
Synthesis and Spectroscopic Analysis of Piperine- and Piperlongumine-Inspired Natural Product Scaffolds and Their Molecular Docking with IL-1β and NF-κB Proteins

 , , , and
, , , and 
Abstract
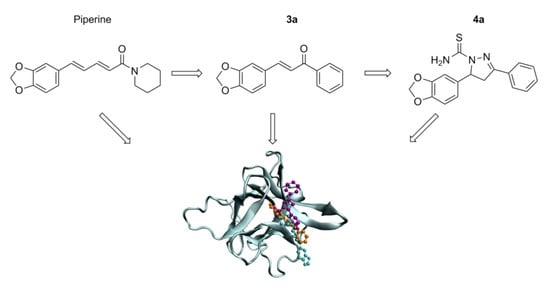
:
1. Introduction
2. Results and Discussion
2.1. Spectroscopic Characterization of Natural Product Inspired Analogues
2.2. Electronic Structures of Piperine and PPL-Inspired Molecules
2.3. Molecular Docking
3. Materials and Methods
3.1. Synthesis of Piperine-Inspired Molecules
Synthesis of Piperonal (2)
Synthesis of (E)-3-(Benzo[d][1,3]dioxol-5-yl)-1-phenylprop-2-en-1-one (3a)
Synthesis of (E)-3-(Benzo[d][1,3]dioxol-5-yl)-1-(3-nitrophenyl)prop-2-en-1-one (3b)
Synthesis of (E)-1-(3-Aminophenyl)-3-(benzo[d][1,3]dioxol-5-yl)prop-2-en-1-one (3c)
Synthesis of 5-(Benzo[d][1,3]dioxol-5-yl)-3-phenyl-4,5-dihydro-1H-pyrazole-1-carbothioamide (4a)
Synthesis of 3-(3-Aminophenyl)-5-(benzo[d][1,3]dioxol-5-yl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (4c)
3.2. Synthesis of PPL-Inspired Molecules
Synthesis of (E)-3-(3,4,5-Trimethoxyphenyl)acrylic acid (7a)
Synthesis of (E)-3-(4-Methoxyphenyl)acrylic acid (7b)
Synthesis of (E)-3-(3,4,5-Trimethoxyphenyl)acryloyl chloride (8a)
Synthesis of (E)-3-(4-Methoxyphenyl)acryloyl chloride (8b)
Synthesis of (E)-1-(3-(3,4,5-Trimethoxyphenyl)acryloyl)piperidin-2-one (9a)
Synthesis of (E)-1-(3-(4-Methoxyphenyl)acryloyl)piperidin-2-one (9b)
Synthesis (E)-6-(3-(3,4,5-Trimethoxyphenyl)acryloyl)cyclohex-2-en-1-one (10)
3.3. Electronic Structure Characterization
3.4. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Collins, I.; Jones, A.M. Diversity-oriented synthetic strategies applied to cancer chemical biology and drug discovery. Molecules 2014, 19, 17221–17255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, D.J. Natural products as leads to potential drugs: An old process or the new hope for drug discovery? J. Med. Chem. 2008, 51, 2589–2599. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, K. Black pepper and its pungent principle-piperine: A review of diverse physiological effects. Crit. Rev. Food Sci. Nutr. 2007, 47, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Umadevi, P.; Deepti, K.; Venugopal, D.V.R. Synthesis, anticancer and antibacterial activities of piperine analogs. Med. Chem. Res. 2013, 22, 5466–5471. [Google Scholar] [CrossRef]
- Venkatasamy, R.; Faas, L.; Young, A.R.; Raman, A.; Hider, R.C. Effects of piperine analogues on stimulation of melanocyte proliferation and melanocyte differentiation. Bioorg. Med. Chem. 2004, 12, 1905–1920. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, D.P.; Pessoa, C.; de Moraes, M.O.; Saker-Neto, N.; Silveira, E.R.; Costa-Lotufo, L.V. Overview of the therapeutic potential of piplartine (piperlongumine). Eur. J. Pharm. Sci. 2013, 48, 453–463. [Google Scholar] [CrossRef]
- Adams, D.J.; Dai, M.; Pellegrino, G.; Wagner, B.K.; Stern, A.M.; Shamji, A.F.; Schreiber, S.L. Synthesis, cellular evaluation, and mechanism of action of piperlongumine analogs. Proc. Natl. Acad. Sci. USA 2012, 109, 15115–15120. [Google Scholar] [CrossRef] [Green Version]
- Piska, K.; Gunia-Krzyżak, A.; Koczurkiewicz, P.; Wójcik-Pszczoła, K.; Pękala, E. Piperlongumine (piplartine) as a lead compound for anticancer agents – Synthesis and properties of analogues: A mini-review. Eur. J. Med. Chem. 2018, 156, 13–20. [Google Scholar] [CrossRef]
- Meghwal, M.; Goswami, T.K. Piper nigrum and piperine: An update. Phyther. Res. 2013, 27, 1121–1130. [Google Scholar] [CrossRef]
- Bang, J.S.; Oh, D.H.; Choi, H.M.; Sur, B.-J.; Lim, S.-J.; Kim, J.Y.; Yang, H.I.; Yoo, M.C.; Hahm, D.-H.; Kim, K.S. Anti-inflammatory and antiarthritic effects of piperine in human interleukin 1β-stimulated fibroblast-like synoviocytes and in rat arthritis models. Arthritis Res. Ther. 2009, 11, R49. [Google Scholar] [CrossRef] [Green Version]
- Ying, X.; Chen, X.; Cheng, S.; Shen, Y.; Peng, L.; Xu, H. Piperine inhibits IL-β induced expression of inflammatory mediators in human osteoarthritis chondrocyte. Int. Immunopharmacol. 2013, 17, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Ying, X.; Yu, K.; Chen, X.; Chen, H.; Hong, J.; Cheng, S.; Peng, L. Piperine inhibits LPS induced expression of inflammatory mediators in RAW 264.7 cells. Cell. Immunol. 2013, 285, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Mathew, A.; Sheeja, M.T.L.; Kumar, K.; Radha, A.T. Design, synthesis and biological evaluation of pyrazole analogues of natural piperine. Hygeia JD Med. 2011, 3, 48–56. [Google Scholar]
- Qu, H.; Lv, M.; Xu, H. Piperine: Bioactivities and structural modifications. Mini Rev. Med. Chem. 2015, 15, 145–156. [Google Scholar] [CrossRef]
- Prasad, S.; Tyagi, A.K. Historical spice as a future drug: Therapeutic potential of piperlongumine. Curr. Pharm. Des. 2016, 22, 4151–4159. [Google Scholar] [CrossRef]
- Wu, Y.; Min, X.; Zhuang, C.; Li, J.; Yu, Z.; Dong, G.; Yao, J.; Wang, S.; Liu, Y.; Wu, S. Design, synthesis and biological activity of piperlongumine derivatives as selective anticancer agents. Eur. J. Med. Chem. 2014, 82, 545–551. [Google Scholar] [CrossRef]
- Ginzburg, S.; Golovine, K.V.; Makhov, P.B.; Uzzo, R.G.; Kutikov, A.; Kolenko, V.M. Piperlongumine inhibits NF-κB activity and attenuates aggressive growth characteristics of prostate cancer cells. Prostate 2014, 74, 177–186. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.-D.; Wang, F.; Dai, F.; Wang, Y.-H.; Lin, D.; Zhou, B. Development and mechanism investigation of a new piperlongumine derivative as a potent anti-inflammatory agent. Biochem. Pharmacol. 2015, 95, 156–169. [Google Scholar] [CrossRef]
- Si, D.; Wang, Y.; Zhou, Y.-H.; Guo, Y.; Wang, J.; Zhou, H.; Li, Z.-S.; Fawcett, J.P. Mechanism of CYP2C9 inhibition by flavones and flavonols. Drug Metab. Dispos. 2009, 37, 629–634. [Google Scholar] [CrossRef] [Green Version]
- Gu, S.M.; Lee, H.P.; Ham, Y.W.; Son, D.J.; Kim, H.Y.; Oh, K.W.; Han, S.-B.; Yun, J.; Hong, J.T. Piperlongumine improves lipopolysaccharide-induced amyloidogenesis by suppressing NF-KappaB pathway. Neuromol. Med. 2018, 20, 312–327. [Google Scholar] [CrossRef] [Green Version]
- Zazeri, G.; Povinelli, A.P.R.; de Lima, F.M.; Cornélio, M.L. Experimental approaches and computational modeling of rat serum albumin and its interaction with piperine. Int. J. Mol. Sci. 2019, 20, 2856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Povinelli, A.P.R.; Zazeri, G.; de Freitas Lima, M.; Cornélio, M.L. Details of the cooperative binding of piperlongumine with rat serum albumin obtained by spectroscopic and computational analyses. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panne, D.; Maniatis, T.; Harrison, S.C. An atomic model of the interferon-β enhanceosome. Cell 2007, 129, 1111–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-Q.; Ghosh, S.; Ghosh, G. A novel DNA recognition mode by the NF-κB p65 homodimer. Nat. Struct. Biol. 1998, 5, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.B.G.; Lawton, M. Drying of organic solvents: Quantitative evaluation of the efficiency of several desiccants. J. Org. Chem. 2010, 75, 8351–8354. [Google Scholar] [CrossRef] [PubMed]
- Bellardita, M.; Loddo, V.; Palmisano, G.; Pibiri, I.; Palmisano, L.; Augugliaro, V. Photocatalytic green synthesis of piperonal in aqueous TiO2 suspension. Appl. Catal. B Environ. 2014, 144, 607–613. [Google Scholar] [CrossRef]
- Elamathi, P.; Chandrasekar, G.; Muthuraman, S.; Kolli, M.K. Pore size engineering of hexagonal mesoporous carbon nitride (HMCN) for high catalytic performance in the synthesis of α, β-unsaturated acid and its derivatives. Appl. Surf. Sci. 2019, 463, 481–491. [Google Scholar] [CrossRef]
- Schmink, J.R.; Kormos, C.M.; Devine, W.G.; Leadbeater, N.E. Exploring the scope for scale-up of organic chemistry using a large batch microwave reactor. Org. Process. Res. Dev. 2010, 14, 205–214. [Google Scholar] [CrossRef]
- Peng, S.; Zhang, B.; Meng, X.; Yao, J.; Fang, J. Synthesis of piperlongumine analogues and discovery of nuclear factor erythroid 2-related factor 2 (Nrf2) activators as potential neuroprotective agents. J. Med. Chem. 2015, 58, 5242–5255. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Gordon, M.S.; Schmidt, M.W. Advances in electronic structure theory: GAMESS a decade later. In Theory and Applications of Computational Chemistry; Elsevier: Amsterdam, The Netherlands, 2005; pp. 1167–1189. [Google Scholar]
- Slater, J.C. A simplification of the Hartree-Fock method. Phys. Rev. 1951, 81, 385. [Google Scholar] [CrossRef]
- Parr, R.G. Density functional theory of atoms and molecules. In Horizons of Quantum Chemistry; Springer: Boston, MA, USA, 1980; pp. 5–15. [Google Scholar]
- Hertwig, R.H.; Koch, W. On the parameterization of the local correlation functional. What is Becke-3-LYP. Chem. Phys. Lett. 1997, 268, 345–351. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A. Potential derived charges using a geodesic point selection scheme. J. Comput. Chem. 1996, 17, 1–18. [Google Scholar] [CrossRef]
- Bode, B.M.; Gordon, M.S. A graphical user interface for GAMESS. J. Mol. Graph. Model. 1998, 16, 133–138. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Povinelli, A.P.R.; Zazeri, G.; Cornélio, M.L. Molecular Mechanism of Flavonoids Using Fluorescence Spectroscopy and Computational Tools. In Flavonoids-A Coloring Model for Cheering Up Life; IntechOpen: London, UK, 2019. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors upon reasonable request. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Amino Acids | |||
|---|---|---|---|---|
| Non-Polar | Positively Charged | Negatively Charged | Polar | |
| Piperine | Leu80, Leu134, Val32, Phe133, Pro131, Trp120 | Lys77 | - | Thr137, Thr79 |
| 3a | Leu82, Leu26, Leu80, Leu134, Phe133, Val132 and Pro131 | Lys77 | Glu25 | Thr79 |
| 4a | Leu80,Leu82, Leu134,Val132, Phe133, Pro78 | - | - | Thr79, Gln81, Tyr24 |
| 3b | Pro2, Pro87, Pro91 | Lys88, Lys92, Lys93, Lys94 | - | Asn89, Tyr90, |
| 3c | Leu80, Leu82, Leu134, Phe133, Pro78, Trp120, Val132, Pro131 | Lys77 | Glu25 | Tyr24, Gln81 |
| 4c | Leu26, Val132, Phe133, Pro78, Leu134, Leu82 Pro131, Leu80, | - | - | Gln81, Tyr24, Thr79 |
| PPL | Val132, Phe133, Gly135, Gly136, Trp120, Leu134, Leu80 | Lys77 | Asp142 | Thr79, Thr137 |
| 9a | Leu80, Trp120, Gly136, Gly135, Phe133, Leu134 | Lys77 | Asp142 | Thr79, Thr137 |
| 10 | Trp120, Leu80, Leu82, Val32, Leu134, Phe133 | Lys77 | Glu25 | Tyr24, Gln81 |
| 9b | Leu82, Leu80, Phe133, Leu134, Trp120, Val132 | Lys77 | Glu25, | Gln81, Tyr24 |
| Molecules | Energy Score kcal/mol (IL-1B) | Energy Score kcal/mol (NF-KB) |
|---|---|---|
| Piperine | −6.08 | −6.08 |
| 3a | −6.04 | −6.53 |
| 3b | −7.44 | −6.24 |
| 3c | −6.25 | −6.07 |
| 4a | −7.18 | −6.64 |
| 4c | −7.22 | −6.4 |
| PPL | −5.83 | −5.36 |
| 9a | −5.38 | −5.85 |
| 10 | −6.19 | −5.72 |
| 9b | −5.62 | −5.87 |
| Molecules | Amino Acids | |||
|---|---|---|---|---|
| Non-Polar | Positively Charged | Negatively Charged | Polar | |
| Piperine | Ala192, Leu194 | Lys195, Arg187, Lys218 | Glu193, Glu282, Asp217 | Asn186, |
| 3a | Val121, Ala129, Cys38, Cys120 | Lys37, Lys122, Arg133 | Glu89 | Tyr36, Gln132 |
| 4a | Ala188, Cys120, Leu154, Val121, Pro189 | Arg187, Lys122, Lys123, His88 | Asp185 | Tyr36, His88, Asn155 |
| 3b | Ala192 | Arg33, Arg187, Lys195, Lys218 | Asp217, Glu193, | Asn186, |
| 3c | Cys120, Leu154, Val121, Pro189, Ala188, Phe34 | His88, Lys123 | - | His88, Tyr36, Asn155, Gln220, Asn190, Asp185 |
| 4c | Pro189, Leu154, His88, Val121, Cys120, Ala188 | Lys123, Lys122, Arg187 | - | Asp185, Tyr36, His88, Asn155, |
| PPL | Cys120, Phe34, Leu154, Ala188, Pro189 | Lys218, Arg187, His88 | Asp185, | Tyr36, Asn155 |
| 9a | Ala188, Ala192, Pro189 | Lys218, Lys195, Arg33, Arg187, Lys194 | Asp217, Glu193 | Asn186, |
| 10 | Ala188, Phe34, Cys120, Leu154, Val121, Pro189 | Lys218, Arg187, His88 | Asp185 | Asn155, His88 |
| 9b | Leu154, Cys120, Val121, | His88, Lys123, Lys122, Arg124 | Asp185 | Tyr36, Asn155 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zazeri, G.; Povinelli, A.P.R.; Le Duff, C.S.; Tang, B.; Cornelio, M.L.; Jones, A.M. Synthesis and Spectroscopic Analysis of Piperine- and Piperlongumine-Inspired Natural Product Scaffolds and Their Molecular Docking with IL-1β and NF-κB Proteins. Molecules 2020, 25, 2841. https://doi.org/10.3390/molecules25122841
Zazeri G, Povinelli APR, Le Duff CS, Tang B, Cornelio ML, Jones AM. Synthesis and Spectroscopic Analysis of Piperine- and Piperlongumine-Inspired Natural Product Scaffolds and Their Molecular Docking with IL-1β and NF-κB Proteins. Molecules. 2020; 25(12):2841. https://doi.org/10.3390/molecules25122841
Chicago/Turabian StyleZazeri, Gabriel, Ana Paula R. Povinelli, Cécile S. Le Duff, Bridget Tang, Marinonio L. Cornelio, and Alan M. Jones. 2020. "Synthesis and Spectroscopic Analysis of Piperine- and Piperlongumine-Inspired Natural Product Scaffolds and Their Molecular Docking with IL-1β and NF-κB Proteins" Molecules 25, no. 12: 2841. https://doi.org/10.3390/molecules25122841