
Imipramine Inhibits Migration and Invasion in Metastatic Castration-Resistant Prostate Cancer PC-3 Cells via AKT-Mediated NF-κB Signaling Pathway



Abstract
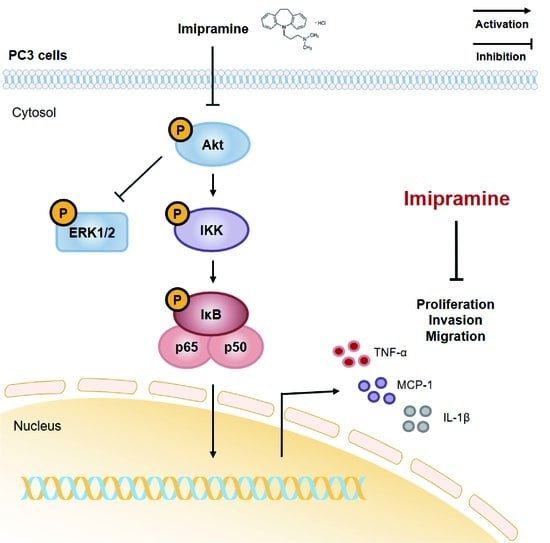
:
1. Introduction
2. Results
2.1. IMI Inhibits Proliferation of PC-3 Cells without Causing Cell Death
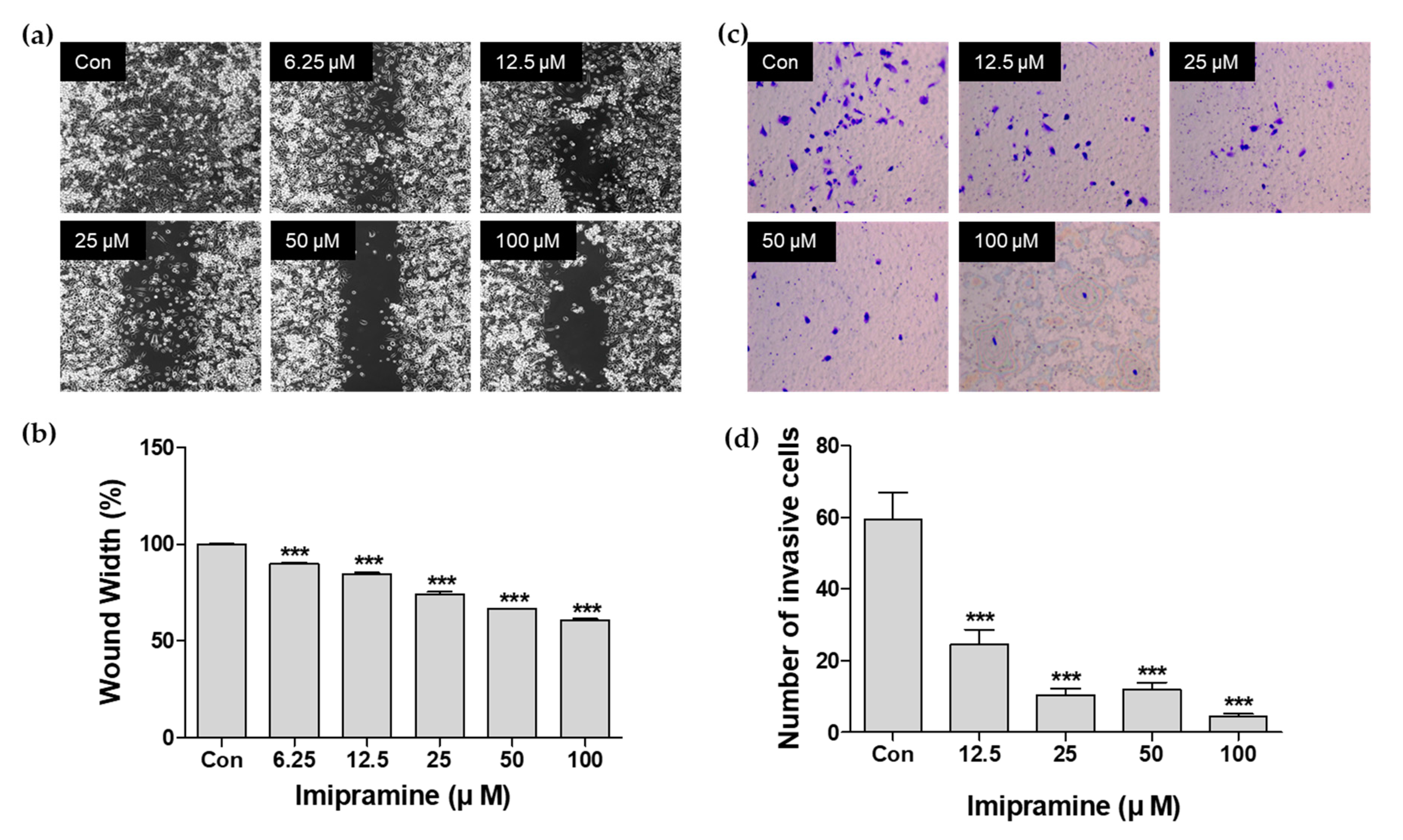
2.2. IMI Inhibits PC-3 Cell Migration
2.3. IMI Inhibits PC-3 Cell Invasion
2.4. IMI Inhibits Phosphorylation of Serine/threonine Protein Kinase (AKT), but Not that of Extracellular Signal-regulated kinase (ERK)1/2 in PC-3 Cells
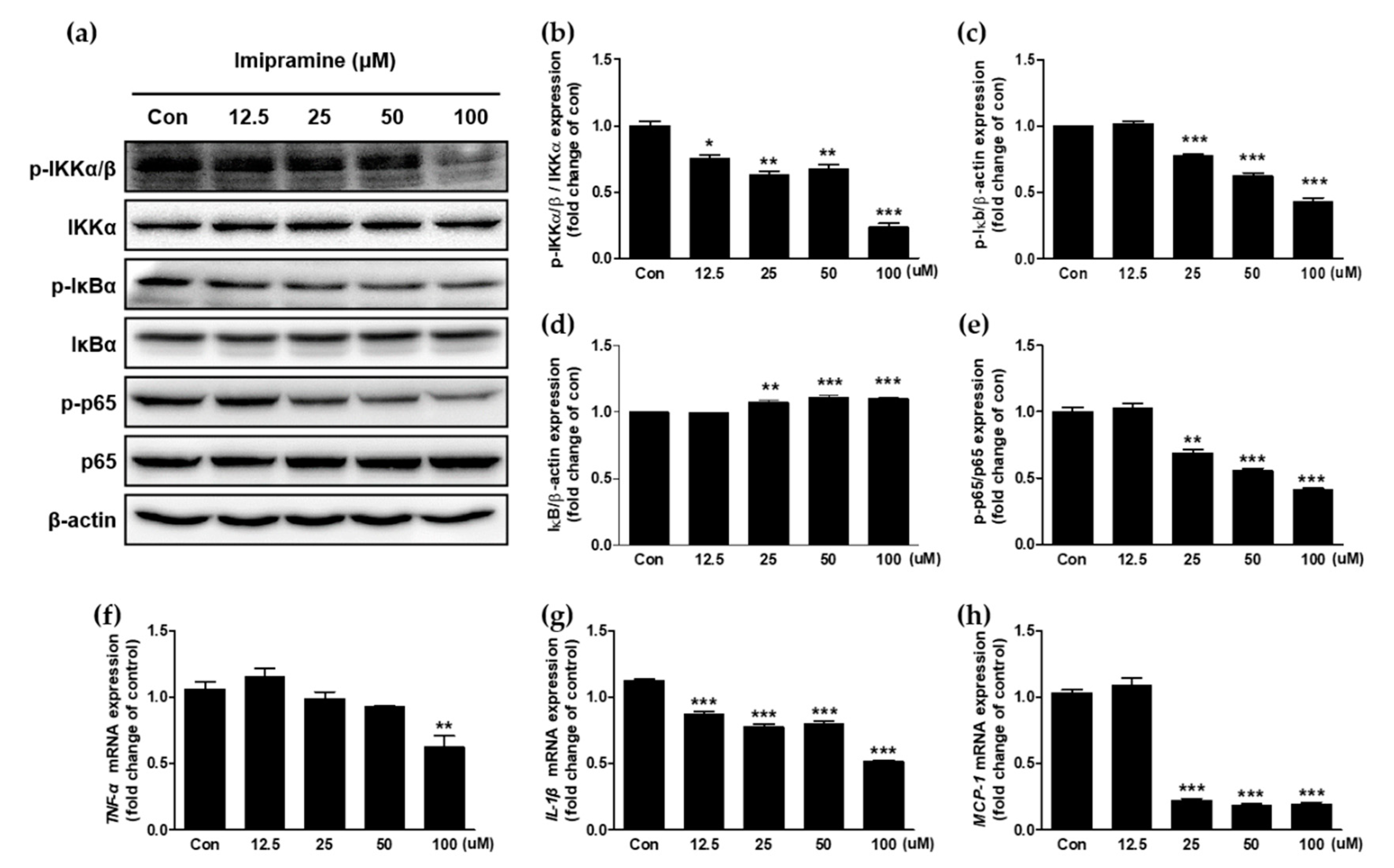
2.5. IMI Attenuates Nuclear Factor Kappa B (NF-κB) Signaling and the Expression of Proinflammatory Cytokines and Chemokines in PC-3 Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Proliferation Assay
4.4. Live/Dead Cell Viability Assay
4.5. Wound Healing Assay
4.6. Cell Invasion Assay
4.7. Western Blot Analysis
4.8. Total RNA Isolation and Quantitative Reverse-Transcription Polymerase Chain Reaction (RT-qPCR)
4.9. Statistics
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, K.-W.; Won, Y.-J.; Hong, S.; Kong, H.-J.; Lee, E.S. Prediction of Cancer Incidence and Mortality in Korea. Cancer Res. Treat. J. Korean Cancer Assoc. 2020, 52, 351. [Google Scholar] [CrossRef] [PubMed]
- Huggins, C. Prostatic cancer treated by orchiectomy: The five year results. J. Am. Med. Assoc. 1946, 131, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Denis, L.; Murphy, G. Overview of phase III trials on combined androgen treatment in patients with metastatic prostate cancer. Cancer 1993, 72, 3888–3895. [Google Scholar] [CrossRef]
- Harris, W.P.; Mostaghel, E.A.; Nelson, P.S.; Montgomery, B. Androgen deprivation therapy: Progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat. Clin. Pract. Urol. 2009, 6, 76–85. [Google Scholar] [CrossRef]
- Seitz, A.K.; Rauscher, I.; Haller, B.; Krönke, M.; Luther, S.; Heck, M.M.; Horn, T.; Gschwend, J.E.; Schwaiger, M.; Eiber, M. Preliminary results on response assessment using 68 Ga-HBED-CC-PSMA PET/CT in patients with metastatic prostate cancer undergoing docetaxel chemotherapy. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 602–612. [Google Scholar] [CrossRef]
- Marques, R.B.; Dits, N.F.; Erkens-Schulze, S.; Van Weerden, W.M.; Jenster, G. Bypass mechanisms of the androgen receptor pathway in therapy-resistant prostate cancer cell models. PLoS ONE 2010, 5, e13500. [Google Scholar] [CrossRef] [Green Version]
- Tannock, I.F.; Osoba, D.; Stockler, M.R.; Ernst, D.S.; Neville, A.J.; Moore, M.J.; Armitage, G.R.; Wilson, J.J.; Venner, P.M.; Coppin, C. Chemotherapy with mitoxantrone plus prednisone or prednisone alone for symptomatic hormone-resistant prostate cancer: A Canadian randomized trial with palliative end points. J. Clin. Oncol. 1996, 14, 1756–1764. [Google Scholar] [CrossRef]
- D’Amico, A.V. US Food and Drug Administration approval of drugs for the treatment of prostate cancer: A new era has begun. J. Clin. Oncol. 2014, 32, 362–364. [Google Scholar] [CrossRef]
- Figg, W.D.; Chau, C.H.; Small, E.J. Drug Management of Prostate Cancer; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Tannock, I.F.; De Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Théodore, C.; James, N.D.; Turesson, I. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef] [Green Version]
- Petrylak, D.P.; Tangen, C.M.; Hussain, M.H.; Lara, P.N., Jr.; Jones, J.A.; Taplin, M.E.; Burch, P.A.; Berry, D.; Moinpour, C.; Kohli, M. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N. Engl. J. Med. 2004, 351, 1513–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bono, J.S.; Oudard, S.; Ozguroglu, M.; Hansen, S.; Machiels, J.-P.; Kocak, I.; Gravis, G.; Bodrogi, I.; Mackenzie, M.J.; Shen, L. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: A randomised open-label trial. Lancet 2010, 376, 1147–1154. [Google Scholar] [CrossRef]
- Wang, S.-M.; Han, C.; Bahk, W.-M.; Lee, S.-J.; Patkar, A.A.; Masand, P.S.; Pae, C.-U. Addressing the side effects of contemporary antidepressant drugs: A comprehensive review. Chonnam. Med. J. 2018, 54, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Guidelines for ATC Classification and DDD Assignment 2020; Norwegian Institute of Public Health: Oslo, Norway, 2019.
- Wu, J.B.; Yin, L.; Shi, C.; Li, Q.; Duan, P.; Huang, J.-M.; Liu, C.; Wang, F.; Lewis, M.; Wang, Y. MAOA-dependent activation of Shh-IL6-RANKL signaling network promotes prostate cancer metastasis by engaging tumor-stromal cell interactions. Cancer Cell 2017, 31, 368–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Luo, J.; Yeh, S.; You, B.; Meng, J.; Chang, P.; Niu, Y.; Li, G.; Lu, C.; Zhu, Y. The MAO inhibitors phenelzine and clorgyline revert enzalutamide resistance in castration resistant prostate cancer. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kocsis, J.H.; Frances, A.J.; Voss, C.; Mann, J.J.; Mason, B.J.; Sweeney, J. Imipramine treatment for chronic depression. Arch. Gen. Psychiatry 1988, 45, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Sindrup, S.H.; Holbech, J.V.; Bach, F.W.; Finnerup, N.B.; Brøsen, K.; Jensen, T.S. The impact of serum drug concentration on the efficacy of imipramine, pregabalin, and their combination in painful polyneuropathy. Clin. J. Pain 2017, 33, 1047–1052. [Google Scholar] [CrossRef]
- Fritz, G.K.; Rockney, R.M.; Yeung, A.S. Plasma levels and efficacy of imipramine treatment for enuresis. J. Am. Acad. Child Adolesc. Psychiatry 1994, 33, 60–64. [Google Scholar] [CrossRef]
- Biber, A.; Durusu, İ.Z.; Özen, C. In vitro anticancer effect of tricyclic antidepressant nortriptyline on multiple myeloma. Turk. J. Biol. 2018, 42, 414–421. [Google Scholar] [CrossRef]
- Jahchan, N.S.; Dudley, J.T.; Mazur, P.K.; Flores, N.; Yang, D.; Palmerton, A.; Zmoos, A.-F.; Vaka, D.; Tran, K.Q.; Zhou, M. A drug repositioning approach identifies tricyclic antidepressants as inhibitors of small cell lung cancer and other neuroendocrine tumors. Cancer Discov. 2013, 3, 1364–1377. [Google Scholar] [CrossRef] [Green Version]
- Barlaz Us, S.; Sogut, F.; Yildirim, M.; Yetkin, D.; Yalin, S.; Yilmaz, S.N.; Comelekoglu, U. Effect of Imipramine on radiosensitivity of Prostate Cancer: An In Vitro Study. Cancer Investig. 2019, 37, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Ueno, L.; Vogt, P.K. Akt-mediated regulation of NFκB and the essentialness of NFκB for the oncogenicity of PI3K and Akt. Int. J. Cancer 2009, 125, 2863–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.; MacLennan, G.T.; Hartman, D.J.; Fu, P.; Resnick, M.I.; Gupta, S. Activation of PI3K-Akt signaling pathway promotes prostate cancer cell invasion. Int. J. Cancer 2007, 121, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Lamoureux, F.; Crafter, C.; Davies, B.R.; Beraldi, E.; Fazli, L.; Kim, S.; Thaper, D.; Gleave, M.E.; Zoubeidi, A. Synergistic targeting of PI3K/AKT pathway and androgen receptor axis significantly delays castration-resistant prostate cancer progression in vivo. Mol. Cancer Ther. 2013, 12, 2342–2355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drake, J.M.; Graham, N.A.; Lee, J.K.; Stoyanova, T.; Faltermeier, C.M.; Sud, S.; Titz, B.; Huang, J.; Pienta, K.J.; Graeber, T.G. Metastatic castration-resistant prostate cancer reveals intrapatient similarity and interpatient heterogeneity of therapeutic kinase targets. Proc. Natl. Acad. Sci. USA 2013, 110, E4762–E4769. [Google Scholar] [CrossRef] [Green Version]
- Ferraldeschi, R.; Rodrigues, D.N.; Riisnaes, R.; Miranda, S.; Figueiredo, I.; Rescigno, P.; Ravi, P.; Pezaro, C.; Omlin, A.; Lorente, D. PTEN protein loss and clinical outcome from castration-resistant prostate cancer treated with abiraterone acetate. Eur. Urol. 2015, 67, 795–802. [Google Scholar] [CrossRef] [Green Version]
- Kimbrough-Allah, M.N.; Millena, A.C.; Khan, S.A. Differential role of PTEN in transforming growth factor β (TGF-β) effects on proliferation and migration in prostate cancer cells. Prostate 2018, 78, 377–389. [Google Scholar] [CrossRef]
- Robertson, B.W.; Bonsal, L.; Chellaiah, M.A. Regulation of Erk1/2 activation by osteopontin in PC3 human prostate cancer cells. Mol. Cancer 2010, 9, 260. [Google Scholar] [CrossRef] [Green Version]
- Serra, V.; Scaltriti, M.; Prudkin, L.; Eichhorn, P.J.; Ibrahim, Y.H.; Chandarlapaty, S.; Markman, B.; Rodriguez, O.; Guzman, M.; Rodriguez, S. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene 2011, 30, 2547–2557. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, W.-Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.-F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. 2015, 35, 600–604. [Google Scholar] [CrossRef]
- Shih, A.; Davis, F.B.; Lin, H.-Y.; Davis, P.J. Resveratrol induces apoptosis in thyroid cancer cell lines via a MAPK-and p53-dependent mechanism. J. Clin. Endocrinol. Metab. 2002, 87, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Rieber, M.; Rieber, M.S. Signalling responses linked to betulinic acid-induced apoptosis are antagonized by MEK inhibitor U0126 in adherent or 3D spheroid melanoma irrespective of p53 status. Int. J. Cancer 2006, 118, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Llorens, F.; Miró, F.A.; Casañas, A.; Roher, N.; Garcia, L.; Plana, M.; Gómez, N.; Itarte, E. Unbalanced activation of ERK1/2 and MEK1/2 in apigenin-induced HeLa cell death. Exp. Cell Res. 2004, 299, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-L.; Wu, L.-J.; Zuo, H.-J.; Tashiro, S.-I.; Onodera, S.; Ikejima, T. Cytochrome c release from oridonin-treated apoptotic A375–S2 cells is dependent on p53 and extracellular signal-regulated kinase activation. J. Pharmacol. Sci. 2004, 96, 155–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, D.; Singh, S.V. Phenethyl isothiocyanate-induced apoptosis in p53-deficient PC-3 human prostate cancer cell line is mediated by extracellular signal-regulated kinases. Cancer Res. 2002, 62, 3615–3619. [Google Scholar]
- Lee, J.; John, T.; Steelman, L.S.; Chappell, W.H.; McCubrey, J.A. Akt inactivates ERK causing decreased response to chemotherapeutic drugs in advanced CaP cells. Cell Cycle 2008, 7, 631–636. [Google Scholar]
- Oeckinghaus, A.; Ghosh, S. The NF-κB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Dan, H.C.; Adli, M.; Baldwin, A.S. Regulation of mammalian target of rapamycin activity in PTEN-inactive prostate cancer cells by IκB kinase α. Cancer Res. 2007, 67, 6263–6269. [Google Scholar] [CrossRef] [Green Version]
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting, J.P.-Y.; Baldwin, A.S. Akt-dependent regulation of NF-κB is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008, 22, 1490–1500. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Shen, S.; Verma, I.M. NF-kappaB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Du, T.; Xu, G.; Lai, Y.; Fan, X.; Chen, X.; Li, W.; Yue, F.; Li, Q.; Liu, L.; et al. Matrine suppresses invasion of castration-resistant prostate cancer cells by downregulating MMP-2/9 via NF-kappa B signaling pathway. Int. J. Oncol. 2017, 50, 640–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Shao, S.; Han, D.; Xu, Y.; Jiao, D.; Wu, J.; Yang, F.; Ge, Y.; Shi, S.; Li, Y.; et al. High mobility group box 1 promotes the epithelial-to-mesenchymal transition in prostate cancer PC3 cells via the RAGE/NF-kappaB signaling pathway. Int. J. Oncol. 2018, 53, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Yang, Q.; Dai, Y.; Guo, W.; Du, H.; Song, L.; Peng, X. Oncogenic miR-210-3p promotes prostate cancer cell EMT and bone metastasis via NF-kappaB signaling pathway. Mol. Cancer 2017, 16, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Wa, Q.; Pan, J.; Peng, X.; Ren, D.; Huang, Y.; Chen, X.; Tang, Y. Downregulation of miR-141-3p promotes bone metastasis via activating NF-kappaB signaling in prostate cancer. J. Exp. Clin. Cancer Res. 2017, 36, 173. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Cai, Z.; Xiao, G.; Keller, E.T.; Mizokami, A.; Yao, Z.; Roodman, G.D.; Zhang, J. Monocyte chemotactic protein-1 mediates prostate cancer–induced bone resorption. Cancer Res. 2007, 67, 3646–3653. [Google Scholar] [CrossRef] [Green Version]
- Michalaki, V.; Syrigos, K.; Charles, P.; Waxman, J. Serum levels of IL-6 and TNF-α correlate with clinicopathological features and patient survival in patients with prostate cancer. Br. J. Cancer 2004, 90, 2312–2316. [Google Scholar] [CrossRef]
- Schulze, J.; Weber, K.; Baranowsky, A.; Streichert, T.; Lange, T.; Spiro, A.S.; Albers, J.; Seitz, S.; Zustin, J.; Amling, M. p65-Dependent production of interleukin-1β by osteolytic prostate cancer cells causes an induction of chemokine expression in osteoblasts. Cancer Lett. 2012, 317, 106–113. [Google Scholar] [CrossRef]
- Lu, Y.; Cai, Z.; Galson, D.L.; Xiao, G.; Liu, Y.; George, D.E.; Melhem, M.F.; Yao, Z.; Zhang, J. Monocyte chemotactic protein-1 (MCP-1) acts as a paracrine and autocrine factor for prostate cancer growth and invasion. Prostate 2006, 66, 1311–1318. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Gene | Primer Sequence | |
|---|---|---|---|
| Forward | Reverse | ||
| Human | TNF-α | CCTCTCTCTAATCAGCCCTCTG | GAGGACCTGGGAGTAGATGAG |
| IL-1β | ATGATGGCTTATTACAGTGGCAA | GTCGGAGATTCGTAGCTGGA | |
| MCP-1 | CAGCCAGATGCAATCAATGCC | TGGAATCCTGAACCCACTTCT | |
| GAPDH | GGAGCGAGATCCCTCCAAAAT | GGCTGTTGTCATACTTCTCATGG | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, E.Y.; Park, J.; Kim, Y.T.; Kim, M.J. Imipramine Inhibits Migration and Invasion in Metastatic Castration-Resistant Prostate Cancer PC-3 Cells via AKT-Mediated NF-κB Signaling Pathway. Molecules 2020, 25, 4619. https://doi.org/10.3390/molecules25204619
Lim EY, Park J, Kim YT, Kim MJ. Imipramine Inhibits Migration and Invasion in Metastatic Castration-Resistant Prostate Cancer PC-3 Cells via AKT-Mediated NF-κB Signaling Pathway. Molecules. 2020; 25(20):4619. https://doi.org/10.3390/molecules25204619
Chicago/Turabian StyleLim, Eun Yeong, Joon Park, Yun Tai Kim, and Min Jung Kim. 2020. "Imipramine Inhibits Migration and Invasion in Metastatic Castration-Resistant Prostate Cancer PC-3 Cells via AKT-Mediated NF-κB Signaling Pathway" Molecules 25, no. 20: 4619. https://doi.org/10.3390/molecules25204619