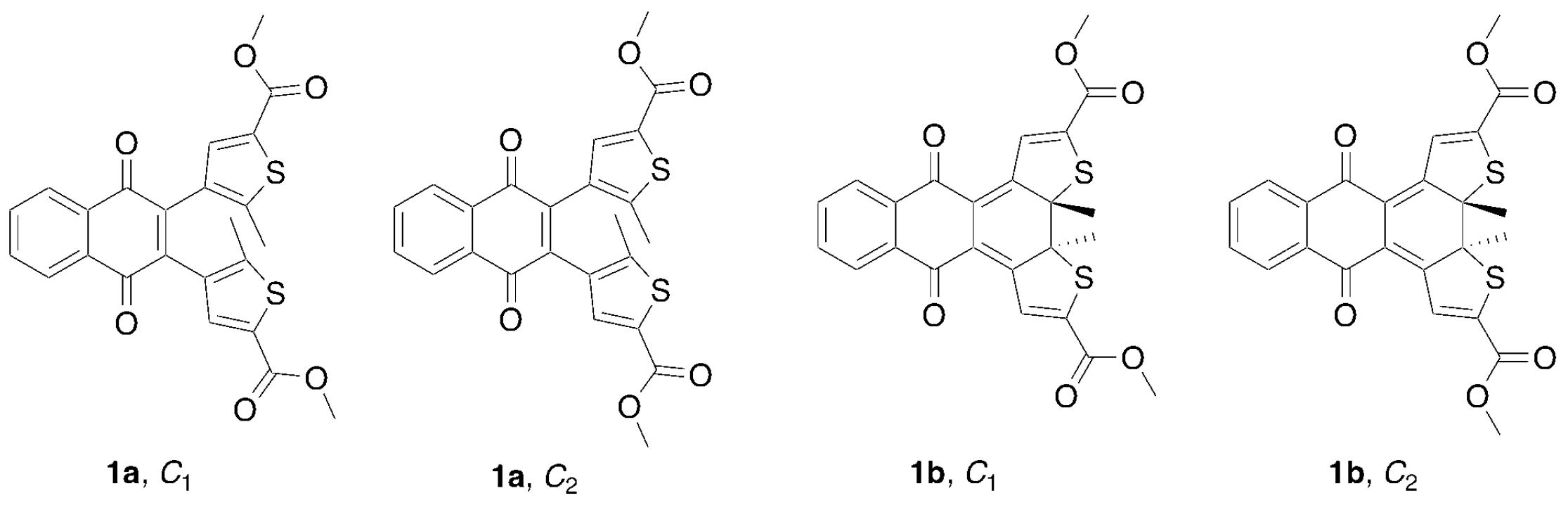
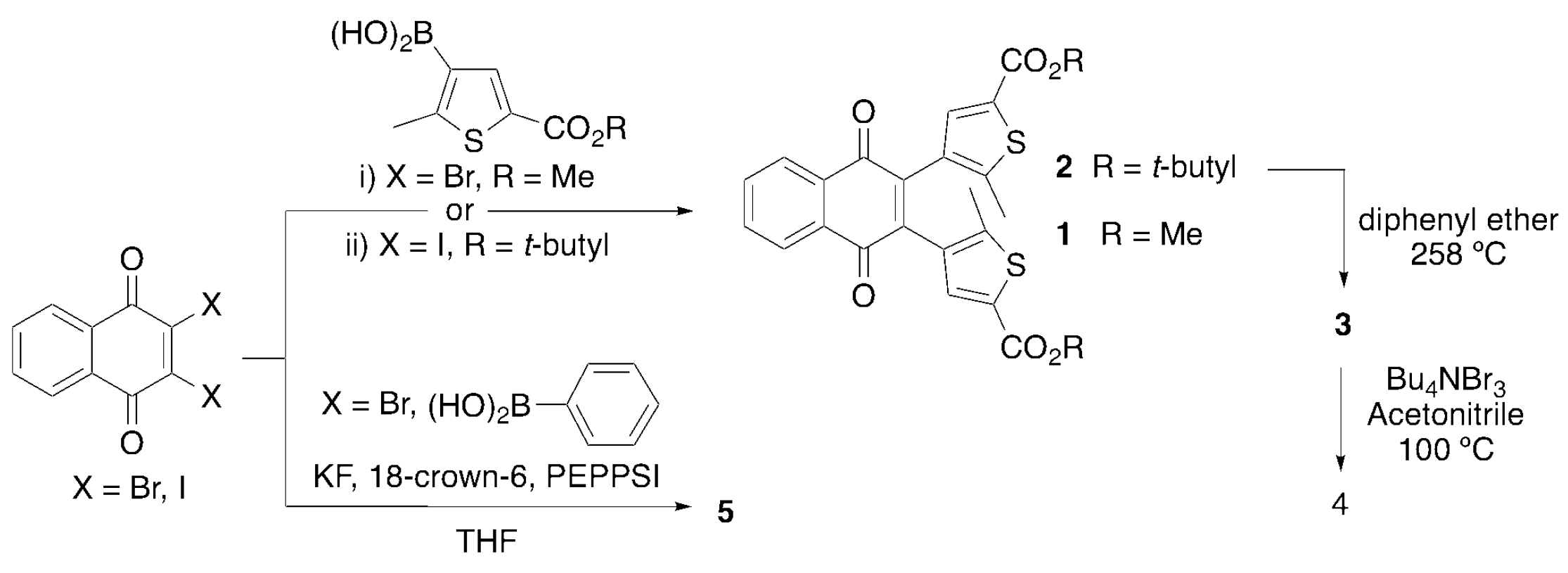
2.1. Synthesis and Characterization
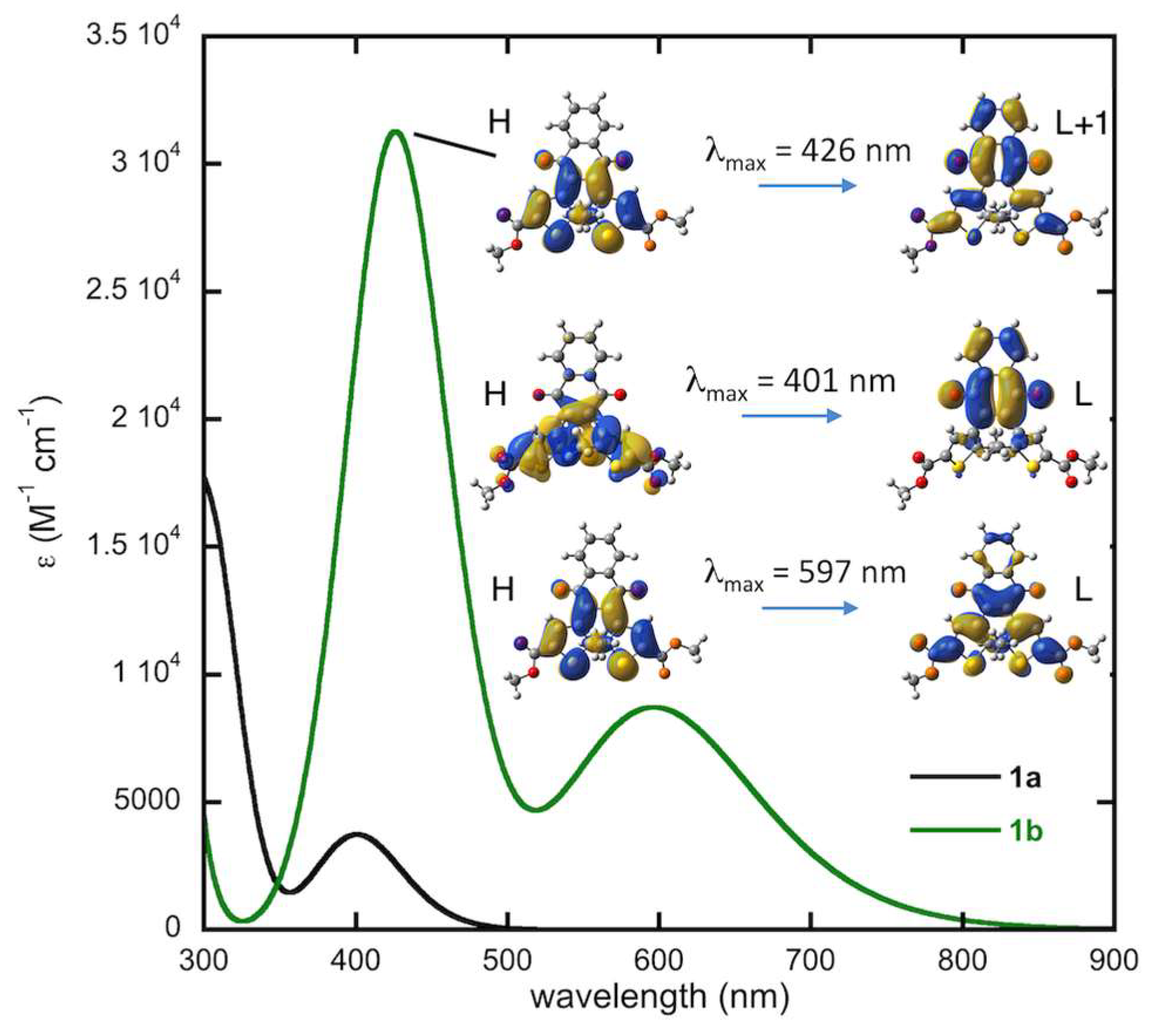
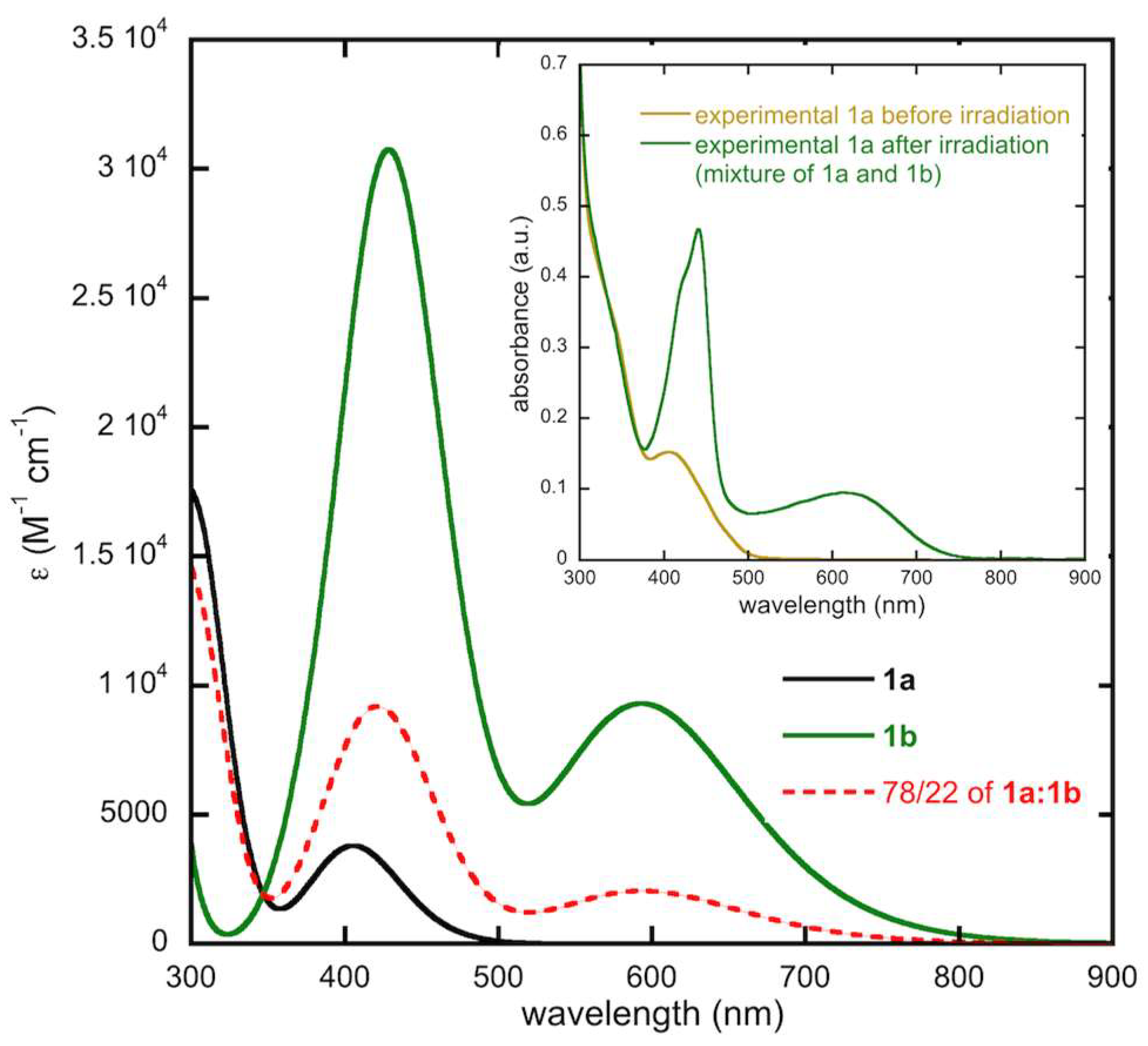
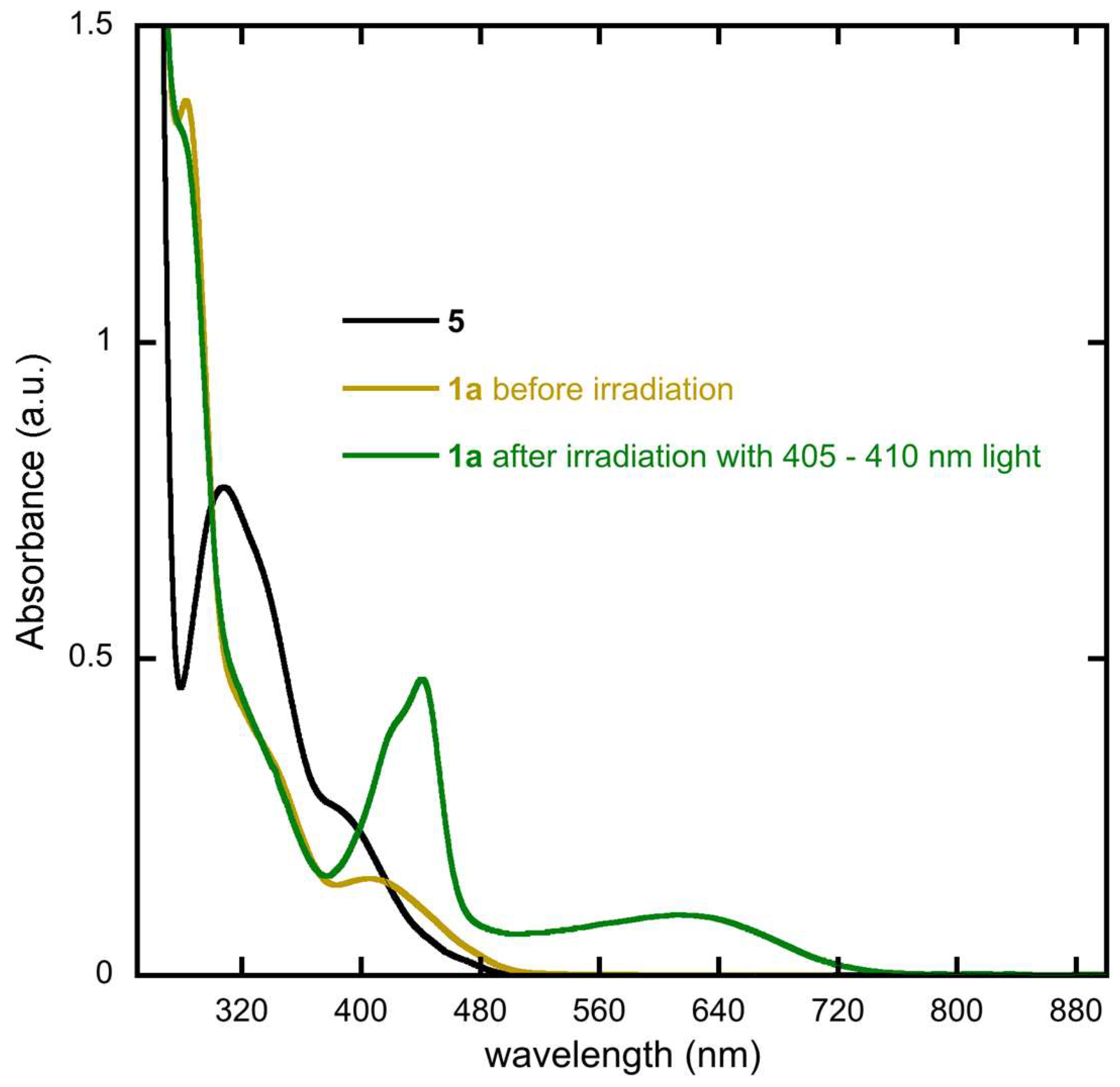
2.2. Optical Studies and Photochromic Properties
2.3. X-Ray Crystallography
2.3.1. Compound 5
2.3.2. Closed form 1b
2.4. Computations
2.4.1. Conformational Analysis and Thermal Stability of Compound 1
2.4.2. Computed Spectra and Molecular Orbital Analysis for Compound 1
2.4.3. Modeling of the Ratio of Open and Closed Forms of 1b
2.4.4. Thermal Stability, Computed Spectra and Molecular Orbital Analysis for Compound 3
2.4.5. Conformational Analysis and Thermal Stability of Compound 4
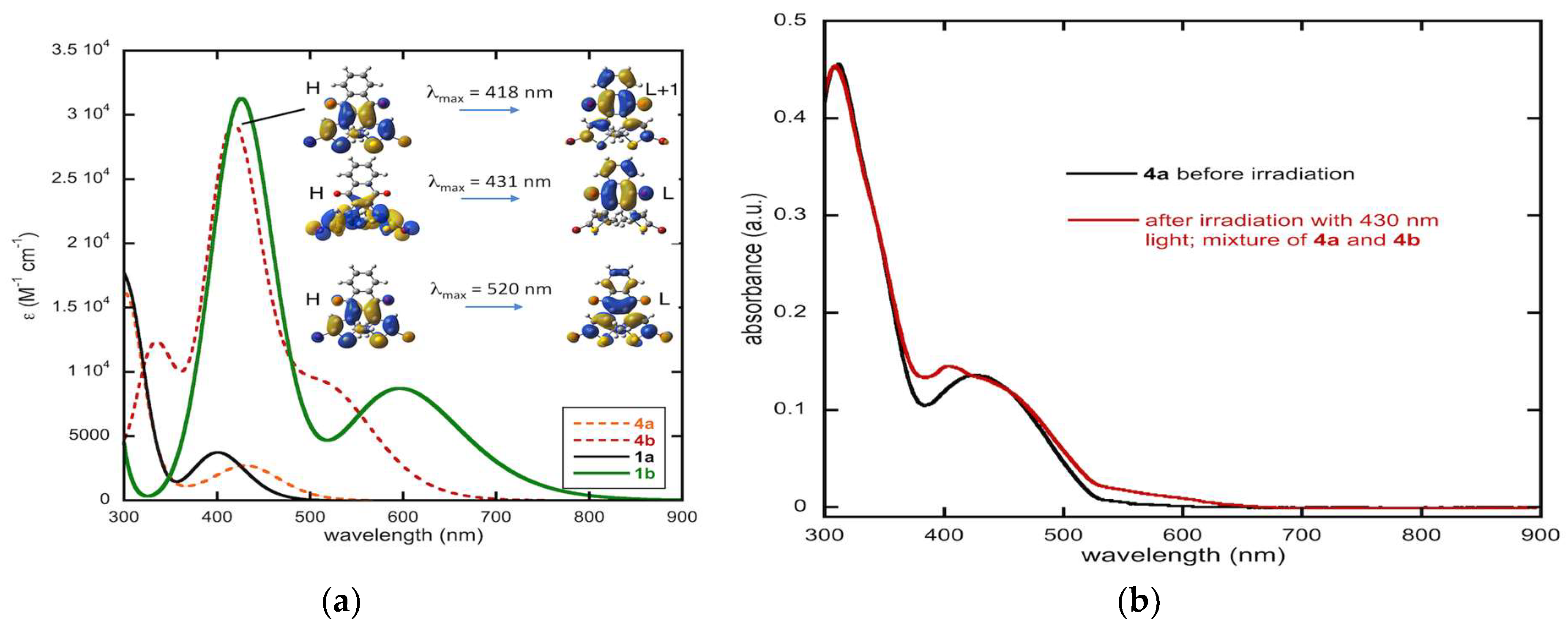
2.4.6. Computed Spectra and Molecular Orbital Analysis for Compound 4
3. Materials and Methods
3.1. General
3.2. Synthesis
3.3. Computational
3.4. X-ray Crystallographic Data Collection and Refinement
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Crano, J.C.; Guglielmetti, R.J. Organic Photochromic and Thermochromic Compounds; Plenum Press: New York, NY, USA, 1999. [Google Scholar]
- Dürr, H.; Bouas-Laurent, H. Photochromism: Molecules and Systems; rev. ed.; Elsevier: Amsterdam, The Netherlands; Boston, MA, USA, 2003. [Google Scholar]
- Davis, D.A.; Hamilton, A.; Yang, J.L.; Cremar, L.D.; Van Gough, D.; Potisek, S.L.; Ong, M.T.; Braun, P.V.; Martinez, T.J.; White, S.R.; et al. Force-induced activation of covalent bonds in mechanoresponsive polymeric materials. Nature 2009, 459, 68–72. [Google Scholar] [CrossRef]
- Huang, Y.; Li, F.; Ye, C.; Qin, M.; Ran, W.; Song, Y. A Photochromic Sensor Microchip for High-performance Multiplex Metal Ions Detection. Sci. Rep. 2015, 5, 9724. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Wang, N.; Pu, S.; Zhang, X.; Liu, G.; Dai, Y. A acid/base gated photochromic and fluorescent sensor based on a diarylethene with a 2-(1H-dithienobenzoimidazole)phenol unit. Dye. Pigment. 2017, 146, 445–454. [Google Scholar] [CrossRef]
- Xie, X.; Mistlberger, G.; Bakker, E. Reversible Photodynamic Chloride-Selective Sensor Based on Photochromic Spiropyran. J. Am. Chem. Soc. 2012, 134, 16929–16932. [Google Scholar] [CrossRef]
- Qin, M.; Huang, Y.; Li, F.; Song, Y. Photochromic sensors: A versatile approach for recognition and discrimination. J. Mater. Chem. C 2015, 3, 9265–9275. [Google Scholar] [CrossRef]
- Lemieux, V.; Branda, N.R. Reactivity-Gated Photochromism of 1,2-Dithienylethenes for Potential Use in Dosimetry Applications. Org. Lett. 2005, 7, 2969–2972. [Google Scholar] [CrossRef] [PubMed]
- Kenji, M.; Irie, M.A. Diarylethene with Two Nitronyl Nitroxides: Photoswitching of Intramolecular Magnetic Interaction. J. Am. Chem. Soc. 2000, 122, 7195–7201. [Google Scholar]
- Matsuda, K.; Matsuo, M.; Irie, M. Photoswitching of intramolecular magnetic interaction using photochromic diarylethene spin coupler: Introduction of thiophene spacer. Chem. Lett. 2001, 5, 436–437. [Google Scholar] [CrossRef]
- Hirshberg, Y. Reversible Formation and Eradication of Colors by Irradiation at Low Temperatures. A Photochemical Memory Model. J. Am. Chem. Soc. 1956, 78, 2304–2312. [Google Scholar] [CrossRef]
- Yao, B.; Wang, Y.; Menke, N.; Lei, M.; Zheng, Y.; Ren, L.; Chen, G.; Chen, Y.; Fan, M. Optical Properties and Applications of Photochromic Fulgides. Mol. Cryst. Liq. Cryst. 2005, 430, 211–219. [Google Scholar] [CrossRef]
- Kawata, S.; Kawata, Y. Three-Dimensional Optical Data Storage Using Photochromic Materials. Chem. Rev. 2000, 100, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Corredor, C.C.; Huang, Z.L.; Belfield, K.D. Two-Photon 3D Optical Data Storage via Fluorescence Modulation of an Efficient Fluorene Dye by a Photochromic Diarylethene. Adv. Mater. 2006, 18, 2910–2914. [Google Scholar] [CrossRef]
- Choi, H.J.; Jeong, K.U.; Chien, L.C.; Lee, M.H. Photochromic 3-dimensional actuator based on an uncrosslinked liquid crystal elastomer. J. Mater. Chem. 2009, 19, 7124–7129. [Google Scholar] [CrossRef]
- Colombier, I.; Spagnoli, S.; Corval, A.; Baldeck, P.L.; Giraud, M.; Léaustic, A.; Yu, P. Strong Photomechanical Effects in Photochromic Organic Microcrystals. Mol. Cryst. Liq. Cryst. 2005, 431, 495–499. [Google Scholar] [CrossRef]
- Koshima, H.; Ojima, N.; Uchimoto, H. Mechanical Motion of Azobenzene Crystals upon Photoirradiation. J. Am. Chem. Soc. 2009, 131, 6890–6891. [Google Scholar] [CrossRef]
- Koshima, H.; Takechi, K.; Uchimoto, H.; Shiro, M.; Hashizume, D. Photomechanical bending of salicylideneaniline crystals. Chem. Commun. 2011, 47, 11423–11425. [Google Scholar] [CrossRef]
- Yu, Y.L.; Nakano, M.; Ikeda, T. Directed bending of a polymer film by light - Miniaturizing a simple photomechanical system could expand its range of applications. Nature 2003, 425, 145. [Google Scholar] [CrossRef]
- Dong, X.; Tong, F.; Hanson, K.M.; Al-Kaysi, R.O.; Kitagawa, D.; Kobatake, S.; Bardeen, C.J. Hybrid Organic–Inorganic Photon-Powered Actuators Based on Aligned Diarylethene Nanocrystals. Chem. Mater. 2019, 31, 1016–1022. [Google Scholar] [CrossRef]
- Morimoto, M.; Irie, M. A Diarylethene Cocrystal that Converts Light into Mechanical Work. J. Am. Chem. Soc. 2010, 132, 14172–14178. [Google Scholar] [CrossRef]
- Szymański, W.; Beierle, J.M.; Kistemaker, H.A.V.; Velema, W.A.; Feringa, B.L. Reversible Photocontrol of Biological Systems by the Incorporation of Molecular Photoswitches. Chem. Rev. 2013, 113, 6114–6178. [Google Scholar] [CrossRef] [Green Version]
- Al-Atar, U.; Fernandes, R.; Johnsen, B.; Baillie, D.; Branda, N.R. A Photocontrolled Molecular Switch Regulates Paralysis in a Living Organism. J. Am. Chem. Soc. 2009, 131, 15966–15967. [Google Scholar] [CrossRef]
- Gemayel, M.E.; Börjesson, K.; Herder, M.; Duong, D.T.; Hutchison, J.A.; Ruzié, C.; Schweicher, G.; Salleo, A.; Geerts, Y.; Hecht, S.; et al. Optically switchable transistors by simple incorporation of photochromic systems into small-molecule semiconducting matrices. Nat. Commun. 2015, 6, 6330. [Google Scholar] [CrossRef] [PubMed]
- Senanayak, S.P.; Sangwan, V.K.; McMorrow, J.J.; Everaerts, K.; Chen, Z.; Facchetti, A.; Hersam, M.C.; Marks, T.J.; Narayan, K.S. Self-Assembled Photochromic Molecular Dipoles for High-Performance Polymer Thin-Film Transistors. ACS Appl. Mater. Interfaces 2018, 10, 21492–21498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.-N.; Leng, B.; Li, Y.-S.; Gao, X.-K. Photoresponsive organic field-effect transistors involving photochromic molecules. Chin. Chem. Lett. 2016, 27, 1319–1329. [Google Scholar] [CrossRef]
- Raymo, F.M.; Giordani, S.; White, A.J.P.; Williams, D.J. Digital Processing with a Three-State Molecular Switch. J. Org. Chem. 2003, 68, 4158–4169. [Google Scholar] [CrossRef]
- Fredrich, S.; Göstl, R.; Herder, M.; Grubert, L.; Hecht, S. Switching Diarylethenes Reliably in Both Directions with Visible Light. Angew. Chem. Int. Ed. 2016, 55, 1208–1212. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Volfova, H.; Mulder, R.J.; Goerigk, L.; Bryant, G.; Riedle, E.; Ritchie, C. Visible-Light-Driven “On”/“Off” Photochromism of a Polyoxometalate Diarylethene Coordination Complex. J. Am. Chem. Soc. 2018, 140, 10482–10487. [Google Scholar] [CrossRef]
- Tang, S.; Song, F.; Lu, M.; Han, K.; Peng, X. Rational design of a visible-light photochromic diarylethene: A simple strategy by extending conjugation with electron donating groups. Sci. China Chem. 2019, 62, 451–459. [Google Scholar] [CrossRef]
- Xi, H.; Zhang, Z.; Zhang, W.; Li, M.; Lian, C.; Luo, Q.; Tian, H.; Zhu, W.-H. All-Visible-Light-Activated Dithienylethenes Induced by Intramolecular Proton Transfer. J. Am. Chem. Soc. 2019, 141, 18467–18474. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, W.; Jin, P.; Xue, J.; Sun, L.; Huang, J.; Zhang, J.; Tian, H. A building-block design for enhanced visible-light switching of diarylethenes. Nat. Commun. 2019, 10, 4232. [Google Scholar] [CrossRef] [Green Version]
- Hanazawa, M.; Sumiya, R.; Horikawa, Y.; Irie, M. Thermally irreversible photochromic systems. Reversible photocyclization of 1,2-bis (2-methylbenzo[b]thiophen-3-yl)perfluorocyclocoalkene derivatives. J. Chem. Soc. Chem. Commun. 1992, 206–207. [Google Scholar] [CrossRef]
- Irie, M.; Sakemura, K.; Okinaka, M.; Uchida, K. Photochromism of Dithienylethenes with Electron-Donating Substituents. J. Org. Chem. 1995, 60, 8305–8309. [Google Scholar] [CrossRef]
- Patel, D.G.; Mitchell, T.B.; Myers, S.D.; Carter, D.A.; Novak, F.A. A Suzuki Approach to Quinone-Based Diarylethene Photochromes. J. Org. Chem. 2020, 85, 2646–2653. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Liebeskind, L.S. A Contribution to the Design of Molecular Switches: Novel Acid-Mediated Ring-Closing−Photochemical Ring-Opening of 2,3-Bis(heteroaryl)quinones (Heteroaryl = Thienyl, Furanyl, Pyrrolyl). J. Am. Chem. Soc. 2001, 123, 7703–7704. [Google Scholar] [CrossRef]
- Boggio-Pasqua, M.; Perrier, A.; Fihey, A.; Jacquemin, D. Modeling diarylethene excited states with ab initio tools: From model systems to large multimers. In Photon-Working Switches; Yokoyama, Y., Nakatani, K., Eds.; Springer: Tokyo, Japan, 2017. [Google Scholar]
- Boggio-Pasqua, M.; Bearpark, M.J.; Robb, M.A. The role of extended conical intersection seams in photochromic systems. In AIP Conference Proceedings, 22 January 2015; American Institute of Physics: College Park, MD, USA, 2015; pp. 453–456. [Google Scholar]
- Robb, M.A. In This Molecule There Must Be a Conical Intersection. In Advances in Physical Organic Chemistry; Williams, I.H., Williams, N.H., Eds.; Academic Press: Cambridge, MA, USA, 2014; Volume 48, pp. 189–228. [Google Scholar]
- Saito, E.; Ako, T.; Kobori, Y.; Tsuda, A. Switching of the π-electronic conjugations in the reduction of a dithienylethene-fused p-benzoquinone. RSC Adv. 2017, 7, 2403–2406. [Google Scholar] [CrossRef] [Green Version]
- Migulin, V.A. A New Synthetic Pathway to Symmetric Bisubstituted Naphthoquinones. Synthesis 2020, 52, 60–68. [Google Scholar] [CrossRef]
- Patel, D.G.; Feng, F.; Ohnishi, Y.-Y.; Abboud, K.A.; Hirata, S.; Schanze, K.S.; Reynolds, J.R. It Takes More Than an Imine: The Role of the Central Atom on the Electron-Accepting Ability of Benzotriazole and Benzothiadiazole Oligomers. J. Am. Chem. Soc. 2012, 134, 2599–2612. [Google Scholar] [CrossRef]
- Yoshida, S.; Kubo, H.; Saika, T.; Katsumura, S. Synthesis of 2,3-Diarylquinone by Palladium Catalyzed Cross-Coupling of Dibromoquinones with Heteroarylstannanes. Chem. Lett. 1996, 25, 139–140. [Google Scholar] [CrossRef]
- Simeth, N.A.; Kneuttinger, A.C.; Sterner, R.; König, B. Photochromic coenzyme Q derivatives: Switching redox potentials with light. Chem. Sci. 2017, 8, 6474–6483. [Google Scholar] [CrossRef] [Green Version]
- Takagi, J.; Sato, K.; Hartwig, J.F.; Ishiyama, T.; Miyaura, N. Iridium-catalyzed C–H coupling reaction of heteroaromatic compounds with bis(pinacolato)diboron: Regioselective synthesis of heteroarylboronates. Tetrahedron Lett. 2002, 43, 5649–5651. [Google Scholar] [CrossRef]
- Ishiyama, T.; Murata, M.; Miyaura, N. Palladium(0)-Catalyzed Cross-Coupling Reaction of Alkoxydiboron with Haloarenes: A Direct Procedure for Arylboronic Esters. J. Org. Chem. 1995, 60, 7508–7510. [Google Scholar] [CrossRef]
- Best, W.M.; Sims, C.G.; Winslade, M. Palladium-Catalysed Cross Coupling of Arylboronic Acids with 2-Chloro-1,4-naphthoquinones: The Synthesis of 2-Aryl- and 2,3-Bisaryl-1,4-naphthoquinones. Aust. J. Chem. 2001, 54, 401–404. [Google Scholar] [CrossRef]
- Tamayo, N.; Echavarren, A.M.; Paredes, M.C. Palladium-catalyzed coupling of 2-bromonaphthoquinones with stannanes: A concise synthesis of antibiotics WS 5995 A and C and related compounds. J. Org. Chem. 1991, 56, 6488–6491. [Google Scholar] [CrossRef]
- Sahariah, B.; Sarma, B.K. Relative orientation of the carbonyl groups determines the nature of orbital interactions in carbonyl–carbonyl short contacts. Chem. Sci. 2019, 10, 909–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, C.A.; Sanders, J.K.M. The nature of pi-pi interactions. J. Am. Chem. Soc. 1990, 112, 5525–5534. [Google Scholar] [CrossRef]
- Martinez, C.R.; Iverson, B.L. Rethinking the term “pi-stacking”. Chem. Sci. 2012, 3, 2191–2201. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, K.; Jensen, F.; Dorigo, A.; Houk, K.N. A spin correction procedure for unrestricted Hartree-Fock and Møller-Plesset wavefunctions for singlet diradicals and polyradicals. Chem. Phys. Lett. 1988, 149, 537–542. [Google Scholar] [CrossRef]
- Yamanaka, S.; Kawakami, T.; Nagao, H.; Yamaguchi, K. Effective exchange integrals for open-shell species by density functional methods. Chem. Phys. Lett. 1994, 231, 25–33. [Google Scholar] [CrossRef]
- Becke, A.D. Density–functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li–F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- APEX3. 2019.1-0: Program for Bruker CCD X-Ray Diffractometer Control; Bruker AXS Inc.: Madison, WI, USA, 2016. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
Sample Availability: Samples of compound 1 are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symmetry | Geometry | ΔE (gas Φ) kJ/mol | ΔE (in DCM) kJ/mol |
|---|---|---|---|
| C1 | Open | 0.0 | 0.0 |
| C2 | Open | 0.8 | 0.4 |
| C1 | Closed | 43.6 | 49.9 |
| C2 | Closed | 43.9 | 50.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, D.G.; Boggio-Pasqua, M.; Mitchell, T.B.; Walton, I.M.; Quigley, W.R.; Novak, F.A. Computational and Crystallographic Examination of Naphthoquinone Based Diarylethene Photochromes. Molecules 2020, 25, 2630. https://doi.org/10.3390/molecules25112630
Patel DG, Boggio-Pasqua M, Mitchell TB, Walton IM, Quigley WR, Novak FA. Computational and Crystallographic Examination of Naphthoquinone Based Diarylethene Photochromes. Molecules. 2020; 25(11):2630. https://doi.org/10.3390/molecules25112630
Chicago/Turabian StylePatel, Dinesh G., Martial Boggio-Pasqua, Travis B. Mitchell, Ian M. Walton, William R. Quigley, and Frank A. Novak. 2020. "Computational and Crystallographic Examination of Naphthoquinone Based Diarylethene Photochromes" Molecules 25, no. 11: 2630. https://doi.org/10.3390/molecules25112630