
m-Carborane as a Novel Core for Periphery-Decorated Macromolecules



Institut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus UAB, 08193 Barcelona, Spain
*
Author to whom correspondence should be addressed.
Molecules 2020, 25(12), 2814; https://doi.org/10.3390/molecules25122814
Submission received: 31 May 2020
/
Revised: 15 June 2020
/
Accepted: 16 June 2020
/
Published: 18 June 2020
(This article belongs to the Special Issue Boron in Catalysis and Materials Chemistry: A Themed Issue in Honor of Professor Todd B. Marder on the Occasion of His 65th Birthday)
Abstract
:Closom-C2B10H12 can perform as a novel core of globular periphery-decorated macromolecules. To do this, a new class of di and tetrabranched m-carborane derivatives has been synthesized by a judicious choice of the synthetic procedure, starting with 9,10-I2-1,7-closo-C2B10H10. The 2a-NPA (sum of the natural charges of the two bonded atoms) value for a bond, which is defined as the sum of the NPA charges of the two bonded atoms, matches the order of electrophilic reaction at the different cluster bonds of the icosahedral o-and m- carboranes that lead to the formation of B-I bonds. As for m-carborane, most of the 2a-NPA values of B-H vertexes are positive, and their functionalization is more challenging. The synthesis and full characterization of dibranched 9,10-R2-1,7-closo-carborane (R = CH2CHCH2, HO(CH2)3, Cl(CH2)3, TsO(CH2)3, C6H5COO(CH2)3, C6H5COO(CH2)3, N3(CH2)3, CH3CHCH, and C6H5C2N3(CH2)3) compounds as well as the tetrabranched 9,10-R2-1,7-R2-closo-C2B10H8 (R = CH2CHCH2, HO(CH2)3) are presented. The X-ray diffraction of 9,10-(HO(CH2)3)2-1,7-closo-C2B10H10 and 9,10-(CH3CHCH)2-1,7-closo-C2B10H10, as well as their Hirshfeld surface analysis and decomposed fingerprint plots, are described. These new reported tetrabranched m-carborane derivatives provide a sort of novel core for the synthesis of 3D radially grown periphery-decorated macromolecules that are different to the 2D radially grown core of the tetrabranched o-carborane framework.
1. Introduction
Icosahedral carborane clusters with empirical formula C2B10H12 can be in three different isomers: 1,2-closo-C2B10H12 (o-carborane), 1,7-closo-C2B10H12 (m-carborane; 1), and 1,12-closo-C2B10H12 (p-carborane). Figure 1 displays a schematic representation of the isomers with their vertexes numbering. Despite their common icosahedral geometry, they display similarities, but also important differences. Among the similarities are the high stabilities and 3D geometrical properties, their very similar 3D aromatic character [1,2] that leads to display great inertia to keep the original scaffold upon electrophilic substitution, their dual-mode as electron-withdrawing through carbon or electron-donating through boron vertexes [3,4,5], their molecular volume that is high compared to rotating benzene [6], and high hydrophobicity [7,8,9]. Among the differences are the dipolar moment and their different reactivity towards boron elimination [8], and the lowest unoccupied molecular orbital (LUMO) geometrical disposition that is responsible for many of the physical properties of the isomers. Among the three of them, the most extensively studied is the o-carborane. Some tips to keep in mind between the three isomers when substitution is sought are: First, the weak acidic Cc-H bond (Cc = cluster C atom) [10] can be deprotonated using a strong base followed by an electrophilic reaction to form the Cc-R bond [8,11]. Second, the B-H hydrogen atoms with the hydridic character on (B(9,12), B(8,10), and B(4,5,7,11)) are subjected to electrophilic substitution to form B-halogen units [8,11] that, if followed by a Kumada cross-coupling reaction, may lead to the introduction of organic moieties to make B-R vertexes [3,4,5,6,7,8]. This procedure does not proceed equally for all B-Hs, for instance, at the B(3,6) vertices, which are the most electron-deficient vertexes, their functionalization does not take place by using the same process at the other cluster’s vertices [8,11]. Substitution on these positions can be achieved via the deboration-capping multistep reaction or via the metallation procedure [11,12,13]. The diverse regioreactivity of o-carborane, has been exploited and adapted to make o-carborane an exceptional core for developing a large variety of multibranched molecules, globular macromolecules, dendrimers (Figure 2b), and so on [14,15,16,17,18]. Moreover, new versatile synthons have been explored through the multi-functionalization of B and Cc atoms jointly, which make the o-carborane clusters an exciting platform for new materials [6,8,18,19,20,21,22,23,24,25].
On the other hand, the reactivity of m-carborane is less studied but for the Cc-H vertexes, which are less acidic as compared to the Cc-H vertexes of the o-isomer [10,26]. Using a similar strategy as for o-carborane, a wide variety of 1-R-1,7-closo-C2B10H11 and 1,7-R2-1,7-closo-C2B10H10 derivatives has been developed [27,28,29,30,31,32,33,34,35]. To some extent, the current state of knowledge of the m-carborane functionalization through the B-H vertexes is in an odd situation. As compared to the o-carborane, a much-limited number of protocols leading to modify the B-H vertexes in the m-cluster have been reported [8,11]. In this context, very few derivatives of m-carborane with a functional group that is bonded to B(9) or B(9) and B(10) synchronously have been described [27,28,29,30,31,32,33,34,35]. The Pd-catalyzed cross-coupling reaction of 9-X-1,7-closo-C2B10H11 and 9,10-X2-1,7-closo-C2B10H10 (X= halogen atom) represents one of these examples of derivatization [34,36,37]. By contrast to the o-carborane, no multibranched m-carborane structures with a general formula 1,7-R2-9,10-R’2-1,7-closo-C2B10H8 have been reported despite the potential of its structure and the relatively high reactivity of the Cc-H bonds that should allow the reaction to a great extent. Notably, as shown in Figure 2, the m-carborane core provides a 3D radially growth core while o-carborane a 2D one.
Consequently, we became interested in introducing organic branches connected to B(9) and B(10) to prepare a new set of 9,10-R2-1,7-closo-C2B10H10 derivatives. In the second part of this paper, we functionalized the two Cc-H of 9,10-(CH2=CHCH2)2-1,7-closo-C2B10H10 to form the quadruped-shaped structure with a general formula 1,7-R2-9,10-R’2-1,7-closo-C2B10H10 which might serve as versatile precursors with free ends for further reaction.
2. Results and Discussion
2.1. Synthesis of di-Branched m-carborane Derivatives at the 9,10 Vertexes
Versatile strategy for the synthesis of the two branches B(9,10) m-carborane derivatives (9,10-R2-1,7-closo-C2B10H10) was achieved by using 9,10-I2-1,7-closo-C2B10H10 as the starting compound.
The synthesis of 9,10-I2-1,7-closo-C2B10H10; (2) has been reported by using two different methodologies: i) the electrophilic iodination reaction of icosahedral closo m-carborane (1) by using a molar Equiv. Of iodine: monochloride, which acts as an electrophilic agent, in the presence of catalytic amounts of aluminum chloride, and ii) using iodine as an electrophilic agent in a very acidic media (HNO3:H2SO4, 1:1). The target compound 2 was obtained in 60% and 87% yield, respectively [38,39]. Our study focused on the synthesis of new Boron disubstituted closo m-carborane derivatives at the 9 and 10 vertexes began with the synthesis of 2 in 87% yield by combining the two reported methods: an equimolar ratio of m-carborane (1): iodine in acidic HNO3:H2SO4 (1:1) solution was left under reflux to react for 3 h (Scheme 1).
To produce the B-C bonds on 1, a useful and general method is the Kumada cross-coupling reaction on B-iodinated m-carborane 2 with Grignard reagents in the presence of Pd(II) and Cu(I) catalysts. To achieve the di-branched m-carborane derivatives at the 9, 10 vertexes, the cross-coupling reaction on 2 was studied using CH2CHCH2MgCl Grignard derivative in the presence of [PdCl2(PPh3)2] and CuI as catalysts to give the 9,10-(CH2CHCH2)2-1,7-closo-C2B10H10 (3) in 95% yield [40].
The terminal olefin groups in 3 are ready for further reactions on them, enabling the m-carborane cluster to become the template for a new type of macromolecules having a rigid head and two appended branches. As a first example of these molecules, compound 3 was converted to 9,10-(HOCH2CH2CH2)2-closo-1,7-C2B10H10, (4) following the hydroboration/oxidation reaction on 3 by using BH3·THF as hydroboration agent and subsequent oxidation with H2O2 in a basic aqueous solution. After workup, crystalline white pure solid, 4, was obtained in 93% yield. The 1H NMR spectrum displays a new broad peak at 3.43 ppm, which supports the presence of the O-H group in 4. Also, the 1H and 13C{1H} NMR spectra revealed that the reaction had proceeded by an anti-Markovnikov addition, therefore having the two hydroxyl groups at terminal positions. No hindered hydroboranes were thus needed for the control of the reaction’s regioselectivity.
These terminal alcohol groups anticipate versatile chemistry for radial growth, given the availability of the terminal hydroxyl groups for further elongation of the chains. Moreover, the Cc–H vertexes on the rigid m-carborane head are ready for derivatization or supramolecular assembly. Then, the m-carborane cluster, as o-carborane does, provides a singular platform for the construction of highly dense multibranched molecules with a wide range of possibilities. Therefore, derivatives of m-carborane with precise patterns of substitution, which are sterically different from the ones of o-carborane but complementary, can be prepared by a judicious choice of the synthetic procedure. Consequently, the substitution of the terminal hydroxyl units in 4 by chloro (5), ester (6), tosyl (7) or azide (8) groups, which enable the branches to grow by a subsequent coupling reaction with nucleophilic agents, was achieved (Scheme 2).
Chlorination in 3 was achieved using SOCl2 and [Nbu4]Cl salt to give 9,10-(ClCH2CHCH2)2-1,7-closo-C2B10H10 (5) in 92% yield.
As an example of the esterification of the terminal alcohol groups, compound 6 was obtained in 90% yield by Steglich esterification [41,42] with benzoic acid using N,N’-dicyclohexylcarbodiimide as a coupling reagent and the N,N-dimethylaminopyridine as a catalyst.
Furthermore, alcohol groups were converted to tosylate groups by performing the reaction of 3 with tosyl chloride, Net3 as a base, and [HNMe3]Cl as a catalyst [43], obtaining 9,10-(TsOCH2CH2CH2)2-1,7-closo-C2B10H10, 7, in 85% yield.
Overall, we have succeeded in the preparation of these new m-carborane derivatives 5–7 thanks to the primary alcohol groups, which undergo chain extension reactions. Owing to the formation of 5, a new way of functionalization is opened to prepare the azide derivative 8, which in turn opens the way to perform the Azide-Alkyne Huisgen Cycloaddition commonly known as the click reaction. Compound 8 was obtained in 81% yield by avigorous stirring of 5, with excess of NaN3 and [Nbu4]Cl in a mixture of toluene and water, at reflux for 24–48 h. An example of the click reaction on 8 was the compound 9 synthesis in 86% yield by simple reaction with phenylaceylene, sodium ascorbate, and hydrated CuSO4 as a catalyst in a mixture of dioxane/water.
2.2. Synthesis of tetra-Branched m-carborane Derivatives at the 1,7,9,10 Vertexes
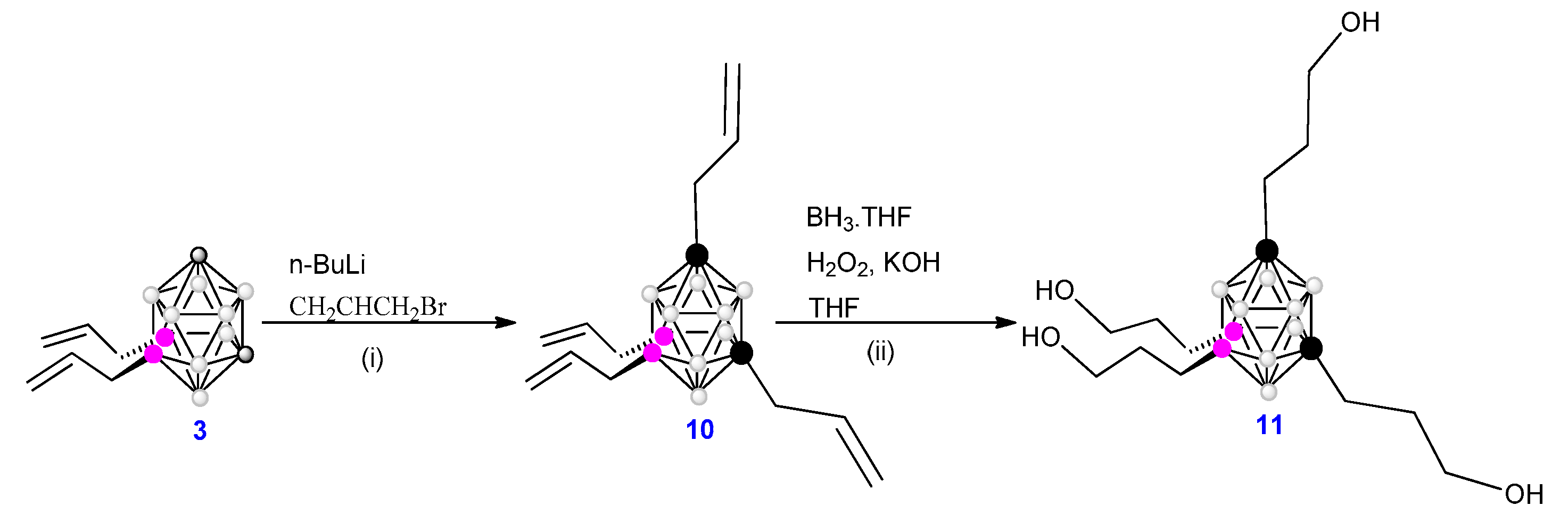
The above described di-branched 9,10-(CH2=CHCH2)2-1,7-closo-C2B10H10 (3) derivative, which still possesses its two Cc-H vertexes are ready for derivatization, offers the possibility to obtain globular icosahedral m-carborane derivatives with four branches as a new dendritic structure (Figure 2) by the incorporation of functional groups at the two carbon vertexes. Consequently, starting with 3 the four branched 1,7-(CH2=CHCH2)2-9,10-(CH2=CHCH2)2-1,7-closo-C2B10H8, 10, is obtained in two steps: i) removing the acidic hydrogen atoms with two equivalents of BuLi and ii) by electrophilic reaction with two equivalents of allylbromide (Scheme 3). From 10, the tetraalcohol 1,7-(OHCH2CH2CH2)2-9,10-(OHCH2CH2CH2)2-1,7-closo-C2B10H8, 11, can be achieved by hydroboration. In the same way, 11 is ready as a core for constructing a tetra-branched m-derivatives using the judicious choice of synthetic procedure (Scheme 3).
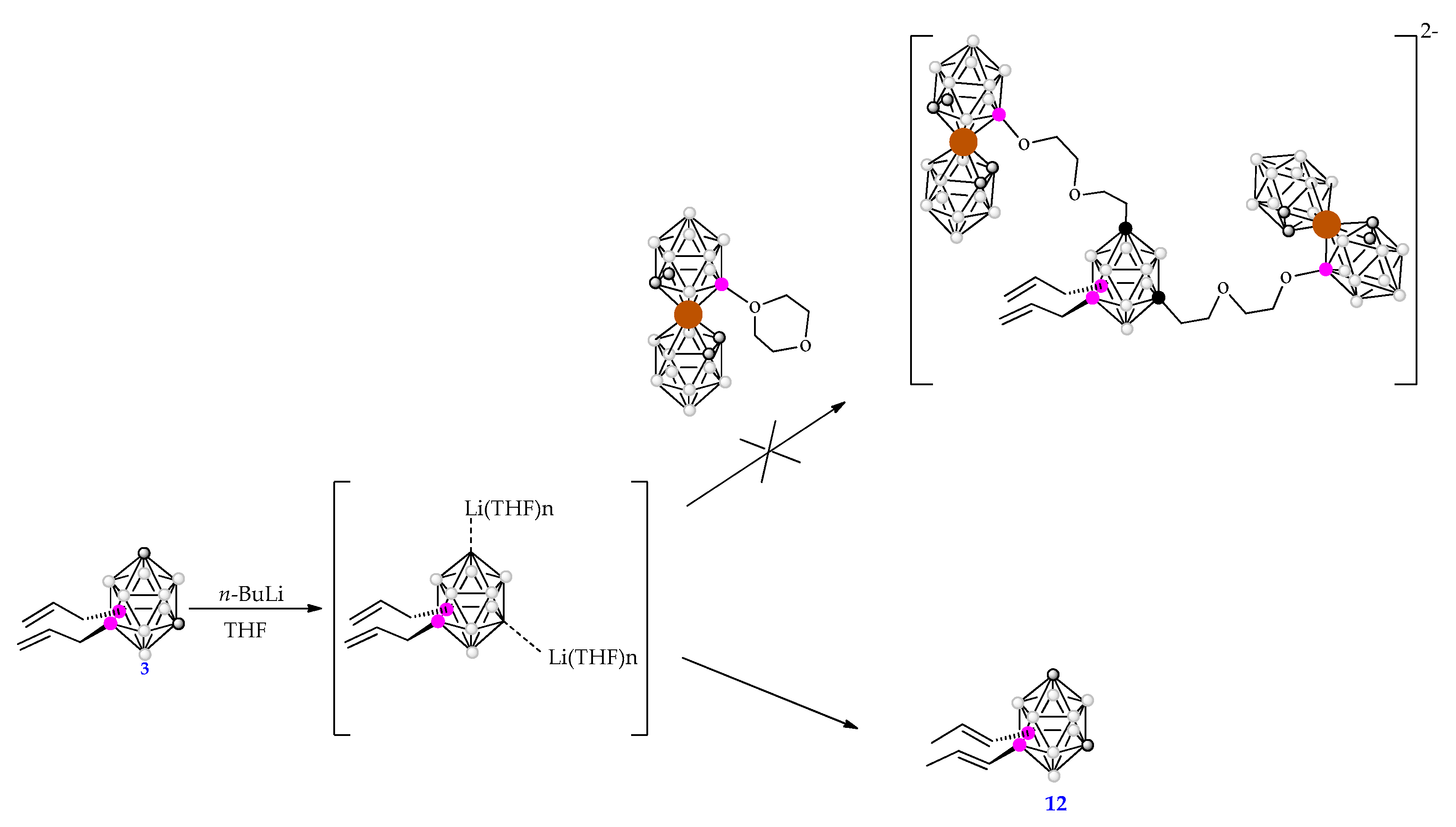
The synthesis of the tetra-substituted dianionic compound [9,10-({3,3’-Co(8’-(OCH2CH2)2-1’,2’-C2B9H10)(1’’,2’’-C2B9H11)}2-1,7-C2B10H8)2− was attempted in THF starting with compound 3 in a two steps reaction: i) the deprotonation of Cc-H vertexes of 3 using two equivalents of n-BuLi to form the intermediate Li2[9,10-(CH2=CHCH2)2-1,7-closo-C2B10H8]] salt and, ii) the nucleophilic attack of this salt to the dioxanate ring of the zwitterion [3,3’-Co-(8-(CH2CH2O)2-1,2-C2B9H10)(1’,2’-C2B9H11)] in the same way as Li2[1,7-closo-C2B10H10]] had performed (see Scheme 4) [44]. Nevertheless, unexpectedly, the synthesis of the dianionic compound was not achieved while the isomerization of allyl branches to propenyl ones took place giving the isomer 9,10-(CH3CH=CH)2-1,7-closo-C2B10H10, 12, in 80% yield (Scheme 4).
The reason for this unexpected reaction can be the comparable acidity of the allyl groups and the Cc-H of the m-carborane unit, which may allow a deprotonation/protonation isomerization of the allyl group as it is well known for allylbenzenes [45]. The pKa value of the unsubstituted carborane clusters, which are insoluble in water, have been determined by two methods [6,10]. The pKa by using Streitwieser’s scale provides the 27.9 value for the isomers m- carborane, while the one obtained by polarography is 24 [46]. Both experimental techniques support that unsubstituted m-carborane is a very weak Brønsted acid [46]. The allyl isomerization of 3 to propenyl in 12, which takes place in THF, is supported by the formation of solvent separated ion pairs that prevent the carboranyl anion to act as a nucleophile. To verify this hypothesis, Density-functional theory (DFT) calculations were performed (details in the S.I.). The proton affinity (PA) of the cluster carbon atom is 332.8 kcal/mol (at B3LYP-D3/6-311+G**, PCM=tetrahydrofuran level of theory), while the proton affinity of allylic carbon atom has a somewhat higher value (342.3 kcal/mol). This moderate difference (∆PA = 9.5 kcal/mol) probably allows for the above-mentioned mechanism. The question arises whether the same process does not occur in the case of the analog o-carborane based compounds [44]. It is known that cluster carbon in m-carborane is more than 1000 times less acidic than its orto isomer [47,48] therefore the difference between the two positions (allylic vs carboranyl) is larger as it was verified by our calculations (∆PA = 18.6 kcal/mol) as well. It should be highlighted that Li+ mediated isomerizations on allyl substituents bonded at the Cc vertexes of the o-carborane cluster was previously demonstrated as well, as Et2O does not tend to induce isomerization, whereas THF or DME produces the propenyl isomer [49]. A similar mechanism should be considered as well.
1H-NMR spectrum of 12 supported the allyl branches isomerization to propenyl ones but, this process was unambiguously proven by X-ray diffraction of 12 from good crystals, which were grown from its acetone solution.
2.3. Characterization of di-Branched m-carborane Derivatives at the 9,10 Vertexes
The electrophilic substitution of the o-carborane led to the formation of the tetrasubstituted 8,9,10,12-I4-1,2-closo-C2B10H8 compound [5,42,43] in which the B-I vertexes reside at the compacted adjacent positions antipodal to the two cluster carbon Cc atoms. Conversely, the iodination electrophilic substitution takes place only at the B(9) and B(10) vertexes of the m-isomer.
We reported that the 2a-NPA value for a bond, defined as the sum of the NPA charges of the two bonded atoms (e.g., B-H or C-H), matches the order of attack on the different cluster’ bonds [50]. Calculated NPA charges of the two bonded atoms (2a-NPA, calculated at B3LYP-D3/6-311+G** level of theory) of ortho- and meta-closo-carborane (present in Table 1) explain the higher accessibility of the B-H vertexes of o-carborane cluster to undergo electrophilic reaction, which drive to the formation of B-I bonds. While in the case of o-carborane there are four negative 2a-NPA values, in the case of m-carborane, there are only two. However, these positions exhibit higher reactivity towards electrophilic agents. Since in the case of m-carborane most of the 2a-NPA values of B-H vertexes are positive, the functionalization of this compound is more challenging. Table 1 shows that the electron density at the B-H vertexes of m-carborane follow a different trend B(9), B(10) >>> B(5), B(12) > B(4), B(6), B(8), B(11) B(2), B(3) than o-carborane, which is B(9,12) > B(8,10) > B(4,5,7,11) > B(3,6) [3,51]. Contrary to the o-carborane that contains two positive natural charges; the m-carborane presents four positive natural charges on BH vertex, which explain the difficulty of the substitution of the B-H vertexes. Figure 3 shows that LUMO in o-carborane is located between the C atoms, whereas it is not the case for m-carborane where it is more disperse. Therefore, the carbon cluster position in the carborane has an important role related to the substitution of the B-H vertexes. Using the electrophilic iodination, it is possible to derivatize only B(9) and B(10) because these boron atoms do not have any connection with the Cc in the meta isomer. On the contrary, for the o- isomer the same procedure allows the attack to all B-H vertexes except B(3) and B(6) that are adjacent to both carbon clusters [52].
The starting (2 and 3) and new compounds (4–9) were fully characterized by 1H, 1H{11B}, 11B, 11B{1H}, 13C{1H}, and 2D COSY 11B {1H}-11B {1H} NMR spectroscopic techniques to be taken as inputs for the discussion of the influence of the substituents at the 9,10 vertexes on the Boron disubstituted closo m-carborane derivatives.
The 11B{1H} NMR spectrum of the parent cluster 1 displays four signals with intensities 2:2:4:2 from low to high field −5.6, −9.5, −12,0 and −15.4 ppm, which corresponds to a weighted average 11B{1H} NMR chemical shift, <δ(11B)> ≈ −10.9 ppm [53]. Conversely, the 11B {1H} NMR spectrum of 2 displays four signals with intensities 2:4:2:2 from low to high field at –3.0, –10.4, –17.0, and –19.5 ppm, which corresponds to a weighted average <δ(11B)> ≈ −12 ppm. The presence of the two iodo groups bonded to the B(9,10) in 2 produces a <δ(11B)> upfield of −1.1 ppm in the 11B NMR. The upfield resonance of the 11B {1H} NMR spectrum of 2 at −19.5 ppm does not split into a doublet in the 11B-NMR spectrum supporting that it corresponds to the B-I at the 9 and 10 vertexes.
11B {1H}-11B {1H} 2D COSY NMR is of enormous use and potential in polyhedral boron chemistry because it provides a way of rapidly assigning 1lB resonances [54,55]. To assign the resonances of compounds 1 and 2 to the different cluster’s vertexes by NMR spectroscopy, the two-dimensional 11B {1H}-11B {1H} COSY NMR spectra of compounds 1 and 2 were run (See Supplementary Information). Once the B(9,10) has been unambiguously assigned in compounds 1 and 2, it is possible to confirm that the substitution of hydrogen by iodo causes significant shielding (−10 ppm) on the boron atoms that support the iodo units. 11B {1H}-11B {1H} 2D COSY NMR spectra of compounds 1 and 2 allow assigning the vertexes’ resonances for 1 and 2 (see Supplementary Information). On the other hand, the assignment of the hydrogen atoms to the respective boron cluster vertexes was done by running the selective irradiation 1H{11B} NMR spectra (Table 2) which confirms the presence of four signals in 1 and only three in 2. Notably, all proton resonances were shifted upfield in the 1H{11B} NMR spectrum which demonstrates the influence of the Iodo groups on all clusters’ vertexes.
The 11B{1H} NMR spectrum of 3 displays four signals with intensities 2:2:4:2 from low to high field at 0.6, −5.4, −12.5, and −19.1 ppm, which corresponds to a weighted average <δ(11B)> of ca. −9.8 ppm. The peak at +0.6 ppm does not split into a doublet in the 11B-NMR spectrum, which supports the substitution of an iodo by carbon from the allyl group, which causes a downfield shift on the boron vertexes. On the other hand, the 1H and 1H{11B} NMR spectra are useful to identify the presence of the organic fragments linked to the carborane cluster. Figure 4a shows the presence of three new signals (area ratio 1:2:2, from low to the high field), which are related to the typical resonances of terminal allyl groups. The protons bonded to the boron vertex (Hd) appear as a doublet (1J(H,H) = 7.7 Hz) at 1.78 ppm. There is an overlap of Ha and Hb resonances, which should appear, each one, as a double doublet, but looks like a triplet at 4.88 ppm. Hc is the most complicated proton of the allyl group because of the presence of four different protons at its neighboring carbon atoms. This appears in the range 5.97–5.82 ppm as a multiplet. The coupling constants of Hc with neighbors is shown in Figure 4b.
Compounds 4, 5, 6, and 7 were also characterized by 1H, 1H{11B}, 11B, 11B{1H} and 13C{1H} NMR spectroscopy.
Table 3 lists the 11B{1H} NMR chemical shifts for the B(9,10) disubstituted m-carborane derivatives while Table 4 summarizes the 1H, 13C{1H } NMR spectra and the stretching frequency of Cc-H in the IR spectra for the B(9,10) disubstituted m-carborane derivatives. The presence of organic branches connected to B(9) and B(10) causes a resonance downfield shift about +11 ppm on these boron atoms. Therefore, the average chemical shift value <δ(11B)> = –10.9 ppm of parent 1 is around –9.6 ppm for 9,10-R2-1,7-closo-C2B10H12 derivatives (R=CH₂=CH–CH₂, HO(CH2)3, Cl(CH2)3, PhCOO(CH2)3, CH3-C6H4-SO3(CH2)3). There is no difference in these two features between the two isomers, ortho and meta.
Table 3 shows a downfield shift (Δδ = +10.1 ppm) of the B(9,10) resonances of 9,10-(allyl)2-1,7-closo-C2B10H10 (3) vs the corresponding B(9,10)-H ones in the parent m-carborane. A similar downfield (Δδ = +10.6 ppm) is reported for the B(9,12) vertexes of 9,12-(allyl)2-1,2-closo-C2B10H10 with respect to the B(9,12)-H vertexes of the parent o-carborane [14]. The 11B{1H} NMR spectrum provides information on the electron density surrounding B atoms in the cluster vertexes, so it can be concluded that the effect of a B-allyl vertex concerning to the former B-H in the 11B{1H} NMR of both isomers is almost the same, Δδ +10.6 ppm and +10.1 ppm, for o- and m-, respectively. However, there is a major difference in the chemical shifts of the B-allyl nuclei of the two isomers: δ = +7.75 pm for 9,12-(allyl)2-1,2-closo-C2B10H10 and δ = +0.6 pm for 9,10-(allyl)2-1,7-closo-C2B10H10. We should remember that B-allyl vertexes are located antipodal to the Cc vertexes in the o- isomer but antipodal to B vertexes in the m-isomer. This fact indicates a quite relevant different electronic surrounding in the B-allyl sites in both isomers, which depends on the atoms’ nature at the antipodal vertexes.
Table 4 summarizes the 1H and 13C{1H } NMR spectra and stretching frequencies of Cc-H bonds in the IR spectra for the reported 9,10-R2-1,7-closo-C2B10H10 derivatives; the presence of the allyl branches at the B(9,10) vertexes produces an upfield of the carbon and hydrogen atoms resonances of the Cc-H concerning to the parent m-carborane in their 1H and 13C{1H } NMR spectra.
In Table 5, the comparison of the influence of the substituents at the B(9,12) in the o-carborane and the B(9,10) in the m-carborane is listed. To notice is that the influence on the chemical shift of the B-halogen (halogen = Cl, Br, I) vertexes in both isomers is the same: iodo is larger than bromo and bromo is larger than chloro. This is due to the i) electronegativity of halogen atoms, which follows the trend Cl > Br > I and ii) π back donation of halogen is I > Br > Cl.
2.4. Characterization of Tetrabranched m-carborane Derivatives at the 1,7,9,10 Vertexes
From the analysis of the 1H NMR spectra of 10, it is seen that the original signal corresponding to the protons linked to the carbon cluster, which appear at 2.83 ppm, vanishes while new signals at 5.58, 5.00 and 2.56 ppm corresponding to the allyl branches on these Cc vertexes are distinguished.
An important influence of the presence of the organic branches linked to the two Cc atoms is observed in the 11B downfield shift of the B(2) and B(3) vertexes that move from –19.1 ppm in 3 to -15.7 ppm in 10. To notice is the upfield shift of <δ(11B)> when moving from 2 to 10: <δ(11B)> = –12.0 ppm in compound 2 (with two B-I and two Cc-H vertexes), <δ(11B)> = -10.9 ppm on the parent m-carborane, <δ(11B)> = –9.8 ppm in 3 (with two B-allyl and two Cc-H vertexes), <δ(11B)> = –8.2 ppm in 10 (with two B-allyl and two Cc-allyl vertexes) (see Table 3). Consequently, the incorporation of organic branches at the cluster vertexes produces a downfield of <δ(11B)> in the 11B NMR while the iodo groups have the opposite effect, supporting that cluster-only total charge is dissimilarly affected by electron-withdrawing substituents than electron-donating ones.
2.5. Structural Description
2.5.1. Crystallographic Studies
A search in the Cambridge Structural Database [57] showed just 3 hits (CUWMUD, TOKCUR and YOZSOV) for 9,10-R2-1,7-closo-C2B10H10 for R = -CCH, -CH2C6H4, and -C6H5, respectively [34,36,37]. In this paper, we contribute with two additional X-ray structures that provide a broader view of the m-carborane derivatives.
To get information in such a family of compounds, good crystals of 9,10-(HOCH2CH2CH2)2-1,7-closo-C2B10H10 (4) and 9,10-(CH3CH=CH)2-1,7-closo-C2B10H10 (12) suitable for X-ray- diffraction were grown from an acetone solution at low temperature. Compound 4 was solved in the triclinic system, with a P 1 space group with four molecules in the asymmetric unit (Z = 4) and all atoms laid on the 1(a) Wyckoff positions. Compound (12) also solved in the triclinic system, but in a different space group (P-1) with two molecules in the asymmetric unit (Z = 2) and all atoms laid in 1(i) Wyckoff position. Figure 5 shows the crystal structures of 4 and 12 with the corresponding atom labels. Table 6 displays all crystallographic data and selected bond distances and angles are in the Supplementary Information.
Compound 4 is the first example of a B(9,10) disubstituted closo 1,7-carborane derivative with a terminal O-H group. Furthermore, compound 12 is the first example of closo m-carborane with branches containing double bonds. For this, the behaviour of the two branches in the crystal network has been studied in detail. Exploring the crystal self-assembly, the presence of H…H short contacts in the range from 1.207 Å to 2.240 Å for compound 4 and equal to 2.252 Å for compound 12 are noticed, which are presented in Figure 6 and Figure 7, respectively. In carborane chemistry, the dihydrogen H…H short contacts are related to the presence of two types of H atoms: the acidic Cc-H and the hydride B-H [58]. The supramolecular structure of 4 has an extensive network of hydrogen bonding due to the presence of the terminal OH groups (Figure 6a,b) with intermolecular distances shorter that sum of Van Der Waals radii (∑vdW) minus 0.8 Å [59] and O-H···O angle values of 163.1° and 177.9°. The crystal packing of 4 is also stabilized by O…O as shown in Figure 6c. Accordingly, three different types of H…H short contacts were observed for 4: C7-H7…H16-O16, O20-H20…H16-O16, and B3-H3…H14-C14.
As expected, the presence of double bonds in 12 has a noticeable role in the stabilization of the supramolecular network (Figure 7). The π electronic effect of the double bond leads to the formation of the π…H-Cc contacts (brown dashed lines), which are substantially shorter than 2.90 Å corresponding to the sum of the van der Waals radii (∑vdW) [60]. The layers of 12 are connected into the final 3D structure through the B3-H3…H15A-C15 bonds due to the acceptor character of the hydrogen-bonded to the boron and the donor character of the hydrogen atoms of the -CH3 group.
2.5.2. Hirshfeld Surface Analysis
The Hirshfeld surface analysis, which is a very valuable method for the analysis of intermolecular contacts that offers a whole-of-the-molecule approach [61], presents three different colours to study the intermolecular interactions in crystal structures. The red colour means the presence of an intermolecular distance shorter that ∑vdW, white colour indicates the presence of intermolecular distances close to ∑vdW and blue colour designates the contacts longer than ∑vdW. Moreover, the shape index on the Hirshfeld surface identify hollows (with shape index < 0) and bumps (with shape index > 0), which are related to the character of each atom; the presence of an acceptor atom is marked by a concavity and the presence of a donor one is marked by a convexity. Therefore, the previous results were corroborated by studying the Hirshfeld surface of both structures using the crystal explorer program [62]. In this respect, Figure 8 presents the dnorm of 4 and 12 to visualize the intermolecular interactions and their contribution towards the supramolecular network. The two-dimensional fingerprint plots, which provide information about the percentage of intermolecular contacts present in the Hirshfeld surface is present in the S.I.
The darkest red area in the dnorm surface of 4 is observed at the end of the molecule, arising from the O…H short contact as presented in Figure 8 and confirmed in the fingerprint plots (See Supplementary Information). Furthermore, the dnorm surface has shown the presence of bright red areas related to the presence of the O…O and H…H short contacts. Because of this packing arrangement, the O atoms at the molecular extremity present an important behaviour on the stability of this molecule by showing close contact values with H atoms of adjacent molecules shorter than ∑vdW.
The dnorm presentation of compound 12 (Figure 8 and S.I.) shows the presence of dark red points related to the classic H…H bonds. The existence of π…H short contacts is observed as a bright red point. The presence of the π acceptor interactions is indicated by the appearance of red concave triangles surrounded by blue ones in the shape index surface (Figure 9a) [63] while the Cc-H donor is confirmed by the blue convex area (Figure 9b) [64].
Despite the presence of many strong intermolecular interactions with contacts shorter than the sum of the van der Waals radii minus 0.80 Å, the H…H interactions are the most dominant with 87.8% and 94.2% in compounds 4 and 10 respectively, as shown in the fingerprint plots (See S.I.). The presence of 10.2% contacts O…O in 4 and 5.8% of π…H short contacts in 12 are also relevant.
3. Materials and Methods
3.1. Experimental Section
Materials and instrumentation: All m-carborane clusters prepared are air-stable. All manipulations were carried out under nitrogen atmosphere. THF and DMF were distilled from sodium benzophenone before use. Reagents were obtained commercially and used as purchased without purification. 1,7-closo-C2B10H12 was obtained from Katchem.
ATR-IR spectra (ν, cm−1) were obtained using the a JASCO FT/IR-4700 spectrometer on a high-resolution (Madrid, Spain). The 1H and 1H{11B} NMR (300.13 MHz), 13C{1H} NMR (75.47 MHz), and 11B and 11B{1H} NMR (96.29 MHz) spectra were recorded on a Bruker ARX300 instrument equipped with the appropriate decoupling accessories (Bruker Biospin, Rheinstetten, Germany)). All NMR spectra were performed in the indicated deuterated solvent at 22 °C. The 11B and 11B{1H} NMR chemical shifts were referenced to external BF3·OEt2, while the 1H, 1H{11B}, and 13C{1H} NMR shifts were referenced to SiMe4. Chemical shifts are reported in units of parts per million downfield from reference, and all coupling constants in Hz.
3.1.1. Synthesis and Characterization of 3
The procedure for the synthesis of 3 was similar to that previously reported [40]. To a stirred solution of 9,10-I2-1,7-closo-C2B10H10 2, (300 mg, 1.34 mmol) in THF (15 mL) cooled to 0 °C in an ice-water bath was added, drop wise, a solution of allylmagnesium chloride in THF (6.06 mL, 1 M, 6.06 mmol). After stirring at room temperature for 30 min, [PdCl2(PPh3)2] (21.28 mg, 4% equiv.) and CuI (5.77 mg, 4% equiv.) were added in a single portion, following which the reaction was heated to reflux overnight. The solvent was removed, and 20 mL of diethyl ether were added to the residue. The excess of Grignard reagent was destroyed by slow addition of dilute HCl. The organic layer was separated from the mixture, and the aqueous layer was extracted with diethyl ether (3 × 10 mL). The combined organic phase was dried over MgSO4, filtered and the solvent removed under reduced pressure. The crude product was dissolved in hexane/chloroform mixture (1:1 by volume, ca. 5 mL) and passed rapidly through a bed of silica. The solvent was removed in a vacuum to give 9,10-(CH2=CHCH2)2-1,7-closo-C2B10H10 3 as a yellowish oil (161.2 mg, 95%). Elemental analysis: calc: %C 42.8, %H 8.7; exp: %C 45.9, %H 8.7. ATR: ν = 3062 (vs, (Cc-H and =CH2)), 2972, 2902 (vs, (=CH- and -CH2-)), 2592 (vs, (B-H)), 1634 (vs, (C=C)), 995, 978, 692 (s, (=CH)). 13C{1H} NMR (75.47 MHz, CDCl3) δ: 139.96 (s, CH2=CHCH2), 112.09 (s, CH2=CHCH2), 52.35 (s, Cc-H), 21.67 (m, CH2=CHCH2). 1H-NMR (300.13 MHz, CDCl3) δ: 5.89 (m, 2H, CH2=CHCH2), 4.88 (m, 4H, CH2=CHCH2), 4.94 (s, 2H, Cc-H), 1.78 (m, 4H, CH2=CHCH2). 1H{11B} NMR (300.13 MHz, CDCl3) δ: 5.89 (m, 2H, CH2=CHCH2), 4.88 (m, 4H, CH2=CHCH2), 4.94 (s, 2H, Cc-H), 2.50 (s, 2H, B(5,12)-H), −2.24 (s, 2H, B(2,3)-H), 2.14 (s, 4H, B(4,6,8,11)-H), 1.77 (d,3J(H,H) = 7.8, 4H, CH2=CHCH2). 11B NMR (96.29 MHz, CDCl3) δ: −0.1 (s, 2B, B(9,10)), −6.2 (d, 1J(B,H) = 160, 2B, B(5,12)), −13.5 (d, 1J(B,H) = 163, 4B, B(4,6,8,11)), 20.3 (d, 1J(B,H) = 180, B(2,3)).
3.1.2. Synthesis and Characterization of 4
To a stirred solution of 9,10-(CH2=CHCH2)2-1,7-closo-C2B10H10, 3, (150 mg, 0.67 mmol) in THF (2 mL) at 0 °C, was added, drop wise, a solution of BH3·THF in THF (1.37 mL, 1 M, 1.37 mmol). The resulting suspension was stirred at 0 °C for 30 min and at room temperature for further 30 min. Then, the reaction mixture was cooled again to 0 °C in an ice-water bath and water (2 mL) was slowly added. When gas evolution had stopped, an aqueous KOH solution (1.68 mL, 3M, 1.94mmol) and subsequently, H2O2 in water (0.20 mL, 35%, 2.30 mmol), were added. Stirring was maintained at room temperature for 1.5 h, after which two liquid phases were observed. The upper organic layer was separated from the mixture and the aqueous layer and washed with THF (3 × 2 mL). The combined organic phase was dried over MgSO4, filtered and the solvent removed in vacuo to give 9,10-(HOCH2CH2CH2)2-1,7-closo-C2B10H10 4. Yield: 151 mg (87%). Elemental analysis: calc: %C 36.9, %H 9.22; exp: %C 36.79, %H 8.2. ATR: ν= 3305 (vs, νs(O-H)), 3038 (vs, Cc-H), 2930, 2886,2850, 2823 (vs, Calkyl-H), 2585 (s, B-H), 1055, 1005, 978 (s, C-O). 13C{1H} NMR (300.13 MHz, (CD3)2CO) δ: 64.20 (s, HOCH2CH2CH2), 52.95 (s, Cc), 32.27 (s, HOCH2CH2CH2), 10.23 (s, HOCH2CH2CH2). 1H-NMR (300.13 MHz, (CD3)2CO) δ: 3.49 (s, 2H, Cc-H), 3.55 (m, 4H, HOCH2CH2CH2), 3.40 (t, 2H, HOCH2CH2CH2), 1.60 (m, 4H, HOCH2CH2CH2), 0.81 (t, 3J(H,H) = 16.7, 4H, HOCH2CH2CH2). 1H{11B} NMR (300.13 MHz, (CD3)2CO) δ: 3.49 (s, 2H, Cc-H), 3.55 (m, 4H, HOCH2CH2CH2), 3.40 (t, 2H, HOCH2CH2CH2), 2.48,2.20,2.08 (s, 8H, B-H), 1.60 (m, 4H, HOCH2CH2CH2), 0.81 (t, 3J(H,H) = 16.7, 4H, HOCH2CH2CH2). 11B NMR (96.29 MHz, (CD3)2CO) δ: 1.9 (s, 2B, B(9,10)), −5.3 (d, 1J(B,H) = 157, 2B, B(5,12)), −12.7 (d, 1J(B,H) = 160, 4B, B(4,6,8,11)), −19.3 (d, 1J(B,H) = 178, 2B, B(2,3)). Colourless good crystals suitable for X-ray diffraction were grown in acetone.
3.1.3. Synthesis and Characterization of 5
To a stirred solution of 9,10-(HOCH2CH2CH2)2-1,7-closo-C2B10H10, 4, (300 mg, 1.14 mmol) and [NBu4]Cl (132.59 mg, 0.478mmol) in dry THF (10mL) at 0 °C, was added SOCl2 dropwise (0.52 mL, 7.076 mmol). The resulting solution was stirred at 0 °C for 1 h and at room temperature overnight. The solvent was removed under reduced pressure, and 8 mL of diethyl ether were added. A solution of Na2CO3 (8 mL, 2 M) was slowly added with stirring. The mixture was thoroughly shaken, and the two layers separated. The aqueous layer was extracted with diethyl ether (3 × 5 mL). Then, the combined organic phase was separated and a solution of HCl (8 mL, 0.1 M) was added, the mixture was thoroughly shaken again. The upper organic layer was separated from the mixture, and the aqueous layer was washed with diethyl ether (3 × 5 mL). Finally, the combined organic phase was dried over MgSO4, filtered and the solvent removed in vacuo to give 9,10-(ClCH2CH2CH2)2-1,7-closo-C2B10H10, 5. Yield: 310 mg (92%). Elemental analysis: calc: %C 32.32, %H 7.40; exp: %C 33.04, %H 7.60. ATR: ν= 3053 (s, Cc-H), 2989, 2972, 2902 (s, Calkyl-H), 2626, 2587 (s, B-H), 1310, 1279 (s, CH2-Cl), 728, 647 (s, C-Cl). 13C{1H} NMR (300.13 MHz, (CD3)2CO) δ: 53.28 (s, Cc), 47.14 (s, ClCH2CH2CH2), 32.98 (s, ClCH2CH2CH2), 11.47 (s, ClCH2CH2CH2). 1H-NMR (300.13 MHz, (CD3)2CO) δ: 3.53 (s, 2H, Cc-H), 3.61 (t, 4H, ClCH2CH2CH2), 1.87 (m, 4H, ClCH2CH2CH2), 0.94 (t, 4H, ClCH2CH2CH2). 1H{11B} NMR (300.13 MHz, (CD3)2CO) δ: 3.53 (s, 2H, Cc-H), 3.61 (t, 4H, ClCH2CH2CH2), 2.51, 2.23, 2.13 (s, 8H, B-H), 1.87 (m,4H, ClCH2CH2CH2), 0.78 (t, 4H, ClCH2CH2CH2). 11B NMR (96.29 MHz, (CD3)2CO) δ: 1.3 (s, 2B, B(9,10)), −5.3 (d, 1J(B,H) = 155, 2B, B(5,12)), −12.6 (d, 1J(B,H) = 159, 4B, B(4,6,8,11)), −19.0 (d, 1J(B,H) = 179, 2B, B(2,3)).
3.1.4. Synthesis and Characterization of 6
To a stirred solution of 9,10-(HOCH2CH2CH2)2-1,7-closo-C2B10H10 (94 mg, 0.361 mmol), 4, 4-N,N-dimethylaminopyridine (97.17 mg, 0.795 mmol), N,N’-dicyclohexylcarbodiimide (164.11 mg, 0.795 mmol) and benzoic acid (97.17 mg, 0.795 mmol) in dry dichloromethane (10 mL). The resulting solution was stirred at room temperature for 1 h. The white precipitate (dicyclohexylurea) is filtered and then an extraction using 10 mL of HCl (1 M) was done. The aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic phase was dried over MgSO4, filtered, and the solvent removed under reduced pressure to give 152 mg (90 %) of 9,10-(C6H5COOCH2CH2CH2)2-1,7-closo-C2B10H10, 6. Elemental analysis: calc: %C 56.41, %H 6.83; exp: %C 55.72, %H 6.81. ATR: ν= 3064 (vs, Cc-H), 2988, 2971, 2904 (vs, Calkyl-H), 2594, 2565 (s, B-H), 1711 (s, vs(C=O)), 1599 (s, C-O). 13C{1H} NMR (300.13 MHz, (CD3)2CO) δ: 165.87 (s, C6H5COOCH2CH2CH2), 132.82 (s, Caryl), 130.70 (s, Caryl), 129.22 (s, Caryl), 128.45 (s, Caryl), 66.71(s, C6H5COOCH2CH2CH2), 53.27 (s, Cc), 25.37 (s, C6H5COOCH2CH2CH2) 10.06 (s, C6H5COOCH2CH2CH2). 1H-NMR (300.13 MHz, (CD3)2CO) δ: 8.03 (d, 1J(H,H) = 8.1, 4H, Haryl), 7.65 (t, 2H, Haryl), 7.53 (m, 4H, aryl), 4.30 (t, 4H, 3J(H,H) = 6.8, C6H5COOCH2CH2CH2), 3.55 (s, 2H, Cc-H), 1.87 (m, 4H, C6H5COOCH2CH2CH2), 0.98 (m, 4H, C6H5COOCH2CH2CH2). 1H{11B} NMR (300.13 MHz, (CD3)2CO) δ: 8.03 (d, 1J(H,H)= 8.1, 4H, Haryl), 7.65 (t, 2H, Haryl), 7.51 (m, 4H, Haryl), 4.30 (t, 3J(H,H) = 6.8, 4H, C6H5COOCH2CH2CH2), 3.55 (s, 2H, Cc-H), 2.52, 2.27, 2.16 (s, 8H, B-H), 1.87 (m, 4H, C6H5COOCH2CH2CH2), 0.98 (m, 4H, C6H5COOCH2CH2CH2). 11B NMR (96.29 MHz, (CD3)2CO) δ: 1.6 (s, 2B, B(9,10)), −5.2 (d, 1J(B,H) = 149, 2B, B(5,12)), −12.5 (d, 1J(B,H) = 158, 4B, B(4,6,8,11)), −19.1 (d, 1J(B,H) = 175, 2B, B(2,3)).
3.1.5. Synthesis and Characterization of 7
To a mixture of 9,10-(HOCH2CH2CH2)2-1,7-closo-C2B10H10, 4, (163 mg, 0.627 mmol) and [HNMe3]Cl (12.76 mg, 0.13 mmol) in 5 mL of dry toluene 1.5 mL of Triethylamine was added. In second flask, the p. toluensulfonyl chloride (363.34 mg, 1.905 mmol) was dissolved in THF, then converted to the first flask at 0ºC. The solvent was evaporated and an extraction using the diethyl ether and water. The organic part was dried over MgSO4, filtered, and the solvent removed under reduced pressure to give 9,10-(CH3-C6H4-SO3-CH2CH2CH2)2-1,7-closo-C2B10H10, 7, (302 mg, 85%). ATR: ν = 3064 (s, (Cc-H)), 2971, 2955, 2895 (s, (Calkyl-H)), 2596, 2565 (s, (B-H)), 1349 (s, (S=O)),1189, 1175 (s, (S-O)), 1097, 981, 954, 918 (s, (C-O)). 13C{1H} NMR (300.13 MHz, (CD3)2CO) δ: 144.77 (s, CH3-C6H4), 133.80 (C6H4-S), 129.94 (s, Caryl), 127.76 (s, Caryl), 72.67 (s, TsOCH2CH2CH2), 53.26 (s, Cc), 37.28 (s, TsOCH2CH2CH2), 20.62 (s, CH3-C6H5), 9.78 (br s,TsOCH2CH2CH2). 1H-NMR (300.13 MHz, (CD3)2CO) δ: 7.82 (d, 4H, 1J(H,H) = 7.2, Haryl), 7.49 (d, 4H,1J(H,H) = 8.0, Haryl), 4.05 (t, 3J(H,H) = 6.6, 4H, TsO-CH2), 3.45 (br s, 2H, Cc-H), 2.47 (s, 6H, CH3C6H4), 1.75 (m, 4H, TsOCH2CH2CH2), 0.77 (m, 4H, TsOCH2CH2CH2). 1H{11B} NMR (300.13 MHz, (CD3)2CO) δ: 7.82 (d, 4H, 1J(H,H) = 7.2, Haryl), 7.49 (d, 4H,1J(H,H)= 8.0, Haryl), 4.05 (t, 3J(H,H)= 6.6, 4H, TsO-CH2), 3.45 (br s, 2H, Cc-H), 2.47 (s, 6H, CH3C6H4), 3.04, 2.03, 2.00 (br s, 8H, BH),1.75 (m, 4H, TsOCH2CH2CH2), 0.77 (m, 4H, TsOCH2CH2CH2). 11B NMR (96.29 MHz, (CD3)2CO) δ: 3.4 (s, 2B, B(9,10)), −5.4 (d, 1J(B,H) = 153, 2B, B(5,12)), −12.7 (d, 1J(B,H) = 155, 4B, B(4,6,8,11)), −19.2 (d, 1J(B,H) = 177, 2B, B(2,3)).
3.1.6. Synthesis and Characterization of 8
To a stirred solution of previously dried 9,10-(ClCH2CH2CH2)2-1,7-closo-C2B10H10, 5, (95 mg, 0.319 mmol) in DMF (10 mL), NaN3 (314.41 mg, 4.83 mmol) was added. At room temperature, the mixture was stirred for 24 h. Then, the solvent was evaporated under vacuum and an extraction with a mixture C6H5CH3-H2O was done. After washing it several times with H2O, the collected organic layer was dried over MgSO4, filtered, and the solvent removed under reduced pressure to give 9,10-(N3CH2CH2CH2)2-1,7-closo-C2B10H10, 8, (80.75 mg, 81%). ATR: ν= 3076 (Cc-H), 2929, 2890 (Calkyl-H), 2591 (B-H), 2095 (C-N). 13C{1H} NMR (300.13 MHz, (CD3)2CO) δ: 53.42 (s, C-N3), 53.26 (Cc-H), 11.13 (m, CH2). 1H-NMR (300.13 MHz, (CD3)2CO) δ: 3.84 (s, 2H, Cc-H), 3.34 (t, 3J(H,H) = 6.90, 4H, N3CH2CH2CH2), 1.69 (m, N3CH2CH2CH2), 0.89 (m, 4H, N3CH2CH2CH2). 1H{11B} NMR (300.13 MHz, (CD3)2CO) δ: 3.84 (s, 2H, Cc-H), 3.34 (t, 3J(H,H) = 6.90, 4H, N3CH2CH2CH2), 2.52, 2.23, 2.13 (br s, BH), 1.69 (m, N3CH2CH2CH2), 0.89 (m, 4H, N3CH2CH2CH2). 11B NMR (96.29 MHz, (CD3)2CO) δ: 1.4 (s, 2B, B(9,10)), −5.3 (d, 1J(B,H)= 157, 2B, B(5,12)), −12.6 (d, 1J(B,H) = 160, 4B, B(4,6,8,11)), −19.1 (d, 1J(B,H) = 180, 2B, B(2,3)).
3.1.7. Synthesis and Characterization of 9
To a solution of 9,10-(N3CH2CH2CH2)2-1,7-closo-C2B10H10, 8, (27 mg, 0.086 mmol) in a mixture of dioxane (2 mL) and distilled H2O (2 mL), phenylacetylene (0.04 mL, 0.36 mmol), sodium ascorbate (17.037 mg, 0.086 mmol) and CuSO4.5H2O (21.47 mg, 0.086 mmol) were added in this order. After 20 min, a yellow solid started to be formed. The reaction was stopped after 3 h, when the walls of the small flask were full yellow solid and the solution was green. Then, the yellow solid was separated and very well dried under vacuum to give 9,10-(C6H5C2N3CH2CH2CH2)2-1,7-closo-C2B10H10, 9, (76 mg, 86%). Elemental analysis: calc: C% 56.23, H% 6.29; exp: %C 56.69, %H 6.88. ATR: ν= 3057 (Cc-H), 2924, 2892, 2852, 2826 (Calkyl-H, CH2N), 2588 (B-H). 13C{1H} NMR (300.13 MHz, (CD3)2SO) δ: 146.69 (C6H5-C-N=N-N), 131.35 (C6H5-C=C-N), 129.34 (C6H5), 128.22, 125.57 (C6H5), 121.68 (C6H5), 54.15 (Cc-H), 52.25 (N-CH2CH2CH2), 30.68 (N-CH2CH2CH2), 11.4 (NCH2CH2CH2). 1H-NMR (300.13 MHz, (CD3)2SO) δ: 8.56 (s, 2H, C=CH-N), 7.85 (d, 4H,1J = 7.7, C6H5), 7.44 (t, 4H, 3J = 7.5, C6H5), 7.32 (t, 2H,3J = 7.1, C6H5), 3.86 (s, 2H, Cc-H), 4.34 (m, 4H, NCH2CH2CH2), 1.86 (m, 4H, NCH2CH2CH2), 0.69 (m, 4H, NCH2CH2CH2). 1H{11B} NMR (300.13 MHz, (CD3)2SO) δ: 8.56 (s, 2H, C=CH-N), 7.85 (d, 4H,1J = 7.7, C6H5), 7.44 (t, 4H, 3J = 7.5, C6H5), 7.32 (t, 2H,3J = 7.1, C6H5), 3.86 (s, 2H, Cc-H), 4.34 (m, 4H, NCH2CH2CH2), 2.38, 2.10, 2.02 (br s, B-H), 1.86 (m, 4H, NCH2CH2CH2), 0.69 (m, 4H, NCH2CH2CH2). 11B and 11B{1H} NMR (96.29 MHz, (CD3)2SO) δ: 1.4 (s, 2B, B(9,10)-C), −5.5 (2B, B(5,12)), −13.2 (4B, B(4,6,8,11)), −19.9 (2B, B(2,3)).
3.1.8. Synthesis and Characterization of 10
To a stirred solution of 9,10-(CH2=CHCH2)2-1,7-closo-C2B10H10 (150 mg, 0.67 mmol) in dry THF (10 mL) at 0 °C were added dropwise n-BuLi in hexane (0.98 mL, 1.5 M, 1.47 mmol), the resulting solution was stirred at 0 °C for 1 h. Then the mixture was cooled at −78°C to add dropwise a solution of CH2=CHCH2-Br in dry THF (1.54 mL, 1 M, 1.54 mmol), and allowed to stir overnight at room temperature. Afterwards, the solvent was removed and 10 mL of diethyl ether and 10 mL of HCl (0.25M) were added to the residue. The organic layer was separated from the mixture, and the aqueous layer was extracted with diethyl ether (3 × 10mL). The combined organic phase was dried over MgSO4, filtered, and the solvent removed under reduced pressure to give 71% of 9,10-(CH2=CHCH2)2-1,7-(CH2=CHCH2)2-closo-C2B10H8 (145 mg). 13C{1H} NMR (300.13 MHz, (CD3)2CO) δ: 140.17 (s, CH2=CH-CH2-B(9,10)), 133.96 (s, CH2=CHCH2-C(1,2)), 117.98 (s, CH2=CH-CH2-B(9,10)), 111.96 (s, CH2=CH-CH2-C(1,2)), 71.8 (s, Cc), 40.97 (s, CH2=CH-CH2-C(1,2)), 21.48 (m, CH2=CH-CH2- B(9,10)). 1H-NMR (300.13 MHz, (CD3)2CO) δ: 5.80 (m, 2H, CH2=CH-CH2-B(9,10)), 5.60 (m, 2H, CH2=CH-CH2-C(1,2)), 4.90 (m, 4H, CH2=CH-CH2-C(1,2)), 4.86 (m, 4H, CH2=CH-CH2-B(9,10)), 2.57 (d, 3J(H-H)=7.3 Hz, 4H, CH2=CH-CH2-C(1,2)), 1.71 (br s, 4H, CH2=CH-CH2-B(9,10)). 1H{11B} NMR (300.13 MHz, (CD3)2CO) δ: 5.80 (m, 2H, CH2=CH-CH2-B(9,10)), 5.60(m, 2H, CH2=CH-CH2-C(1,7)), 4.90 (m, 4H, CH2=CHCH2-C(1,7)), 4.86 (m, 4H, CH2=CHCH2-B(9,10)), 2.57 (d, 3J(H,H) = 7.3 Hz, 4H, CH2=CHCH2-C(1,7)), 2.17 (brs, 2H, B(5,12)-H), 2.12 (brs, 4H, B(4,6,8,11)-H), 1.87 (brs, 2H, B(2,3)-H), 1.71 (brs, 4H, CH2=CH-CH2-B(9,10)). 11B-NMR (96.29 MHz, (CD3)2CO) δ: 0.8 (s, 2B, B(9,10)), −5.4 (d, 1J(B,H) = 154, 2B, B(5,12)), −10.2 (d, 1J(B,H) = 159, 4B, B(4,6,8,11)), −15.9 (d, 1J(B,H) =174, 2B, B(2,3)).
3.1.9. Synthesis and Characterization of 11
To a stirred solution of 9,10-(CH2=CHCH2)2-1,7-(CH2=CHCH2)2-closo-C2B10H8, 3, (130 mg, 0.24 mmol) in THF (3.5 mL) at 0 °C, was added, drop wise, a solution of BH3·THF in THF (1.765 mL, 1M, 1.765 mmol). The resulting suspension was stirred at 0 °C for 30 min and at room temperature for further 30 min. Then, the reaction mixture was cooled again to 0 °C in an ice-water bath and water (1mL) was slowly added. When gas evolution had stopped, an aqueous KOH solution (0.583 mL, 3 M, 1.75 mmol) and subsequently, H2O2 in water (0.3 mL, 35%), were added. Stirring was maintained at room temperature for 1.5 h, after which two liquid phases were observed. The upper organic layer was separated from the mixture and the aqueous layer. Then, it was washed with THF (3 × 2 mL). The combined organic phase was dried over MgSO4, filtered and the solvent removed in vacuo to give 9,10-(HOCH2CH2CH2)2-1,7-(HOCH2CH2CH2)2-closo-C2B10H8.Yield: 119mg (74%). Elemental analysis: calc: C% 44.7, H% 9.6; exp: %C 45.1, %H 9.8. 13C{1H} NMR (300.13 MHz, (CD3)2CO) δ: 66.04 (s, HOCH2CH2CH2-C(1,7)), 64.18 (s, HOCH2CH2CH2-C(1,7), HOCH2CH2CH2-B(9,10)), 60.52 (s, HOCH2CH2CH2-C(1,7)), 33.38 (s, HOCH2CH2CH2-C(1,7), 33.19 (s, HOCH2CH2CH2-B(9,10)), 23.83 (s, HOCH2CH2CH2B(9,10)). 1H{11B} NMR (300.13 MHz, (CD3)2CO) δ: 3.67–3.51 (m, 12H, HOCH2CH2CH2), 1.59 (m, 8H, HOCH2CH2CH2), 0.99–0.81 (m, 12H, HOCH2CH2CH2). 11B-NMR (96.29 MHz, (CD3)2CO) δ: 1.2 (s, 2B, B(9,10)), −5.6 (d,1J(B,H) = 147, 2B, B(5,12)), −10.6 (d, 1J(B,H) = 150, 4B, B(4,6,8,11)), −15.8 (d, 1J(B,H) = 175, 2B, B(2,3)).
3.1.10. Synthesis and Characterization of 12
To a stirred solution of 9,10-(CH2=CHCH2)2-1,7-closo-C2B10H10, 3, (20 mg, 0.09 mmol) in THF (3 mL) at 0 °C, was added, drop wise, a solution of BuLi (0.18 mmol, 1.6 M, 0.12 mL). The resulting suspension was stirred at 0 °C for 30 min and at room temperature for further 30 min. In another flask and under nitrogen, 8-{3,3’-Co(8-C4H8O2-1,2-C2B9H10)(1’,2’-C2B9H11) (0.18 mmol, 77 mg) was dissolved in 8 mL of THF. Then, the new solution was transferred to the suspension mixture and the reaction mixture stirred 2 h under reflux at inert atmosphere. The solvent was evaporated, and an extraction took place. The organic layer was evaporated to give a mixture of white and orange compounds. The 8-{3,3’-Co(8-C4H8O2-1,2-C2B9H10)(1’,2’-C2B9H11) was recuperated and the new isomer 9,10-(CH3CH=CH)2-1,7-closo-C2B10H10, 12, was obtained with 80% of yield (16 mg). ATR: ν = 3046 (Cc-H), 2998–2849 (m, υs(CH3, =CH)), 2623, 2592 (vs, υs(B-H)), 1634, 1442 (vs, υs(C=C)), 978 (s, υas(-CH=CH-)). 1H-NMR (300.13 MHz, CDCl3) δ: 5.90 (m, 2H, CH3CH=CH), 5.55 (m, 2H, CH3CH=CH), 2.82 (s, 2H, Cc-H), 1.78 (dd, 3J(Ha,Hb) = 6.3, 4J(Ha,Hc) = 1.6, 6H, CH3CH=CH). 1H{11B} NMR (300.13 MHz, CDCl3) δ: 5.90 (m, 2H, CH3CH=CH), 5.57 (m, 2H, CH3CH=CH), 2.82 (s, 2H, Cc-H), 2.50 (s, 2H, B(5,12)-H), 2.34 (s, 2H, B(2,3)-H), 2.19 (s, 4H, B(4,6,8,11)-H), 1.78 (d, 6H, CH3CH=CH). 11B-NMR (96.29 MHz, (CD3)2CO) δ: −0.5 (s, 2B, B(9,10)), −5.8 (d,1J(B,H) = 159, 2B, B(5,12)), −12.5 (d, 1J(B,H) = 163, 4B, B(4,6,8,11)), −19.8 (d, 1J(B,H) = 180, B(2,3)). Good crystals suitable for X-ray diffraction were grown in acetone.
3.2. X-ray Structure Determinations of 9,10-(HOCH2CH2CH2)2-1,7-closo-C2B10H10, 4 and 9,10-(CH3CH=CH)2-1,7-closo-C2B10H10, 12
Single-crystal data collections for 4 and 12 were performed with an Bruker D8 QUEST ECO three-circle diffractometer system equipped with a Ceramic x-ray tube (Mo Kα, λ = 0.71076 Å) and a doubly curved silicon crystal Bruker Triumph monochromator (Bruker, Karlsruhe, Germany). The structures were solved by direct methods and refined on F2 by the SHELXL97 program [65]. The non-hydrogen atoms were refined with anisotropic displacement parameters. The hydrogen atoms were treated as riding atoms using the SHELXL97 default parameters. The crystallographic, structure refinement, and bond parameters for 4 and 10 are reported in CIF-files deposited at CCDC with the reference numbers CCDC 2004945 and 2004946. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, U.K.; fax: +44-1223-336033; or e-mail: [email protected]).
3.3. Hirshfield Surface Analysis
The Hirshfeld surface analyses were run using the CIF format by the CrystalExplorer program [62]. Hirshfeld surface analysis help to recognize the strong and weak intermolecular interactions area and the nature of these interactions from the electron distribution. The dnorm (normalized contact distance) is given by the Equation (1):
where di is from the Hirshfeld surface to the nearest atom outside-external, de from the Hirshfeld surface to the nearest internal atom, and rvdw is the Van Der Walls radii of the atom
4. Conclusions
All allyl di and tetrabranched derivatives of the m-carborane framework have been synthesized. The starting 9,10-(allyl)2-1,7-closo-carborane compound was made by Kumada cross-coupling reaction on 9,10-I2-1,7-closo-carborane with allyl Grignard reagent in the presence of Pd(II) and Cu(I) as catalysts. These olefin groups have led to a variety of functional groups, alcohol, chloro, tosyl, and azide that have permitted to produce esters and 1,2,3-triazoles by the azide-alkyne cycloaddition, as examples of reactions that show the wide possibilities of this globular icosahedral m-carborane to act as a novel core for periphery-decorated macromolecules. Importantly, the four branches in the tetrabranched m-carborane derivatives are located in two perpendicular planes and are coplanar in the o-carborane isomer. This difference provides novel cores for 3D and 2D radially grown periphery-decorated macromolecules, respectively. Unexpectedly, the isomerization of B-allyl to B-propenyl vertexes in 9,10-(allyl)2-1,7-closo-C2B10H10 was observed in THF. DFT calculation studies conclude that the comparable acidity of the allyl groups and the Cc-H of the m-carborane unit allows a deprotonation/protonation isomerization of the allyl group as it is well known for allylbenzenes. X-ray crystal structures of 9,10-(OHCH2CH2CH2)2-1,7-closo-C2B10H10 and 9,10-(CH3CHCH)2-1,7-closo-C2B10H10 compounds show an extensive network of hydrogen bonding and π···H-Cc contacts, respectively, due to the presence of alcohol and olefin groups that have been analyzed by Hirshfeld surfaces and decomposed fingerprint plots.
Supplementary Materials
The following are available online, Spectroscopic characterization of compounds 1–12. Figures S1–S78 with IR and NMR spectra and crystal packing of 4 and 12. Table S1–S4 containing the bond lengths and bond angles of crystals 4 and 12; S5-S11 with XYZ coordinates and total energies of the investigated systems.
Author Contributions
Conceptualization, C.V.; methodology, C.V., and I.B.; computational Studies, Z.K.; writing—original draft preparation, I.B., and C.V.; writing—review and editing, I.B., C.V., F.T., and Z.K.; supervision, C.V.; project administration, C.V.; funding acquisition, C.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Spanish MINECO, grant number CTQ2016-75150-R, and Generalitat de Catalunya, grant number 2017 SGR 1720. C.V. and Z.K. thanks European Union’s Horizon 2020 Marie Skłodowska-Curie grant agreement MSCA-IF-2016-751587.
Acknowledgments
Dedicated to Todd Marder, who significantly contributed to the Boron chemistry, on his 65th birthday.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Poater, J.; Solà, M.; Viñas, C.; Teixidor, F. π Aromaticity and Three-Dimensional Aromaticity: Two sides of the Same Coin? Angew. Chem. Int. Ed. 2014, 53, 12191–12195. [Google Scholar] [CrossRef] [PubMed]
- Poater, J.; Viñas, C.; Bennour, I.; Escayola, S.; Solà, M.; Teixidor, F. Too Persistent to Give Up: Aromaticity in Boron Clusters Survives Radical Structural Changes. J. Am. Chem. Soc. 2020, 142, 9396–9407. [Google Scholar] [CrossRef] [PubMed]
- Teixidor, F.; Barbera, G.; Vaca, A.; Kivekäs, R.; Sillanpää, R.; Oliva, J.; Viñas, C. Are Methyl Groups Electron-Donating or Electron-Withdrawing in Boron Clusters? Permethylation of o-Carborane. J. Am. Chem. Soc. 2005, 127, 10158–10159. [Google Scholar] [CrossRef] [PubMed]
- Teixidor, F.; Núñez, R.; Viñas, C.; Sillanpää, R.; Kivekäs, R. A discrete P···I-I center dot center dot center dot P assembly: The large influence of weak interactions on the P-31 NMR spectra of phosphane-diiodine complexes. Angew. Chem. Int. Ed. 2000, 39, 4290–4292. [Google Scholar] [CrossRef]
- Spokoyny, A.M.; Machan, C.W.; Clingerman, D.J.; Rosen, M.S.; Wiester, M.J.; Kennedy, R.D.; Stern, C.L.; Sarjeant, A.A.; Mirkin, C.A. A coordination chemistry dichotomy for icosahedral carborane-based ligands. Nat. Chem. 2011, 3, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Scholz, M.; Hey-Hawkins, E. Carbaboranes as Pharmacophores: Properties, Synthesis, and Application Strategies. Chem. Rev. 2011, 111, 7035–7062. [Google Scholar] [CrossRef]
- Plesek, J. Potential applications of the boron cluster compounds. Chem. Rev. 1992, 92, 269–278. [Google Scholar] [CrossRef]
- Grimes, R.N. Carboranes, 3rd ed.; Elsevier Inc.: New York, NY, USA, 2016. [Google Scholar]
- Issa, F.; Kassiou, M.; Rendina, L.M. Boron in Drug Discovery: Carboranes as Unique Pharmacophores in Biologically Active Compounds. Chem. Rev. 2011, 111, 5701–5722. [Google Scholar] [CrossRef]
- Hermansson, K.; Wojcik, M.; Sjoberg, S. o-, m-, and p-carboranes and their anions: Ab initio calculations of structures, electron affinities, and acidities. Inorg. Chem. 1999, 38, 6039–6048. [Google Scholar] [CrossRef]
- Olid, D.; Núñez, R.; Viñas, C.; Teixidor, F. Methods to produce B-C, B-P, B-N and B-S bonds in boron clusters. Chem Soc Rev. 2013, 42, 3318–3336. [Google Scholar] [CrossRef]
- Quan, Y.; Xie, Z.W. Controlled functionalization of o-carborane via transition metal catalyzed B-H activation. Chem. Rev. 2019, 48, 3660–3673. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zheng, H.; Li, J.; Xu, F.; Zhao, J.; Yan, H. Selective Catalytic B−H Arylation of o-Carboranyl Aldehydes by a Transient Directing Strategy. J. Am. Chem. Soc. 2017, 139, 14511–14517. [Google Scholar] [CrossRef] [PubMed]
- Teixidor, F.; Sillanpää, R.; Pepiol, A.; Lupu, M.; Viñas, C. Synthesis of Globular Precursors. Chem. Eur. J. 2015, 21, 12778–12786. [Google Scholar] [CrossRef] [PubMed]
- Teixidor, F.; Pepiol, A.; Viñas, C. Synthesis of Periphery-Decorated and Core-Initiated Borane Polyanionic Macromolecules. Chem. Eur. J. 2015, 21, 10650–10653. [Google Scholar] [CrossRef]
- Kelemen, Z.; Pepiol, A.; Lupu, M.; Sillanpää, R.; Hänninen, M.M.; Teixidor, F.; Viñas, C. Icosahedral carboranes as scaffolds for congested regioselective polyaryl compounds: the distinct distance tuning of C–C and its antipodal B–B. Chem. Commun. 2019, 55, 8927–8930. [Google Scholar] [CrossRef] [Green Version]
- Janczak, S.; Olejniczak, A.; Balabańska, S.; Chmielewski, M.K.; Lupu, M.; Viñas, C.; Lesnikowski, Z.J. Boron Clusters as a Platform for New Materials: Synthesis of Functionalized o-Carborane (C2B10H12) Derivatives Incorporating DNA Fragments. Chem. Eur. J. 2015, 21, 15118–15122. [Google Scholar] [CrossRef]
- Kaniowski, D.; Ebenryter-Olbinska, K.; Kulik, K.; Janczak, S.; Maciaszek, A.; Bednarska-Szczepaniak, K.; Nawrot, B.; Lesnikowski, Z. Boron clusters as a platform for new materials: composites of nucleic acids and oligofunctionalized carboranes (C2B10H12) and their assembly into functional nanoparticles. Nanoscale 2020, 12, 103–114. [Google Scholar] [CrossRef]
- Ochi, J.; Tanaka, K.; Chujo, Y. Recent Progress in the Development of Solid-State Luminescent o-Carboranes with Stimuli Responsivity. Angew. Chem. Int. Ed. 2020. [Google Scholar] [CrossRef]
- Wang, S.; Blaha, C.; Santos, R.; Huynh, T.; Hayes, T.R.; Beckford-Vera, D.R.; Blecha, J.E.; Hong, A.S.; Fogarty, M.; Hope, T.A.; et al. Synthesis and Initial Biological Evaluation of Boron-Containing Prostate-Specific Membrane Antigen Ligands for Treatment of Prostate Cancer Using Boron Neutron Capture Therapy. Mol. Pharmaceutics. 2019, 16, 3831–3841. [Google Scholar] [CrossRef]
- Hosmane, N.S. Boron Science. In New Technologies and Applications; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Hosmane, N.S.; Eagling, R. Handbook of Boron Chemistry in Organometallics, Catalysis, Materials and Medicine; World Science Publishers: Hackensack, NJ, USA, 2018. [Google Scholar]
- Hey-Hawkins, E.; Viñas Teixidor, C. Boron-Based Compounds: Potential and Emerging applications in Medicine; John Wiley & Sons Ltd: Chichester, UK, 2018. [Google Scholar]
- Fisher, S.P.; Tomich, A.W.; Lovera, S.O.; Kleinsasser, J.F.; Guo, J.; Asay, M.J.; Nelson, H.M.; Lavallo, V. Nonclassical Applications of closo-Carborane Anions: From Main Group Chemistry and Catalysis to Energy Storage. Chem. Rev. 2019, 119, 8262–8290. [Google Scholar] [CrossRef]
- Barth, R.F.; Vicente, M.G.H.; Harling, O.K.; Kiger, W.S.; Riley, K.J.; Binns, P.J.; Wagner, F.M.; Suzuki, M.; Aihara, T.; Kato, I.; et al. Current status of boron neutron capture therapy of high grade gliomas and recurrent head and neck cancer. Radiat. Oncol. 2012, 7, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashin, A.N.; Butin, K.P.; Stanko, V.I.; Beletskaya, I.P. Acidity of ortho-, meta-, and para-barenes. Russ. Chem. Bull. 1969, 18, 1775–1777. [Google Scholar] [CrossRef]
- Gan, L.; Fonquernie, P.G.; Light, M.E.; Norjmaa, G.; Ujaque, G.; Choquesillo-Lazarte, D.; Fraile, J.; Teixidor, F.; Viñas, C.; Planas, J.G. A Reversible Phase Transition of 2D Coordination Layers by B–H∙∙∙Cu(II) Interactions in a Coordination Polymer. Molecules 2019, 24, 3204. [Google Scholar] [CrossRef] [Green Version]
- Cabrera-González, J.; Chaari, M.; Teixidor, F.; Viñas, C.; Núñez, R. Blue Emitting Star-Shaped and Octasilsesquioxane-Based Polyanions Bearing Boron Clusters. Photophysical and Thermal Properties. Molecules 2020, 25, 1210. [Google Scholar] [CrossRef] [Green Version]
- Mori, S.; Takagaki, R.; Fujii, S.; Urushibara, K.; Tanatani, A.; Kagechika, H. Novel Non-steroidal Progesterone Receptor Ligands Based on m-Carborane Containing a Secondary Alcohol: Effect of Chirality on Ligand Activity. Chem. Pharm. Bull. 2017, 65, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Eleazer, B.J.; Smith, M.D.; Popov, A.A.; Peryshkov, D.V. Expansion of the (BB) Ru metallacycle with coinage metal cations: Formation of B-M-Ru-B (M = Cu, Ag, Au) dimetalacyclodiboryls. Chem. Sci. 2018, 9, 2601–2608. [Google Scholar] [CrossRef] [Green Version]
- Eleazer, B.J.; Smith, M.D.; Popov, A.A. Peryshkov, Dmitry V. Rapid reversible borane to boryl hydride exchange by metal shuttling on the carborane cluster surface. Chem. Sci. 2017, 9, 2601–2608. [Google Scholar] [CrossRef] [Green Version]
- Eleazer, B.J.; Smith, M.D.; Peryshkov, D.V. Metal- and Ligand-Centered Reactivity of meta-Carboranyl-Backbone Pincer Complexes of Rhodium. Organometallics 2016, 35, 106–112. [Google Scholar] [CrossRef]
- Dziedzic, R.M.; Martin, J.L.; Axtell, J.C.; Saleh, L.M.A.; Ong, T.-C.; Yang, Y.-F.; Messina, M.S.; Rheingold, A.L.; Houk, K.N.; Spokoyny, A.M. Cage-Walking: Vertex Differentiation by Palladium-Catalyzed Isomerization of B(9)-Bromo-meta-Carborane. J. Am. Chem. Soc. 2017, 139, 7729–7732. [Google Scholar] [CrossRef]
- Himmelspach, A.; Finze, M. Dicarba-closo-dodecaboranes with One and Two Ethynyl Groups Bonded to Boron. Eur. J. Inorg. Chem. 2010, 2012–2024. [Google Scholar] [CrossRef]
- Ohta, K.; Endo, Y. Chemistry of boron clusters, carboranes synthesis, structure and application for molecular construction. J. Synth. Org. Chem. Jpn. 2007, 65, 320–333. [Google Scholar] [CrossRef]
- Bayer, M.J.; Herzog, A.; Diaz, M.; Harakas, G.A.; Lee, H.; Knobler, C.B.; Hawthorne, M.F. The Synthesis of Carboracycles Derived from B,B_Bis(aryl) Derivatives of Icosahedral ortho-Carborane. Chem. Eur. J. 2003, 9, 2732–2744. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Chizhevsky, I.T.; Mortimer, M.D.; Chen, W.; Knobler, C.B.; Johnson, S.E.; Gomez, F.A.; Hawthorne, M.F. Carboracycles: Macrocyclic Compounds Composed of Carborane Icosahedra Linked by Organic Bridging Groups. Inorg. Chem. 1996, 35, 5417–5426. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Jiang, W.; Zinn, A.A.; Knobler, C.B.; Hawthorne, M.F. Facile Electrophilic Iodination of Icosahedral Carboranes. Synthesis of Carborane Derivatives with Boron-Carbon Bonds via the Palladium-Catalyzed Reaction of Diiodocarboranes with Grignard Reagents. Inorg. Chem. 1995, 34, 2095–2100. [Google Scholar] [CrossRef]
- Fox, M.A. Icosahedral Carborane Derivatives. Ph.D. Thesis, Durham University, Durham, UK, 1991. Available online: https://www.dur.ac.uk/chemistry/fox.group/publications/phd.thesis/ (accessed on 17 June 2020).
- Ol’shevskaya, V.A.; Makarenkov, A.V.; Kononova, E.G.; Peregudov, A.S.; Lyssenko, K.A.; Kalinin, V.N. An efficient synthesis of carboranyl tetrazoles via alkylation of 5-R-1H-tetrazoles with allylcarboranes. Polyhedron 2016, 115, 128–136. [Google Scholar] [CrossRef]
- Neises, B.; Steglich, W. Simple Method for the Esterification of Carboxylic Acids. Angew. Chem. Int. Ed. 1978, 17, 522–524. [Google Scholar] [CrossRef]
- Neises, B.; Steglich, W. Esterification of carboxylic acids with dicyclohexylcarbodiimide/4-dimethylaminopyridine: tert-butyl ethyl fumarate. Org. Synth. 1985, 63, 183. [Google Scholar]
- Whitaker, D.T.; Whitaker, K.S.; Johnson, C.R.; Haas, J. p-Toluenesulfonyl Chloride In Encyclopedia of Reagents for Organic Synthesis; John Wiley: New York, NY, USA, 2006. [Google Scholar]
- Farràs, P.; Cioran, A.M.; Šίcha, V.; Teixidor, F.; Štίbr, B.; Grüner, B.; Viñas, C. Toward the Synthesis of High Boron Content Polyanionic Multicluster Macromolecules. Inorg. Chem. 2009, 48, 8210–8219. [Google Scholar] [CrossRef]
- Hassam, M.; Taher, A.; Arnott, G.E.; Green, I.R.; van Otterlo, W.A.L. Isomerization of Allylbenzenes. Chem. Rev. 2015, 115, 5462–5569. [Google Scholar] [CrossRef]
- Reutov, O.A.; Beletskaya, I.P.; Butkin, K.P. CH-Acids; Pergamon Press: Oxford, UK, 1978; pp. 13–14, 29, 34, 123–124. [Google Scholar]
- Shatenshtein, A.I.; Zakharkin, L.I.; Petrov, E.S.; Yakovleva, E.A.; Yakushin, F.S.; Matic-Vukmirovic, Z.B.; Isaeva, G.G.; Kalinin, V.N. The equilibrium and kinetic acidities of isomeric carborane methines. J. Organomet. Chem. 1970, 23, 313–322. [Google Scholar] [CrossRef]
- Petrov, E.S.; Yakovleva, E.A.; Isaeva, G.G.; Kalinin, V.N.; Zakharkin, L.I.; Shatenshtein, A.I. Thermodynamic and kinetic acidity of the CH bonds of certain ortho- and meta-barenes. Russ. Chem. Bull. 1969, 18, 1576–1582. [Google Scholar] [CrossRef]
- Popescu, A.-R.; Musteti, A.D.; Ferrer-Ugalde, A.; Viñas, C.; Nüñez, R.; Teixidor, F. Influential Role of Ethereal Solvent on Organolithium Compounds: The Case of Carboranyllithium. Chem. Eur. J. 2012, 18, 3174–3184. [Google Scholar] [CrossRef]
- Farràs, P.; Viñas, C.; Teixidor, F. Preferential chlorination vertices in cobaltabisdicarbollide anions. Substitution rate correlation with site charges computed by the two atoms natural population analysis method (2a-NPA). J. Organomet. Chem. 2013, 747, 119–125. [Google Scholar]
- Potenza, J.A.; Lipscomb, W.M.; Vickers, G.D.; Schroeder, H. Order of Electrophilic Substitution in 1,2-Dicarbaclovododecaborane(12) and Nuclear Magnetic Resonance Assignment. J. Am. Chem. Soc. 1966, 88, 628–629. [Google Scholar] [CrossRef]
- Barberà, G.; Vaca, A.; Teixidor, F.; Sillanpää, R.; Kivekaäs, R.; Viñas, C. From Mono- to Poly-Substituted Frameworks: A Way of Tuning the Acidic Character of Cc-H in o-Carborane Derivatives. Chem. Eur. J. 2009, 15, 9755–9763. [Google Scholar]
- Cowie, J.; Reid, B.D.; Watmough, J.M.S.; Welch, A.J. Steric effects in heteroboranes. Part 7: The synthesis and characterisation of arene-ruthenium complexes of C-substituted carbaboranes. Molecular structures of 1-Ph-3-(mes)-3,1,2-closo-RuC2B9H10 (mes = C6H3-1,3,5) and 1-Ph-2-Me-3-(p-cym)-3,1,2-closo-RuC2B9H9 (p-cym = C6H4Me-1-iPr-4), the latter showing an incipient deformation. J. Organomet. Chem. 1994, 481, 283–293. [Google Scholar]
- Venable, T.L.; Hutton, W.C.; Grimes, R.N. Atom connectivities in polyhedral boranes elucidated via two-dimensional J-correlated boron-11-boron-11 FT NMR: a general method. J. Am. Chem. Soc. 1982, 104, 4716–4717. [Google Scholar] [CrossRef]
- Venable, T.L.; Hutton, W.C.; Grimes, R.N. Two-dimensional boron-11-boron-11 nuclear magnetic resonance spectroscopy as a probe of polyhedral structure: Application to boron hydrides, carboranes, metallaboranes, and metallacarboranes. J. Am. Chem. Soc. 1984, 106, 29–37. [Google Scholar] [CrossRef]
- Todd, L.J.; Siedle, A.R.; Bodner, G.M.; Kahl, S.B.; Hickey, J.P. An NMR Study of Icosahedral Heteroatom Borane Derivatives. J. Magn. Reson. 1976, 23, 301–311. [Google Scholar] [CrossRef]
- Bruno, J.; Cole, J.C.; Edgington, P.R.; Kessler, M.; Macrae, C.F.; McCabe, P.; Pearson, J.; Taylor, R. New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Crystallogr. 2002, B58, 389–397. [Google Scholar] [CrossRef]
- Fox, M.A.; Hughes, A.K. Cage C-H⋯X interactions in solid-state structures of icosahedral carboranes. Coord. Chem. Rev. 2004, 248, 457–476. [Google Scholar] [CrossRef]
- Pauling, L. The Nature of the Chemical Bond, 3rd ed.; Cornell University Press: Ithaca, NY, USA, 1960. [Google Scholar]
- Nishio, M. CH/π hydrogen bonds in crystals. Cryst. Eng. Comm. 2004, 6, 130–158. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17; The University of Western Australia: Perth, Australia, 2017. [Google Scholar]
- Wolff, S.K.; Grimwood, D.J.; McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer 3.0; University of Western Australia: Perth, Australia, 2007. [Google Scholar]
- Pangajavalli, S.; Ranjithkumar, R.; Ramaswamy, S. Structural, Hirshfeld, Spectroscopic, Quantum Chemical and Molecular docking Studies on 6b′,7′,8′,9′-Tetrahydro-2H,6′H-spiro[acenaphthylene-1,11′-chromeno[3,4-a]pyrrolizine]-2,6′(6a′H,11a′H)-dione. J. Mol. Struct. 2020, 1209, 127921. [Google Scholar] [CrossRef]
- Bojarska, J.; Remko, M.; Fruziński, A.; Maniukiewicz, W. The experimental and theoretical landscape of a new antiplatelet drug ticagrelor: Insight into supramolecular architecture directed by C-H…F, π...π and C-H...π interactions. J. Mol. Struct. 2018, 1154, 290–300. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are available from the authors. |
Figure 1.
Icosahedral 1,2-closo-C2B10H12, 1,7-closo-C2B10H12 (1) and 1,12-closo-C2B10H12 isomers with their vertexes numbering. Dark circles are Cc-H vertexes and grey ones are B-H vertexes.
Figure 1.
Icosahedral 1,2-closo-C2B10H12, 1,7-closo-C2B10H12 (1) and 1,12-closo-C2B10H12 isomers with their vertexes numbering. Dark circles are Cc-H vertexes and grey ones are B-H vertexes.

Figure 2.
Schematic view of the two types of radially expanded tetrabranched core for constructing dendritic structures. Circles colour: dark grey correspond to Cc-H bonds, black to C atoms, pink to Boron atoms and grey to B-H vertexes in the tetra-branched clusters.
Figure 2.
Schematic view of the two types of radially expanded tetrabranched core for constructing dendritic structures. Circles colour: dark grey correspond to Cc-H bonds, black to C atoms, pink to Boron atoms and grey to B-H vertexes in the tetra-branched clusters.

Scheme 1.
Synthesis of 9,10-(CH2=CHCH2)2-1,7-closo-C2B10H10 (3). Dark circles are Cc-H vertexes, pink circles are boron atoms, and grey circles are B-H vertexes.
Scheme 1.
Synthesis of 9,10-(CH2=CHCH2)2-1,7-closo-C2B10H10 (3). Dark circles are Cc-H vertexes, pink circles are boron atoms, and grey circles are B-H vertexes.

Scheme 2.
Derivatization reactions on 9,10-(CH2=CHCH2)2-1,7-closo-C2B10H10 3. Dark circles are Cc-H vertexes, pink circles are boron atoms, and grey circles are B-H vertexes.
Scheme 2.
Derivatization reactions on 9,10-(CH2=CHCH2)2-1,7-closo-C2B10H10 3. Dark circles are Cc-H vertexes, pink circles are boron atoms, and grey circles are B-H vertexes.

Scheme 3.
(i) Deprotonation reaction on Cc-H of B(9,10)-disubstituted m-carborane derivative 3 with n-BuLi followed by nucleophilic substitution with allyl bromide. (ii) Hydroboration/oxidation process on terminal olefinic groups in 10 by using BH3·THF, H2O2 in basic aqueous solution (KOH) to obtain 11. Dark circles are Cc-H vertexes, pink circles are boron atoms, black circles are Cc atoms and grey circles are B-H vertexes.
Scheme 3.
(i) Deprotonation reaction on Cc-H of B(9,10)-disubstituted m-carborane derivative 3 with n-BuLi followed by nucleophilic substitution with allyl bromide. (ii) Hydroboration/oxidation process on terminal olefinic groups in 10 by using BH3·THF, H2O2 in basic aqueous solution (KOH) to obtain 11. Dark circles are Cc-H vertexes, pink circles are boron atoms, black circles are Cc atoms and grey circles are B-H vertexes.

Scheme 4.
Top: Designed a synthetic reaction to achieve the dianionic species. Bottom: Achieved reaction was the isomerization of 9,10-(CH2=CHCH2)2-1,7-closo-C2B10H10, to 9,10-(CH3CH=CH)2-1,7-closo-C2B10H10. Dark circles are Cc-H vertexes, pink circles are boron atoms, and grey circles are B-H vertexes.
Scheme 4.
Top: Designed a synthetic reaction to achieve the dianionic species. Bottom: Achieved reaction was the isomerization of 9,10-(CH2=CHCH2)2-1,7-closo-C2B10H10, to 9,10-(CH3CH=CH)2-1,7-closo-C2B10H10. Dark circles are Cc-H vertexes, pink circles are boron atoms, and grey circles are B-H vertexes.

Figure 3.
Comparison of the HOMO and LUMO orbitals of the o- and m-carborane.

Figure 4.
(a) The 1H {11B} spectrum of 3 in CDCl3. (b) Hc allyl resonance as well as the schematic coupling between Hc protons and the protons of the allyl branches with the corresponding coupling constant values.
Figure 4.
(a) The 1H {11B} spectrum of 3 in CDCl3. (b) Hc allyl resonance as well as the schematic coupling between Hc protons and the protons of the allyl branches with the corresponding coupling constant values.

Figure 5.
ORTEP presentation of 9,10-(HOCH2CH2CH2)2-1,7-closo-C2B10H10 (4) and 9,10-(CH3CH=CH)2-1,7-closo-C2B10H10 (12) showing the atom numbering and displacement. Ellipsoids are at 30% and 50% probability level, respectively.
Figure 5.
ORTEP presentation of 9,10-(HOCH2CH2CH2)2-1,7-closo-C2B10H10 (4) and 9,10-(CH3CH=CH)2-1,7-closo-C2B10H10 (12) showing the atom numbering and displacement. Ellipsoids are at 30% and 50% probability level, respectively.

Figure 6.
Network presentation of 4 showing all intermolecular contacts as dashed lines: (a) H…H, (b) O…H and (c) O…O (H are omitted for clarity).
Figure 6.
Network presentation of 4 showing all intermolecular contacts as dashed lines: (a) H…H, (b) O…H and (c) O…O (H are omitted for clarity).

Figure 7.
Network presentation of 12 showing all intermolecular contacts as dashed lines: (a) C-H…H-B and (b) Cc-H…π interactions.
Figure 7.
Network presentation of 12 showing all intermolecular contacts as dashed lines: (a) C-H…H-B and (b) Cc-H…π interactions.

Figure 8.
Presentation of close contacts for 4 (on left) and 12 (on right) through the dnorm.

Figure 9.
Shape index presentation of 12 showing the red concave and the blue convex areas, which correspond to (a) H…π and (b) π …H, respectively.
Figure 9.
Shape index presentation of 12 showing the red concave and the blue convex areas, which correspond to (a) H…π and (b) π …H, respectively.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Theoretical calculations of natural charges, 2a-NPA charges, and cumulative build-up of the cluster-only total charge (CTC) of ortho-closo and meta-closo carborane. See Figure 1 for the numbering of the clusters’ vertexes.
Table 1.
Theoretical calculations of natural charges, 2a-NPA charges, and cumulative build-up of the cluster-only total charge (CTC) of ortho-closo and meta-closo carborane. See Figure 1 for the numbering of the clusters’ vertexes.
| o-closo-C2B10H12 | m-closo-C2B10H12 | ||||
|---|---|---|---|---|---|
| NPA | 2a-NPA | NPA | 2a-NPA | ||
| C(1) | −0.498 | −0.198 | C(1) | −0.654 | −0.354 |
| C(2) | −0.498 | −0.198 | B(2) | 0.151 | 0.215 |
| B(3) | 0.159 | 0.213 | B(3) | 0.151 | 0.215 |
| B(4) | 0.000 | 0.054 | B(4) | 0.001 | 0.070 |
| B(5) | 0.000 | 0.069 | B(5) | 0.023 | 0.087 |
| B(6) | 0.159 | 0.213 | B(6) | 0.001 | 0.070 |
| B(7) | 0.000 | 0.069 | C(7) | −0.654 | −0.354 |
| B(8) | −0.165 | −0.087 | B(8) | 0.001 | 0.070 |
| B(9) | −0.140 | −0.067 | B(9) | −0.165 | −0.087 |
| B(10) | −0.165 | −0.087 | B(10) | −0.165 | −0.087 |
| B(11) | 0.000 | 0.054 | B(11) | 0.001 | 0.070 |
| B(12) | −0.140 | −0.067 | B(12) | 0.023 | 0.087 |
| CTC | −1.288 | - | CTC | −1.286 | - |
Table 2.
The 11B{1H} and 1H{11B} chemical shifts of icosahedral compounds 1 and 2. Spectra were recorded in (CD3)2CO. See Figure 1 for vertexes numbering.
Table 2.
The 11B{1H} and 1H{11B} chemical shifts of icosahedral compounds 1 and 2. Spectra were recorded in (CD3)2CO. See Figure 1 for vertexes numbering.
| 1,7-closo-C2B10H12 | 9,10-I2-1,7-closo-C2B10H10 | |||
|---|---|---|---|---|
| 11B{1H} (ppm) | 1H{11B} (ppm) | 11B{1H} (ppm) | 1H{11B} (ppm) | |
| B(5,12) | −6.6 | 2.27 | −4.1 | 2.76 |
| B(9,10) | −10.5 | 2.10 | −20.7 | - |
| B(4,6,8,11) | −13.3 | 2.19 | −11.9 | 2.93 |
| B(5,12) | −17.0 | 2.64 | −18.8 | 3.15 |
Table 3.
11B{1H} NMR chemical shifts (in ppm) of di-branched (2–9 and 12) and tetra-branched (10 and 11) m-carborane derivatives. <δ(11B)> corresponds to the weighted average 11B{1H} NMR spectrum (in ppm). Spectra were recorded in (CD3)2CO and referenced to external BF3.Et2O unless noted otherwise: #(CD3)2SO.
Table 3.
11B{1H} NMR chemical shifts (in ppm) of di-branched (2–9 and 12) and tetra-branched (10 and 11) m-carborane derivatives. <δ(11B)> corresponds to the weighted average 11B{1H} NMR spectrum (in ppm). Spectra were recorded in (CD3)2CO and referenced to external BF3.Et2O unless noted otherwise: #(CD3)2SO.
| R | B(9,10) | Δ | B(5,12) | B(4,6,8,11) | B(2,3) | <δ(11B)> | |
|---|---|---|---|---|---|---|---|
| 1 | H | −9.5 | - | −5.6 | −11.9 | −15.4 | −10.9 |
| 2 | 9,10-I2 | −19.4 | −9.9 | −3.0 | −10.4 | −17.0 | −12.0 |
| 3 | 9,10-(CH₂=CH–CH₂)2 | 0.6 | +10.1 | −5.4 | −12.5 | −19.1 | −9.8 |
| 4 | 9,10-(HO(CH2)3)2 | 1.8 | +11.3 | −5.3 | −12.7 | −19.4 | −9.7 |
| 5 | 9,10-(Cl(CH2)3)2 | 1.3 | +10.8 | −5.2 | −12.5 | −19.0 | −9.6 |
| 6 | 9,10-(PhCOO(CH2)3)2 | 1.6 | +11.1 | −5.2 | −12.5 | −19.0 | −9.5 |
| 7 | 9,10-(CH3-C6H4-SO3(CH2)3)2 | 1.2 | +10.7 | −5.3 | −12.6 | −19.1 | −9.7 |
| 8 | 9,10-(N3(CH2)3)2 | 1.4 | +10.9 | −5.3 | −12.6 | −19.1 | −9.6 |
| 9 | 9,10-(C6H5C2N3(CH2)3)2 # | 1.4 | +10.9 | −5.5 | −12.4 | −19.0 | −9.6 |
| 10 | 1,7,9,10-(CH₂=CH–CH₂)4 | 0.8 | +10.3 | −5.3 | −10.2 | −15.9 | −8.2 |
| 11 | 1,7,9,10-(HO(CH2)3)4 | 1.4 | +10.9 | −5.5 | −10.5 | −15.7 | −8.2 |
| 12 | 9,10-(CH3CH=CH)2 | −0.5 | +9.0 | −5.8 | −12.5 | −19.8 | −10.1 |
Table 4.
Chemical shift of 1H and 13C{1H} NMR spectra (in ppm) and stretching frequencies of Cc-H (in cm−1) in the IR spectra for the 9,10-R2-1,7-closo-C2B10H10 derivatives. NMR spectra were run in (CD3)2CO unless noted otherwise: * CDCl3 and #(CD3)2SO.
Table 4.
Chemical shift of 1H and 13C{1H} NMR spectra (in ppm) and stretching frequencies of Cc-H (in cm−1) in the IR spectra for the 9,10-R2-1,7-closo-C2B10H10 derivatives. NMR spectra were run in (CD3)2CO unless noted otherwise: * CDCl3 and #(CD3)2SO.
| R | δ1H(Cc-H) | Δδ1H | δ13C(Cc-H) | Δδ13C(Cc-H) | ν(Cc-H) | |
|---|---|---|---|---|---|---|
| 1 | H | 3.65 2.91 * | - - | 56.17 55.23 * | - | |
| 2 | I | 4.11 | 0.46 | 58.18 | +2.01 | - |
| 3 | CH₂=CH–CH₂- | 3.50 | −0.15 | 52.35* | −2.88* | 3062 |
| 4 | HO(CH2)3- | 3.46 | −0.19 | 52.95 | −3.22 | 3038 |
| 5 | Cl(CH2)3- | 3.54 | −0.11 | 53.28 | −2.89 | 3063 |
| 6 | PhCOO(CH2)3- | 3.55 | −0.10 | 53.27 | −2.90 | 3064 |
| 7 | CH3-C6H4-SO3-(CH2)3- | 3.48 | −0.17 | 53.26 | −2.91 | 3064 |
| 8 | N3(CH2)3- | 3.54 | −0.11 | 53.26 | −2.91 | 3065 |
| 9 | C6H5C2N3(CH2)3- | 3.86 # | 0.21 | Not observed | Not observed | 3057 |
| 12 | CH3CH=CH- | 2.82 * | 0.09 | Not observed | Not observed | 3046 |
Table 5.
1H- and 13C{1H} NMR chemical shift values (in ppm) of Cc-H vertexes for several 9,12-R2-1,2-closo-C2B10H10 and 9,10-R2-1,7-closo-C2B10H10 derivatives. NMR spectra were run in *(CD3)2CO or #CDCl3.
Table 5.
1H- and 13C{1H} NMR chemical shift values (in ppm) of Cc-H vertexes for several 9,12-R2-1,2-closo-C2B10H10 and 9,10-R2-1,7-closo-C2B10H10 derivatives. NMR spectra were run in *(CD3)2CO or #CDCl3.
| δ1H(Cc-H) | Δδ1H | δ13C(Cc-H) | Δδ13C | |
|---|---|---|---|---|
| 1,2-closo-C2B10H12 | 3.56 # 4.40 * | - - | 54.46 # 56.20 * | - - |
| 9,12-I2-1,2-closo-C2B10H10 [39] | 4.00 # | +0.44 # | 52.23 # | −2.23 # |
| 9,12-Br2-1,2-closo-C2B10H10 [56] | 4.78 * | +0.38 * | 48.50 * | −7.70 * |
| 9,12-(CH₂=CH–CH₂)2-1,2-closo-C2B10H10 [14] | 3.42 # | −0.14 # | 48.26 # | −6.2 # |
| 9,12-(OHCH₂CH2CH₂)2-1,2-closo-C2B10H10 [14] | 4.30 * | −0.10 * | 49.17 * | −7.03 * |
| 1,7-closo-C2B10H10 | 2.91 # 3.63 * | - - | 55.23 # 56.17 * | - - |
| 9,10-I2-1,7-closo-C2B10H10 [39] | 3.16 # 4.08 * | +0.40 # +0.32 * | - 58.18 * | - +2.01 * |
| 9,10-Cl2-1,7-closo-C2B10H10 [56] | 3.76 * | +0.13 * | 51.60 * | −4.57 * |
| 9,10-Br2-1,7-closo-C2B10H10 [56] | 3.88 * | +0.25 * | 54.00 * | −2.17 * |
| 9,10-(CH₂=CH–CH₂)2-1,7-closo-C2B10H10 | 2.84 # 3.50 * | −0.07 # −0.13 * | 52.35 # - | −2.88 # - |
| 9,10-(OHCH2CH2CH2)2-1,7-closo-C2B10H10 | 3.49 * | −0.14 * | 52.95 * | −3.22 * |
Table 6.
Crystal data and structure refinement.
| Compound | 4 | 12 |
|---|---|---|
| Empirical formula | C8H24B10O2 | C8H20B10 |
| Formula weight(g/mol) | 260.37 | 224.34 |
| Temperature (K) | 100(2) | |
| Wavelength Å | 0.71073 | |
| Crystal system | Triclinic | |
| Space group | P 1 | P -1 |
| Unit cell dimensions | a = 10.7502(13)Å b = 11.1738(13)Å c = 14.2934(16)Å α = 73.346(5)° β = 75.220(5)° γ = 72.062(5)° | a = 6.9248(11)Å b = 7.4642(11)Å c = 13.454(2)Å α = 90.768(4)° β = 94.188(4)° γ = 95.488(4)° |
| Volume Å3 | 1538.5(3) | 690.23(18) |
| Z | 4 | 2 |
| ρcal (g/cm3 ) | 1.124 | 1.079 |
| Absorption coefficient (mm−1) | 0.062 | 0.049 |
| F (000) | 552 | 236 |
| Theta range for data collection | 2.74 to 28.30° | 2.96 to 27.52° |
| Index ranges | −14<=h<=14 −14<=k<=14 −19<=l<=19 | −9<=h<=8 −9<=k<=9 −17<=l<=17 |
| Refinement method | Full-matrix least-squares on F2 | |
| Final R indices [I > 2σ(I)] | 7879 data; I>2σ(I) R1 = 0.1499 wR2 = 0.3573 all data R1 = 0.2389 wR2 = 0.4145 | 2428 data I>2σ(I) R1 = 0.0530, wR2 = 0.1302 all data R1 = 0.0745 wR2 = 0.1444 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bennour, I.; Teixidor, F.; Kelemen, Z.; Viñas, C. m-Carborane as a Novel Core for Periphery-Decorated Macromolecules. Molecules 2020, 25, 2814. https://doi.org/10.3390/molecules25122814
AMA Style
Bennour I, Teixidor F, Kelemen Z, Viñas C. m-Carborane as a Novel Core for Periphery-Decorated Macromolecules. Molecules. 2020; 25(12):2814. https://doi.org/10.3390/molecules25122814
Chicago/Turabian StyleBennour, Ines, Francesc Teixidor, Zsolt Kelemen, and Clara Viñas. 2020. "m-Carborane as a Novel Core for Periphery-Decorated Macromolecules" Molecules 25, no. 12: 2814. https://doi.org/10.3390/molecules25122814