
The Dynamics of the Neuropeptide Y Receptor Type 1 Investigated by Solid-State NMR and Molecular Dynamics Simulation

 ,
,  , and
, and
Abstract
:
1. Introduction
2. Results
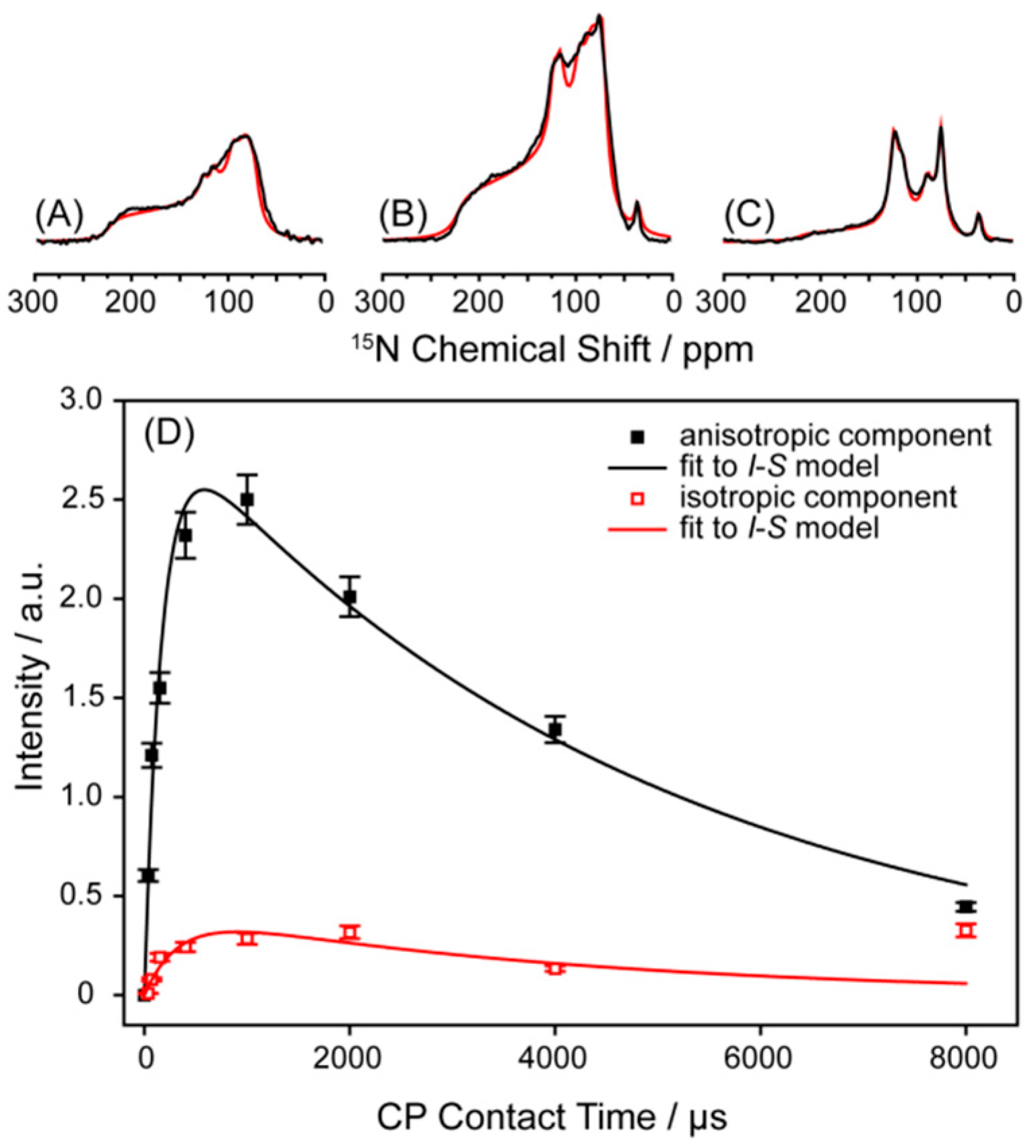
2.1. Static 15N-NMR Spectroscopy on Y1R in Liquid Crystalline Membranes
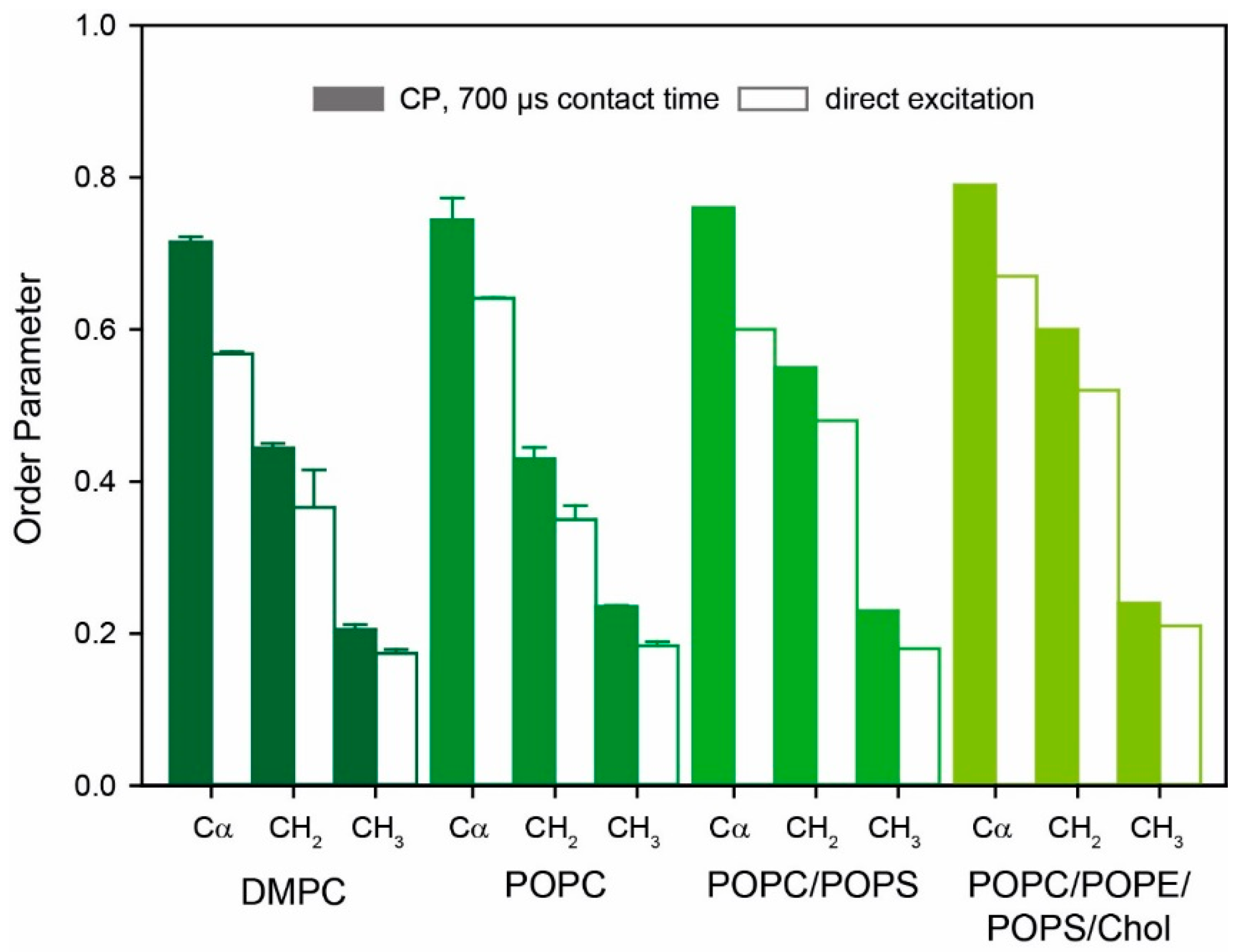
2.2. 13C-NMR Studies of the Molecular Dynamics of Y1R by DipShift Experiments
2.3. Molecular Dynamics of Y1R in the Presence of the Agonist and Coupled to Arr3-3A
2.4. Molecular Dynamics Simulations of Y1R in the Absence and in the Presence of the Antagonist UR-MK299
3. Discussion
4. Materials and Methods
4.1. Materials and NPY Synthesis
4.2. Y1R Expression
4.3. Arr3-3A Expression
4.4. Y1R Sample Preparation
4.5. NMR Experiments
4.6. MD Simulations
Author Contributions
Funding
Conflicts of Interest
References
- Zernia, G.; Huster, D. Collagen dynamics in articular cartilage under osmotic pressure. NMR Biomed. 2006, 19, 1010–1019. [Google Scholar] [CrossRef] [PubMed]
- Mogilner, A.; Keren, K. The Shape of Motile Cells. Curr. Biol. 2009, 19, R762–R771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huster, D.; Arnold, K.; Gawrisch, K. Investigation of lipid organization in biological membranes by two-dimensional nuclear overhauser enhancement spectroscopy. J. Phys. Chem. B 1999, 103, 243–251. [Google Scholar] [CrossRef]
- White, S.H.; Ladokhin, A.S.; Jayasinghe, S.; Hristova, K. How membranes shape protein structure. J. Biol. Chem. 2001, 276, 32395–32398. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.F. Soft Matter in lipid–protein interactions. Annu. Rev. Biophys. 2017, 46, 379–410. [Google Scholar] [CrossRef] [Green Version]
- Henzler-Wildman, K.A.; Kern, D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Kobilka, B. The structural basis of G-protein-coupled receptor signaling. Angew. Chem. Int. Ed. Engl. 2013, 52, 6380–6388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, I.; Ueda, T.; Kofuku, Y.; Eddy, M.T.; Wüthrich, K. GPCR drug discovery: Integrating solution NMR data with crystal and cryo-EM structures. Nat. Rev. Drug Discov. 2019, 18, 59–82. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Kofuku, Y.; Okude, J.; Imai, S.; Shiraishi, Y.; Shimada, I. Function-related conformational dynamics of G protein–coupled receptors revealed by NMR. Biophys. Rev. 2019, 11, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Scheerer, P.; Park, J.H.; Hildebrand, P.W.; Kim, Y.J.; Krauss, N.; Choe, H.-W.; Hofmann, K.P.; Ernst, O.P. Crystal structure of opsin in its G-protein-interacting conformation. Nature 2008, 455, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.E.; Zhang, Y.; Hu, H.; Suomivuori, C.-M.; Kadji, F.M.N.; Aoki, J.; Kumar, K.K.; Fonseca, R.; Hilger, D.; Huang, W.; et al. Conformational transitions of a neurotensin receptor 1–Gi1 complex. Nature 2019, 572, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.G.F.; Choi, H.J.; Rosenbaum, D.M.; Kobilka, T.S.; Thian, F.S.; Edwards, P.C.; Burghammer, M.; Ratnala, V.R.P.; Sanishvili, R.; Fischetti, R.F.; et al. Crystal structure of the human beta(2) adrenergic G-protein-coupled receptor. Nature 2007, 450, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Han, S.; Keller, M.; Kaiser, A.; Bender, B.J.; Bosse, M.; Burkert, K.; Kögler, L.M.; Wifling, D.; Bernhardt, G.; et al. Structural basis of ligand binding modes at the neuropeptide Y Y1 receptor. Nature 2018, 556, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Staus, D.P.; Hu, H.; Robertson, M.J.; Kleinhenz, A.; Wingler, L.M.; Capel, W.D.; Latorraca, N.R.; Lefkowitz, R.J.; Skiniotis, G. Structure of the M2 muscarinic receptor-ß-arrestin complex in a lipid nanodisc. Nature 2020, 579, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Masureel, M.; Qu, Q.; Janetzko, J.; Inoue, A.; Kato, H.E.; Robertson, M.J.; Nguyen, K.C.; Skiniotis, G.; Kobilka, B.K. Structure of the neurotensin receptor 1 in complex with ß-arrestin 1. Nature 2020, 579, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.A.; Motoshima, H.; Fox, B.A.; Le Trong, I.; Teller, D.C.; Okada, T.; Stenkamp, R.E.; et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000, 289, 739–745. [Google Scholar] [CrossRef] [Green Version]
- Shiimura, Y.; Horita, H.; Hamamoto, A.; Asada, H.; Hirata, K.; Tanaka, M.; Mori, K.; Uemura, T.; Kobayashi, T.; Iwata, S.; et al. Structure of an antagonist-bound ghrelin receptor reveals possible ghrelin recognition mode. Nat. Commun. 2020, 11, 4160. [Google Scholar] [CrossRef]
- Rasmussen, S.G.; DeVree, B.T.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D.; et al. Crystal structure of the b2 adrenergic receptor-Gs protein complex. Nature 2011, 477, 549–555. [Google Scholar] [CrossRef] [Green Version]
- Tian, H.; Fürstenberg, A.; Huber, T. Labeling and single-molecule methods to monitor G protein-coupled receptor dynamics. Chem. Rev. 2016, 117, 186–245. [Google Scholar] [CrossRef]
- Imai, S.; Yokomizo, T.; Kofuku, Y.; Shiraishi, Y.; Ueda, T.; Shimada, I. Structural equilibrium underlying ligand-dependent activation of ß2-adrenoreceptor. Nat. Chem. Biol. 2020, 16, 430–439. [Google Scholar] [CrossRef]
- Isogai, S.; Deupi, X.; Opitz, C.; Heydenreich, F.M.; Tsai, C.J.; Brueckner, F.; Schertler, G.F.; Veprintsev, D.B.; Grzesiek, S. Backbone NMR reveals allosteric signal transduction networks in the ß1-adrenergic receptor. Nature 2016, 530, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Frei, J.N.; Broadhurst, R.W.; Bostock, M.J.; Solt, A.; Jones, A.; Gabriel, F.; Tandale, A.; Shrestha, B.; Nietlispach, D. Conformational plasticity of ligand-bound and ternary GPCR complexes studied by 19F NMR of the ß1-adrenergic receptor. Nat. Commun. 2020, 11, 669. [Google Scholar] [CrossRef] [PubMed]
- Eddy, M.T.; Lee, M.-Y.; Gao, Z.; Didenko, T.; Horst, R.; Audet, M.; Stanczak, P.; McClary, K.M.; Han, G.W.; Jacobson, K.A.; et al. Allosteric coupling of drug binding and intracellular signaling in the A2A adenosine receptor. Cell 2018, 172, 68–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krug, U.; Gloge, A.; Schmidt, P.; Becker-Baldus, J.; Bernhard, F.; Kaiser, A.; Montag, C.; Gauglitz, M.; Vishnivetskiy, S.A.; Gurevich, V.V.; et al. The conformational equilibrium of the neuropeptide Y2 Receptor in bilayer membranes. Angew. Chem. Int. Ed. 2020, 59. [Google Scholar] [CrossRef]
- Pope, A.L.; Sanchez-Reyes, O.B.; South, K.; Zaitseva, E.; Ziliox, M.; Vogel, R.; Reeves, P.J.; Smith, S.O. A conserved proline hinge mediates helix dynamics and activation of rhodopsin. Structure 2020, 28, 1004–1013. [Google Scholar] [CrossRef]
- Hofmann, K.P.; Scheerer, P.; Hildebrand, P.W.; Choe, H.-W.; Park, J.H.; Heck, M.; Ernst, O.P. A G protein-coupled receptor at work: The rhodopsin model. Trends Biochem. Sci. 2009, 34, 540–552. [Google Scholar] [CrossRef]
- Nygaard, R.; Frimurer, T.M.; Holst, B.; Rosenkilde, M.M.; Schwartz, T.W. Ligand binding and micro-switches in 7TM receptor structures. Trends Pharmacol. Sci. 2009, 30, 249–259. [Google Scholar] [CrossRef]
- Ye, L.; Van Eps, N.; Zimmer, M.; Ernst, O.P.; Prosser, R.S. Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature 2016, 533, 265–268. [Google Scholar] [CrossRef]
- Liu, J.J.; Horst, R.; Katritch, V.; Stevens, R.C.; Wüthrich, K. Biased signaling pathways in beta2-adrenergic receptor characterized by 19F NMR. Science 2012, 335, 1106–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nygaard, R.; Zou, Y.; Dror, R.O.; Mildorf, T.J.; Arlow, D.H.; Manglik, A.; Pan, A.C.; Liu, C.W.; Fung, J.J.; Bokoch, M.P.; et al. The dynamic process of beta (2)-adrenergic receptor activation. Cell 2013, 152, 532–542. [Google Scholar] [CrossRef] [Green Version]
- Deupi, X.; Kobilka, B. Energy landscapes as a tool to integrate GPCR structure, dynamics, and function. Physiology 2010, 25, 293–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frauenfelder, H.; Sligar, S.G.; Wolynes, P.G. The energy landscapes and motions of proteins. Science 1991, 254, 1598–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, A.G.; Williams, J.; McDermott, A.E. Nuclear magnetic resonance studies of biopolymer dynamics. J. Phys. Chem. 1996, 100, 13293–13310. [Google Scholar] [CrossRef]
- Good, D.B.; Wang, S.; Ward, M.E.; Struppe, J.; Brown, L.S.; Lewandowski, J.R.; Ladizhansky, V. Conformational dynamics of a seven transmembrane helical protein anabaena sensory rhodopsin probed by solid-state NMR. J. Am. Chem. Soc. 2014, 136, 2833–2842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, P.; Thomas, L.; Müller, P.; Scheidt, H.A.; Huster, D. The G-protein-coupled neuropeptide Y receptor type 2 is highly dynamic in lipid membranes as revealed by solid-state NMR spectroscopy. Chem. A Eur. J. 2014, 20, 4986–4992. [Google Scholar] [CrossRef] [PubMed]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR dynamics: Structures in motion. Chem. Rev. 2017, 117, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Arlow, D.H.; Maragakis, P.; Mildorf, T.J.; Pan, A.C.; Xu, H.; Borhani, D.W.; Shaw, D.E. Activation mechanism of the b2-adrenergic receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 18684–18689. [Google Scholar] [CrossRef] [Green Version]
- Kohlhoff, K.J.; Shukla, D.; Lawrenz, M.; Bowman, G.R.; Konerding, D.E.; Belov, D.; Altman, R.B.; Pande, V.S. Cloud-based simulations on Google Exacycle reveal ligand modulation of GPCR activation pathways. Nat. Chem. 2014, 6, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Rose, A.S.; Elgeti, M.; Zachariae, U.; Grubmüller, H.; Hofmann, K.P.; Scheerer, P.; Hildebrand, P.W. Position of transmembrane helix 6 determines receptor G protein coupling specificity. J. Am. Chem. Soc. 2014, 136, 11244–11247. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Bhattacharya, S.; Tate, C.G.; Grisshammer, R.; Vaidehi, N. Structural dynamics and thermostabilization of neurotensin receptor 1. J. Phys. Chem. B 2015, 119, 4917–4928. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Zhao, X.; Watts, A. G-protein-coupled receptor structure, ligand binding and activation as studied by solid-state NMR spectroscopy. Biochem. J. 2013, 450, 443–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbell, W.L.; Altenbach, C.; Hubbell, C.M.; Khorana, H. Rhodopsin structure, dynamics, and activation: A perspective from crystallography, site-directed spin labeling, sulfhydryl reactivity, and disulfide cross-linking. Adv. Protein Chem. 2003, 63, 243–290. [Google Scholar] [CrossRef] [PubMed]
- Pedragosa-Badia, X.; Stichel, J.; Beck-Sickinger, A.G. Neuropeptide Y receptors: How to get subtype selectivity. Front. Endocrinol. 2013, 4, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, L.; Kahr, J.; Schmidt, P.; Krug, U.; Scheidt, H.A.; Huster, D. The dynamics of the G protein-coupled neuropeptide Y2 receptor in monounsaturated membranes investigated by solid-state NMR spectroscopy. J. Biomol. NMR 2015, 61, 347–359. [Google Scholar] [CrossRef]
- Schrottke, S.; Kaiser, A.; Vortmeier, G.; Els-Heindl, S.; Worm, D.; Bosse, M.; Schmidt, P.; Scheidt, H.A.; Beck-Sickinger, A.G.; Huster, D. Expression, functional characterization, and solid-state NMR investigation of the G protein-coupled GHS receptor in bilayer membranes. Sci. Rep. 2017, 7, srep46128. [Google Scholar] [CrossRef] [Green Version]
- Pacull, E.M.; Sendker, F.; Bernhard, F.; Scheidt, H.A.; Schmidt, P.; Huster, D.; Krug, U. Integration of cell-free expression and solid-state NMR to investigate the dynamic properties of different sites of the growth hormone secretagogue receptor. Front. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Park, S.H.; Casagrande, F.; Das, B.B.; Albrecht, L.; Chu, M.; Opella, S.J. Local and global dynamics of the G protein-coupled receptor CXCR1. Biochemistry 2011, 50, 2371–2380. [Google Scholar] [CrossRef] [Green Version]
- Kolodziejski, W.; Klinowski, J. Kinetics of cross-polarization in solid-state NMR: A guide for chemists. Chem. Rev. 2002, 102, 613–628. [Google Scholar] [CrossRef]
- Rothwell, W.P.; Waugh, J.S. Transverse relaxation of dipolar coupled spin systems under rf irradiation: Detecting motions in solids. J. Chem. Phys. 1981, 74, 2721–2732. [Google Scholar] [CrossRef]
- Munowitz, M.G.; Griffin, R.G.; Bodenhausen, G.; Huang, T.H. Two-dimensional rotational spin-echo nuclear magnetic resonance in solids: Correlation of chemical shift and dipolar interactions. J. Am. Chem. Soc. 1981, 103, 2529–2533. [Google Scholar] [CrossRef]
- Huster, D.; Xiao, L.; Hong, M. Solid-state NMR investigation of the dynamics of the soluble and membrane-bound colicin in channel-forming domain. Biochemistry 2001, 40, 7662–7674. [Google Scholar] [CrossRef]
- Barré, P.; Zschörnig, O.; Arnold, K.; Huster, D. Structural and dynamical changes of the bindin B18 peptide upon binding to lipid membranes. A solid-state NMR study. Biochemistry 2003, 42, 8377–8386. [Google Scholar] [CrossRef] [PubMed]
- Huster, D.; Arnold, K.; Gawrisch, K. Strength of Ca2+ binding to retinal lipid membranes: Consequences for lipid organization. Biophys. J. 2000, 78, 3011–3018. [Google Scholar] [CrossRef] [Green Version]
- Manglik, A.; Kobilka, B.K. The role of protein dynamics in GPCR function: Insights from the β2AR and rhodopsin. Curr. Opin. Cell Biol. 2014, 27, 136–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, A.; Reuther, G.; Roark, M.B.; Tan, K.-T.; Waldmann, H.; Feller, S.E.; Huster, D. Backbone conformational flexibility of the lipid modified membrane anchor of the human N-Ras protein investigated by solid-state NMR and molecular dynamics simulation. Biochim. Biophys. Acta (BBA)-Biomembr. 2010, 1798, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Götz, A.; Mylonas, N.; Högel, P.; Silber, M.; Heinel, H.; Menig, S.; Vogel, A.; Feyrer, H.; Huster, D.; Luy, B.; et al. Modulating hinge flexibility in the APP transmembrane domain alters gamma-secretase cleavage. Biophys. J. 2019, 116, 2103–2120. [Google Scholar] [CrossRef]
- Clore, G.M.; Szabo, A.; Bax, A.; Kay, L.E.; Driscoll, P.C.; Gronenborn, A.M. Deviations from the simple two-parameter model-free approach to the interpretation of nitrogen-15 nuclear magnetic relaxation of proteins. J. Am. Chem. Soc. 1990, 112, 4989–4991. [Google Scholar] [CrossRef]
- Lipari, G.; Szabo, A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theorie and range of validity. J. Am. Chem. Soc. 1982, 104, 4546–4559. [Google Scholar] [CrossRef]
- Lipari, G.; Szabo, A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. J. Am. Chem. Soc. 1982, 104, 4559–4570. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Huster, D. Backbone dynamics of bacteriorhodopsin as studied by 13 C solid-state NMR spectroscopy. Eur. Biophys. J. 2003, 32, 578–584. [Google Scholar] [CrossRef]
- Yang, J.; Aslimovska, L.; Glaubitz, C. Molecular dynamics of proteorhodopsin in lipid bilayers by solid-state NMR. J. Am. Chem. Soc. 2011, 133, 4874–4881. [Google Scholar] [CrossRef] [PubMed]
- Reuther, G.; Tan, K.-T.; Vogel, A.; Nowak, C.; Arnold, K.; Kuhlmann, J.; Waldmann, H.; Huster, D. The lipidated membrane anchor of full length N-ras protein shows an extensive dynamics as revealed by solid-state NMR spectroscopy. J. Am. Chem. Soc. 2006, 128, 13840–13846. [Google Scholar] [CrossRef]
- Soubias, O.; Niu, S.-L.; Mitchell, D.C.; Gawrisch, K. Lipid−rhodopsin hydrophobic mismatch alters rhodopsin helical content. J. Am. Chem. Soc. 2008, 130, 12465–12471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheidt, H.A.; Meyer, T.; Nikolaus, J.; Baek, D.J.; Haralampiev, I.; Thomas, L.; Bittman, R.; Herrmann, A.; Müller, P.; Huster, D. Cholesterol’s aliphatic side chain structure modulates membrane properties. Angew. Chem. Int. Ed. 2013, 52, 12848–12851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celver, J.; Vishnivetskiy, S.A.; Chavkin, C.; Gurevich, V.V. Conservation of the phosphate-sensitive elements in the Arrestin family of proteins. J. Biol. Chem. 2002, 277, 9043–9048. [Google Scholar] [CrossRef] [Green Version]
- Deazevedo, E.R.; Saalwächter, K.; Pascui, O.; De Souza, A.A.; Bonagamba, T.J.; Reichert, D. Intermediate motions as studied by solid-state separated local field NMR experiments. J. Chem. Phys. 2008, 128, 104505. [Google Scholar] [CrossRef] [Green Version]
- Bowman, G.R. Accurately modeling nanosecond protein dynamics requires at least microseconds of simulation. J. Comput. Chem. 2015, 37, 558–566. [Google Scholar] [CrossRef]
- Rosenbaum, D.M.; Cherezov, V.; Hanson, M.A.; Rasmussen, S.G.F.; Thian, F.S.; Kobilka, T.S.; Choi, H.J.; Yao, X.J.; Weis, W.I.; Stevens, R.C.; et al. GPCR engineering yields high-resolution structural insights into beta(2)-adrenergic receptor function. Science 2007, 318, 1266–1273. [Google Scholar] [CrossRef] [Green Version]
- Elgeti, M.; Rose, A.S.; Bartl, F.J.; Hildebrand, P.W.; Hofmann, K.-P.; Heck, M. Precision vs Flexibility in GPCR signaling. J. Am. Chem. Soc. 2013, 135, 12305–12312. [Google Scholar] [CrossRef]
- Ma, X.; Hu, Y.; Batebi, H.; Heng, J.; Xu, J.; Liu, X.; Niu, X.; Li, H.; Hildebrand, P.W.; Jin, C.; et al. Analysis of β(2)AR-G(s) and β(2)AR-G(i) complex formation by NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2020, 117, 23096–23105. [Google Scholar] [CrossRef]
- Heberle, J.; Fitter, J.; Sass, H.J.; Büldt, G. Bacteriorhodopsin: The functional details of a molecular machine are being resolved. Biophys. Chem. 2000, 85, 229–248. [Google Scholar] [CrossRef]
- Kaiser, A.; Müller, P.; Zellmann, T.; Scheidt, H.A.; Thomas, L.; Bosse, M.; Meier, R.; Meiler, J.; Huster, D.; Beck-Sickinger, A.G.; et al. Unwinding of the C-terminal residues of neuropeptide Y is critical for Y2 receptor binding and activation. Angew. Chem. Int. Ed. 2015, 54, 7446–7449. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, P.; Bender, B.J.; Kaiser, A.; Gulati, K.; Scheidt, H.A.; Hamm, H.E.; Meiler, J.; Beck-Sickinger, A.G.; Huster, D. Improved in vitro folding of the Y2 G protein-coupled receptor into bicelles. Front. Mol. Biosci. 2018, 4, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, P.; Berger, C.; Scheidt, H.A.; Berndt, S.; Bunge, A.; Beck-Sickinger, A.G.; Huster, D. A reconstitution protocol for the in vitro folded human G protein-coupled Y2 receptor into lipid environment. Biophys. Chem. 2010, 150, 29–36. [Google Scholar] [CrossRef]
- Schmidt, P.; Lindner, D.; Montag, C.; Berndt, S.; Beck-Sickinger, A.G.; Rudolph, R.; Huster, D. Prokaryotic expression, in vitro folding, and molecular pharmacological characterization of the neuropeptide Y receptor type 2. Biotechnol. Prog. 2009, 25, 1732–1739. [Google Scholar] [CrossRef]
- Vishnivetskiy, S.A.; Zhan, X.; Chen, Q.; Iverson, T.M.; Gurevich, V.V. Arrestin expression in E. coli and purification. Curr. Protoc. Pharmacol. 2014, 67, 2.11.1–2.11.19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudolph, R.; Lilie, H. In vitro folding of inclusion body proteins. FASEB J. 1996, 10, 49–56. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, A.A.; Opella, S.J. Bicelle samples for solid-state NMR of membrane proteins. Nat. Protoc. 2007, 2, 2332–2338. [Google Scholar] [CrossRef] [PubMed]
- Bennett, A.E.; Rienstra, C.M.; Auger, M.; Lakshmi, K.V.; Griffin, R.G. Heteronuclear decoupling in rotating solids. J. Chem. Phys. 1995, 103, 6951–6958. [Google Scholar] [CrossRef]
- Hong, M.; Gross, J.D.; Griffin, R.G. Site-resolved determination of peptide torsion angle F from relative orientations of backbone N-H and C-H bonds by solid-state NMR. J. Phys. Chem. 1997, 101, 5869–5874. [Google Scholar] [CrossRef]
- Bielecki, A.; Kolbert, A.; Levitt, M. Frequency-switched pulse sequences: Homonuclear decoupling and dilute spin NMR in solids. Chem. Phys. Lett. 1989, 155, 341–346. [Google Scholar] [CrossRef]
- Ismer, J.; Rose, A.S.; Tiemann, J.K.S.; Goede, A.; Preissner, R.; Hildebrand, P.W. SL2: An interactive webtool for modeling of missing segments in proteins. Nucleic Acids Res. 2016, 44, W390–W394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, J.A.D.; Pastor, R.W. Update of the CHARMM All-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Hermans, J. Hydrophilicity of cavities in proteins. Proteins Struct. Funct. Bioinform. 1996, 24, 433–438. [Google Scholar] [CrossRef]
- Liu, W.; Chun, E.; Thompson, A.A.; Chubukov, P.; Xu, F.; Katritch, V.; Han, G.W.; Roth, C.B.; Heitman, L.H.; Ijzerman, A.P.; et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 2012, 337, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Parker, M.S.; Crowley, W.R.; Parker, S.L. Differences in cation sensitivity of ligand binding to Y1 and Y2 subtype of neuropeptide Y receptor of rat brain. Eur. J. Pharmacol. 1996, 318, 193–200. [Google Scholar] [CrossRef]
- Parker, M.S.; Parker, S.L.; Kane, J.K. Internalization of neuropeptide Y Y1 and Y5 and of pancreatic polypeptide Y4 receptors is inhibited by lithium in preference to sodium and potassium ions. Regul. Pept. 2004, 118, 67–74. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI membrane builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI membrane builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Jo, S.; MacKerell, A.D.; Klauda, J.B.; Im, W. CHARMM-GUI input generator for NAMD, Gromacs, Amber, Openmm, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. Biophys. J. 2016, 110, 641a. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Im, W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS ONE 2007, 2, e880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Patel, D.S.; Ståhle, J.; Park, S.J.; Kern, N.R.; Kim, S.; Lee, J.; Cheng, X.; Valvano, M.A.; Holst, O.; et al. CHARMM-GUI membrane builder for complex biological membrane simulations with glycolipids and lipoglycans. J. Chem. Theory. Comput. 2019, 15, 775–786. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2009, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Tiemann, J.K.S.; Guixà-González, R.; Hildebrand, P.W.; Rose, A.S. MDsrv: Viewing and sharing molecular dynamics simulations on the web. Nat. Methods 2017, 14, 1123–1124. [Google Scholar] [CrossRef]
Sample Availability: Samples are available for collaborations upon request from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cα | CH2 | CH3 | ||||
|---|---|---|---|---|---|---|
| Excitation Scheme | CP, 700 µs | Direct Excitation | CP, 700 µs | Direct Excitation | CP, 700 µs | Direct Excitation |
| Y1R (apo) | 0.72 ± 0.01 * | 0.57 ± 0.01 | 0.44 ± 0.01 | 0.37 ± 0.05 | 0.21 ± 0.01 | 0.18 ± 0.01 |
| Y1R + NPY | 0.78 ± 0.03 | 0.61 ± 0.02 | 0.45 ± 0.03 | 0.32 ± 0.04 | 0.23 ± 0.01 | 0.20 ± 0.01 |
| Y1R + NPY + Arr3-3A | 0.86 ± 0.05 | 0.75 ± 0.14 | 0.55 ± 0.04 | 0.42 ± 0.04 | 0.24 ± 0.01 | 0.19 ± 0.01 |
| Cα | CH2 | CH3 | ||||
|---|---|---|---|---|---|---|
| Excitation Scheme | CP, 700 µs | Direct Excitation | CP, 700 µs | Direct Excitation | CP, 700 µs | Direct Excitation |
| POPC | 0.74 ± 0.03 * | 0.64 ± 0.01 | 0.43 ± 0.02 | 0.35 ± 0.02 | 0.24 ± 0.01 | 0.18 ± 0.01 |
| POPC/POPS (8/2, mol/mol) | 0.76 | 0.60 | 0.55 | 0.48 | 0.23 | 0.18 |
| POPC/POPE/POPS/cholesterol (8/2, mol/mol) | 0.79 | 0.67 | 0.60 | 0.52 | 0.24 | 0.21 |
| POPC + UR-MK299 | 0.76 ± 0.02 | 0.63 ± 0.01 | 0.42 ± 0.01 | 0.34 ± 0.01 | 0.23 ± 0.02 | 0.18 ± 0.01 |
| Structural Element | Subs-Segment | Amino Acids |
|---|---|---|
| NTER | 1 | 18–24 |
| 2 | 24–30 | |
| 3 | 30–36 | |
| Helix 1 | Extracellular | 38–47 |
| Intracellular | 49–67 | |
| ICL1 | 1 | 68–70 |
| 2 | 70–73 | |
| Helix 2 | Intracellular | 75–91 |
| Extracellular | 95–103 | |
| ECL1 | 1 | 104–106 |
| 2 | 106–109 | |
| Helix 3 | Extracellular | 111–116 |
| Intracellular | 119–138 | |
| ICL2 | 1 | 144–147 |
| 2 | 147–151 | |
| Helix 4 | intracellular | 153–170 |
| Extracellular | 172–176 | |
| ECL2 | 1 | 177–181 |
| 2 | 181–186 | |
| 3 | 186–191 | |
| 4 | 191–196 | |
| 5 | 196–201 | |
| 6 | 201–204 | |
| Helix 5 | Extracellular | 205–218 |
| Intracellular | 223–233 | |
| ICL3 | 1 | 245–248 |
| 2 | 248–251 | |
| Helix 6 | Intracellular | 264–277 |
| Extracellular | 279–288 | |
| ECL3 | 1 | 289–292 |
| 2 | 292–295 | |
| Helix 7 | Extracellular | 297–314 |
| Intracellular | 316–323 | |
| Helix 8 | - | 326–334 |
| CTER | - | 335–339 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vogel, A.; Bosse, M.; Gauglitz, M.; Wistuba, S.; Schmidt, P.; Kaiser, A.; Gurevich, V.V.; Beck-Sickinger, A.G.; Hildebrand, P.W.; Huster, D. The Dynamics of the Neuropeptide Y Receptor Type 1 Investigated by Solid-State NMR and Molecular Dynamics Simulation. Molecules 2020, 25, 5489. https://doi.org/10.3390/molecules25235489
Vogel A, Bosse M, Gauglitz M, Wistuba S, Schmidt P, Kaiser A, Gurevich VV, Beck-Sickinger AG, Hildebrand PW, Huster D. The Dynamics of the Neuropeptide Y Receptor Type 1 Investigated by Solid-State NMR and Molecular Dynamics Simulation. Molecules. 2020; 25(23):5489. https://doi.org/10.3390/molecules25235489
Chicago/Turabian StyleVogel, Alexander, Mathias Bosse, Marcel Gauglitz, Sarah Wistuba, Peter Schmidt, Anette Kaiser, Vsevolod V. Gurevich, Annette G. Beck-Sickinger, Peter W. Hildebrand, and Daniel Huster. 2020. "The Dynamics of the Neuropeptide Y Receptor Type 1 Investigated by Solid-State NMR and Molecular Dynamics Simulation" Molecules 25, no. 23: 5489. https://doi.org/10.3390/molecules25235489