
Design, Synthesis and Functional Analysis of Cyclic Opioid Peptides with Dmt-Tic Pharmacophore

 , , and
, , and
Abstract
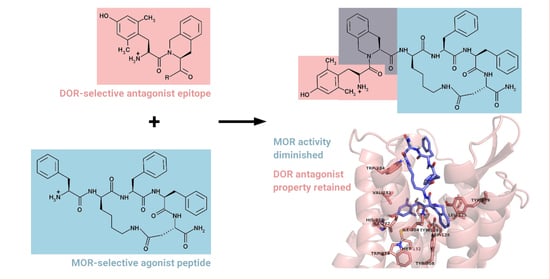
:
1. Introduction
2. Results
2.1. Chemistry
2.2. Biological Evaluation
2.3. Docking Studies
3. Experimental
3.1. General
3.2. Peptide Synthesis
3.3. Enzymatic Stability
3.4. Cell Culture
3.5. Calcium Mobilization Assay
3.6. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Waldhoer, M.; Bartlett, S.E.; Whistler, J.L. Opioid receptors. Annu. Rev. Biochem. 2004, 73, 953–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balogh, M.; Zádori, Z.S.; Lázár, B.; Karádi, D.; László, S.; Mousa, S.A.; Hosztafi, S.; Zádor, F.; Riba, P.; Schäfer, M.; et al. The Peripheral versus central antinociception of a novel opioid agonist: Acute inflammatory pain in rats. Neurochem. Res. 2018, 43, 1250–1257. [Google Scholar] [PubMed]
- Bailey, C.P.; Connor, M. Opioids: Cellular mechanisms of tolerance and physical dependence. Curr. Opin. Pharmacol. 2005, 5, 60–68. [Google Scholar] [CrossRef]
- Brock, C.; Olesen, S.S.; Olesen, A.E.; Frøkjaer, J.B.; Andresen, T.; Drewes, A.M. Opioid-induced bowel dysfunction: Pathophysiology and management. Drugs 2012, 72, 1847–1865. [Google Scholar] [CrossRef] [PubMed]
- Law, P.Y.; Reggio, P.H.; Loh, H.H. Opioid receptors: Toward separation of analgesic from undesirable effects. Trends Biochem. Sci. 2013, 38, 275–282. [Google Scholar] [PubMed] [Green Version]
- Wang, H.B.; Zhao, B.; Zhong, Y.Q.; Li, K.C.; Li, Z.Y.; Wang, Q.; Lu, Y.J.; Zhang, Z.N.; He, S.Q.; Zheng, H.C.; et al. Coexpression of delta- and mu-opioid receptors in nociceptive sensory neurons. Proc. Natl. Acad. Sci. USA. 2010, 107, 13117–13122. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Pan, Z.Z. Synaptic mechanism for functional synergism between delta- and mu-opioid receptors. J. Neurosci. 2010, 30, 4735–4745. [Google Scholar]
- Ananthan, S. Opioid ligands with mixed mu/delta opioid receptor interactions: An emerging approach to novel analgesics. AAPS J. 2006, 8, E118–E125. [Google Scholar]
- Zhu, Y.; King, M.A.; Schuller, A.G.; Nitsche, J.F.; Reidl, M.; Elde, R.P.; Unterwald, E.; Pasternak, G.W.; Pintar, J.E. Retention of supraspinal delta-like analgesia and loss of morphine tolerance in delta opioid receptor knockout mice. Neuron 1999, 24, 243–252. [Google Scholar]
- Abdelhamid, E.E.; Sultana, M.; Portoghese, P.S.; Takemori, A.E. Selective blockage of delta opioid receptors prevents the development of morphine tolerance and dependence in mice. J. Pharmacol. Exp. Ther. 1991, 258, 299–303. [Google Scholar]
- Fundytus, M.E.; Schiller, P.W.; Shapiro, M.; Weltrowska, G.; Coderre, T.J. Attenuation of morphine tolerance and dependence with the highly selective delta-opioid receptor antagonist TIPP [psi]. Eur. J. Pharmacol. 1995, 286, 105–108. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. Designing multiple ligands-medicinal chemistry strategies and challenges. Curr. Pharm. Des. 2009, 15, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Dietis, N.; Guerrini, R.; Calo, G.; Salvadori, S.; Rowbotham, D.J.; Lambert, D.G. Simultaneous targeting of multiple opioid receptors: A strategy to improve side-effect profile. Br. J. Anaesth. 2009, 103, 38–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiller, P.W. Bi- or multifunctional opioid peptide drugs. Life Sci. 2010, 86, 598–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvoracsko, S.; Stefanucci, A.; Novellino, E.; Mollica, A. The design of multitarget ligands for chronic and neuropathic pain. Future Med. Chem. 2015, 7, 2469–2483. [Google Scholar] [CrossRef] [PubMed]
- Mollica, A.; Costante, R.; Stefanucci, A.; Pinnen, F.; Luisi, G.; Pieretti, S.; Borsodi, A.; Bojnik, E.; Benyhe, S. Hybrid peptides endomorphin-2/DAMGO: Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2013, 68, 167–177. [Google Scholar] [CrossRef]
- Turnaturi, R.; Aricò, G.; Ronsisvalle, G.; Parenti, C.; Pasquinucci, L. Multitarget opioid ligands in pain relief: New players in an old game. Eur. J. Med. Chem. 2016, 108, 211–228. [Google Scholar] [CrossRef]
- Wtorek, K.; Piekielna-Ciesielska, J.; Janecki, T.; Janecka, A. The search for opioid analgesics with limited tolerance liability. Peptides 2020, 130, 170331. [Google Scholar] [CrossRef]
- Schiller, P.W.; Nguyen, T.M.; Weltrowska, G.; Wilkes, B.C.; Marsden, B.J.; Lemieux, C.; Chung, N.N. Differential stereochemical requirements of mu vs. delta opioid receptors for ligand binding and signal transduction: Development of a class of potent and highly delta-selective peptide antagonists. Proc. Natl. Acad. Sci. USA 1992, 89, 11871–11875. [Google Scholar] [CrossRef] [Green Version]
- Schiller, P.W.; Fundytus, M.E.; Merovitz, L.; Weltrowska, G.; Nguyen, T.M.; Lemieux, C.; Chung, N.N.; Coderre, T.J. The opioid mu agonist/delta antagonist DIPP-NH2 [Psi] produces a potent analgesic effect, no physical dependence, and less tolerance than morphine in rats. J. Med. Chem. 1999, 42, 3520–3526. [Google Scholar] [CrossRef]
- Bryant, S.D.; Jinsmaa, Y.; Salvadori, S.; Okada, Y.; Lazarus, L.H. Dmt and opioid peptides: A potent alliance. Biopolymers 2003, 71, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Giulianotti, M.A.; McLaughlin, J.P.; Shao, J.; Debevec, G.; Maida, L.E.; Geer, P.; Cazares, M.; Misler, J.; Li, L.; et al. Synthesis and biological evaluations of novel endomorphin analogues containing α-hydroxy-β-phenylalanine (AHPBA) displaying mixed μ/δ opioid receptor agonist and δ opioid receptor antagonist activities. Eur. J. Med. Chem. 2015, 92, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Balboni, G.; Guerrini, R.; Salvadori, S.; Bianchi, C.; Rizzi, D.; Bryant, S.D.; Lazarus, L.H. Evaluation of the Dmt-Tic pharmacophore: Conversion of a potent delta-opioid receptor antagonist into a potent delta agonist and ligands with mixed properties. J. Med. Chem. 2002, 45, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Salvadori, S.; Trapella, C.; Fiorini, S.; Negri, L.; Lattanzi, R.; Bryant, S.D.; Jinsmaa, Y.; Lazarus, L.H.; Balboni, G. A new opioid designed multiple ligand derived from the micro opioid agonist endomorphin-2 and the delta opioid antagonist pharmacophore Dmt-Tic. Bioorg. Med. Chem. 2007, 15, 6876–6881. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, M. Recent advances in selective opioid receptor agonists and antagonists. Med. Res. Rev. 2004, 24, 182–212. [Google Scholar] [CrossRef]
- Janecka, A.; Kruszynski, R. Conformationally restricted peptides as tools in opioid receptor studies. Curr. Med. Chem. 2005, 12, 471–481. [Google Scholar] [CrossRef]
- Keresztes, A.; Borics, A.; Tóth, G. Recent advances in endomorphin engineering. Chem. Med. Chem. 2010, 5, 1176–1196. [Google Scholar] [CrossRef]
- Gendron, L.; Cahill, C.M.; von Zastrow, M.; Schiller, P.W.; Pineyro, G. Molecular pharmacology of δ-opioid receptors. Pharmacol. Rev. 2016, 68, 631–700. [Google Scholar] [CrossRef] [Green Version]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Manglik, A.; Venkatakrishnan, A.J.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C.; et al. Structural insights into µ-opioid receptor activation. Nature 2015, 524, 315–321. [Google Scholar] [CrossRef] [Green Version]
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H.; et al. Structure of the µ-opioid receptor-Gi protein complex. Nature 2018, 558, 547–552. [Google Scholar] [PubMed]
- Granier, S.; Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Weis, W.I.; Kobilka, B.K. Structure of the δ-opioid receptor bound to naltrindole. Nature 2012, 485, 400–404. [Google Scholar]
- Fenalti, G.; Zatsepin, N.A.; Betti, C.; Giguere, P.; Han, G.W.; Ishchenko, A.; Liu, W.; Guillemyn, K.; Zhang, H.; James, D.; et al. Structural basis for bifunctional peptide recognition at human δ-opioid receptor. Nat. Struct. Mol. Biol. 2015, 22, 265–268. [Google Scholar] [PubMed] [Green Version]
- Claff, T.; Yu, J.; Blais, V.; Patel, N.; Martin, C.; Wu, L.; Han, G.W.; Holleran, B.J.; Van der Poorten, O.; White, K.L.; et al. Elucidating the active δ-opioid receptor crystal structure with peptide and small-molecule agonists. Sci. Adv. 2019, 5, eaax9115. [Google Scholar]
- Perlikowska, R.; do-Rego, J.C.; Cravezic, A.; Fichna, J.; Wyrebska, A.; Toth, G.; Janecka, A. Synthesis and biological evaluation of cyclic endomorphin-2 analogs. Peptides 2010, 31, 339–345. [Google Scholar]
- Perlikowska, R.; Piekielna, J.; Gentilucci, L.; Marco, R.D.; Cerlesi, M.C.; Calo’, G.; Artali, R.; Tömböly, C.; Kluczyk, A.; Janecka, A. Synthesis of mixed MOR/KOR efficacy cyclic opioid peptide analogs with antinociceptive activity after systemic administration. Eur. J. Med. Chem. 2016, 109, 276–286. [Google Scholar]
- Piekielna, J.; Kluczyk, A.; Gentilucci, L.; Cerlesi, M.C.; Calo’, G.; Tomböly, C.; Łapiński, K.; Janecki, T.; Janecka, A. Ring size in cyclic endomorphin-2 analogs modulates receptor binding affinity and selectivity. Org. Biomol. Chem. 2015, 13, 6039–6046. [Google Scholar]
- Piekielna, J.; Perlikowska, R.; do-Rego, J.C.; do-Rego, J.L.; Cerlesi, M.C.; Calo, G.; Kluczyk, A.; Łapiński, K.; Tömböly, C.; Janecka, A. Synthesis of mixed opioid affinity cyclic endomorphin-2 analogues with fluorinated phenylalanines. ACS Med. Chem. Lett. 2015, 6, 579–583. [Google Scholar]
- Kolesinska, B.; Rozniakowski, K.K.; Fraczyk, J.; Relich, I.; Papini, A.M.; Kaminski, Z.J. The effect of counterion and tertiary amine on the efficiency of N-triazinylammonium sulfonates in solution and solid-phase peptide synthesis. Eur. J. Org. Chem. 2015, 2, 401–408. [Google Scholar]
- Chmiel, T.; Mieszkowska, A.; Kempińska-Kupczyk, D.; Kot-Wasik, A.; Namieśnik, J.; Mazerska, Z. The impact of lipophilicity on environmental processes, drug delivery and bioavailability of food components. Microchem. J. 2019, 146, 393–406. [Google Scholar]
- Eisenried, A.; Peter, J.; Lerch, M.; Schüttler, J.; Jeleazcov, C. HPLC-MS/MS based time course analysis of morphine and morphine-6-glucoronide in ICU patients. J. Chromatogr. Sep. Tech. 2017, 8, 368–372. [Google Scholar]
- Ma, Q.; Ye, L.; Liu, H.; Shi, Y.; Zhou, N. An overview of Ca2+ mobilization assays in GPCR drug discovery. Expert Opin. Drug Discov. 2017, 12, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Caers, J.; Peymen, K.; Suetens, N.; Temmerman, L.; Janssen, T.; Schoofs, L.; Beets, I. Characterization of G protein-coupled receptors by a fluorescence-based calcium mobilization assay. J. Vis. Exp. 2014, 89, e51516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camarda, V.; Calo’, G. Chimeric G proteins in fluorimetric calcium assays: Experience with opioid receptors. Methods Mol. Biol. 2013, 937, 293–306. [Google Scholar]
- Perlikowska, R.; Piekielna, J.; Fichna, J.; do-Rego, J.C.; Toth, G.; Janecki, T.; Janecka, A. Pharmacological properties of novel cyclic pentapeptides with µ-opioid receptor agonist activity. Med. Chem. 2014, 10, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Wtorek, K.; Artali, R.; Piekielna-Ciesielska, J.; Koszuk, J.; Kluczyk, A.; Gentilucci, L.; Janecka, A. Endomorphin-2 analogs containing modified tyrosines: Biological and theoretical investigation of the influence on conformation and pharmacological profile. Eur. J. Med. Chem. 2019, 179, 527–536. [Google Scholar] [CrossRef]
- Kenakin, T.P. Orthosteric drug antagonism. In A Pharmacology Primer, 4th ed.; Kenakin, T.P., Ed.; Academic Press: San Diego, CA, USA; pp. 119–154.
Sample Availability: Samples of the compoundsare available from the authors in very small quantities. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Sequence | Ring Size | Monomer/ Dimer Ratio | Yield [%] | Enzymatic Stability Area [%] a |
|---|---|---|---|---|---|
| 1 | Dmt-Tic-c[d-Lys-Phe-Phe-Asp]NH2 | 17 | 2.9 | 28 | 97.88 ± 0.83 |
| 2 | Dmt-Tic-c[d-Lys-Phe-d-2Nal-Asp]NH2 | 17 | 2.8 | 25 | 98.17 ± 0.23 |
| 3 | Dmt-Tic-c[d-Lys-Phe-2,4F2-Phe-Asp]NH2 | 17 | 3.1 | 25 | 95.19 ± 0.86 |
| 4 | Dmt-Tic-c[d-Dap-Phe-Phe-Asp]NH2 | 14 | 1.2 | 18 | 96.21 ± 0.89 |
| 5 | Dmt-Tic-c[d-Lys-Phe-Asp]NH2 | 14 | 0.4 | 11 | 96.76 ± 0.91 |
| 6 | Dmt-Tic-c[d-Lys-Phe-Asp]-Tic-Dmt-NH2 | 14 | 1.6 | 19 | 97.61 ± 1.20 |
| Peptide | MOR | DOR | KOR | |||
|---|---|---|---|---|---|---|
| pEC50 (CL95%) | α ± SEM | pEC50 (CL95%) | α ± SEM | pEC50 (CL95%) | α ± SEM | |
| dermorphin | 8.66 ± 0.10 | 1.00 | inactive | inactive | ||
| DPDPE | inactive | 7.32 ± 0.18 | 1.00 | inactive | ||
| dynorphin A | 6.67 ± 0.50 | 0.83 ± 0.10 | 7.73 ± 0.27 | 0.99 ± 0.04 | 9.04 ± 0.09 | 1.00 |
| 1 | 6.18 ± 0.51 | 0.15 ± 0.02 | inactive | 6.31 ± 0.59 | 0.20 ± 0.06 | |
| 2 | 6.21 ± 0.5 | 0.37 ± 0.05 | inactive | inactive | ||
| 3 | inactive | inactive | inactive | |||
| 4 | inactive | inactive | inactive | |||
| 5 | 6.09 ± 0.17 | 0.3 ± 0.03 | inactive | inactive | ||
| 6 | 6.48 ± 0.49 | 0.17 ± 0.20 | inactive | inactive | ||
| No | Sequence | pKB(CL95%) |
|---|---|---|
| 1 | Dmt-Tic-c[d-Lys-Phe-Phe-Asp]NH2 | 7.37 ± 0.29 |
| 2 | Dmt-Tic-c[d-Lys-Phe-d-2Nal-Asp]NH2 | 7.55 ± 0.32 |
| 3 | Dmt-Tic-c[d-Lys-Phe-2,4F2-Phe-Asp]NH2 | 9.17 ± 0.35 |
| 4 | Dmt-Tic-c[d-Dap-Phe-Phe-Asp]NH2 | 8.61 ± 0.15 |
| 5 | Dmt-Tic-c[d-Lys-Phe-Asp]NH2 | 9.28 ± 0.34 |
| 6 | Dmt-Tic-c[d-Lys-Phe-Asp]Tic-Dmt-NH2 | 8.96 ± 0.28 |
| naltrindole | 9.89 ± 0.12 |
| No | Sequence | Predicted Receptor Affinity (Ki/pM) | |||
|---|---|---|---|---|---|
| DOR | |||||
| Active State | Inactive State | ||||
| cis | trans | cis | trans | ||
| 1 | Dmt-Tic-c[d-Lys-Phe-Phe-Asp]NH2 | 3.9 | 159.0 | 6500 | 211.5 |
| 2 | Dmt-Tic-c[d-Lys-Phe-d-2Nal-Asp]NH2 | 681.9 | 7170 | 9660 | 380.0 |
| 3 | Dmt-Tic-c[d-Lys-Phe-2,4F2-Phe-Asp]NH2 | 7920 | 4930 | 4010 | 1150 |
| 4 | Dmt-Tic-c[d-Dap-Phe-Phe-Asp]NH2 | 2640 | 1190 | 9040 | 6030 |
| 5 | Dmt-Tic-c[d-Lys-Phe-Asp]NH2 | 1870 | 5700 | 3200 | 9640 |
| 6 | Dmt-Tic-c[d-Lys-Phe-Asp]Tic-Dmt-NH2 | 1460 | 3310 | 520.5 | 540.8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarkar, A.; Adamska-Bartlomiejczyk, A.; Piekielna-Ciesielska, J.; Wtorek, K.; Kluczyk, A.; Borics, A.; Janecka, A. Design, Synthesis and Functional Analysis of Cyclic Opioid Peptides with Dmt-Tic Pharmacophore. Molecules 2020, 25, 4260. https://doi.org/10.3390/molecules25184260
Sarkar A, Adamska-Bartlomiejczyk A, Piekielna-Ciesielska J, Wtorek K, Kluczyk A, Borics A, Janecka A. Design, Synthesis and Functional Analysis of Cyclic Opioid Peptides with Dmt-Tic Pharmacophore. Molecules. 2020; 25(18):4260. https://doi.org/10.3390/molecules25184260
Chicago/Turabian StyleSarkar, Arijit, Anna Adamska-Bartlomiejczyk, Justyna Piekielna-Ciesielska, Karol Wtorek, Alicja Kluczyk, Attila Borics, and Anna Janecka. 2020. "Design, Synthesis and Functional Analysis of Cyclic Opioid Peptides with Dmt-Tic Pharmacophore" Molecules 25, no. 18: 4260. https://doi.org/10.3390/molecules25184260