
Theoretical Studies of Photophysical Properties of D−π−A−π−D-Type Diketopyrrolopyrrole-Based Molecules for Organic Light-Emitting Diodes and Organic Solar Cells

Abstract
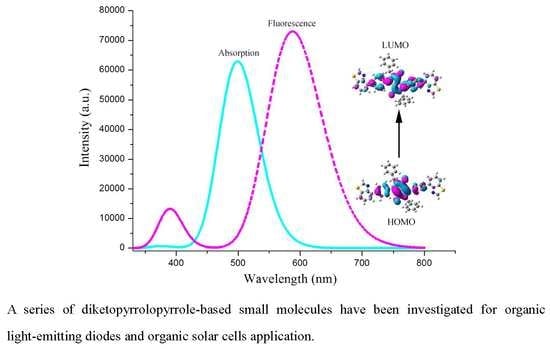
:
1. Introduction
2. Results and Discussion
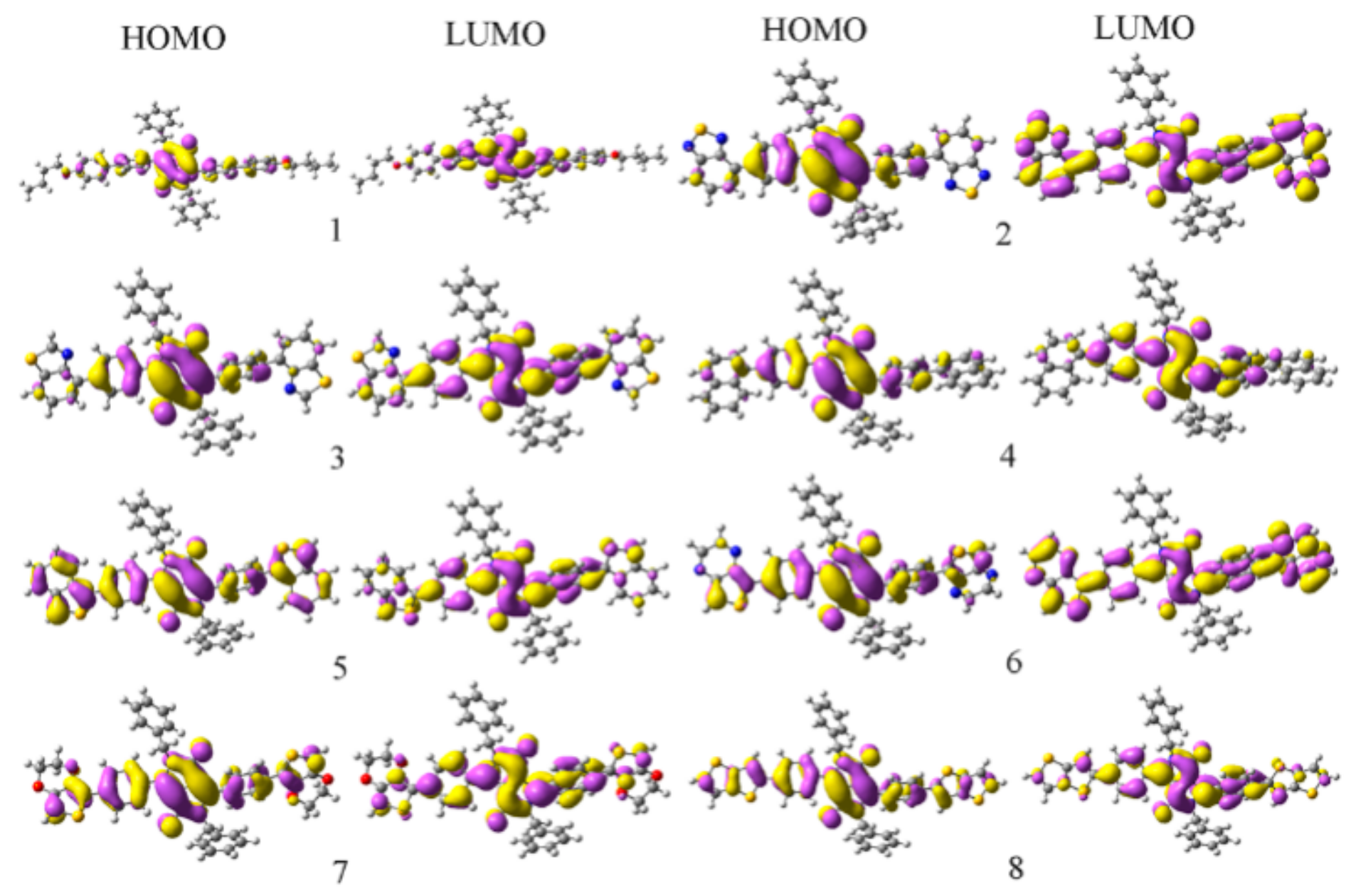
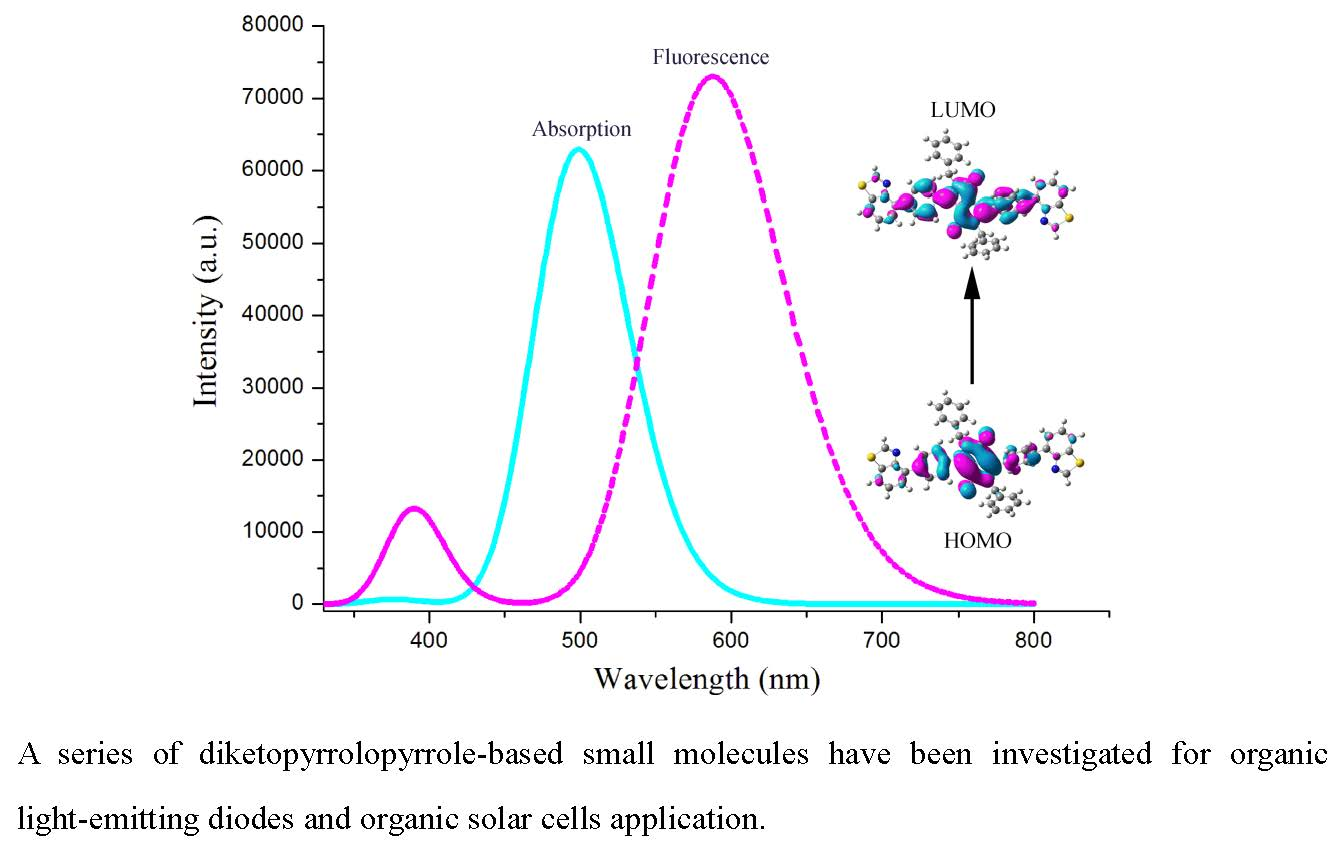
2.1. Frontier Molecular Orbitals
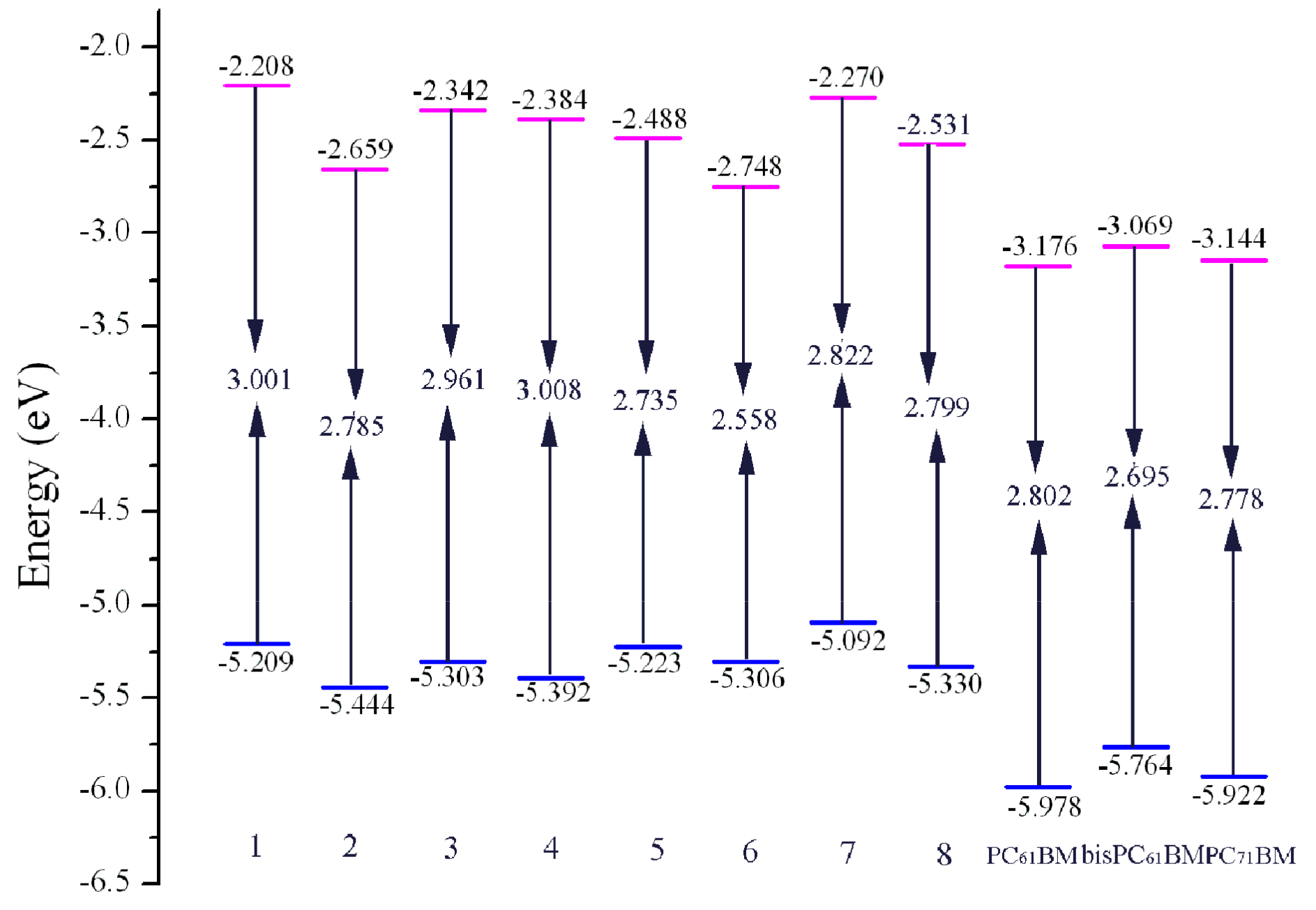
2.2. Match between Donor and Acceptor Material
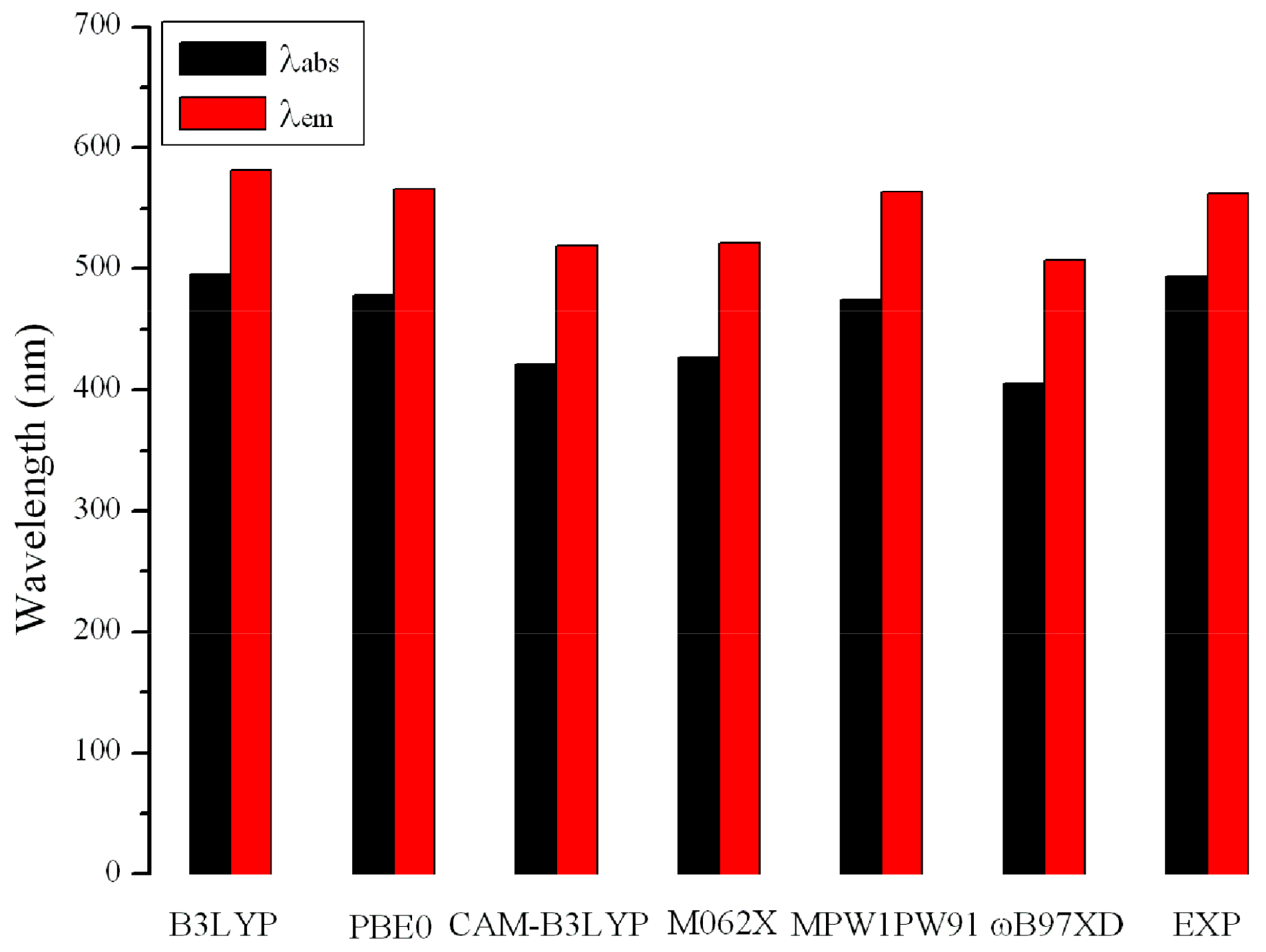
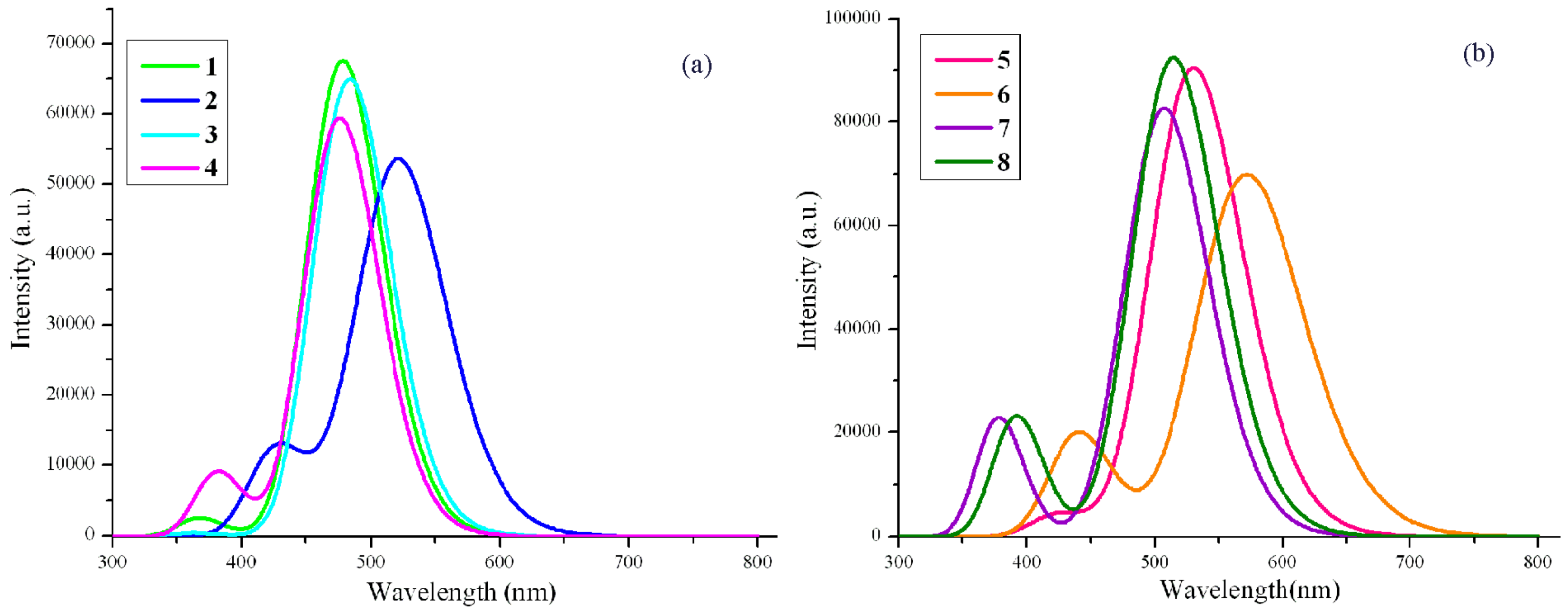
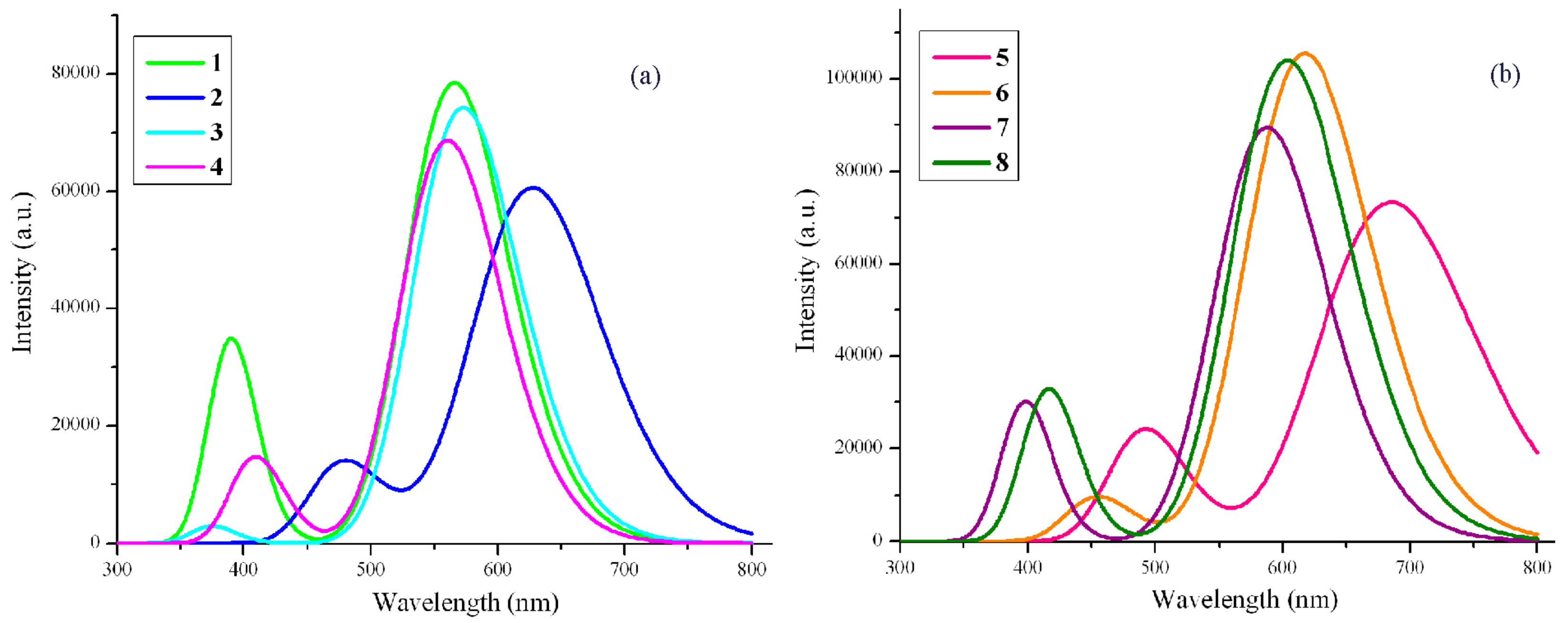
2.3. Absorption and Fluorescent Properties
2.4. Reorganization Energies and Stabilities
3. Materials and Methods
Computational Methods
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| OSCs | Organic solar cells |
| OLEDs | Organic light-emitting diodes |
| FETs | Field effect transistors |
| ΔEL-L | Downhill energetic driving force |
| DPP | Diketopyrrolopyrrole |
| ICT | Intramolecular charge transfer |
| DFT | Density function theory |
| TD-DFT | Time dependent density function theory |
| FMOs | Frontier molecular orbital energies |
| HOMO | Highest occupied molecular orbital |
| LUMO | Lowest unoccupied molecular orbital |
References
- Zhao, J.; Li, Y.; Yang, G.; Jiang, K.; Lin, H.; Ade, H.; Ma, W.; Yan, H. Efficient organic solar cells processed from hydrocarbon solvents. Nat. Energy 2016, 1, 1–7. [Google Scholar] [CrossRef]
- Fan, B.; Ying, L.; Zhu, P.; Pan, F.; Liu, F.; Chen, J.; Huang, F.; Cao, Y. All-polymer solar cells based on a conjugated polymer containing siloxane-functionalized side chains with efficiency over 10%. Adv. Mater. 2017, 29, 1703906. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Zhang, Y.; Wan, X.; Li, C.; Zhang, X.; Wang, Y.; Ke, X.; Xiao, Z.; Ding, L.; Xia, R.; et al. Organic and solution-processed tandem solar cells with 17.3% efficiency. Science 2018, 361, 1094–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, Y.; Zhang, H.L.; Zhan, X. Non-fullerene all-small molecule organic solar cells. ACS Energy Lett. 2019, 2, 2717–2722. [Google Scholar]
- Cui, Y.; Yao, H.; Hong, L.; Zhang, T.; Xu, Y.; Xian, K.; Gao, B.; Qin, J.; Zhang, J.; Wei, Z.; et al. Achieving over 15% efficiency in organic photovoltaic cells via copolymer design. Adv. Mater. 2019, 31, 1808356. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.D.; Ran, N.A.; Heiber, M.C.; Nguyen, T.Q. Small is powerful: Recent progress in solution-processed small molecule solar cells. Adv. Energy Mater. 2017, 7, 1602242. [Google Scholar] [CrossRef]
- Han, L.; Wang, C.; Ren, A.; Liu, Y.; Liu, P. Structural and optical properties of triphenylamin-substituted anthracene derivatives. J. Mol. Sci. 2013, 29, 146–151. [Google Scholar]
- Sahu, D.; Tsai, C.H.; Wei, H.Y.; Ho, K.C.; Chang, F.C.; Chu, C.W. Synthesis and applications of novel low bandgap star-burst molecules containing a triphenylamine core and dialkylated diketopyrrolopyrrole arms for organic photovoltaics. J. Mater. Chem. 2012, 22, 7945–7953. [Google Scholar] [CrossRef]
- Coughlin, J.E.; Henson, Z.B.; Welch, G.C.; Bazan, G.C. Design and synthesis of molecular donors for solution-processed high-efficiency organic solar cells. Acc. Chem. Res. 2014, 47, 257–270. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, M.; Chen, H.; Wu, F.; Cao, Z.; Zhang, L.; Tan, S. Efficient polymer solar cells based on terpolymers with a broad absorption range of 300–900 nm. J. Mater. Chem. A 2014, 2, 5218–5223. [Google Scholar] [CrossRef]
- Koster, L.; Shaheen, S.; Hummelen, J. Pathways to a New Efficiency Regime for Organic Solar Cells. Adv. Energy Mater. 2012, 2, 1246–1253. [Google Scholar] [CrossRef]
- Coffey, D.; Larson, B.; Hains, A.; Whitaker, J.; Kopidakis, N.; Boltalina, O.; Strauss, S.; Rumbles, G. An optimal driving force for converting excitons into free carriers in excitonic solar cells. J. Phys. Chem. C 2012, 116, 8916–8923. [Google Scholar] [CrossRef]
- Song, X.; Gasparini, N.; Baran, D. The influence of solvent additive on polymer solar cells employing fullerene and non-fullerene acceptors. Adv. Electron. Mater. 2017, 2, 1700358. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Gasparini, N.; Nahid, M.M.; Chen, H.; Macphee, S.M.; Zhang, W.; Norman, V.; Zhu, C.; Bryant, D.; Ade, H.; et al. A highly crystalline fused-ring n-type small molecule for non-fullerene acceptor based organic solar cells and field-effect transistors. Adv. Funct. Mater. 2018, 28, 1802895. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Zhao, X.; Shen, Y.; Wang, M.; Wang, L.; Meier, H.; Cao, D. Diketopyrrolopyrrole based D-π-A- π-D type small organic molecules as hole transporting materials for perovskite solar cells. J. Energy Chem. 2018, 27, 1175–1182. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Niu, Q.; Sun, T.; Li, Y.; Li, T.; Liu, H. The synthesis and properties of linear A–π–D–π–A type organic small molecule containing diketopyrrolopyrrole terminal units. Spectrochim. Acta A 2017, 183, 172–176. [Google Scholar] [CrossRef]
- Zhang, S.; Sun, T.; Xu, Z.; Li, T.; Li, Y.; Niu, Q.; Liu, H. Novel biphenylene-diketopyrrolopyrrole-based A-π-D-π–A molecule: Synthesis, optical, electrochemical and electronical properties. Tetrahedron. Lett. 2017, 58, 2779–2783. [Google Scholar] [CrossRef]
- Liu, H.; Jia, H.; Wang, L.; Wu, Y.; Zhan, C.; Fu, H.; Yao, J. Intermolecular electron transfer promoted by directional donor–acceptor attractions in self-assembled diketopyrrolopyrrole-thiophenefilms. Phys. Chem. Chem. Phys. 2012, 14, 14262–14269. [Google Scholar] [CrossRef]
- Wang, M.; Wang, H.; Yokoyama, T.; Liu, X.; Huang, Y.; Zhang, Y.; Nguyen, T.Q.; Aramaki, S.; Bazan, G.C. High open circuit voltage in regioregular narrow band gap polymer solar cells. J. Am. Chem. Soc. 2014, 136, 12576–12579. [Google Scholar] [CrossRef]
- Wang, E.; Mammo, W.; Andersson, M.R. 25th Anniversary article: Isoindigo-based polymers and small molecules for bulk heterojunction solar cells and field effect transistors. Adv. Mater. 2014, 26, 1801–1826. [Google Scholar] [CrossRef]
- Chen, S.; Xiao, L.; Zhu, X.; Peng, X.; Wong, W.K.; Wong, W.Y. Solution-processed new porphyrin-based small molecules as electron donors for highly efficient organic photovoltaics. Chem. Commun. 2015, 51, 14439–14442. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.; Tamayo, A.B.; Dang, X.D.; Zalar, P.; Seo, J.H.; Garcia, A.; Tantiwiwat, M.; Nguyen, T.Q. Nanoscale phase separation and high photovoltaic efficiency in solution-processed, small-molecule bulk heterojunction solar cells. Adv. Funct. Mater. 2009, 19, 3063–3069. [Google Scholar] [CrossRef]
- Lin, Y.; Wang, Y.; Wang, J.; Hou, J.H.; Li, Y.F.; Zhu, D.B.; Zhan, X. A star-shaped perylene diimide electron acceptor for high-performance organic solar cells. Adv. Mater. 2014, 26, 5137–5142. [Google Scholar] [CrossRef]
- Liu, Y.; Mu, C.; Jiang, K.; Zhao, J.; Li, Y.; Zhang, L.; Li, Z.; Lai, J.Y.L.; Hu, H.; Ma, T.; et al. A tetraphenylethylene corebased 3D sructure small molecular acceptor enabling efficient non-fullerene organic solar cells. Adv. Mater. 2015, 27, 1015–1020. [Google Scholar] [CrossRef] [PubMed]
- Mazzio, K.A.; Yuan, M.J.; Okamoto, K.; Luscombe, C.K. Oligoselenophene Derivatives Functionalized with a Diketopyrrolopyrrole Core for Molecular Bulk Heterojunction Solar Cells. ACS Appl. Mater. Interfaces 2011, 3, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.L.; Li, A.; Fan, B.; Xue, F.; Adachi, C.; Ouyang, J. Phenanthrene-functionalized 3,6-dithiophen-2-yl-2,5-dihydropyrrolo[3,4–c]pyrrole-1,4-diones as donor molecules for solution-processed organic photovoltaic cells. Sol. Energy Mater. Sol. Cell. 2011, 95, 2516–2523. [Google Scholar] [CrossRef]
- Deng, J.; Chen, J.; Tao, Q.; Yan, D.; Fu, Y.; Tan, H. Improved photovoltaic performance of 2,7-pyrene based small molecules via the use of 3-carbazole as terminal unit. Tetrahedron 2018, 74, 3989–3995. [Google Scholar] [CrossRef]
- Lin, Y.; Cheng, P.; Li, Y.; Zhan, X.A. A 3D star-shaped non-fullerene acceptor for solution-processed organic solar cells with a high open-circuit voltage of 1.18 V. Chem. Commun. 2012, 48, 4773–4775. [Google Scholar] [CrossRef]
- Bürckstümmer, H.; Weissenstein, A.; Bialas, D.; Würthner, F. Synthesis and characterization of optical and redox properties of bithiophene-functionalized diketopyrrolopyrrole chromophores. J. Org. Chem. 2011, 76, 2426–2432. [Google Scholar] [CrossRef]
- Huang, Y.F.; Chang, S.T.; Wu, K.Y.; Wu, S.L.; Ciou, G.T.; Chen, C.Y.; Liu, C.L.; Wang, C.L. Influences of conjugation length on organic field-effect transistor performances and thin film structures of diketopyrrolopyrrole-oligomers. ACS Appl. Mater. Interfaces 2018, 10, 8869–8876. [Google Scholar] [CrossRef]
- Ha, J.S.; Kim, K.H.; Choi, D.H. 2,5-Bis(2-octyldodecyl)pyrrolo[3,4-c]pyrrole-1,4-(2H,5H)-dione-based donor-acceptor alternating copolymer bearing 5,5′-di(thiophen-2-yl)-2,2′-biselenophene exhibiting 1.5 cm2·v−1·s−1hole mobility in thin-film transistors. J. Am. Chem. Soc. 2011, 133, 10364–10367. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Wu, S.; Xing, W.; Shen, H.; Xiang, L.; Liang, Y.; Xu, W.; Zhu, D. Diketopyrrolopyrrole based small molecular semiconductors containing thiazole units for solution-processed n-channel thin-film transistors. Dye Pigment. 2019, 163, 707–714. [Google Scholar] [CrossRef]
- Ponsot, F.; Bucher, L.; Desbois, N.; Rousselin, Y.; Mondal, P.; Devillers, C.H.; Romieu, A.; Gros, C.P.; Singhalc, R.; Sharma, G.D. A bacteriochlorin-diketopyrrolopyrrole triad as a donor for solution-processed bulk heterojunction organic solar cells. J. Mater. Chem. C 2019, 7, 9655–9664. [Google Scholar] [CrossRef]
- Song, X.; Gasparini, N.; Nahid, M.M.; Paleti, S.H.K.; Li, C.; Li, W.; Ade, H.; Baran, D. Efficient DPP donor and nonfullerene acceptor organic solar cells with high photon-to-current ratio and low energetic loss. Adv. Funct. Mater. 2019, 29, 1902441. [Google Scholar] [CrossRef]
- He, X.; Yin, L.; Li, Y. Efficient design and structural modifications for tuning the photoelectric properties of small molecule acceptors in organic solar cells. New J. Chem. 2019, 43, 6577–6586. [Google Scholar] [CrossRef]
- Zhao, C.; Guo, Y.; Zhang, Y.; Yan, N.; You, S.; Li, W. Diketopyrrolopyrrole-based conjugated materials for non-fullerene organic solar cells. J. Mater. Chem. A 2019, 7, 10174–10199. [Google Scholar] [CrossRef]
- Roland, T.; Heyer, E.; Liu, L.; Ruff, E.; Ludwigs, S.; Ziessel, R.; Haacke, S. Unsymmetrical donor-acceptor-acceptor-π-donor type benzothiadiazole-based small molecule for a solution processed bulk heterojunction organic solar cell. ACS Appl. Mater. Interfaces 2015, 7, 10283–10292. [Google Scholar]
- Heyer, E.; Ziessel, R. Panchromatic push-pull dyes of elongated form from triphenylamine, diketopyrrolopyrrole, and tetracyanobutadiene modules. Synlett 2015, 26, 2109–2116. [Google Scholar]
- Kuwabara, J.; Yamagata, T.; Kanbara, T. Solid-state structure and op tical properties of highly fluorescent diketopyrrolopyrrole derivatives synthesized by cross-coupling reaction. Tetrahedron 2010, 66, 3736–3741. [Google Scholar] [CrossRef] [Green Version]
- Knupfer, M. Exciton binding energies in organic semiconductors. Appl. Phys. A 2003, 77, 623–626. [Google Scholar] [CrossRef]
- Hill, I.G.; Kahn, A.; Soos, Z.G.; Pascal, R.A., Jr. Charge-separation energy in films of π-conjugated organic molecules. Chem. Phys. Lett. 2000, 327, 181–188. [Google Scholar] [CrossRef]
- Li, J.; Ong, K.H.; Lim, S.L.; Ng, G.M.; Tan, H.S.; Chen, Z.K. A random copolymer based on dithienothiophene and diketopyrrolopyrrole units for high performance organic solar cells. Chem. Commun. 2011, 47, 9480–9482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.S.; Jo, W.H. A strategy to enhance both Voc and Jsc of A-D-A type small molecules based on diketopyrrolopyrrole for high efficient organic solar cells. Org. Electron. 2013, 14, 1621–1628. [Google Scholar] [CrossRef]
- Stuart, A.C.; Tumbleston, J.R.; Zhou, H.; Li, W.; Liu, S.; Ade, H.; You, W. Fluorine substituents reduce charge recombination and drive structure and morphology development in polymer solar cells. J. Am. Chem. Soc. 2013, 135, 1806–1815. [Google Scholar] [CrossRef]
- Kim, H.; Lee, H.; Seo, D.; Jeong, Y.; Cho, K.; Lee, J.; Lee, Y. Regioregular low bandgap polymer with controlled thieno[3,4-b]thiophene orientation for highefficiency polymer solar cells. Chem. Mater. 2015, 27, 3102–3107. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Adamo, C.; Barone, V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models. J. Chem. Phys. 1998, 108, 664–675. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M.H.H. Toward reliable density functional methods without adjustable parameters: The PBE0 model. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Marcus, R.A. Chemical and electrochemical electron-transfer theory. Annu. Rev. Phys. Chem. 1964, 15, 155–196. [Google Scholar] [CrossRef]
- Marcus, R.A. Electron transfer reactions in chemistry. Theory and experiment. Rev. Mod. Phys. 1993, 65, 599–610. [Google Scholar] [CrossRef] [Green Version]
- Köse, M.E.; Mitchell, W.J.; Kopidakis, N.; Chang, C.H.; Shaheen, S.E.; Kim, K.; Rumbles, G. Theoretical studies on conjugated phenyl-cored thiophene dendrimers for photovoltaic applications. J. Am. Chem. Soc. 2007, 129, 14257–14270. [Google Scholar] [CrossRef]
- Pearson, R.G. Absolute electronegativity and absolute hardness of Lewis acids and bases. J. Am. Chem. Soc. 1985, 107, 6801–6806. [Google Scholar] [CrossRef]
- Start, M.S. Epoxidation of alkenes by peroxyl radicals in the gas phase: Structure–activity relationships. J. Phys. Chem. A 1997, 101, 8296–8301. [Google Scholar] [CrossRef]
- Chaudhry, A.R.; Ahmed, R.; Irfan, A.; Muhammad, S.; Shaari, A.; Al-Sehemi, A.G. Influence of push–pull configuration on the electro-optical and charge transport properties of novel naphtho-difuran derivatives: A DFT study. RSC Adv. 2014, 4, 48876–48887. [Google Scholar] [CrossRef]
- Irfan, A.; Zhang, J. Effect of one ligand substitution on charge transfer and optical properties in mer-Alq3: A theoretical study. Theor. Chem. Acc. 2009, 124, 339–344. [Google Scholar] [CrossRef]
- Chaudhry, A.R.; Ahmed, R.; Irfan, A.; Shaari, A.; Al-Sehemi, A.G. Effects of electron withdrawing groups on transfer integrals, mobility, electronic and photo-phys- ical properties of naphtho[2,1-b:6,5-b]difuran derivatives: A theoretical study. Sci. Adv. Mater. 2014, 6, 1727–1739. [Google Scholar] [CrossRef]
Sample Availability: Not available. |
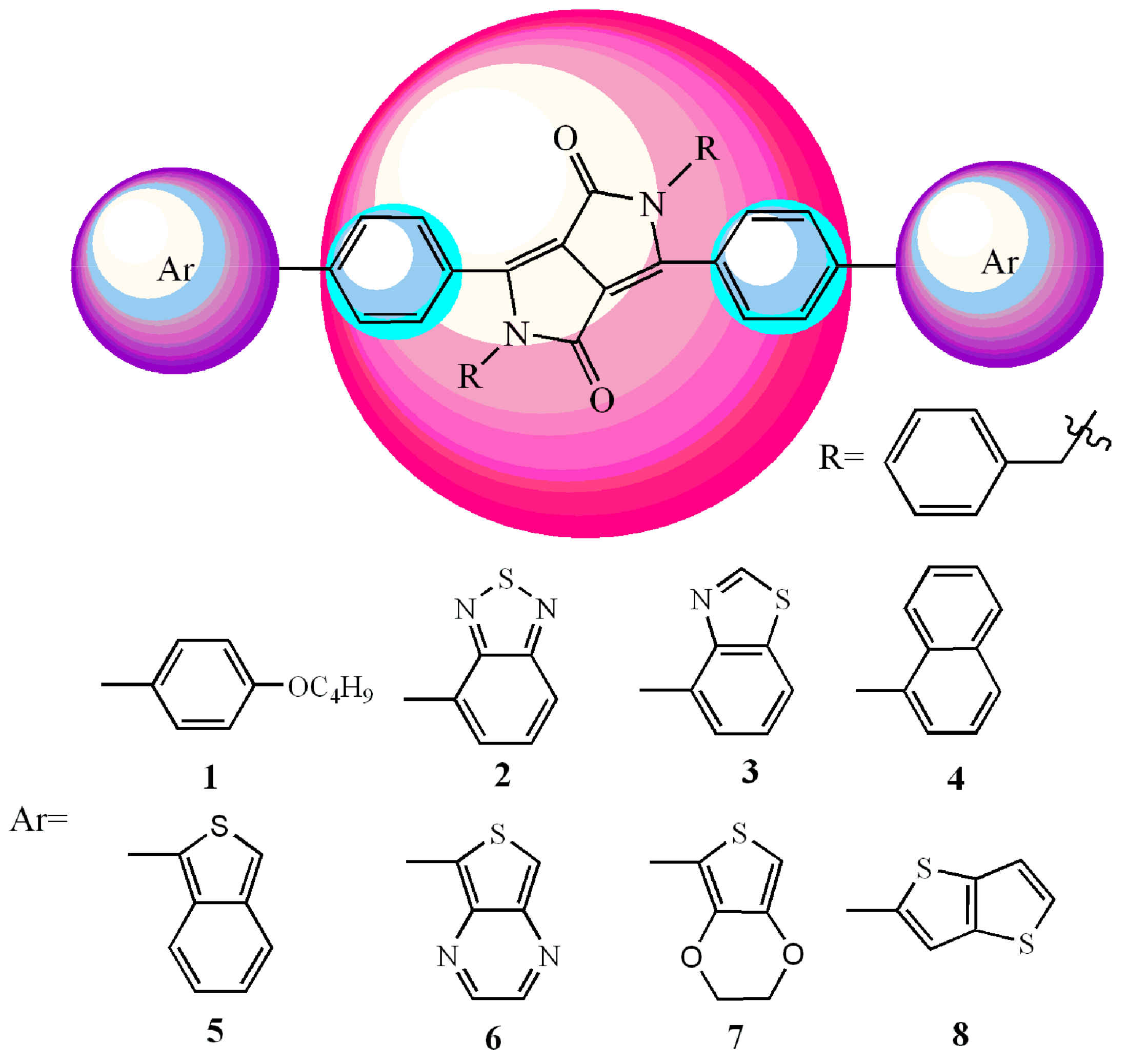


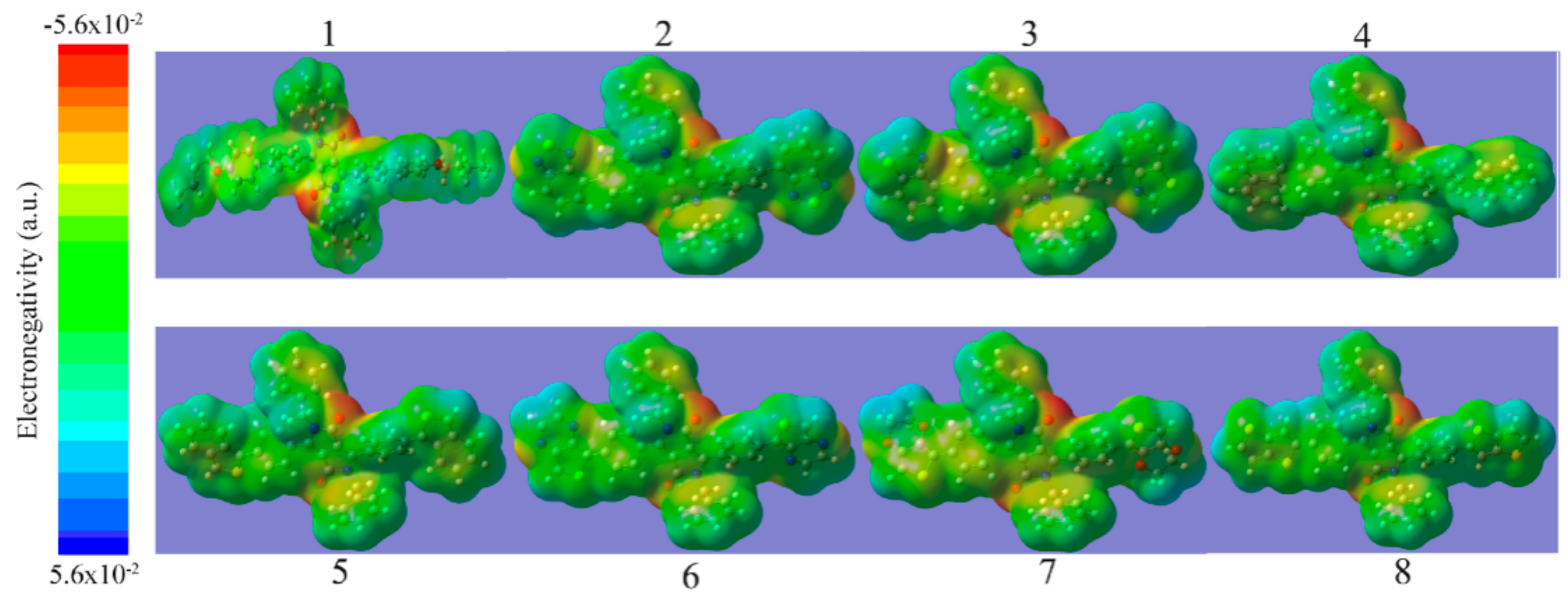

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | HOMOs | LUMOs | ||||
|---|---|---|---|---|---|---|
| DPP a | CB b | Ar c | DPP | CB | Ar | |
| 1 | 71.0 | 19.2 | 9.8 | 66.1 | 28.5 | 5.4 |
| 2 | 75.3 | 19.0 | 5.7 | 33.9 | 21.1 | 45.0 |
| 3 | 75.5 | 19.4 | 5.1 | 60.2 | 30.3 | 9.5 |
| 4 | 73.1 | 19.0 | 7.9 | 64.3 | 30.2 | 5.6 |
| 5 | 47.3 | 19.7 | 33.0 | 54.8 | 29.8 | 15.5 |
| 6 | 63.9 | 21.3 | 14.9 | 28.3 | 21.1 | 50.6 |
| 7 | 61.2 | 22.6 | 16.3 | 58.2 | 30.4 | 11.5 |
| 8 | 61.3 | 21.0 | 17.7 | 56.3 | 31.1 | 12.5 |
| Species | ΔEL-L a | ΔEL-L b | ΔEL-L c |
|---|---|---|---|
| 1 | 0.968 | 0.861 | 0.936 |
| 2 | 0.517 | 0.410 | 0.485 |
| 3 | 0.834 | 0.727 | 0.802 |
| 4 | 0.792 | 0.685 | 0.760 |
| 5 | 0.688 | 0.581 | 0.656 |
| 6 | 0.428 | 0.321 | 0.396 |
| 7 | 0.906 | 0.799 | 0.874 |
| 8 | 0.645 | 0.538 | 0.613 |
| Species | λabs (nm) | f | Assignment |
|---|---|---|---|
| 1 | 478.2 | 0.93 | HOMO → LUMO (0.71) |
| 2 | 521.6 | 0.74 | HOMO → LUMO (0.70) |
| 3 | 483.6 | 0.90 | HOMO → LUMO(0.71) |
| 4 | 475.9 | 0.82 | HOMO → LUMO(0.71) |
| 5 | 530.3 | 1.25 | HOMO → LUMO (0.71) |
| 6 | 571.9 | 0.96 | HOMO → LUMO(0.70) |
| 7 | 507.2 | 1.14 | HOMO → LUMO(0.71) |
| 8 | 514.3 | 1.28 | HOMO → LUMO(0.70) |
| Exp a | 494 |
| Species | λflu | f | Assignment |
|---|---|---|---|
| 1 | 565.6 | 1.08 | HOMO ← LUMO (0.71) |
| 2 | 627.9 | 0.83 | HOMO ← LUMO (0.71) |
| 3 | 572.4 | 1.03 | HOMO ← LUMO(0.71) |
| 4 | 560.3 | 0.95 | HOMO ← LUMO(0.71) |
| 5 | 685.5 | 1.01 | HOMO ← LUMO (0.71) |
| 6 | 617.1 | 1.46 | HOMO ← LUMO(0.71) |
| 7 | 587.6 | 1.23 | HOMO ← LUMO(0.71) |
| 8 | 603.5 | 1.44 | HOMO ← LUMO(0.71) |
| Exp a | 562 |
| Species | λh | λe | η |
|---|---|---|---|
| 1 | 0.401 | 0.343 | 2.284 |
| 2 | 0.374 | 0.240 | 2.174 |
| 3 | 0.386 | 0.322 | 2.277 |
| 4 | 0.364 | 0.315 | 2.305 |
| 5 | 0.325 | 0.260 | 2.104 |
| 6 | 0.356 | 0.222 | 2.017 |
| 7 | 0.392 | 0.278 | 2.180 |
| 8 | 0.380 | 0.263 | 2.145 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, R.; Zhang, X.; Xiao, W. Theoretical Studies of Photophysical Properties of D−π−A−π−D-Type Diketopyrrolopyrrole-Based Molecules for Organic Light-Emitting Diodes and Organic Solar Cells. Molecules 2020, 25, 667. https://doi.org/10.3390/molecules25030667
Jin R, Zhang X, Xiao W. Theoretical Studies of Photophysical Properties of D−π−A−π−D-Type Diketopyrrolopyrrole-Based Molecules for Organic Light-Emitting Diodes and Organic Solar Cells. Molecules. 2020; 25(3):667. https://doi.org/10.3390/molecules25030667
Chicago/Turabian StyleJin, Ruifa, Xiaofei Zhang, and Wenmin Xiao. 2020. "Theoretical Studies of Photophysical Properties of D−π−A−π−D-Type Diketopyrrolopyrrole-Based Molecules for Organic Light-Emitting Diodes and Organic Solar Cells" Molecules 25, no. 3: 667. https://doi.org/10.3390/molecules25030667