
A SF5 Derivative of Triphenylphosphine as an Electron-Poor Ligand Precursor for Rh and Ir Complexes


Department of Chemistry, Humboldt–Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany
*
Author to whom correspondence should be addressed.
Molecules 2020, 25(17), 3977; https://doi.org/10.3390/molecules25173977
Submission received: 11 August 2020
/
Revised: 25 August 2020
/
Accepted: 26 August 2020
/
Published: 1 September 2020
(This article belongs to the Special Issue Boron in Catalysis and Materials Chemistry: A Themed Issue in Honor of Professor Todd B. Marder on the Occasion of His 65th Birthday)
Abstract
:The synthesis of the triarylphosphine, P(p-C6H4SF5)3 containing a SF5 group, has been achieved. The experimental and theoretical studies showed that P(p-C6H4SF5)3 is a weaker σ-donor when compared with other substituted triarylphosphines, which is consistent with the electron-withdrawing effect of the SF5 moiety. The studies also revealed a moderate air stability of the phosphine. The σ-donor capabilities of P(p-C6H4SF5)3 were estimated from the phosphorus-selenium coupling constant in SeP(p-C6H4SF5)3 and by DFT calculations. The behavior of P(p-C6H4SF5)3 as ligand has been investigated by the synthesis of the iridium and rhodium complexes [MCl(COD){P(p-C6H4SF5)3}], [MCl(CO)2{P(p-C6H4SF5)3}2] (M = Ir, Rh), or [Rh(µ-Cl)(COE){P(p-C6H4SF5)3}]2, and the molecular structures of [IrCl(COD){P(p-C6H4SF5)3}] and [Rh(µ-Cl)(COE){P(p-C6H4SF5)3}]2 were determined by single X-ray diffraction. The structures revealed a slightly larger cone angle for P(p-C6H4SF5)3 when compared to other para-substituted triarylphosphines.
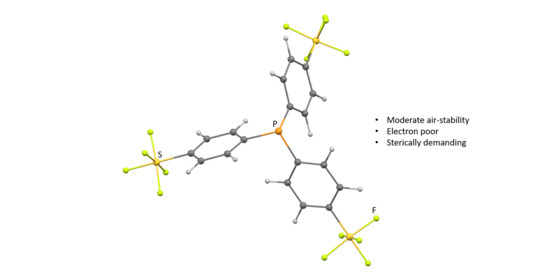
1. Introduction
Phosphines are one of the most important and widely used ligands in homogeneous catalysis and organometallic chemistry due to the extensive options to modify their electronic and steric properties by varying their substitution pattern [1,2,3,4]. Phosphines are σ-donor and π-acceptor ligands and the choice of the phosphorus-bound entities can effectively tune their electronic characteristics. Thus, the electronic, but also steric properties of triarylphosphines are highly dependent on the employed aryl groups [1,2,3,4].
Triarylphosphines bearing fluorine atoms or fluorinated substituents are characterized by a higher steric demand and a lower basicity than the non-halogenated counterparts which might lead to changes in the reactivity [5]. Thus, complexes bearing fluorinated triarylphosphines have been widely investigated over the last years [5,6,7,8,9,10,11,12,13]. In particular, arylphosphines containing a CF3 group have been studied for catalytic processes such as hydroformylation [14], C-C coupling [13,15,16,17] as well as hydroalkylation reactions [18]. They present an alternative to fluoroarylphosphines with respect to the increased electron-withdrawing properties of the phosphine.
Another alternative and fascinating chemically and thermally rather stable group is the SF5 group [19,20], which displays a large σ-withdrawing nature (F = 0.56) and π-withdrawing ability (R = 0.12) based on the Swann-Lupton constants. In addition, phenyl groups with SF5 moieties at both meta and para positions exhibit a stronger electron-withdrawing effect according to Hammett constants (σm = 0.61, σp = 0.68), when compared with a fluorine substituent (F = 0.45, R = −0.39, σm = 0.34, σp = 0.06) or the trifluoromethyl group (F = 0.38, R = 0.16, σm = 0.43, σp = 0.54) [21]. Despite the increasing interest on SF5-containing compounds described in literature and their applications as building blocks in many fields such as agrochemical, medicinal, or materials chemistry [22,23,24,25,26,27,28,29,30], SF5 derivatized ligands in transition metal complexes are still rare [31,32,33,34,35,36,37,38,39,40,41]. Examples mainly focus on C^N cyclometalated ligands which contain the SF5 moiety in their phenyl rings in order to stabilize iridium(III) [34,35,36,37] or platinum(II) [38] complexes with optoelectronic properties.
Herein, we present the synthesis and characterization of a SF5 derivative of triphenylphosphine P(p-C6H4SF5)3 (1). Its air stability and electronic properties have been estimated and the derivatives O=P(p-C6H4SF5)3 (2) and Se=P(p-C6H4SF5)3 (3) were prepared. In addition, the rhodium and iridium complexes [MCl(COD){P(p-C6H4SF5)3}] (M = Ir (4), Rh (5)), [MCl(CO)2{P(p-C6H4SF5)3}2] (M = Ir (8), Rh (9)) or [Rh(µ-Cl)(COE){P(p-C6H4SF5)3}]2 (6) were synthesized.
2. Results and Discussion
2.1. Synthesis and Air-Stability of P(p-C6H4SF5)3 (1)
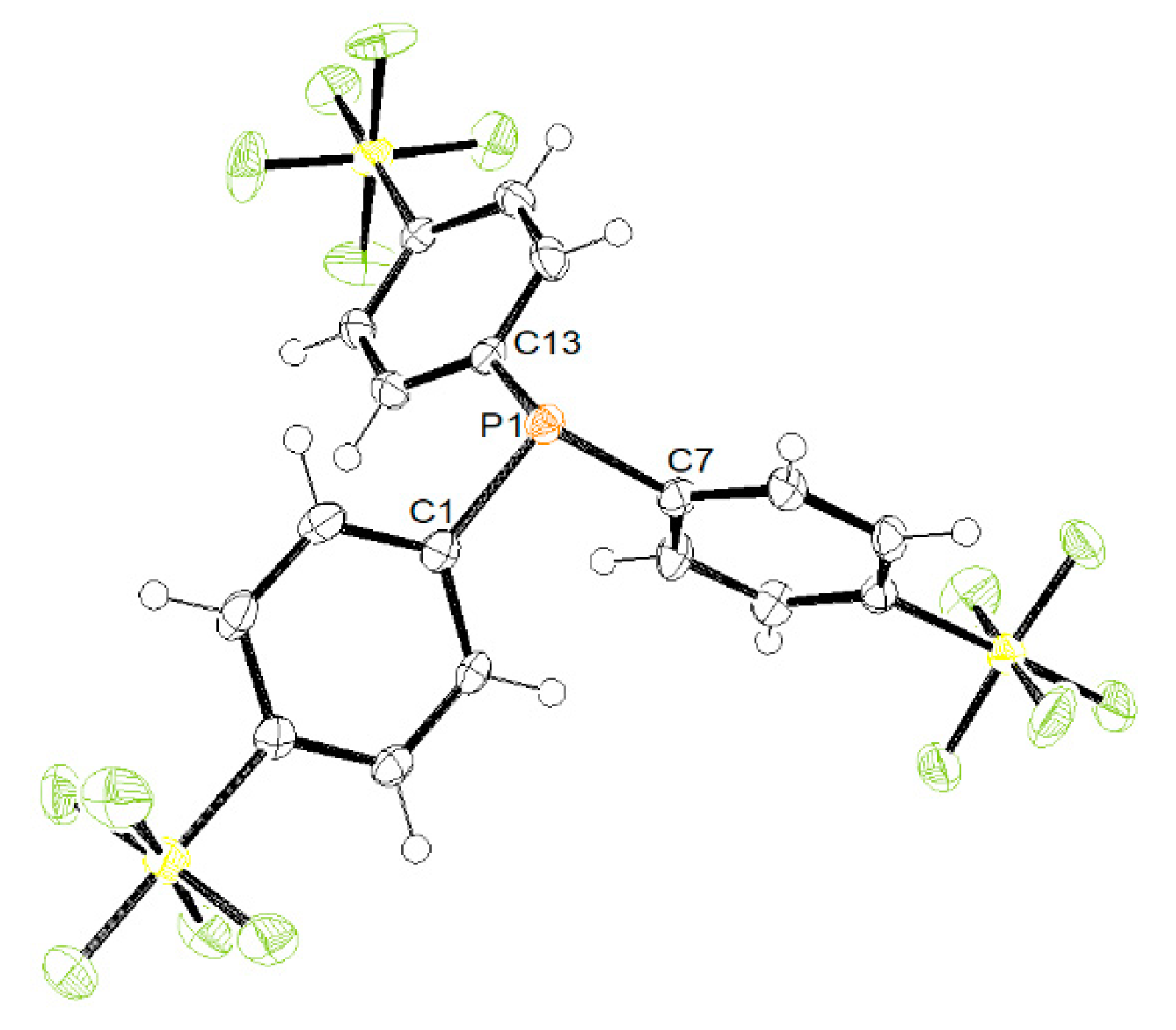
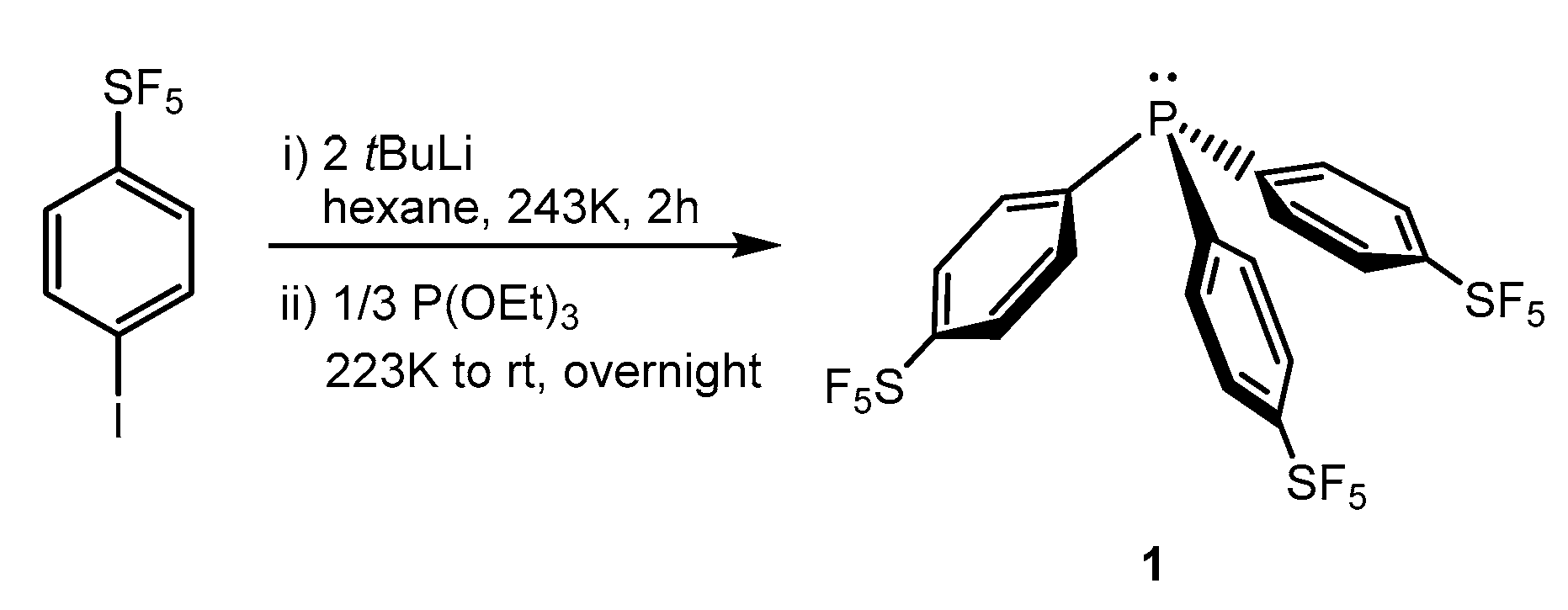
Treatment of 4-iodophenylsulfur pentafluoride with an excess of tBuLi and a subsequent reaction with triethylphosphite afforded the tris(p-pentafluorosulfanylphenyl)phosphine (1) in 39% isolated yield (Scheme 1). Compound 1 shows in the 19F NMR spectrum the characteristic signal pattern for the SF5 moiety [42], a doublet at 62.6 ppm corresponding to the four equatorial equivalent fluorine atoms and a pentet at 83.7 ppm for the axial fluorine with coupling constants of 150 Hz. The 31P{1H} NMR spectrum shows a singlet at −7.8 ppm, whereas the GC/MS gave a mass peak of m/z 640.
Single crystals of compound 1, which were suitable for X-ray crystallography were obtained from concentrated solutions in benzene. The molecular structure is shown in Figure 1. Compound 1 shows a trigonal pyramidal arrangement at the phosphorus atom when taking the electron lone-pair into account. The C–P–C angles are comparable to the ones reported for triphenylphosphine, where only one angle is slightly larger (101.72(1)° for PPh3 vs. 99.18(10)° for 1) than the others [43].
In order to test the air-stability of the phosphine 1, a solution of 1 in toluene-d8 was left under air for 2 weeks. Seventy-eight percent of the phosphine 1 was then converted into phosphine oxide 2 (Scheme 2). In the solid state 41% of conversion was achieved after a month of air exposure. These data indicate a higher stability of 1 towards air when compared with the CF3 analogue, which is fully oxidized in the solid state after one month [44]. 31P{1H} NMR data of the solution as well as liquid injection field desorption/ionization mass spectrometry (LIFDI-MS) confirm the formation of the oxide with a shift of the signal in the 31P{1H} NMR to lower field (δ = 21.09 ppm) and a mass peak of m/z 656.
Crystals of 2 suitable for X-ray diffraction analysis were obtained from the reaction solution (Figure 2). The compound shows the expected tetrahedral arrangement with a C–P–C mean angle of 106.6°, a O–P–C mean angle of 112.2° and the P–O bond length of 1.4867(14) Å. All of the data are consistent with these of other reported triarylphosphine oxides derivatives [45].
In order to further compare the air-stability of phosphine 1 with other triarylphosphines, DFT calculations were performed. It has been suggested previously that the steric demand of the phosphines, but also the SOMO energy of radical cations of phosphines can be correlated with their air-stability [46]. Thus, a radical cation with a SOMO at lower energy would be more prone to react with dioxygen generating the corresponding phosphine oxide [46]. Therefore, the energy of the SOMO of different triarylphosphines radical cations was calculated using the CAM-B3LYP functional (Table 1). The phosphine 1 has a SOMO energy of −12.22 eV, which is lower than the energy of other electron-withdrawing triarylphosphines, and lower than the one of the air-stable triphenylphoshine (−11.11 eV). The data are in accordance with the observed moderate air sensitivity of phosphine 1.
2.2. Estimation of the Donor Properties
Different methods have been reported to determine the electronic properties of phosphines. The σ-donor ability increases when the s-character of the lone pair of the phosphine decreases, which is associated with a higher energy level of the HOMO [47,48]. Thus, DFT studies have been performed in order to calculate the energy level of the HOMO of compound 1 and compare it with other triarylphosphines which were also calculated. The data indicate that compound 1 (−8.69 eV) is a less pronounced σ-donor than most of the calculated phosphines (Table 2). It is worth noting that P(p-C6H4CF3)3 (−8.17 eV) seems, according to this data, to be a better σ-donor than 1, whereas P(m-C6H3(CF3)2)3 is a weaker σ-donor (−8.78 eV).
The HOMO energy level and, therefore, the s-character of the lone pair is also related to the phosphorus-selenium coupling constant of the corresponding selenide. This has been commonly used to experimentally estimate the σ-donor abilities of a broad range of phosphines [49,50,51]. Thus, for more electron-withdrawing substituents a larger coupling is expected [51]. Taking this into account, phosphine 1 was reacted with selenium and after 3d, a full conversion to SeP(p-C6H4SF5)3 (3) was observed (Scheme 2). The 31P{1H} NMR spectrum shows a singlet at δ 32.5 ppm with selenium satellites and a phosphorus-selenium coupling constant of 792 Hz. Correspondingly, the resonance in the 77Se NMR spectrum at −273.3 ppm appears as a doublet with the same coupling constant. Among the data for the phosphines shown in Table 2 only P(m-C6H3(CF3)2)3 shows a larger coupling constant of 802 Hz, which is consistent with the lower HOMO energy values.
Another common method to determine the electronic properties of ligands is the calculation of the Tolman’s electronic parameter TEP [52]. This method consists in the analysis of the frequency of the A1 carbonyl vibration mode of [Ni(CO)3L] complexes, which will decrease due to the back-donation into the CO π* orbitals when the ligand L is a better donor. While Tolman experimentally determined the parameter for a broad range of phosphines [52], DFT studies have demonstrated the correlation between the calculated and experimental values for different ligands L [53,54]. Thus, the calculated values determined in this work correlate well with the experimentally obtained for PPh3 and P(p-C6H4Me)3 (Δexp-calc ≈ 5 cm−1) (Table 2) [52]. The data also indicate that the phosphine 1 might be a slightly more electron-withdrawing phosphine than P(p-C6H4CF3)3, but somewhat weaker than P(m-C6H3(CF3)2)3, although the calculated values are very close.
2.3. Synthesis of Iridium and Rhodium Complexes
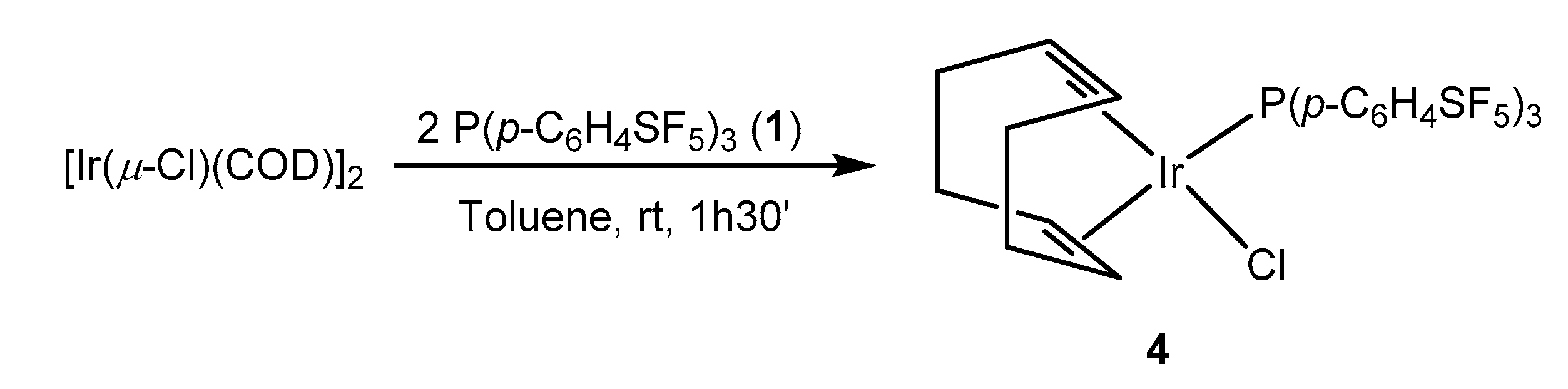
Treatment of the binuclear iridium complex [Ir(µ-Cl)(COD)]2 (COD = 1,5-cyclooctadiene) with two equivalents of the phosphine 1 in toluene yielded the iridium(I) complex [IrCl(COD){P(p-C6H4SF5)3}] (4) (Scheme 3). The same reactivity was reported for other phosphines such as P(p-C6H4CF3)3 [56]. The structure of 4 is supported by the 31P{1H} NMR spectrum, which shows a resonance at δ 21.7 ppm and the 1H NMR spectrum with two resonances at δ 5.64 and 2.45 ppm corresponding to the olefinic protons of the COD ligand in a trans arrangement to the phosphine and the chlorido ligands. The LIFDI mass spectrometry reveals a mass peak of m/z 976.
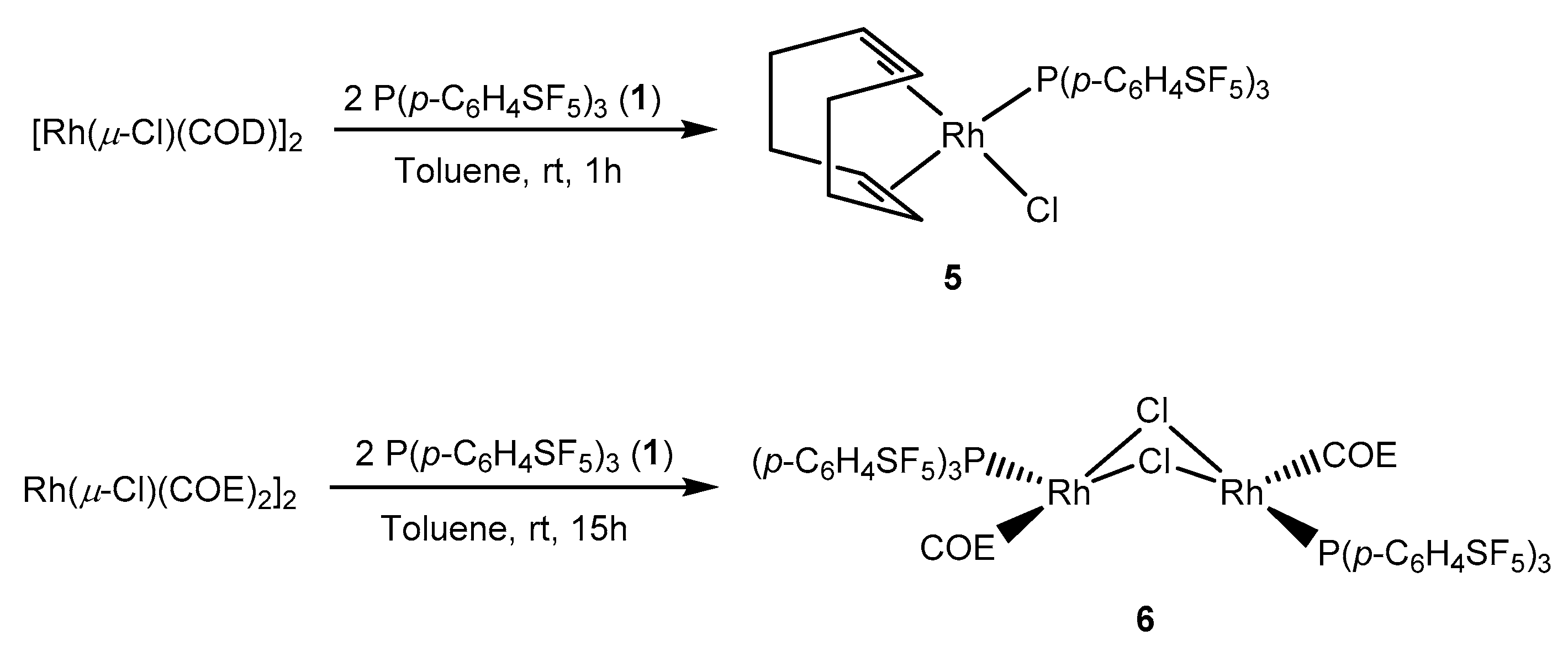
Analogously, [Rh(µ-Cl)(COD)]2 reacted with two equivalents of the phosphine 1 affording the rhodium(I) complex [RhCl(COD){P(p-C6H4SF5)3}] (5) (Scheme 4). The 31P{1H} NMR spectrum of 5 depicts a doublet at δ 31.5 ppm with a rhodium-phosphorus coupling constant of 155.9 Hz. The coupling constant of 5 is 1.8 Hz higher than the one for [RhCl(COD){P(p-C6H4CF3)3}] (1JP-Rh = 154.1 Hz) [57]. As for the iridium complex 4, 1H NMR spectrum of complex 5 shows two resonances for the olefinic protons of the cyclooctadiene ligand at δ 5.67 and 3.12 ppm which are also consistent with the CF3 derivative [57].
In contrast, when [Rh(µ-Cl)(COE)2]2 (COE = cis-cyclooctene) was used as binuclear starting compound, the reaction with two equivalents of phosphine 1 selectively provided trans-[Rh(µ-Cl)(COE){P(p-C6H4SF5)3}]2 (6) (Scheme 5). The dimeric nature of the product is supported by the 31P{1H} NMR data revealing a signal at δ 55.5 ppm with an increased value of the rhodium-phosphorus coupling constant (1JP-Rh = 194.5 Hz). This coupling constant value is slightly larger than for other trans-[Rh(µ-Cl)(COE)(L)]2 complexes, for which L is a σ-donor phosphine (1JP-Rh = 183–188Hz) [58,59].
2.4. Steric Properties of 1
Steric properties of phosphines is another important feature when studying a ligand. The Tolman cone angle [52] of a phosphine is a widely used parameter to describe the steric effects in phosphines and can be estimated not only from molecular models but also from molecular structures determined by X-ray crystallography [60].
The molecular structures of the complexes 4 and 6 were obtained by X-ray diffraction analysis from concentrated C6H6 solutions (Figure 3 and Figure 4). In the case of complex 4, the iridium center displays a square planar coordination geometry when considering COD as a bidentate ligand with an Ir–P bond distance of 2.2888(12) Å, which is slightly shorter than the corresponding separation in [IrCl(COD){P(C6H5)3}] (2.3172(9) Å) [61].
The Tolman cone angle of compound 1 can be estimated to be 150.8° as the average value for the complexes 4 and 6, and by considering van der Waals radii of 1.20 Å and 1.47 Å for the H and F nuclei, respectively [62]. For the calculations, the algorithm reported by Müller and Mingos was used and the metal-phosphorus distances were fixed to 2.28 Å [60]. The obtained value is slightly larger than for other triarylphosphines with a substituent at the para position P(p-C6H4X)3 (X = NMe2, Me, OMe, F, CF3), all of which have a cone angle of 145° [52,63].
2.5. Reactivity of Complexes 4 and 5 towards CO
Bubbling of CO into a dichloromethane solution of [IrCl(COD){P(p-C6H4SF5)3}] (4) resulted in a partial conversion of 4 to yield a complex, for which we suggest the structure [IrCl(CO)(COD){P(p-C6H4SF5)3}] (7). However, when a degassed solution was treated with CO gas, full conversion of complex 4 was observed and a dark unidentified precipitate was formed. In the solution, the formation of a complex containing both COD and CO ligands without the presence of phosphine 1 was detected. Unfortunately, no further identification of this complex was possible. In addition, two products bearing the phosphine ligand 1 in a 1:1 ratio were obtained. Thus complex [IrCl(CO)(COD){P(p-C6H4SF5)3}] (7) was formed together with a second complex, the analytical data of which are consistent with the structure [IrCl(CO)2{P(p-C6H4SF5)3}2] (8). To support the structural assignments further, 13C labeled carbon monoxide was reacted with a solution of complex 4 to give 7′ and 8′. However, the formation of the unknown complex as well as the mixture of products was avoided and a full conversion of 4 into complexes 8 or 8′ was achieved, when the reaction was performed in presence of one equivalent of phosphine 1 (Scheme 5).
Complexes 7 and 8 show singlet resonances for the phosphine ligands at δ 3.65 and 0.84 ppm, respectively, in the 31P{1H} NMR spectrum. In addition, the signals for the olefinic protons of COD in complex 7 appear at δ 4.25 and 3.91 ppm, which would correspond to the protons trans to the CO and Cl ligands, respectively, suggesting that the phosphine ligand is in a apical position. The 31P{1H} NMR spectrum for the mixture of complexes 7′ and 8′ showed the two resonances at δ 3.52 and 0.84 ppm as a doublet (2JP-C = 13.7 Hz) and a triplet (2JP-C = 13.5 Hz), respectively. Note that the values of the carbon-phosphorus coupling constants are consistent with cis arrangements [64,65]. On the other hand, in the 13C{1H} NMR spectrum a doublet (δ = 175.6 ppm, complex 7′) and a triplet (δ = 179.6 ppm, complex 8′) with similar coupling constants are observed for the carbonyl ligands.
The trigonal bipyramid proposed for complex 8 is supported by the IR data in the solid state. The IR spectrum of complex 8 shows two bands at 1940 and 1986 cm−1 for the symmetric and the asymmetric stretching bands of the CO ligands, which shift to 1896 and 1933 cm−1 for 8′ suggesting that the CO ligands are in an equatorial position (see Figure S39). This data for 8 are in the same range as the ones observed for [IrCl(CO)2(PPh3)2] [66].
For comparison, [RhCl(COD){P(p-C6H4SF5)3}] (5) was also reacted with CO or 13CO in the presence of one equivalent of phosphine 1 to give complexes, for which we suggest the structures trans,trans-[RhCl(CO)2{P(p-C6H4SF5)3}2] (9) and trans,trans-[RhCl(13CO)2{P(p-C6H4SF5)3}2] (9′) (Scheme 5). The NMR data of 9 showed a broad band in the 31P{1H} NMR spectrum at room temperature at δ 28.6 ppm. When the sample was measured at −70 °C, the coupling to rhodium was observed (1JP-Rh = 130 Hz). As also found for the PPh3 analogue [67], the coupling to carbon in the 13C labeled complex was not observed even not at −70 °C. The 13C{1H} NMR spectrum revealed a Rh-C coupling constant of 72 Hz in the doublet at 186.5 ppm. Finally, in the IR spectrum, one unique band was observed for the CO ligands at 1992 and 1945 cm−1 for the complexes 9 and 9′, respectively (see Figure S40). This is consistent with data for [RhCl(CO)2(PPh3)2], where only one stretching band was observed for CO at 1990 cm−1 and a square pyramidal structure with the chlorido ligand in the apical position as the most probable structure was proposed [67].
3. Materials and Methods
3.1. General Procedures, Methods and Materials
All experiments were carried out under an atmosphere of argon by Schlenk techniques. Solvents were dried by the usual procedures [68] and, prior to use, distilled under argon. The rhodium and iridium complexes [Rh(µ-Cl)(COE)2]2 and [Ir(µ-Cl)(COD)]2 were prepared as described in the literature [69,70]. All reagents were obtained from commercial sources. Unless stated, NMR spectra were recorded at room temperature on a Bruker DPX 300 (Bruker BioSpin, Rheinstetten, Germany,) or a Bruker Avance 300 spectrometer (Bruker BioSpin, Rheinstetten, Germany). 1H and 13C{1H} signals are referred to residual solvent signals, those of 31P{1H} to external 85% H3PO4, the 19F NMR spectra to external CFCl3 and the 77Se NMR spectra to external SePh2 (δ = 414 ppm). 1H and 13C{1H} NMR signal assignments were confirmed by 1H{31P}, 1H,1H COSY, 1H,13C HMQC and 1H,13C HMBC NMR experiments. Mass spectra were measured with a Micromass Q–Tof–2 instrument equipped with a Linden LIFDI source (Linden CMS GmbH, Weyhe, Germany). GC/MS analyses were performed with an Agilent 6890N gas–phase chromatograph (Shimadzu, Berlin, Germany) equipped with an Agilent 5973 Network mass selective detector at 70eV. Infrared spectra were recorded with the Platinum ATR module of a Bruker FT-IR Alpha II spectrometer (Bruker Optics, Leipzig, Germany) equipped with an ATR unit (diamond). NMR spectra are included as Supplementary Material (Figures S1–S38).
3.2. Synthesis of Tris-(p-pentafluorosulfanylphenyl)phosphine (1)
4-iodophenylsulfur pentafluoride (250 mg, 0.76 mmol) was dissolved in 10 mL of hexane at 243 K. Then, two equivalents of tert–buthyllithium (1.7 M in pentanes, 1.52 mmol, 0.9 mL) was added dropwise to the solution and the reaction mixture was stirred for 2 h at 243 K. Afterwards, the mixture was cooled down to 223 K and triethylphosphite (0.25 mmol, 45 µL) was added slowly. The mixture was stirred while warming up overnight. The volatiles were removed under vacuum, toluene (2 × 10 mL) was added and the product extracted. The solvent was removed from the extract and the beige solid obtained dried under vacuum. Yield: 190 mg (39%).
GC–MS (toluene): Calculated (m/z) for [M]: 640.43; found: 640. 31P{1H} NMR (121.5 MHz, C6D6): δ = −7.8 (s) ppm. 1H NMR (300.1 MHz, C6D6): δ = 7.26 (dm, 3J(H,H) = 8.7 Hz, 6H, m–CH); 6.74 (dd, 3J(H,H) = 8.6, 2J(H,P) = 6.8 Hz, 6H, o–CH) ppm. 19F NMR (282.4 MHz, C6D6): δ = 83.7 (p, 2J(F,F) = 150 Hz, 1F, SF5); 62.6 (d, 2J(F,F) = 150 Hz, 4F, SF5) ppm. 13C{1H} NMR (75.4 MHz, CD2Cl2): 155.5–154.1 (m, Cq–SF5); 140.6 (d, 1J(C,P) = 15.4 Hz, P–Cq); 134.5 (d, 2J(C,P) = 20.9 Hz, o–CH); 126.8 (dp, 3J(C,P) = 6.8 Hz, 3J(C,Feq) = 4.3 Hz, m–CH) ppm.
3.3. Formation of Tris-(p-pentafluorosulfanylphenyl)phosphine oxide (2)
Method a: Tris-(p-pentafluorosulfanylphenyl)phosphine 1 (20 mg, 0.03 mmol) was dissolved in toluene-d8 (0.4 mL) and the solution was exposed under air. The conversion was followed by NMR spectroscopy, and after 2 weeks 78% conversion was observed.
Method b: Tris-(p-pentafluorosulfanylphenyl)phosphine 1 (20 mg, 0.03 mmol) was exposed under air in an open vial. After 1 month, a 41% conversion to compound 2 was found.
LIFDI (toluene-d8): Calculated (m/z) for [M]: 656.4; found: 656. 31P{1H} NMR (121.5 MHz, toluene–d8): δ = 21.1 (s) ppm. 1H NMR (300.1 MHz, toluene–d8): δ = 7.30 (dd, 3J(H,H) = 8.3; 3J(H,P) = 2.2 Hz, 6H, m-CH); 7.21 (dd, 2J(H,P) = 10.9, 3J(H,H) = 8.3 Hz, 6H, o-CH) ppm. 19F NMR (282.4 MHz, toluene-d8): δ = 82.7 (p, 2J(F,F) = 150 Hz, 1F, SF5); 62.3 (d, 2J(F,F) = 150 Hz, 4F, SF5) ppm.
3.4. Synthesis of Tris-(p-pentafluorosulfanylphenyl)phosphine Selenide (3)
Tris-(p-pentafluorosulfanylphenyl)phosphine 1 (50 mg, 0.08 mmol) was dissolved in toluene (10 mL) and one equivalent of selenium (6 mg, 0.08 mmol) was added. Then, the reaction mixture was stirred at 343 K for 3 days. The reaction solution was filtered and the volatiles were removed from the filtrate. The dark solid was dried in vacuum. Yield: 54 mg (94%).
LIFDI (toluene-d8): Calculated (m/z) for [M]: 719.39; found: 719. 31P{1H} NMR (202.4 MHz, toluene–d8): δ = 32.5 (s + sat, 1J(P,Se) = 791.8 Hz) ppm. 77Se NMR (95.4 MHz, toluene-d8): δ = −273.3 (d, 1J(Se,P) = 792.3 Hz) ppm. 1H NMR (500.1 MHz, toluene-d8): δ = 7.27 (dd, 2J(H,P) = 12.8; 3J(H,H) = 8.7 Hz, 6H, o-CH); 7.20 (dd, 3J(H,H) = 8.8, 3J(H,P) = 2.2 Hz, 6H, m-CH) ppm. 19F NMR (470.6 MHz, toluene-d8): δ = 82.4 (p, 2J(F,F) = 150 Hz, 1F, SF5); 62.3 (d, 2J(F,F) = 150 Hz, 4F, SF5) ppm.
3.5. Synthesis of [IrCl(COD){P(p-C6H4SF5)3}] (4)
[Ir(µ-Cl)(COD)]2 (100 mg, 0.15 mmol) (COD = cyclooctadiene) was dissolved in toluene (7 mL) and a solution of P(p-C6H4SF5)3 (1) (192 mg, 0.30 mmol) in 5 mL of toluene was added slowly. After stirring for 1 h 30′, the volatiles were removed under vacuum and a dark red solid was obtained. The solid was washed with hexane (3 × 5 mL) and dried in vacuum. Yield: 117 mg (80%).
LIFDI-TOF-MS (toluene): Calculated (m/z) for [M]+: 976.28; found: 976. 31P{1H} NMR (121.5 MHz, C6D6): δ = 21.7 (s) ppm. 1H NMR (300.1 MHz, C6D6): δ = 7.41–7.23 (m, 12H, Ph); 5.64 (s br, 2H, =CH trans to P); 2.45 (s br, 2H, =CH trans to Cl); 2.16–1.84 (m, 2H, CH2); 1.82–1.57 (m, 2H, CH2); 1.54–1.29 (m, 2H, CH2); 1.17–0.85 (m, 2H, CH2) ppm. 19F NMR (282.4 MHz, C6D6): δ = 83.0 (p, 2J(F,F) = 150 Hz, 1F, SF5); 62.4 (d, 2J(F,F) = 150 Hz, 4F, SF5) ppm.
3.6. Synthesis of [RhCl(COD){P(p-C6H4SF5)3}] (5)
[Rh(µ-Cl)(COD)]2 (50 mg, 0.10 mmol) was dissolved in toluene (5 mL) and P(p-C6H4SF5)3 (1) (130 mg, 0.20 mmol) was added to the solution. After stirring for 1h15′, the volatiles were removed under vacuum and a yellow solid was obtained. The solid was washed with cold hexane (2 × 4 mL) and finally dried in vacuum. The NMR spectra of the yellow solid confirmed the formation of complex 5. Yield: 162 mg (92%).
31P{1H} NMR (121.5 MHz, C6D6): δ = 31.5 (d, 1J(P,Rh) = 155.9 Hz) ppm. 1H NMR (300.1 MHz, C6D6): δ = 7.89–7.78 (m, 12H, Ph); 5.67 (s br, 2H, =CH trans to P); 3.12 (s br, 2H, =CH trans to Cl); 2.55–2.35 (m, 4H, CH2); 2.25–1.94 (m, 4H, CH2) ppm. 19F NMR (282.4 MHz, C6D6): δ = 82.33 (p, 2J(F,F) = 150 Hz, 1F, SF5); 61.89 (d, 2J(F,F) = 150 Hz, 4F, SF5) ppm. 13C{1H} NMR (75.4 MHz, CD2Cl2): 156.4–155.0 (m, Cq–SF5); 135.7 (m, overlapped with CH signals, P–Cq); 135.6 (d, 2J(C,P) = 12.8 Hz, o–CH); 126.4 (dp, 3J(C,P) = 9.6 Hz, 3J(C,Feq) = 4.8 Hz, m–CH); 108.7 (dd, 1J(C,Rh) = 12.0 Hz; 2J(C,P) = 7.2 Hz, =CH trans to P); 72.2 (d, 1J(C,Rh) = 12.8 Hz, =CH trans to Cl); 33.44, 33.43 and 29.2 (all s, CH2) ppm.
3.7. Synthesis of [Rh(µ-Cl)(COE){P(p-C6H4SF5)3}]2 (6)
[Rh(µ-Cl)(COE)2]2 (50 mg, 0.07 mmol) (COE = cyclooctene) was dissolved in toluene (7 mL) and a solution of P(p-C6H4SF5)3 (1) (90 mg, 0.14 mmol) in 5 mL of toluene was added slowly. Instantly, the solution turned red and after stirring for 1 day, the volatiles were removed under vacuum. The red solid obtained was dried in vacuum. Yield: 123 mg (99%)
31P{1H} NMR (121.5 MHz, C6D6): δ = 54.4 (d, 1J(P,Rh) = 194.5 Hz) ppm. 1H NMR (300.1 MHz, C6D6): δ = 7.39–7.28 (m, 6H, m-CH); 7.54-7.40 (m, 6H, o-CH); 2.66–2.20 (m, 6H, COE); 1.55–1.15 (m, 8H, COE) ppm. 19F NMR (282.4 MHz, C6D6): δ = 83.2 (p, 2J(F,F) = 150 Hz, 1F, SF5); 62.4 (d, 2J(F,F) = 150 Hz, 4F, SF5) ppm.
3.8. Reaction of [IrCl(COD){P(p-C6H4SF5)3}] (4) with CO. Formation of [IrCl(CO)(COD){P(p-C6H4SF5)3}] (7) and [IrCl(CO)2{P(p-C6H4SF5)3}2] (8)
In a Young NMR tube, a solution of [IrCl(COD){P(p-C6H4SF5)3}] (4) (25 mg, 0.03 mmol) in CD2Cl2 (0.4 mL) was cooled to 77 K, degassed and treated with CO. After 10′ the solution turned dark brown and a black precipitate was formed. The NMR analysis showed full conversion to yield the complexes 7 and 8 in a 1:1 ratio together with an unknown iridium complex bearing no phosphine ligand.
NMR data for 7: 31P{1H} NMR (121.5 MHz, CD2Cl2): δ = 3.65 (s) ppm. 1H NMR (300.1 MHz, CD2Cl2): δ 7.90–7.82 (m, 12H, Ph); 4.25 (s br, 2H, =CH trans to CO); 3.91 (s br, 2H, =CH trans to Cl); 2.95 (s br, 4H, CH2); 2.79–2.53 (m, 2H, CH2); 2.06-1.83 (m, 2H, CH2) ppm. 19F NMR (282.4 MHz, CD2Cl2): δ = 82.0 (p, 2J(F,F) = 150 Hz, 1F, SF5); 61.9 (d, 2J(F,F) = 150 Hz, 4F, SF5) ppm.
When the reaction was performed with 13CO, the complexes 7′ and 8′ were obtained.
Selected NMR data for 7′: 31P{1H} NMR (121.5 MHz, CD2Cl2): δ = 3.65 (d, 2J(P,C) = 13.7 Hz) ppm. 13C{1H} NMR (75.4 MHz, CD2Cl2): 176.0 (t, 2J(C,P) = 13.6 Hz, 13CO); 135.6-135.2 (m, P–Cq and o–CH);127.1–126.6 (m, m–CH) ppm.
NMR data for the unknown complex: 1H NMR (300.1 MHz, CD2Cl2): δ 4.79 (s br, 4H, =CH); 2.79–2.53 (m, 8H, CH2) ppm. 13C{1H} NMR (75.4 MHz, CD2Cl2): 169.7 (s br, CO); 80.4 (s, =CH); 33.7 (s, CH2) ppm.
3.9. Independent Formation of [IrCl(CO)2{P(p-C6H4SF5)3}2] (8)
In a Young NMR tube, complex [IrCl(COD){P(p-C6H4SF5)3}] (4) (25 mg, 0.03 mmol) was dissolved in C6D6 (0.4 mL) and the phosphine 1 (19 mg, 0.03 mmol) was added. Then, the solution was cooled to 77 K, degassed and treated with CO. After 10 min, the solution turned orange and a yellow precipitate was formed. The solid was filtered off, washed with C6D6 (2 × 0.25 mL) and dried under vacuum. Yield: 34 mg (85%).
31P{1H} NMR (121.5 MHz, CD2Cl2): δ = 0.84 (s) ppm. 1H NMR (300.1 MHz, CD2Cl2): δ 7.92 (dm, 3J(H,H) = 8.6 Hz, m–CH); 7.79 (dd, 3J(H,H) = 8.6, 2J(H,P) = 5.8 Hz, o–CH) ppm. 19F NMR (282.4 MHz, CD2Cl2): δ = 81.7 (p, 2J(F,F) = 150 Hz, 1F, SF5); 61.8 (d, 2J(F,F) = 150 Hz, 4F, SF5) ppm. 13C{1H} NMR (75.4 MHz, CD2Cl2): 156.1 (p, 1J(C,P) = 18.5 Hz, Cq–SF5); 135.0 (m, overlapped with CH signals, P–Cq); 134.9 (d, 2J(C,P) = 12.8 Hz, o–CH); 126.5 (dp, 3J(C,P) = 10.0 Hz, 3J(C,Feq) = 4.9 Hz, m–CH) ppm. The resonance for the CO ligand was not observed. IR (ATR): 1986 (C≡O), 1940 (C≡O), 824 (S–F) cm−1.
When the reaction was performed with 13CO, the complex 8′ was obtained.
Selected NMR data: 31P{1H} NMR (121.5 MHz, CD2Cl2): δ = 0.84 (t, 2J(P,C) = 13.5 Hz) ppm. 13C{1H} NMR (75.4 MHz, CD2Cl2): 179.6 (t, 2J(C,P) = 13.6 Hz, 13CO) ppm. IR (ATR): 1933 (C≡O), 1896 (C≡O), 824 (S–F) cm−1.
3.10. Formation of Trans,trans-[RhCl(CO)2{P(p-C6H4SF5)3}2] (9)
Complex [RhCl(COD){P(p-C6H4SF5)3}] (5) (20 mg, 0.02 mmol) was dissolved in C6D6 (0.4 mL) and the phosphine 1 (14 mg, 0.02 mmol) was added. Then, the solution was cooled to 77 K, degassed and treated with CO. After 10 min, the solution turned clear and a yellow precipitate was formed. The solid was filtered off and washed with C6D6 (2 × 0.25 mL) and dried under vacuum Yield: 29 mg (89%).
31P{1H} NMR (121.5 MHz, acetone-d6): δ = 27.6 (s, br) ppm. 31P{1H} NMR (121.5 MHz, 243 K, acetone-d6): δ = 29.15 (d, br, 2J(P,Rh) ≈ 130 Hz) ppm. 1H NMR (300.1 MHz, acetone-d6): δ 8.22–8.00 (m, m–CH + o–CH) ppm. 19F NMR (282.4 MHz, 243 K acetone-d6): δ = 82.9 (p, 2J(F,F) = 148 Hz, 1F, SF5); 61.6 (d, 2J(F,F) = 148 Hz, 4F, SF5) ppm. 13C{1H} NMR (75.4 MHz, acetone-d6): 156.1–154.7 (m, Cq–SF5); 136.3 (s br, P–Cq + o–CH); 127.0 (s br, m–CH) ppm. The resonance for the CO ligand was not observed. IR (ATR): 1992 (C≡O), 828 (S–F) cm−1.
When the reaction was performed with 13CO, complex 9′ was obtained.
Selected NMR data: 31P{1H} NMR (121.5 MHz, acetone-d6): δ = 28.62 (s, br) ppm. 31P{1H} NMR (121.5 MHz, 203 K, acetone-d6): δ = 29.49 (d, br, 2J(P,Rh) ≈ 130 Hz) ppm. 13C{1H} NMR (75.4 MHz, acetone-d6): 187.4 (t, 1J(C,Rh) = 73.0 Hz, 13CO) ppm. 13C{1H} NMR (75.4 MHz, 203 K, acetone-d6): 187.1 (d, 1J(C,Rh) = 72.6 Hz, 13CO) ppm. IR (ATR): 1945 (C≡O), 824 (S–F) cm−1.
3.11. X-ray Diffraction Analysis
For the structure determination, the data collection was performed with a BRUKER APEX-II CCD diffractometer (Bruker AXS, Karlsruhe, Germany) using Mo-Kα radiation (λ = 0.71073 Å). Multi-scan absorption corrections implemented in SADABS [71] were applied to the data. The structures were solved by intrinsic phasing method (SHELXT-2014) [72] and refined by full–matrix least square procedures based on F2 with all measured reflections (SHELXL-2018) [73], with anisotropic temperature factors for all non-hydrogen atoms. In complex 4·0.5C6H6, C(sp3) atoms of COD ligand were treated with ISOR while in complex 6·C6H6, all the carbon atoms of the solvent molecule were treated with EADP. All carbon bound hydrogen atoms were added geometrically and refined by using a riding model. CCDC 2021751–2021754 contain the supplementary crystallographic data. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E–mail: [email protected]).
Crystal Data for1·C6H6. C18H12F15PS3·C6H6, MW 718.53, triclinic, space group P, a: 10.0772(6) Å, b: 10.2132(6) Å, c: 15.1197(12) Å, α: 70.446(3)°, β: 70.770(4)°, γ = 85.849(3)°, V = 1383.31(16) Å3, Z = 2, Dcalc: 1.725 mg m−3, T = 103(2) K, μ = 0.443 mm−1. 28,056 measured reflections (2θ: 2.142–26.416°), 5658 unique (Rint = 0.0533). Final agreement factors were R1 = 0.0402 (I > 2σ(I)) and wR2 = 0.0862.
Crystal Data for2·2C6H6. C30H24F15OPS3·2C6H6, MW 812.64, triclinic, space group P, a: 7.7689(7) Å, b: 12.3481(10) Å, c: 17.6929(17) Å, α: 106.915(3)°, β: 90.217(4)°, γ = 92.514(3)°, V = 1622.1(3) Å3, Z = 2, Dcalc: 1.664 mg m−3, T = 102(2) K, μ = 0.391 mm−1. 65,957 measured reflections (2θ: 2.374–26.846°), 6935 unique (Rint = 0.0577). Final agreement factors were R1 = 0.0388 (I > 2σ(I)) and wR2 = 0.0890.
Crystal Data for4·0.5C6H6. C26H24ClF15IrPS3·0.5C6H6, MW 1015.30, monoclinic, space group P21/c, a: 16.8589(11) Å, b: 17.1921(11) Å, c: 11.8495(8) Å, β: 101.744(2)°, V = 3362.6(4) Å3, Z = 2, Dcalc: 2.006 mg m−3, T = 100(2) K, μ = 4.390 mm−1. 41,026 measured reflections (2θ: 2.264–26.436°), 6901 unique (Rint = 0.0505). Final agreement factors were R1 = 0.0340 (I > 2σ(I)) and wR2 = 0.0763.
Crystal Data for6·C6H6. C52H52Cl2F30Rh2P2S6·C6H6, MW 1856.06, monoclinic, space group C2/c, a: 19.8800(13) Å, b: 11.6762(7) Å, c: 29.2074(19) Å, β: 96.426(2)°, V = 6737.1(7) Å3, Z = 4, Dcalc: 1.830 mg m−3, T = 103(2) K, μ = 0.926 mm−1. 62,530 measured reflections (2θ: 2.182–24.840°), 5816 unique (Rint = 0.1005). Final agreement factors were R1 = 0.0431 (I > 2σ(I)) and wR2 = 0.0910.
3.12. Computational Details
Calculations were run using the Gaussian 09 (Revision D.01, Gaussian, Inc., Wallingford, CT, USA) program package [74]. In the case of phosphines and phosphine radical cations the CAM-B3LYP functional was used and 6-311G(d,p) basis set were employed for all atoms. For the nickel complexes, the B3LYP functional was chosen. Nickel was described with RECPs and the associated LANL2DZ basis sets [75] while the ligands were described with 6-31G(d,p). All calculated structures were identified as minima (no negative eigenvalues). All xyz coordinates are included as Supplementary Material (Tables S1–S3).
4. Conclusions
An unprecedented phosphine bearing SF5 groups, P(p-C6H4SF5)3 (1), was successfully synthesized. It exhibits a moderate air stability, which is corroborated experimentally and by theoretical methods. The HOMO energy level and the phosphorus-selenium coupling constant of the synthesized phosphine selenide indicate that 1 is a weaker donor than P(p-C6H4CF3)3 and very close to P(m-C6H4(CF3)2)3. The calculated Tolman electronic parameter are consistent with an electron-withdrawing character. The molecular structures of iridium and rhodium complexes allowed for an estimation of the cone angle, which is slightly larger than the one for P(p-C6H4CF3)3. Finally, reactivity studies of [MCl(COD){P(p-C6H4SF5)3}] (M = Ir (4), Rh (5)) towards CO revealed a preference for the formation of the diphosphine complexes [MCl(CO)2{P(p-C6H4SF5)3}2] (M = Ir (8), Rh (9)) instead of [MCl(CO)2{P(p-C6H4SF5)3}]. Overall, the presence of SF5 groups in the phosphine 1 qualifies it as an electron-poor and sterically demanding phosphine. This might provide new opportunities for applications in catalysis.
Supplementary Materials
The Supplementary Materials are available online.
Author Contributions
Conceptualization, T.B.; methodology, S.H. and M.T.; investigation, S.H., M.T., R.H. and R.L.; writing—original draft preparation, M.T.; writing—review and editing, M.T. and T.B.; supervision, T.B.; funding acquisition, T.B. All authors have read and agreed to the published version of the manuscript.
Funding
We acknowledge the CRC 1349 funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation; Gefördert durch die Deutsche Forschungsgemeinschaft–Projektnummer 387284271–SFB 1349). The APC was funded by MDPI.
Acknowledgments
The authors want to thank Philipp Wittwer and Mike Ahrens for helpful discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Crabtree, R.H. The Organometallic Chemistry of the Transition Metals, 6th ed.; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2014. [Google Scholar]
- Hartwig, J.F. Organotransition Metal Chemistry: From Bonding to Catalysis; University Science Books: Sausalito, CA, USA, 2010. [Google Scholar]
- McAuliffe, C.A.; Levason, W. Phosphine, Arsine and Stibine Complexes of the Transition Elements; Elsevier Scientific Pub. Co.: Amsterdam, The Netherlands, 1999. [Google Scholar]
- Pignolet, L.H. Homogeneous Catalysis with Metal Phosphine Complexes; Plenum Press: New York, NY, USA, 1983. [Google Scholar]
- Pollock, C.L.; Saunders, G.C.; Smyth, E.C.M.S.; Sorokin, V.I. Fluoroarylphosphines as ligands. J. Fluor. Chem. 2008, 129, 142–166. [Google Scholar] [CrossRef]
- Clarke, M.L.; Ellis, D.; Mason, K.L.; Orpen, A.G.; Pringle, P.G.; Wingad, R.L.; Zaher, D.A.; Baker, R.T. The electron-poor phosphines P{C6H3(CF3)2-3,5}3 and P(C6F5)3 do not mimic phosphites as ligands for hydroformylation. A comparison of the coordination chemistry of P{C6H3(CF3)2-3,5}3 and P(C6F5)3 and the unexpectedly low hydroformylation activity of their rhodium complexes. Dalton Trans. 2005, 7, 1294–1300. [Google Scholar]
- Fawcett, J.; Hope, E.G.; Kemmitt, R.D.; Paige, D.R.; Russell, D.R.; Stuart, A.M.; Stuart, M. Platinum group metal complexes of arylphosphine ligands containing perfluoroalkyl ponytails; crystal structures of [RhCl2(η5-C5Me5){P(C6H4C6F13-4)3}] and cis- and trans-[PtCl2{P(C6H4C6F13-4)3}2]. J. Chem. Soc. Dalton Trans. 1998, 22, 3751–3764. [Google Scholar] [CrossRef]
- Hope, E.G.; Kemmitt, R.D.W.; Paige, D.R.; Stuart, A.M.; Wood, D.R.W. Synthesis and coordination chemistry of meta-perfluoroalkyl-derivatised triarylphosphines. Polyhedron 1999, 18, 2913–2917. [Google Scholar] [CrossRef]
- Corcoran, C.; Fawcett, J.; Friedrichs, S.; Holloway, J.H.; Hope, E.G.; Russell, D.R.; Saunders, G.C.; Stuart, A.M. Structural and electronic impact of fluorine in the ortho positions of triphenylphosphine and 1,2-bis(diphenylphosphino)ethane; a comparison of 2,6-difluorophenyl- with pentafluorophenyl-phosphines. J. Chem. Soc. Dalton Trans. 2000, 2, 161–172. [Google Scholar] [CrossRef]
- Croxtall, B.; Fawcett, J.; Hope, E.G.; Stuart, A.M. Synthesis and coordination chemistry of ortho-perfluoroalkyl-derivatised triarylphosphines. J. Chem. Soc. Dalton Trans. 2002, 4, 491–499. [Google Scholar] [CrossRef]
- Saunders, G.C. Structural and electronic properties of tris(4-trifluoromethyltetrafluorophenyl)phosphine. J. Fluor. Chem. 2015, 180, 15–20. [Google Scholar] [CrossRef]
- Uson, R.; Oro, L.A.; Fernandez, M.J. Preparation, reactions and catalytic activity of complexes of the type [Ir(COD){P(p-RC6H4)3}2]A (R = Cl, F, H, CH3 or CH3O.; A = ClO4− or B(C6H5)4−). J. Organomet. Chem. 1980, 193, 127–133. [Google Scholar] [CrossRef]
- Matsubara, K.; Fujii, T.; Hosokawa, R.; Inatomi, T.; Yamada, Y.; Koga, Y. Fluorine-Substituted Arylphosphine for an NHC-Ni(I) System, Air-Stable in a Solid State but Catalytically Active in Solution. Molecules 2019, 24, 3222. [Google Scholar] [CrossRef] [Green Version]
- Moser, W.R.; Papile, C.J.; Brannon, D.A.; Duwell, R.A.; Weininger, S.J. The mechanism of phosphine-modified rhodium-catalyzed hydroformylation studied by CIR-FTIR. J. Mol. Catal. 1987, 41, 271–292. [Google Scholar] [CrossRef]
- Chen, C.; Wang, Y.; Shi, X.; Sun, W.; Zhao, J.; Zhu, Y.-P.; Liu, L.; Zhu, B. Palladium-Catalyzed C-2 and C-3 Dual C–H Functionalization of Indoles: Synthesis of Fluorinated Isocryptolepine Analogues. Org. Lett. 2020, 22, 4097–4102. [Google Scholar] [CrossRef] [PubMed]
- Paterson, A.J.; Dunås, P.; Rahm, M.; Norrby, P.-O.; Kociok-Köhn, G.; Lewis, S.E.; Kann, N. Palladium Catalyzed Stereoselective Arylation of Biocatalytically Derived Cyclic 1,3-Dienes: Chirality Transfer via a Heck-Type Mechanism. Org. Lett. 2020, 22, 2464–2469. [Google Scholar] [CrossRef] [PubMed]
- Jakab, A.; Dalicsek, Z.; Holczbauer, T.; Hamza, A.; Pápai, I.; Finta, Z.; Timári, G.; Soós, T. Superstable Palladium(0) Complex as an Air- and Thermostable Catalyst for Suzuki Coupling Reactions. Eur. J. Org. Chem. 2015, 2015, 60–66. [Google Scholar] [CrossRef]
- Cheng, L.; Li, M.-M.; Wang, B.; Xiao, L.-J.; Xie, J.-H.; Zhou, Q.-L. Nickel-catalyzed hydroalkylation and hydroalkenylation of 1,3-dienes with hydrazones. Chem. Sci. 2019, 10, 10417–10421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheppard, W.A. The Electrical Effect of the Sulfur Pentafluoride Group. J. Am. Chem. Soc. 1962, 84, 3072–3076. [Google Scholar] [CrossRef]
- Hansch, C.; Muir, R.M.; Fujita, T.; Maloney, P.P.; Geiger, F.; Streich, M. The Correlation of Biological Activity of Plant Growth Regulators and Chloromycetin Derivatives with Hammett Constants and Partition Coefficients. J. Am. Chem. Soc. 1963, 85, 2817–2824. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Savoie, P.R.; Welch, J.T. Preparation and Utility of Organic Pentafluorosulfanyl-Containing Compounds. Chem. Rev. 2015, 115, 1130–1190. [Google Scholar] [CrossRef]
- Lentz, D.; Seppelt, K. The -SF5, -SeF5, and -TeF5 Groups in Organic Chemistry. In Chemistry of Hypervalent Compounds; Akiba, K.-Y., Ed.; Wiley-VCH: New York, NY, USA, 1999; pp. 295–325. [Google Scholar]
- Gard, G.L. Recent Milestones in SF5-Chemistry. Chim. Oggi 2009, 27, 10–13. [Google Scholar]
- Altomonte, S.; Zanda, M. Synthetic chemistry and biological activity of pentafluorosulphanyl (SF5) organic molecules. J. Fluor. Chem. 2012, 143, 57–93. [Google Scholar] [CrossRef] [Green Version]
- Kanishchev, O.S.; Dolbier, W.R. Chapter One—SF5-Substituted Aromatic Heterocycles. In Advances in Heterocyclic Chemistry; Scriven, E.F.V., Ramsden, C.A., Eds.; Academic Press: Cambridge, MA, USA, 2016; Volume 120, pp. 1–42. [Google Scholar]
- Chan, J.M.W. Pentafluorosulfanyl group: An emerging tool in optoelectronic materials. J. Mater. Chem. C 2019, 7, 12822–12834. [Google Scholar] [CrossRef]
- Beier, P. Synthesis and reactivity of novel sulfur pentafluorides—Effect of the SF5 group on reactivity of nitrobenzenes in nucleophilic substitution. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 212–215. [Google Scholar] [CrossRef]
- Beier, P. Pentafluorosulfanylation of Aromatics and Heteroaromatics. In Emerging Fluorinated Motifs; Ma, J.-A., Cahard, D., Eds.; Wiley-VCH: Weinheim, Germany, 2020; Volume 2, pp. 551–570. [Google Scholar]
- Haufe, G. Pentafluorosulfanylation of Aliphatic Substrates. In Emerging Fluorinated Motifs; Ma, J.-A., Cahard, D., Eds.; Wiley-VCH: Weinheim, Germany, 2020; Volume 2, pp. 571–609. [Google Scholar]
- Damerius, R.; Leopold, D.; Schulze, W.; Seppelt, K. Strukturen von SF5-substituierten Metallkomplexen. Z. Anorg. Allg. Chem. 1989, 578, 110–118. [Google Scholar] [CrossRef]
- Henkel, T.; Klauck, A.; Seppelt, K. Pentafluoro-λ6-sulfanylacetylene complexes of cobalt. J. Organomet. Chem. 1995, 501, 1–6. [Google Scholar] [CrossRef]
- Preugschat, D.; Thrasher, J.S. Pentacarbonylchrom-Komplexe SF5-substituierter Isocyanide. Z. Anorg. Allg. Chem. 1996, 622, 1411–1414. [Google Scholar] [CrossRef]
- Shavaleev, N.M.; Xie, G.; Varghese, S.; Cordes, D.B.; Slawin, A.M.Z.; Momblona, C.; Ortí, E.; Bolink, H.J.; Samuel, I.D.W.; Zysman-Colman, E. Green Phosphorescence and Electroluminescence of Sulfur Pentafluoride-Functionalized Cationic Iridium(III) Complexes. Inorg. Chem. 2015, 54, 5907–5914. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.-F.; Luo, X.-F.; Yan, Z.-P.; Wu, Z.-G.; Zhao, Y.; Zheng, Y.-X.; Zuo, J.-L. Syntheses, Crystal Structures, and Photoluminescence of a Series of Iridium(III) Complexes Containing the Pentafluorosulfanyl Group. Organometallics 2019, 38, 3553–3559. [Google Scholar] [CrossRef]
- Pal, A.K.; Henwood, A.F.; Cordes, D.B.; Slawin, A.M.Z.; Samuel, I.D.W.; Zysman-Colman, E. Blue-to-Green Emitting Neutral Ir(III) Complexes Bearing Pentafluorosulfanyl Groups: A Combined Experimental and Theoretical Study. Inorg. Chem. 2017, 56, 7533–7544. [Google Scholar] [CrossRef]
- Groves, L.M.; Schotten, C.; Beames, J.; Platts, J.A.; Coles, S.J.; Horton, P.N.; Browne, D.L.; Pope, S.J.A. From Ligand to Phosphor: Rapid, Machine-Assisted Synthesis of Substituted Iridium(III) Pyrazolate Complexes with Tuneable Luminescence. Chem. A Eur. J. 2017, 23, 9407–9418. [Google Scholar] [CrossRef] [Green Version]
- Henwood, A.F.; Webster, J.; Cordes, D.; Slawin, A.M.Z.; Jacquemin, D.; Zysman-Colman, E. Phosphorescent platinum(ii) complexes bearing pentafluorosulfanyl substituted cyclometalating ligands. RSC Adv. 2017, 7, 25566–25574. [Google Scholar] [CrossRef] [Green Version]
- Berg, C.; Braun, T.; Laubenstein, R.; Braun, B. Palladium-mediated borylation of pentafluorosulfanyl functionalized compounds: The crucial role of metal fluorido complexes. Chem. Commun. 2016, 52, 3931–3934. [Google Scholar] [CrossRef] [PubMed]
- Golf, H.R.A.; Reissig, H.-U.; Wiehe, A. Synthesis of SF5-Substituted Tetrapyrroles, Metalloporphyrins, BODIPYs, and Their Dipyrrane Precursors. J. Org. Chem. 2015, 80, 5133–5143. [Google Scholar] [CrossRef] [PubMed]
- Berry, A.D.; De Marco, R.A. Reaction of pentafluoro[(trifluoromethyl)acetylenyl]sulfur with nickel tetracarbonyl. Inorg. Chem. 1982, 21, 457–458. [Google Scholar] [CrossRef]
- Sergeeva, T.A.; Dolbier, W.R. A New Synthesis of Pentafluorosulfanylbenzene. Org. Lett. 2004, 6, 2417–2419. [Google Scholar] [CrossRef]
- Dunne, B.J.; Orpen, A.G. Triphenylphosphine: A redetermination. Acta Cryst. 1991, C47, 345–347. [Google Scholar] [CrossRef]
- Eapen, K.C.; Tamborski, C. The synthesis of tris-(trifluoromethylphenyl)phosphines and phosphine oxides. J. Fluor. Chem. 1980, 15, 239–243. [Google Scholar] [CrossRef]
- See, R.F.; Dutoi, A.D.; Fettinger, J.C.; Nicastro, P.J.; Ziller, J.W. The crystal structures of (p-ClPh)3PO and (p-OMePh)3PO, including an analysis of the P-O bond in triarylphosphine oxides. J. Chem. Crystallogr. 1998, 28, 893–898. [Google Scholar] [CrossRef]
- Stewart, B.; Harriman, A.; Higham, L.J. Predicting the Air Stability of Phosphines. Organometallics 2011, 30, 5338–5343. [Google Scholar] [CrossRef]
- Dunne, B.J.; Morris, R.B.; Orpen, A.G. Structural systematics. Part 3. Geometry deformations in triphenylphosphine fragments: A test of bonding theories in phosphine complexes. J. Chem. Soc. Dalton Trans. 1991, 653–661. [Google Scholar] [CrossRef]
- Palau, C.; Berchadsky, Y.; Chalier, F.; Finet, J.-P.; Gronchi, G.; Tordo, P. Tris(monochlorophenyl)- and Tris(dichlorophenyl)phosphines: Molecular Geometry, Anodic Behavior, and ESR Studies. J. Phys. Chem. 1995, 99, 158–163. [Google Scholar] [CrossRef]
- Chevykalova, M.N.; Manzhukova, L.F.; Artemova, N.V.; Luzikov, Y.N.; Nifant’ev, I.E.; Nifant’ev, E.E. Electron-donating ability of triarylphosphines and related compounds studied by 31P NMR spectroscopy. Russ. Chem. Bull. 2003, 52, 78–84. [Google Scholar] [CrossRef]
- Howell, J.S.; Lovatt, J.; Yates, P.; Gottlieb, H.; Hursthouse, M.; Light, M. Effect of fluorine and trifluoromethyl substitution on the donor properties and stereodynamical behaviour of triarylphosphines. J. Chem. Soc. Dalton Trans. 1999, 17, 3015–3028. [Google Scholar] [CrossRef]
- Allen, D.W.; Taylor, B.F. The chemistry of heteroarylphosphorus compounds. Part 15. Phosphorus-31 nuclear magnetic resonance studies of the donor properties of heteroarylphosphines towards selenium and platinum(II). J. Chem. Soc. Dalton Trans. 1982, 1, 51–54. [Google Scholar] [CrossRef]
- Tolman, C.A. Steric effects of phosphorus ligands in organometallic chemistry and homogeneous catalysis. Chem. Rev. 1977, 77, 313–348. [Google Scholar] [CrossRef]
- Gusev, D.G. Donor Properties of a Series of Two-Electron Ligands. Organometallics 2009, 28, 763–770. [Google Scholar] [CrossRef]
- Perrin, L.; Clot, E.; Eisenstein, O.; Loch, J.; Crabtree, R.H. Computed Ligand Electronic Parameters from Quantum Chemistry and Their Relation to Tolman Parameters, Lever Parameters, and Hammett Constants. Inorg. Chem. 2001, 40, 5806–5811. [Google Scholar] [CrossRef]
- Kawaguchi, S.-I.; Minamida, Y.; Okuda, T.; Sato, Y.; Saeki, T.; Yoshimura, A.; Nomoto, A.; Ogawa, A. Photoinduced Synthesis of P-Perfluoroalkylated Phosphines from Triarylphosphines and Their Application in the Copper-Free Cross-Coupling of Acid Chlorides and Terminal Alkynes. Adv. Synth. Catal. 2015, 357, 2509–2519. [Google Scholar] [CrossRef]
- Ma, X.-Y.; Wang, K.; Zhang, L.; Li, X.-J.; Li, R.-X. Selective Hydrogenation of Avermectin Catalyzed by Iridium-Phosphine Complexes. Chin. J. Chem. 2007, 25, 1503–1507. [Google Scholar] [CrossRef]
- Tiburcio, J.; Bernès, S.; Torrens, H. Electronic and steric effects of triarylphosphines on the synthesis, structure and spectroscopical properties of mononuclear rhodium(I)–chloride complexes. Polyhedron 2006, 25, 1549–1554. [Google Scholar] [CrossRef]
- Naaktgeboren, A.J.; Nolte, R.J.M.; Drenth, W. Phosphorus-31 nuclear magnetic resonance studies of polymer-anchored rhodium(I) complexes. J. Am. Chem. Soc. 1980, 102, 3350–3354. [Google Scholar] [CrossRef]
- Canepa, G.; Brandt, C.D.; Werner, H. Mono- and Dinuclear Rhodium(I) and Rhodium(III) Complexes with the Bulky Phosphine 2,6-Me2C6H3CH2CH2PtBu2, Including the First Structurally Characterized Cis-Configurated Dicarbonyl Compound, cis-[RhCl(CO)2(PR3)]. Organometallics 2004, 23, 1140–1152. [Google Scholar] [CrossRef]
- Müller, T.E.; Mingos, D.M.P. Determination of the Tolman cone angle from crystallographic parameters and a statistical analysis using the crystallographic data base. Transit. Met. Chem. 1995, 20, 533–539. [Google Scholar] [CrossRef]
- Reyna-Madrigal, A.; Ortiz-Pastrana, N.; Paz-Sandoval, M.A. Cyclooctadiene iridium complexes with phosphine and pentadienyl ligands. J. Organomet. Chem. 2019, 886, 13–26. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Joerg, S.; Drago, R.S.; Sales, J. Reactivity of Phosphorus Donors. Organometallics 1998, 17, 589–599. [Google Scholar] [CrossRef]
- Ortega-Moreno, L.; Fernández-Espada, M.; Moreno, J.J.; Navarro-Gilabert, C.; Campos, J.; Conejero, S.; López-Serrano, J.; Maya, C.; Peloso, R.; Carmona, E. Synthesis, properties, and some rhodium, iridium, and platinum complexes of a series of bulky m-terphenylphosphine ligands. Polyhedron 2016, 116, 170–181. [Google Scholar] [CrossRef]
- Von Hahmann, C.N.; Talavera, M.; Xu, C.; Braun, T. Reactivity of 3,3,3-Trifluoropropyne at Rhodium Complexes: Development of Hydroboration Reactions. Chem. A Eur. J. 2018, 24, 11131–11138. [Google Scholar] [CrossRef]
- Vaska, L. Reversible Combination of Carbon Monoxide with a Synthetic Oxygen Carrier Complex. Science 1966, 152, 769–771. [Google Scholar] [CrossRef]
- Sanger, A.R. Five-coordinate dicarbonyl complexes of rhodium(I): [RhX(CO)2(PPh3)2] (X = Cl, Br, I). Can. J. Chem. 1985, 63, 571–575. [Google Scholar] [CrossRef] [Green Version]
- Perrin, D.D.; Armarego, W.L.F. Purification of Laboratory Chemicals, 3rd ed.; Butterworth/Heinemann: London/Oxford, UK, 1988. [Google Scholar]
- Herde, J.-L.; Lambert, J.C.; Senoff, C.V. Cyclooctene and 1,5-Cyclooctadiene Complexes of Iridium. Inorg. Synth. 1974, 15, 18–20. [Google Scholar]
- Van der Ent, A.; Onderdelinden, A.L. Chlorobis(cyclooctene)rhodium(I) and -iridium(I) Complexes. Inorg. Synth. 1973, 14, 92–95. [Google Scholar]
- Sheldrick, G.M. SADABS, Program for Empirical Absorption Correction of Area Detector Data, May 2014; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. SHELXT-2014, Program for the Solution of Crystal Structures from X-ray Data; University of Göttingen: Göttingen, Germany, 2013. [Google Scholar]
- Sheldrick, G.M. SHELXL-2018, Program for the Refinement of Crystal Structures from X-ray Data; University of Göttingen: Göttingen, Germany, 2018. [Google Scholar]
- Frisch, M.J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |
Scheme 1.
Synthesis of the phosphine 1.

Figure 1.
ORTEP representation of 1 with thermal ellipsoids drawn at 50% probability level. The C6H6 molecule in the asymmetric unit has been omitted for clarity. Selected bond lengths [Å] and bond angles [deg]: P1–C1 1.829(2), P1–C7 1.833(2), P1–C13 1.831(2), C1–P1–C7 103.21(10), C7–P1–C13 99.18(10), C13–P1–C1 103.08(10).
Figure 1.
ORTEP representation of 1 with thermal ellipsoids drawn at 50% probability level. The C6H6 molecule in the asymmetric unit has been omitted for clarity. Selected bond lengths [Å] and bond angles [deg]: P1–C1 1.829(2), P1–C7 1.833(2), P1–C13 1.831(2), C1–P1–C7 103.21(10), C7–P1–C13 99.18(10), C13–P1–C1 103.08(10).

Scheme 2.
Formation of the phosphine oxide 2 and the phosphine selenide 3.

Figure 2.
ORTEP representation of 2 with thermal ellipsoids drawn at 50% probability level. The two C6H6 molecules contained in the asymmetric unit have been omitted for clarity. Selected bond lengths [Å] and bond angles [deg]: P1–O1 1.4867(14), P1–C1 1.8086(19), P1–C7 1.8086(19), P1–C13 1.8019(19), C1–P1–C7 107.19(8), C7–P1–C13 106.28(8), C13–P1–C1 106.35(9), O1–P1–C1 113.23(8), O1–P1–C7 111.35(8), O1–P1–C13 112.02(8).
Figure 2.
ORTEP representation of 2 with thermal ellipsoids drawn at 50% probability level. The two C6H6 molecules contained in the asymmetric unit have been omitted for clarity. Selected bond lengths [Å] and bond angles [deg]: P1–O1 1.4867(14), P1–C1 1.8086(19), P1–C7 1.8086(19), P1–C13 1.8019(19), C1–P1–C7 107.19(8), C7–P1–C13 106.28(8), C13–P1–C1 106.35(9), O1–P1–C1 113.23(8), O1–P1–C7 111.35(8), O1–P1–C13 112.02(8).

Scheme 3.
Synthesis of the iridium(I) complex 4.

Scheme 4.
Synthesis of the rhodium(I) complexes 5 and 6.
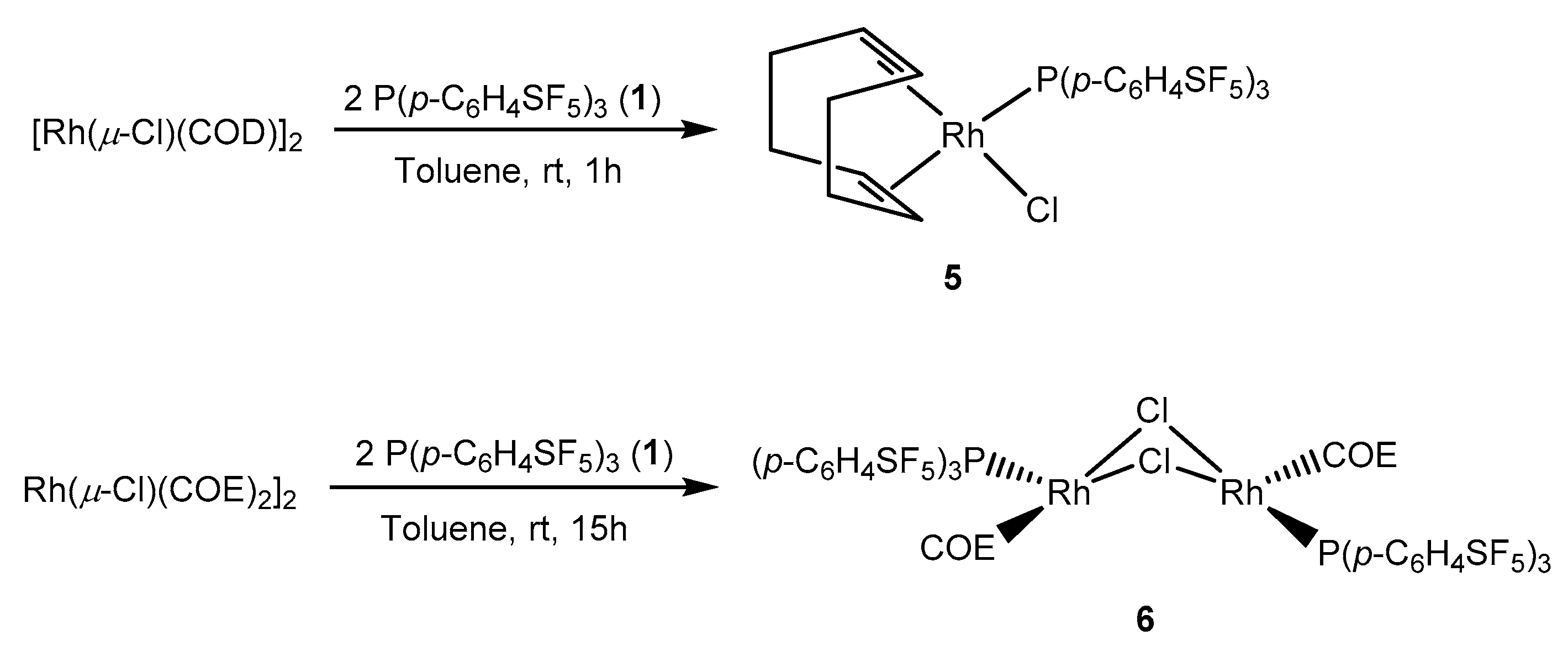
Figure 3.
ORTEP representation of 4 with thermal ellipsoids drawn at 50% probability level. Hydrogen atoms and the additional C6H6 molecule in the unit cell have been omitted for clarity. Note that some disorder is observed in COD ligand. Selected bond lengths [Å] and bond angles [deg]: Ir1–P1 2.2888(12), Ir1–Cl1 2.4070(10), P1–C1 1.831(5), P1–C7 1.834(5), P1–C13 1.838(5), Cl1–Ir1–P1 90.27(4), Ir1–P1–C1 118.23(15), Ir1–P1–C7 118.61(15), Ir1–P1–C13 108.27(16), C1–P1–C7 100.7(2), C7–P1–C13 104.2(2), C13–P1–C1 105.3(2).
Figure 3.
ORTEP representation of 4 with thermal ellipsoids drawn at 50% probability level. Hydrogen atoms and the additional C6H6 molecule in the unit cell have been omitted for clarity. Note that some disorder is observed in COD ligand. Selected bond lengths [Å] and bond angles [deg]: Ir1–P1 2.2888(12), Ir1–Cl1 2.4070(10), P1–C1 1.831(5), P1–C7 1.834(5), P1–C13 1.838(5), Cl1–Ir1–P1 90.27(4), Ir1–P1–C1 118.23(15), Ir1–P1–C7 118.61(15), Ir1–P1–C13 108.27(16), C1–P1–C7 100.7(2), C7–P1–C13 104.2(2), C13–P1–C1 105.3(2).

Figure 4.
ORTEP representation of 6 with thermal ellipsoids drawn at 50% probability level. Hydrogen atoms and the C6H6 molecule in the unit cell have been omitted for clarity. Selected bond lengths [Å] and bond angles [deg]: Rh1–P1 2.1930(11), Rh1–Cl1 2.3911(11), Rh1–Cl1i 2.4368(11), Rh1–C19 2.120(4), Rh1–C20 2.131(4), P1–C1 1.827(4), P1–C7 1.842(4), P1–C13 1.825(4), Cl1–Rh1–Cl1i 80.57(4), Cl1–Rh1–P1 89.66(4), Cl1i–Rh1–P1 166.80(4), P1–Rh1–C19 94.48(12), P1–Rh1–C20 93.38(12), Rh1–P1–C1 113.38(14), Rh1–P1–C7 111.20(14), Rh1–P1–C13 120.94(14), C1–P1–C7 103.73(19), C7–P1–C13 103.51(19), C13–P1–C1 102.26(19).
Figure 4.
ORTEP representation of 6 with thermal ellipsoids drawn at 50% probability level. Hydrogen atoms and the C6H6 molecule in the unit cell have been omitted for clarity. Selected bond lengths [Å] and bond angles [deg]: Rh1–P1 2.1930(11), Rh1–Cl1 2.3911(11), Rh1–Cl1i 2.4368(11), Rh1–C19 2.120(4), Rh1–C20 2.131(4), P1–C1 1.827(4), P1–C7 1.842(4), P1–C13 1.825(4), Cl1–Rh1–Cl1i 80.57(4), Cl1–Rh1–P1 89.66(4), Cl1i–Rh1–P1 166.80(4), P1–Rh1–C19 94.48(12), P1–Rh1–C20 93.38(12), Rh1–P1–C1 113.38(14), Rh1–P1–C7 111.20(14), Rh1–P1–C13 120.94(14), C1–P1–C7 103.73(19), C7–P1–C13 103.51(19), C13–P1–C1 102.26(19).

Scheme 5.
Reactivity of the complexes 4 and 5 towards CO in presence of phosphine 1.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
DFT calculated SOMO energies (eV) of triarylphosphines radical cations [PAr3]•+ (CAM-B3LYP/6-311G(d,p)).
Table 1.
DFT calculated SOMO energies (eV) of triarylphosphines radical cations [PAr3]•+ (CAM-B3LYP/6-311G(d,p)).
| Ar Group | SOMO | Ar Group | SOMO |
|---|---|---|---|
| p-C6H4SF5 | −12.22 | 3,4,5-C6H2F3 | –11.93 |
| p-C6H4CF3 | −11.82 | p-C6H4Me | –10.62 |
| m-C6H3(CF3)2 | –12.36 | p-C6H4OMe | –10.01 |
| p-C6H4F | –11.21 | C6H5 | −11.11 |
Table 2.
Calculated HOMO energies and TEP values of triarylphosphines (PAr3) and experimental 77Se NMR data of the corresponding selenides.
Table 2.
Calculated HOMO energies and TEP values of triarylphosphines (PAr3) and experimental 77Se NMR data of the corresponding selenides.
| Ar Group | HOMO (eV) | TEP (cm−1) [a] | 1JSe-P (Hz) |
|---|---|---|---|
| p-C6H4SF5 | −8.69 | 2072.6 | 792 |
| p-C6H4CF3 | −8.17 | 2069.3 | 765 [b] |
| m-C6H3(CF3)2 | −8.78 | 2075.2 | 802 [b] |
| p-C6H4F | −7.58 | n.d. | 741 [b] |
| 3,4,5-C6H2F3 | −8.36 | n.d. | 792 [c] |
| p-C6H4Me | −7.10 | 2061.7 (2166.7) | 715 [b] |
| p-C6H4OMe | −6.78 | n.d. | 710 [b] |
| C6H5 | −7.34 | 2063.5 (2068.9) | 733 [b] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Talavera, M.; Hinze, S.; Braun, T.; Laubenstein, R.; Herrmann, R. A SF5 Derivative of Triphenylphosphine as an Electron-Poor Ligand Precursor for Rh and Ir Complexes. Molecules 2020, 25, 3977. https://doi.org/10.3390/molecules25173977
AMA Style
Talavera M, Hinze S, Braun T, Laubenstein R, Herrmann R. A SF5 Derivative of Triphenylphosphine as an Electron-Poor Ligand Precursor for Rh and Ir Complexes. Molecules. 2020; 25(17):3977. https://doi.org/10.3390/molecules25173977
Chicago/Turabian StyleTalavera, Maria, Silke Hinze, Thomas Braun, Reik Laubenstein, and Roy Herrmann. 2020. "A SF5 Derivative of Triphenylphosphine as an Electron-Poor Ligand Precursor for Rh and Ir Complexes" Molecules 25, no. 17: 3977. https://doi.org/10.3390/molecules25173977