
Structure-Based Discovery of Dual-Target Hits for Acetylcholinesterase and the α7 Nicotinic Acetylcholine Receptors: In Silico Studies and In Vitro Confirmation

 ,
,
Abstract
:
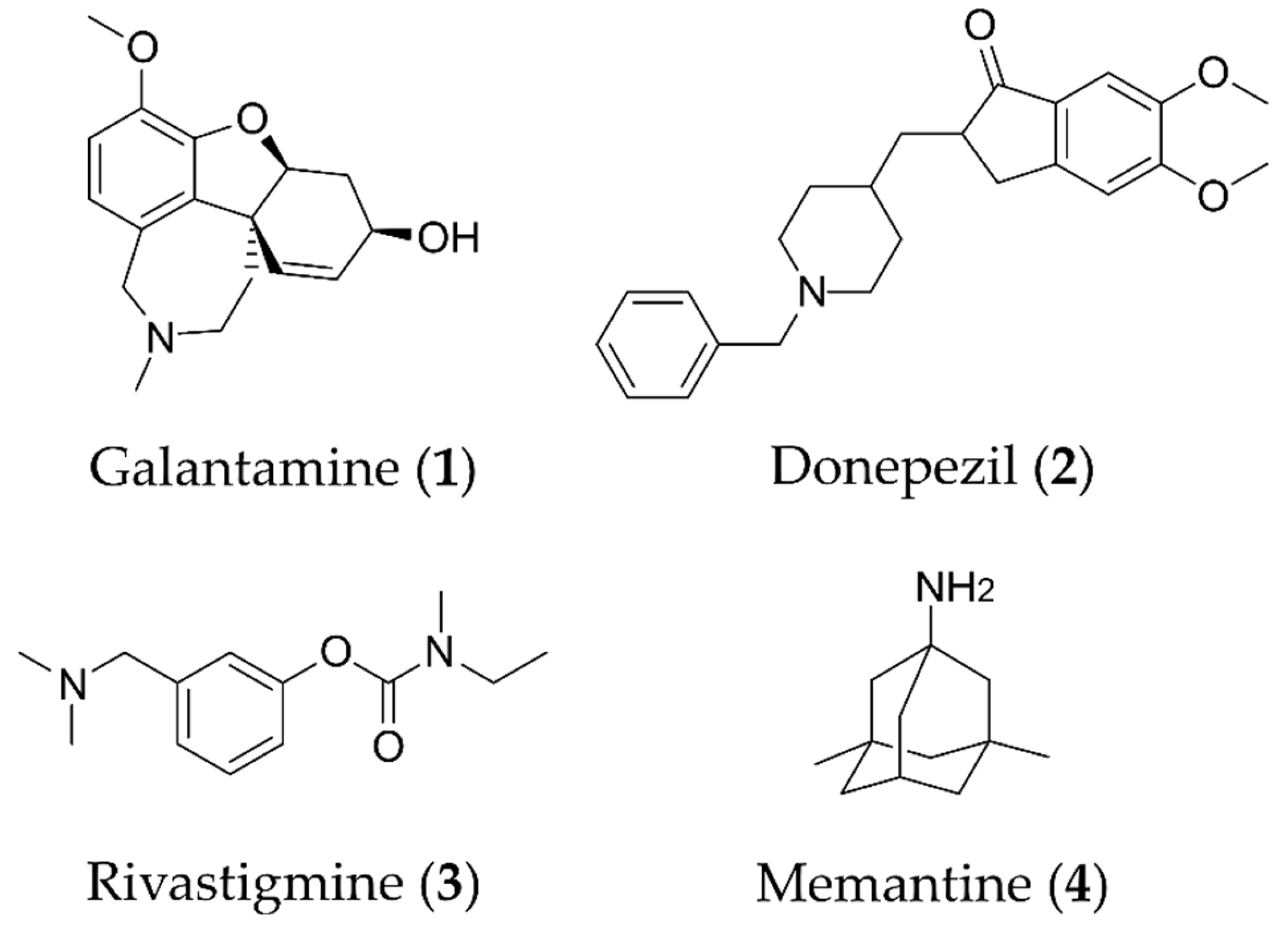
1. Introduction
2. Results
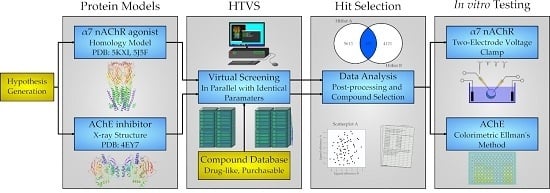
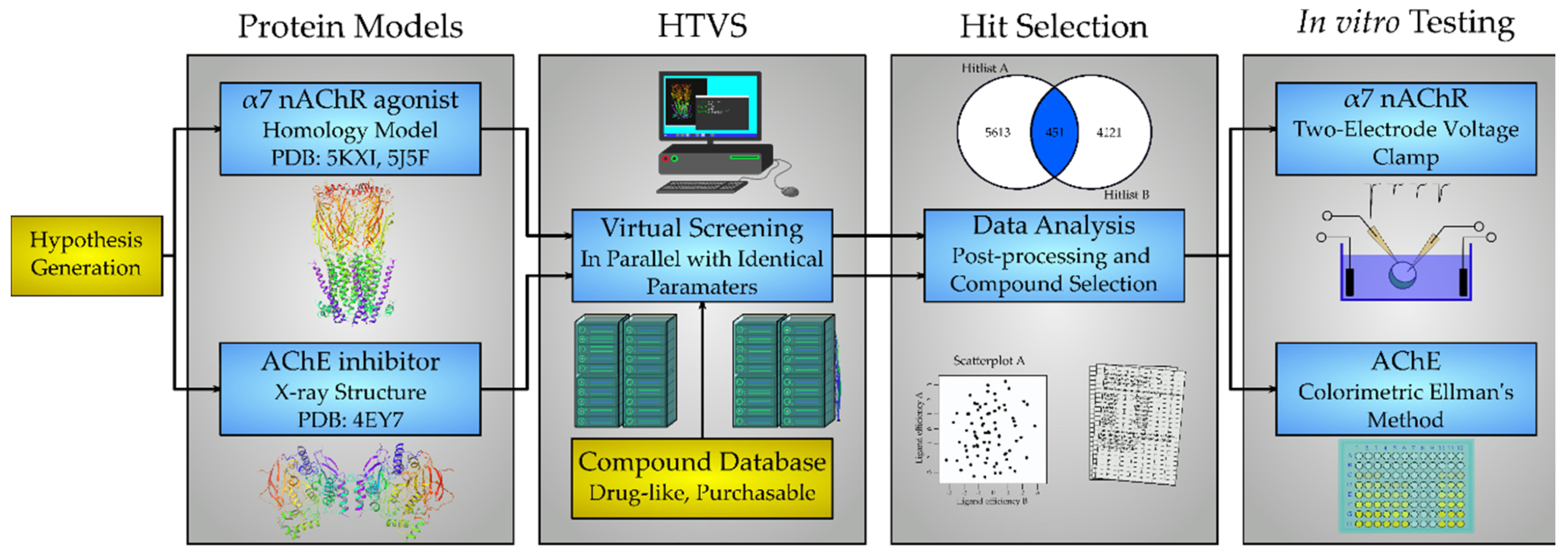
2.1. Protein Structures and Homology Modeling
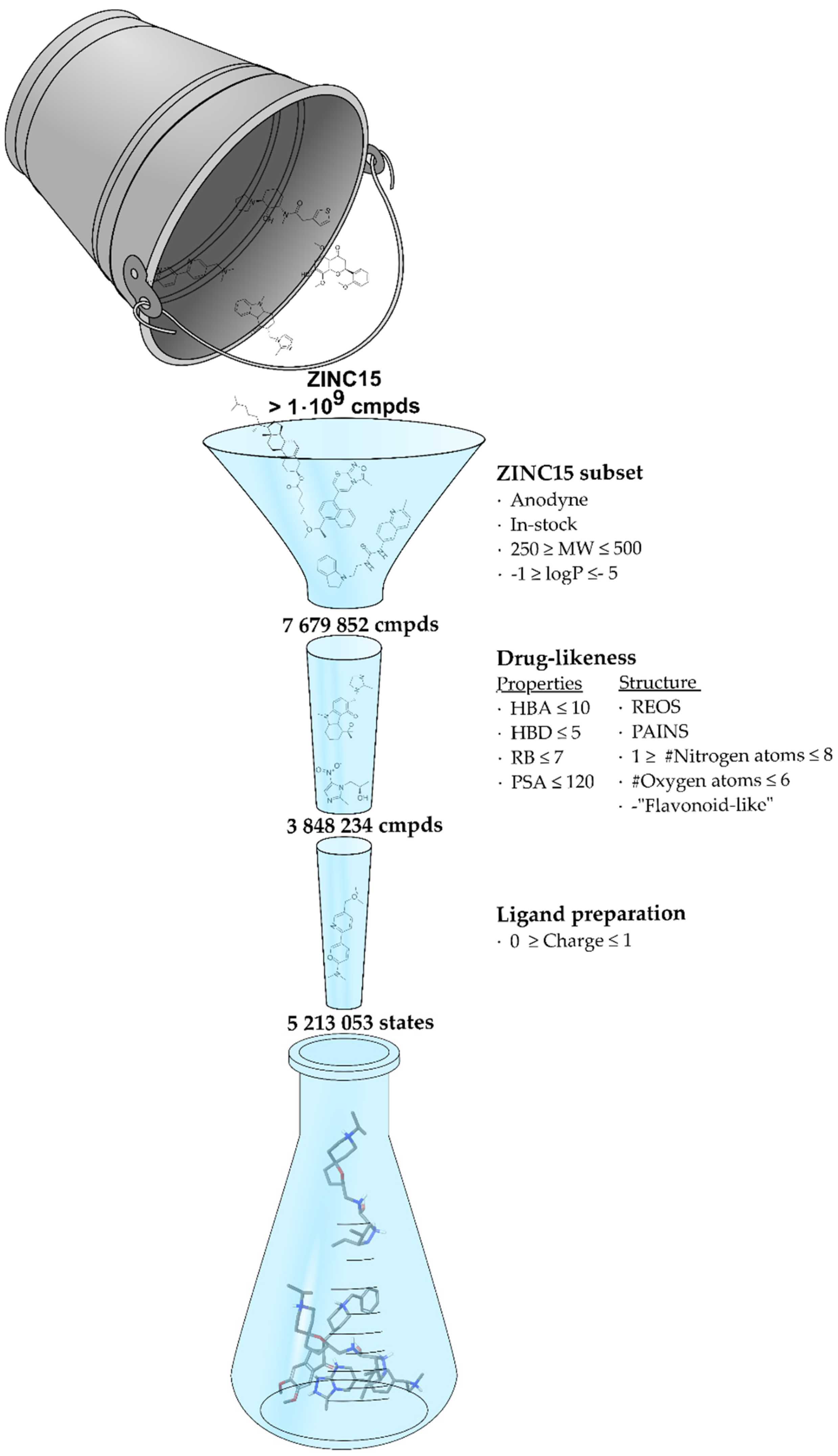
2.2. Screening Database
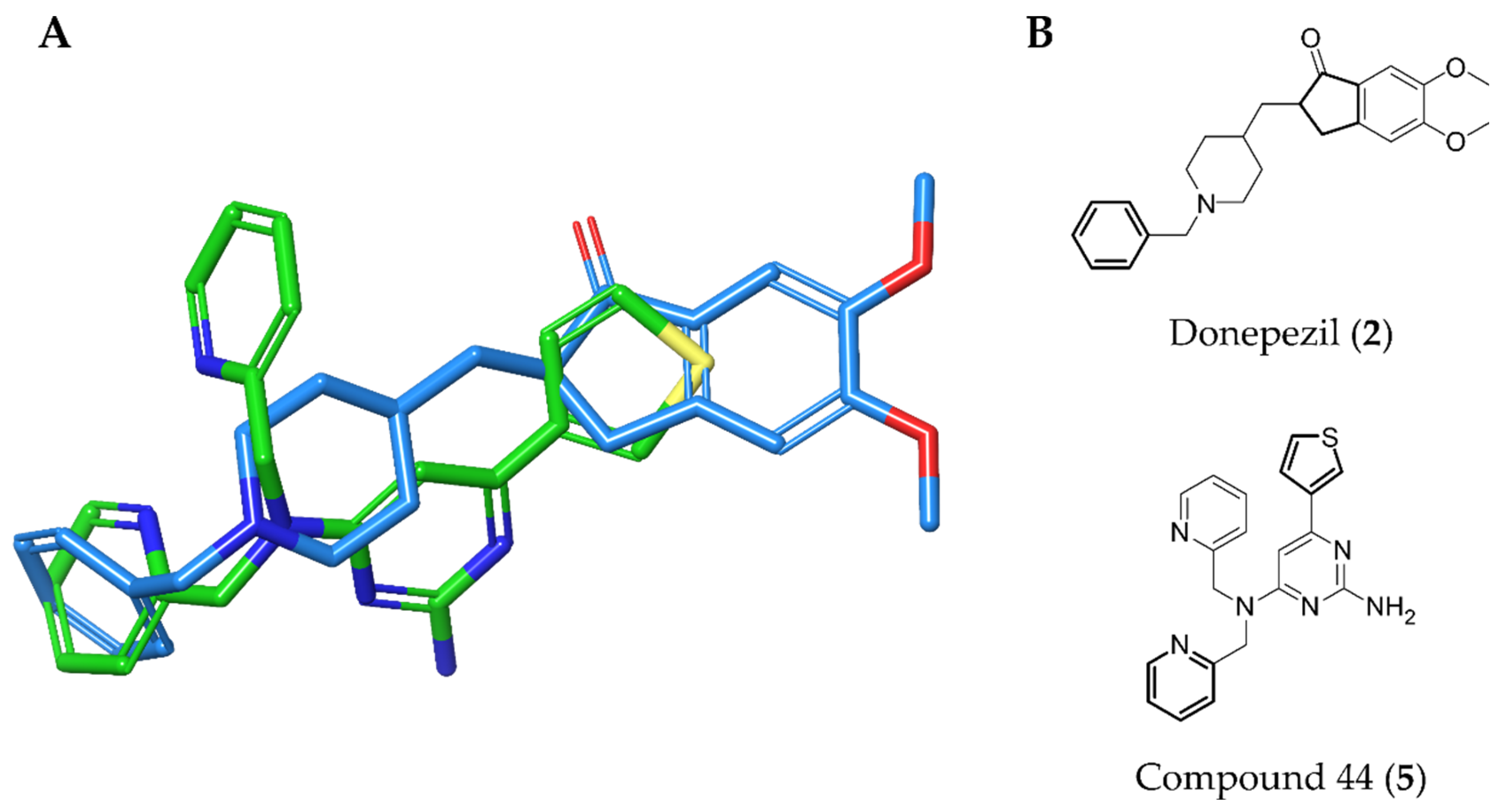
2.3. Virtual Screening
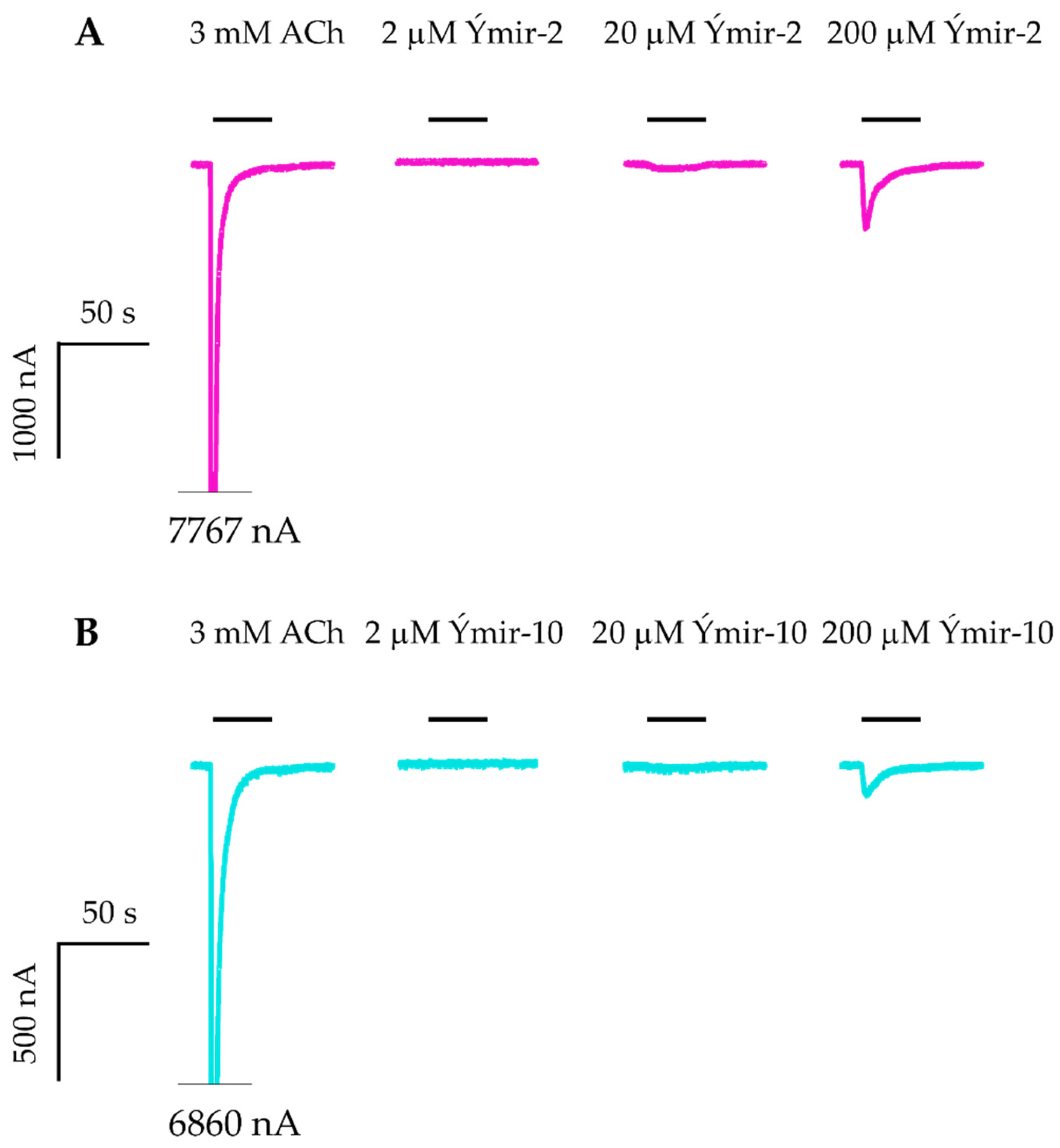
2.4. AChE and nAChR α7 Activity Testing
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Protein Models
4.2.1. nAChR α7 Homology Model
4.2.2. AChE Model
4.3. Screening Library
4.4. Virtual Screening
4.5. AChE Activity Testing
4.6. nAChR α7 Activity Testing
4.6.1. Molecular Biology
4.6.2. Expression of α7 nAChR in Xenopus laevis Oocytes
4.6.3. Oocyte Electrophysiology
4.7. Data Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Model. Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, C.; World Alzheimer. Report 2018: The state of the art of dementia research: New frontiers. ADI 2018, 15, 1473. [Google Scholar]
- Perl, D.P. Neuropathology of Alzheimer’s disease. Mt. Sinai J. Med. 2010, 77, 32–42. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R. Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Del Tredici, K.; et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Cerejeira, J.; Lagarto, L.; Mukaetova-Ladinska, E.B. Behavioral and psychological symptoms of dementia. Front. Neurol. 2012, 3, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Linde, R.M.; Dening, T.; Stephan, B.C.; Prina, A.M.; Evans, E.; Brayne, C. Longitudinal course of behavioural and psychological symptoms of dementia: Systematic review. Br. J. Psychiatry 2016, 209, 366–377. [Google Scholar] [CrossRef]
- Zemek, F.; Drtinova, L.; Nepovimova, E.; Sepsova, V.; Korabecny, J.; Klimes, J. Kuca, K. Outcomes of Alzheimer’s disease therapy with acetylcholinesterase inhibitors and memantine. Expert Opin. Drug Saf. 2014, 13, 759–774. [Google Scholar]
- Allain, H.; Bentue-Ferrer, D.; Tribut, O.; Gauthier, S.; Michel, B.F.; Drieu-La Rochelle, C. Alzheimer’s disease: The pharmacological pathway. Fundam. Clin. Pharmacol. 2003, 17, 419–428. [Google Scholar] [CrossRef]
- Tan, C.C.; Yu, J.T.; Wang, H.F.; Tan, M.S.; Meng, X.F.; Wang, C.; Jiang, T.; Zhu, X.C.; Tan, L. Efficacy and safety of donepezil, galantamine, rivastigmine, and memantine for the treatment of Alzheimer’s disease: A systematic review and meta-analysis. J. Alzheimers Dis. 2014, 41, 615–631. [Google Scholar] [CrossRef]
- Schmitt, B.; Bernhardt, T.; Moeller, H.J.; Heuser, I.; Frolich, L. Combination therapy in Alzheimer’s disease: A review of current evidence. CNS Drugs 2004, 18, 827–844. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, S.; Kishi, T.; Iwata, N. Combination Therapy with Cholinesterase Inhibitors and Memantine for Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Int. J. Neuropsychopharmacol. 2015, 18, P859–P860. [Google Scholar] [CrossRef] [PubMed]
- Namzaric (Memantine Hydrochloride Extended-Release/Donepezil Hydrochloride) Capsules; FDA: Silver Spring, MD, USA, 2014.
- Kumar, A.; Singh, A.; Ekavali. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef] [PubMed]
- Rosini, M. Polypharmacology: The rise of multitarget drugs over combination therapies. Future Med. Chem. 2014, 6, 485–487. [Google Scholar] [CrossRef]
- Van der Schyf, C.J.; Geldenhuys, W.J. Multimodal drugs and their future for Alzheimer’s and Parkinson’s disease. Int. Rev. Neurobiol. 2011, 100, 107–125. [Google Scholar] [PubMed]
- Rosini, M.; Simoni, E.; Caporaso, R.; Minarini, A. Multitarget strategies in Alzheimer’s disease: Benefits and challenges on the road to therapeutics. Future Med. Chem. 2016, 8, 697–711. [Google Scholar] [CrossRef]
- Talevi, A. Multi-target pharmacology: Possibilities and limitations of the “skeleton key approach” from a medicinal chemist perspective. Front. Pharmacol. 2015, 6, 205. [Google Scholar] [CrossRef] [Green Version]
- Michalska, P.; Buendia, I.; Barrio, L.D.; Leon, R. Novel Multitarget Hybrid Compounds for the Treatment of Alzheimer’s Disease. Curr. Top. Med. Chem. 2017, 17, 1027–1043. [Google Scholar] [CrossRef]
- Zhou, S.; Li, Y.; Hou, T. Feasibility of using molecular docking-based virtual screening for searching dual target kinase inhibitors. J. Chem. Inf. Model. 2013, 53, 982–996. [Google Scholar] [CrossRef] [PubMed]
- McKie, S.A. Polypharmacology: In silico methods of ligand design and development. Future Med. Chem. 2016, 8, 579–602. [Google Scholar] [CrossRef] [PubMed]
- Kowal, N.M.; Indurthi, D.C.; Ahring, P.K.; Chebib, M.; Olafsdottir, E.S.; Balle, T. Novel approach for the search for chemical scaffolds with activity at both acetylcholinesterase and the alpha 7 nicotinic acetylcholine receptor: A perspective on scaffolds with dual activity for the treatment of neurodegenerative disorders. Molecules 2019, 24, 446. [Google Scholar] [CrossRef] [Green Version]
- Zoli, M.; Pistillo, F.; Gotti, C. Diversity of native nicotinic receptor subtypes in mammalian brain. Neuropharmacology 2015, 96, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Feather-Stone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef]
- Morales-Perez, C.L.; Noviello, C.M.; Hibbs, R.E. X-ray structure of the human alpha4beta2 nicotinic receptor. Nature 2016, 538, 411–415. [Google Scholar] [CrossRef] [Green Version]
- Kaczanowska, K.; Camacho Hernandez, G.A.; Bendiks, L.; Kohs, L.; Cornejo-Bravo, J.M.; Harel, M.; Finn, M.G.; Taylor, P. Substituted 2-aminopyrimidines selective for alpha7-nicotinic acetylcholine receptor activation and association with acetylcholine Binding Proteins. J. Am. Chem. Soc. 2017, 139, 3676–3684. [Google Scholar] [CrossRef]
- Shen, M.Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [Green Version]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B., 3rd; de Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Calpha geometry: Phi, psi and Cbeta deviation. Proteins 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15-Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Walters, W.P.; Murcko, A.A.; Murcko, M.A. Recognizing molecules with drug-like properties. Curr. Opin. Chem. Biol. 1999, 3, 384–387. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef]
- da Silva Rocha, S.F.L.; Olanda, C.G.; Fokoue, H.H.; Sant’Anna, C.M.R. Virtual Screening Techniques in Drug Discovery: Review and Recent Applications. Curr. Top. Med. Chem. 2019, 19, 1751–1767. [Google Scholar] [CrossRef]
- Celie, P.H.; van Rossum-Fikkert, S.E.; van Dijk, W.J.; Brejc, K.; Smit, A.B.; Sixma, T.K. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron 2004, 41, 907–914. [Google Scholar] [CrossRef] [Green Version]
- UniProt, C. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [Green Version]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 10 January 2018).
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Metropolis, N.; Ulam, S. The Monte Carlo method. J. Am. Stat. Assoc 1949, 44, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [Green Version]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [Green Version]
- Timmermann, D.B.; Gronlien, J.H.; Kohlhaas, K.L.; Nielsen, E.O.; Dam, E.; Jorgensen, T.D.; Ahring, P.K.; Peters, D.; Holst, D.; Christensen, J.K.; et al. An allosteric modulator of the α7 nicotinic acetylcholine receptor possessing cognition-enhancing properties in vivo. J. Pharmacol. Exp. Ther. 2007, 323, 294–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza, N.R.; Larsen, J.S.; Mathiasen, C.; Jacobsen, T.A.; Munro, G.; Erichsen, H.K.; Nielsen, A.N.; Troelsen, K.B.; Nielsen, E.O.; Ahring, P.K. NS11394 [3′-[5-(1-hydroxy-1-methyl-ethyl)-benzoimidazol-1-yl]-biphenyl-2-carbonitrile], a unique subtype-selective GABAA receptor positive allosteric modulator: In vitro actions, pharmacokinetic properties and in vivo anxiolytic efficacy. J. Pharmacol. Exp. Ther. 2008, 327, 954–968. [Google Scholar] [CrossRef] [PubMed]
- Kowal, N.M.; Ahring, P.K.; Liao, V.W.Y.; Indurti, D.C.; Harvey, B.S.; O’Connor, S.M.; Chebib, M.; Olafsdottir, E.S.; Balle, T. Galantamine is not a positive allosteric modulator of human α4β2 or α7 nicotinic acetylcholine receptors. Br. J. Pharmacol. 2018, 175, 2911–2925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Not available. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. ID | Structure | AChE G-Score a Norm. LE b %Inhibition c,d IC50 (µM)(pIC50 ± SEM) | α7 nAChR G-Score a Norm. LE b %Activation c %Inhibition c | Compd. ID | Structure | AChE G-Score a Norm. LE b %Inhibition c,d IC50 (µM) (pIC50 ± SEM) | α7 nAChR G-Score a Norm. LE b %Activation c %Inhibition c |
|---|---|---|---|---|---|---|---|
| 6 |  | –12.30 –2.11 76.1 ± 0.9% IC50 = 58.47 (4.23 ± 0.02) | –13.85 –1.71 0.4 ± 0.4% 47.2 ± 3% | 14 | 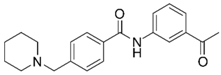 | –13.04 –1.06 100% IC50 = 1.10 (5.96 ± 0.02) | –15.56 –1.09 0.3 ± 0.3% 60.0 ± 1% |
| Ýmir-2 (7)f | 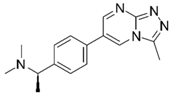 | –14.90 –2.63 98.4 ± 0.9% IC50 = 2.58 (5.59 ± 0.02) | –11.50 –0.65 7.0 ± 0.9% - | Ýmir-10 (15) | 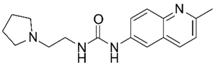 | –11.79 –1.18 66.6 ± 1.7% IC50 = 114.70 (3.94 ± 0.02) | –13.13 –0.94 2.3 ± 0.4% - |
| 8 | 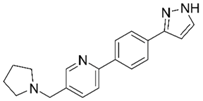 | –12.34 –1.19 100%d - | –16.35 –1.60 0.3 ± 0.3% 59.0 ± 5% | 16 |  | –11.81 –0.50 100%d - - | –16.16 –1.08 1.0 ± 0.0% 90.7 ± 1% |
| 9 |  | –11.91 –1.23 45.9 ± 3.8%d - - | –15.40 –1.54 1.0 ± 0.6% 66.7 ± 4% | 17 |  | –10.22 –0.59 22.9 ± 1.1% - - | –13.35 –1.00 - - |
| 10 | 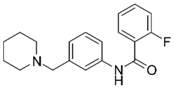 | –12.11 –1.10 90.8 ± 0.2% IC50 = 11.28 (4.95 ± 0.02) | –15.93 –1.50 0.3 ± 0.3% 85.0 ± 2% | 18 |  | –12.92 –0.86 91.6 ± 2.2% IC50 = 17.88 4.75 ± 0.02 | –14.49 –0.71 0.7 ± 0.3% 68.7 ± 1% |
| 11 | 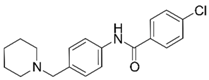 | –12.32 –1.18 100%d - - | –15.16 –1.30 0.7 ± 0.3% 73.3 ± 6% | Ýmir-14 (19) | 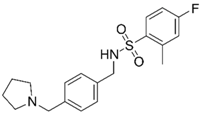 | –12.48 –0.88 100% IC50 = 1.32 (5.88 ± 0.01) | –13.13 –0.52 0.7 ± 0.3% 96.0 ± 1% |
| 12 | 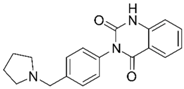 | –12.75 –1.14 63.7 ± 4.4%d - - | –15.61 –1.25 0.3 ± 0.3% 81.7 ± 1% | 20f |  | –13.45 –0.87 86.0 ± 0.1% IC50 = 33.47 (4.48 ± 0.02) | –13.71 –0.42 0.3 ± 0.3% 94.3 ± 1% |
| 13 | 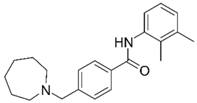 | –13.20 –1.12 82.5 ± 1.1% IC50 = 30.24 (4.52 ± 0.02) | –15.60 –1.10 0.7 ± 0.7% 68.0 ± 2% | Ýmir-16 (21) |  | –12.48 –0.71 87.7 ± 0.6% IC50 = 7.76 (5.10 ± 0.02) | –13.27 –0.44 0.3 ± 0.3% 97.3 ± 1% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oddsson, S.; Kowal, N.M.; Ahring, P.K.; Olafsdottir, E.S.; Balle, T. Structure-Based Discovery of Dual-Target Hits for Acetylcholinesterase and the α7 Nicotinic Acetylcholine Receptors: In Silico Studies and In Vitro Confirmation. Molecules 2020, 25, 2872. https://doi.org/10.3390/molecules25122872
Oddsson S, Kowal NM, Ahring PK, Olafsdottir ES, Balle T. Structure-Based Discovery of Dual-Target Hits for Acetylcholinesterase and the α7 Nicotinic Acetylcholine Receptors: In Silico Studies and In Vitro Confirmation. Molecules. 2020; 25(12):2872. https://doi.org/10.3390/molecules25122872
Chicago/Turabian StyleOddsson, Sebastian, Natalia M. Kowal, Philip K. Ahring, Elin S. Olafsdottir, and Thomas Balle. 2020. "Structure-Based Discovery of Dual-Target Hits for Acetylcholinesterase and the α7 Nicotinic Acetylcholine Receptors: In Silico Studies and In Vitro Confirmation" Molecules 25, no. 12: 2872. https://doi.org/10.3390/molecules25122872