Synthesis of Functionalized Cannabilactones
by
 , , and
, , and
Yingpeng Liu
1, 
Thanh C. Ho
1,
Mohammed Baradwan
1,
Maria Pascual Lopez-Alberca
1,
Christos Iliopoulos-Tsoutsouvas
1,
Spyros P. Nikas
1,* and
Alexandros Makriyannis
1,2,* 1
Center for Drug Discovery and Department of Pharmaceutical Sciences, Northeastern University, Boston, MA 02115, USA
2
Departments of Chemistry and Chemical Biology, Northeastern University, Boston, MA 02115, USA
*
Authors to whom correspondence should be addressed.
Molecules 2020, 25(3), 684; https://doi.org/10.3390/molecules25030684
Submission received: 6 December 2019
/
Revised: 1 February 2020
/
Accepted: 1 February 2020
/
Published: 6 February 2020
(This article belongs to the Special Issue Cannabinergic Ligands: Chemistry, Pharmacology and Therapeutic Potential)
Abstract
:A new approach to synthesize cannabilactones using Suzuki cross-coupling reaction followed by one-step demethylation-cyclization is presented. The two key cannabilactone prototypes AM1710 and AM1714 were obtained selectively in high overall yields and in a lesser number of synthetic steps when compared to our earlier synthesis. The new approach expedited the synthesis of cannabilactone analogs with structural modifications at the four potential pharmacophoric regions.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Two cannabinoid receptors (CB1 and CB2) belonging to the superfamily of G-protein-coupled receptors (GPCRs) are well characterized and play a vital role in multiple (patho)physiological functions [1,2]. Their widespread distribution and potential therapeutics have made CB1 and CB2 attractive targets for the treatment of pain, inflammation, glaucoma, cancer, and central nervous system (CNS) disorders [1,3,4]. The CB1 receptor is abundantly expressed in the CNS and mediates most of the cannabinoid psychotropic and behavioral effects. The CB2 receptor is predominantly expressed in immune cells and in peripheral tissues and organs. In the brain, this receptor is detectable at very low levels; however, it may be induced under inflammatory situations [5,6,7]. CB2 receptor activation does not produce the unwanted CNS mediated side effects associated with CB1, but it does have therapeutic potential in neurodegenerative, fibrotic, and inflammatory diseases [8,9]. For these reasons, the development of CB2 selective agonists has received significant attention from both pharmaceutical industry and academic institutions. The recent breakthrough work on the crystal structures of the antagonist- and agonist-bound CB1 and CB2 should aid in the future rational design of CB2 selective agonists with improved biopharmaceutical profiles [10,11,12,13,14,15].
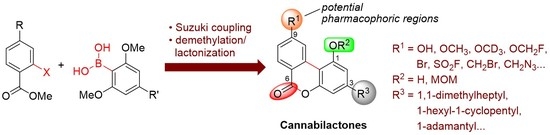
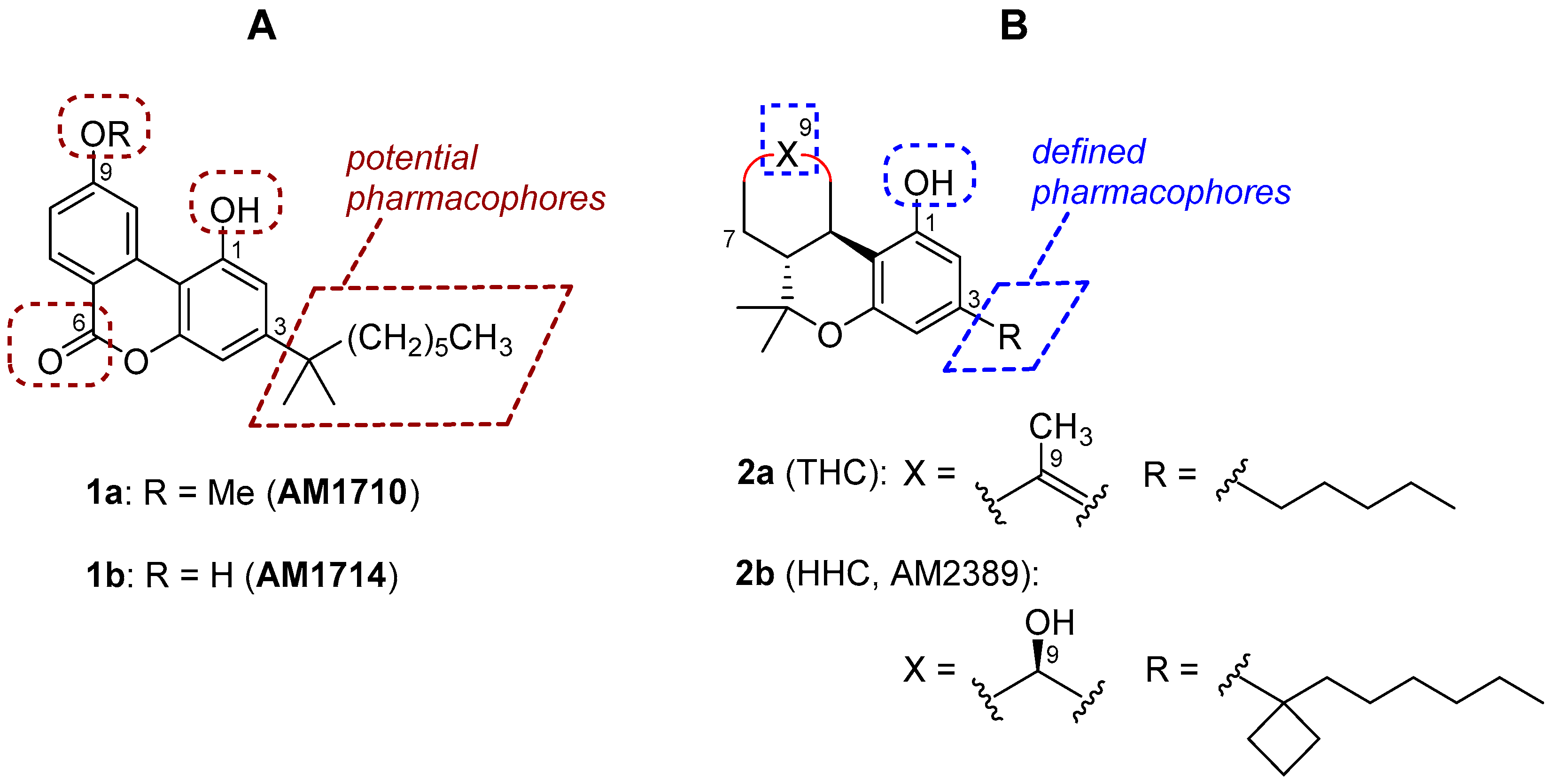
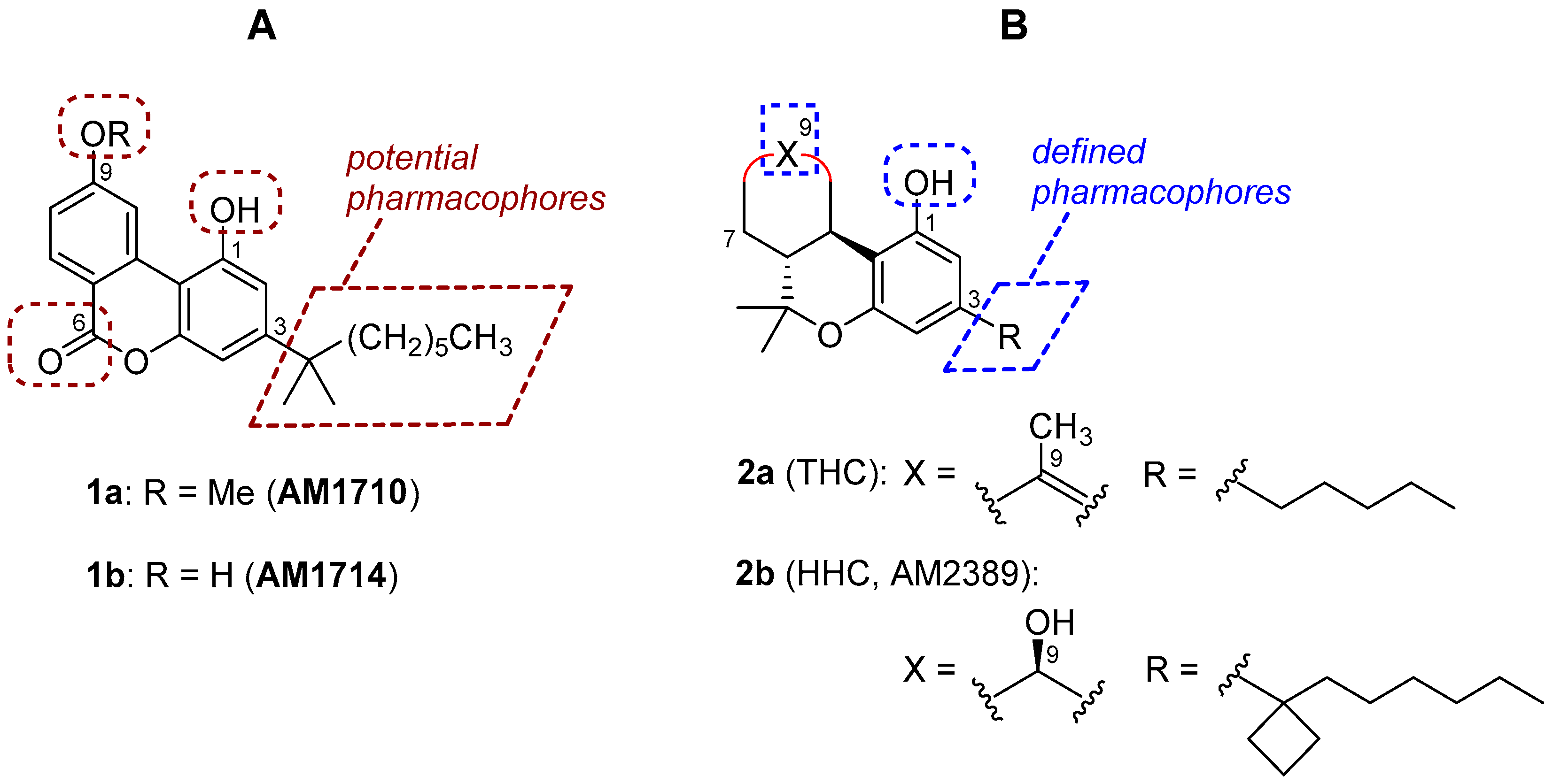
In earlier work including a preceding article of this special issue, we have identified the tricyclic cannabilactone chemical structure as a novel template for developing CB2 selective agonists [16,17]. Two first-generation cannabilactone analogs, namely AM1710 and AM1714 (1a and 1b, Figure 1), exhibited potent peripheral analgesic activity in several animal models of inflammatory and neuropathic pain [16,18,19]. Additionally, AM1710 was found to suppress chemotherapy-induced neuropathic pain and delay the development of antinociceptive tolerance to morphine in rodents [20]. The cannabilactone template has some structural similarities with the psychoactive cannabis constituent (−)-Δ9/Δ8-tetrahydrocannabinol (THC, 2a, Figure 1). Thus, both prototypes have a tricyclic ring system carrying a hydroxy group at C1 and a hydrophobic alkyl chain at C3. However, we can also observe structural dissimilarities between the two templates. In the CB2 selective cannabilactone template, the six-membered lactone contributes to the overall aromatic character, thus making the molecules a robust tricyclic system in which all three rings are coplanar [16]. In contrast, the tricyclic ring system in THC and the related hexahydrocannabinols (HHCs, for example 2b (AM2389) [21], Figure 1) is not coplanar, and these compounds bind both at CB1 and CB2 and behave as mixed CB1/CB2 agonists [21,22,23,24]. It should also be noted that the pharmacophoric regions (Figure 1) of the classical tricyclic THC/HHC scaffold are well defined through extensive structure-activity relationship (SAR) studies [21,22]. However, the pharmacophoric regions of the cannabilactone scaffold have not been well explored. Taking together all the above, we have decided to revisit the cannabilactone prototype and explore in more detail the four potential pharmacophoric regions, including the phenolic hydroxyl at C1, the side chain at C3, the C6 keto group, and the substituents at C9 (see Figure 1). The long-term goal of this project is to develop second-generation cannabilactone analogs with optimized pharmacodynamic and pharmacokinetic properties. Toward this end, the development of a general and effective synthesis of functionalized cannabilactones was required.
2. Results and Discussion
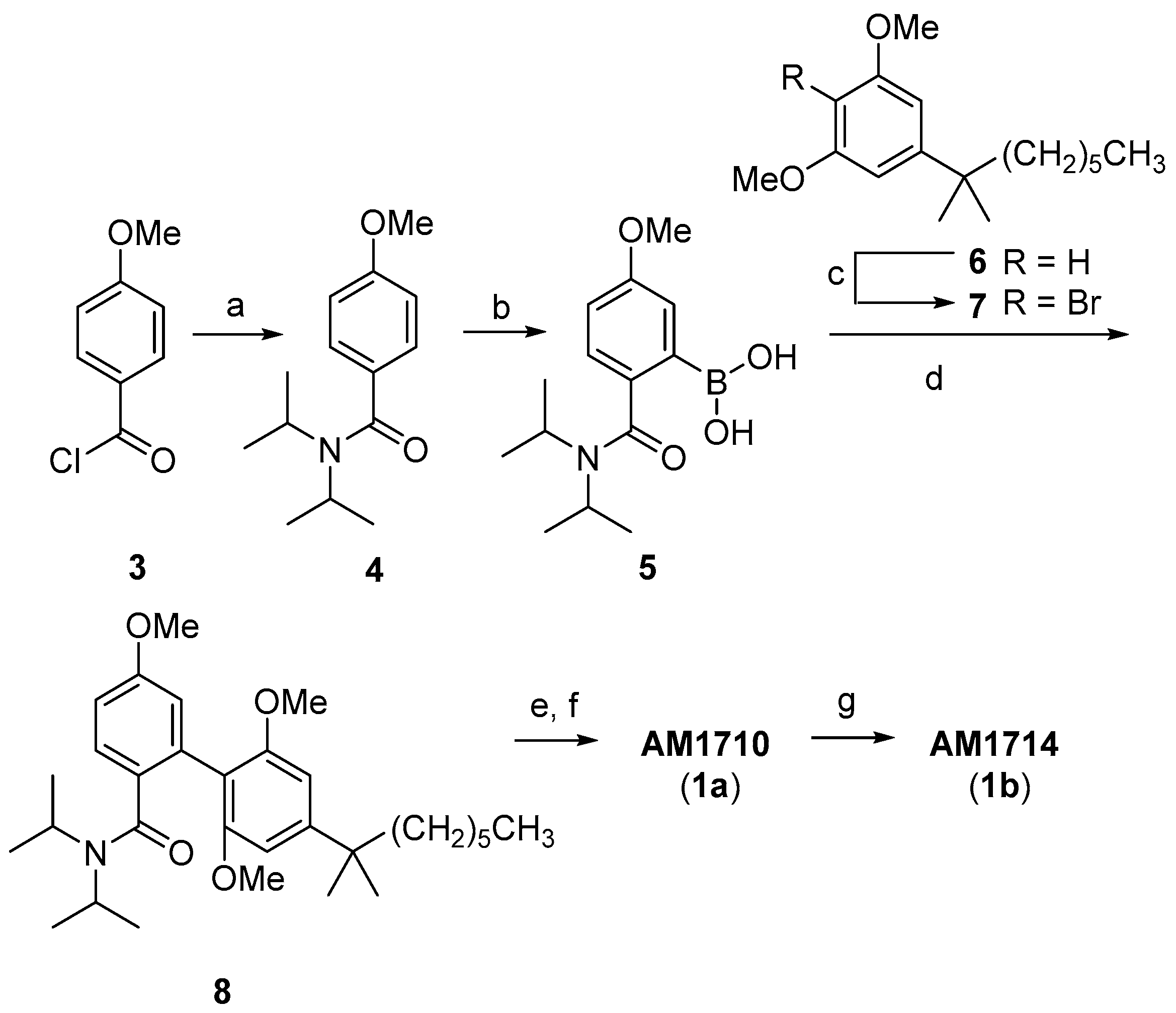
In our earlier work [16], AM1710 and AM1714 were prepared in six to seven synthetic steps and in 38% and 27% overall yields, respectively. In this method, the Suzuki coupling reaction [25] of (2-(diisopropylcarbamoyl)-5-methoxyphenyl)boronic acid (5) with 2-bromo-5-(1′,1′-dimethylheptyl)-1,3-dimethoxybenzene (7) is the key synthetic step (Scheme 1) [16]. Both boronic acid 5 and aryl bromide 7 are not commercially available. As shown in Scheme 1, 5 was prepared from 4-methoxybenzoyl chloride (3) through amide formation and subsequent ortho lithiation–boronation reactions. This lengthens by two steps the overall synthesis of AM1710 and AM1714. Synthesis of the other component of the Suzuki coupling, the aryl bromide 7, requires a careful monobromination of the synthetically feasible resorcinol dimethyl ether 6 using toxic bromine. Additionally, this earlier approach has some limitations regarding its application to the synthesis of functionalized cannabilactone analogs, especially those with modifications at C9. This is because the required boronic acids or their synthetic precursors are not commercially available, and their synthesis would require additional synthetic steps.
Herein, we report a general and effective approach for the synthesis of cannabilactones including AM1710 and AM1714 as well as their analogs with structural modifications at the potential pharmacophoric regions (Scheme 2). The new approach reverses the partners of the Suzuki cross-coupling reaction to take advantage of the large variety of commercially available aryl halides (bromides and iodides, 9). The preparation of resorcinol boronic acids 10 is straightforward, while the use of bromine is avoided [27]. It is also worthy to note that the presence of the methyl carboxylate in the biphenyl intermediates 11 allows the subsequent demethylation–lactonization reactions to occur in a single operation and provides the final products in high overall yields. This demethylation–lactonization process could be carried out in the presence of either BBr3 or the milder reagent 9-iodo-9-BBN [23]. Unlike the carboxylate biphenyls 11, the conversion of diisopropyl amide biphenyl intermediates, for example 8 (Scheme 1), to the final product lactones requires two to three synthetic steps. Due to these modifications the new synthesis of AM1710 and AM1714 proceeded in only three steps and in 55–58% overall yields. This is a significant improvement compared to the earlier method (six to seven steps, 27–38% overall yields). Furthermore, the new synthetic approach allowed us to synthesize various cannabilactone analogs with structural modifications at the C3 side chain, C9/C11, and C1 positions. Some C6 derivatives of cannabilactone were also prepared.
The details of the synthesis of AM1710 and AM1714 are summarized in Scheme 3a. Cannabilactones AM1710 and AM1714 were prepared starting from commercially available 1-(1,1-dimethylheptyl)-3,5-dimethoxybenzene (6). Ortho-lithiation of 6 followed by in situ boronation with trimethyl borate, and hydrolysis of the resulting boronate ester intermediate led to boronic acid 13 in 88% yield. Twenty-five Suzuki cross-coupling of 13 with commercially available 2-bromo-4-methoxybenzoate (14) using tetrakis(triphenylphosphophine)palladium(0) as catalyst and cesium carbonate in dimethoxyethane/water under microwave irradiation at 110 °C for 45 min gave biphenyl compound 15 in 84% yield. AM1710 and AM1714 were obtained selectively in high yield from biphenyl 15. The exposure of 15 to excess boron tribromide in dichloromethane at reflux for 2 days led to the cleavage of all methyl ether groups and lactonization in a single step that resulted in the formation of AM1714 directly in 74% yield. The use of stoichiometric 9-iodo-9-BBN at room temperature in place of boron tribromide gave AM1710 in 78% yield from 15. During this transformation, cleavage of the methoxy group of the electron-poor aromatic C-ring bearing a carboxyester group did not occur.
Cannabilactone analogs with modifications at the C3 side chain were prepared following the general method used for the synthesis of AM1710 (Scheme 3b–f). The design of these analogs was influenced by our earlier SAR work on the C3 side chain of tricyclic tetrahydrocannabinols (THCs) and hexahydrocannabinols (HHCs) [21,23,24,28,29,30,31,32]. The addition of the larger cyclopentyl ring at C1′ was attempted to explore the effect of the size of the benzylic substituents. Incorporation of a double bond at C2′ imposes additional conformational restrictions on the flexible side chain. Groups at the terminal carbon atom will probe the potency while they enhance the polar characteristics of the molecule. The adamantyl group represents a conformationally fixed and sterically demanding side chain. For the synthesis of these cannabilactone analogs, the synthesis of resorcinol dimethyl ether precursors carrying suitable substituents at C5 is required. Thus, resorcinol dimethyl ethers 16 [24], 20 [24], 24 [21], and 29 [33] were synthesized following the procedures previously reported by our group, while 2-(3,5-dimethoxyphenyl)adamantane (34) was prepared from 2,6-dimethoxyphenol (41) as follows. Friedel–Crafts alkylation of 41 (Scheme 3g) with 1-adamantanol in the presence of methanesulfonic acid gave 42 in 58% yield. The exposure of 42 to N-phenyltrifluoromethanesulfonimide formed the phenolic triflate 43 in high yield (95%). Reductive cleavage of the phenolic triflate 43 in the presence of PdCl2, 1,3-bis(diphenylphosphino)propane (dppp), and polymethylhydrosiloxane (PHMS) gave 34 in 92% yield [31]. With resorcinol dimethyl ethers 16, 20, 24, 29, and 34 in hand, we follow the three steps used in the new synthesis of AM1710 to obtain cannabilactones 19, 23, 27, 32, and 39 in good to excellent overall yields. These steps include the formation of boronic acids from the respective resorcinol dimethyl ethers, the Suzuki coupling of boronic acids with either commercially available aryl bromide 14 or triisopropylsilyl (TIPS)-protected bromide 37, and finally, the one-step demethylation/cyclization. Note that the phenoxy group in 26 and 31 (Scheme 3d,e) was also replaced with the iodo group upon exposure of these biphenyls to 9-iodo-9-BBN reagent. This enables the incorporation of functional groups and/or pharmaceutically favorable moieties at the terminal carbon atom of the side chain of the cannabilactone prototype. For example, the exposure of iodide 27 to sodium cyanide in dimethyl sulfoxide gave cannabilactone 28 in 90% yield, whereas the treatment of 32 with silver nitrate in acetonitrile led to 33 in 91% yield. Furthermore, silyl ether 39, when treated with tetrabutylammonium fluoride (TBAF) in THF, formed the C3 adamantyl cannabilactone 40 in 90% yield. This analog is also an example of dual modification of the cannabilactone prototype at C3 and C11 positions.
Cannabilactone analogs with modifications at the C9/C11 positions were also prepared in the same manner (Scheme 4). Earlier SAR data indicate that the C9/C11 position of the tricyclic cannabinoids (i.e., THCs, HHCs) is another pharmacophore that can affect the ligand’s binding affinity for CB1 and CB2 receptors and pharmacological potency [28,30,31,34]. Thus, in this work, we incorporate various functional groups at C9 of the cannabilactone template, including the hydroxyl and deuterated methoxy groups. This is expected to probe the potential hydrogen bonding interactions between the ligand and the receptor and/or to modify the polar/pharmacokinetic profiles of the analogs [35]. Deuterium atoms are introduced to the C9 methoxy group, which is a potential labile site, in order to increase the metabolic stability. Cannabilactones bearing C11 alkoxide groups with different bulkiness (OMe, OEt, and Oi-Pr) were prepared to explore the stereoelectronic requirements for activity of the ether group seen in AM1710. Functionalized cannabilactones carrying the electrophilic sulfonyl fluoride moiety [36], or the photoactivatable azido group [31,37] (yields nitrene upon UV irradiation) offer opportunities for developing potentially tight/irreversible binders for the receptor [2]. The synthesis of these cannabilactones requires the use of aryl halides containing an appropriate moiety that would be transformed into the desired functional group at the C9/C11 position of the final product. Aryl bromides 44, 36, and 53 are commercially available. Bromide 47 was prepared from 44 in good yields via O-alkylation, while bromide 37 was prepared through TIPS protection of 36 (Scheme 3f). Following the approach typified by the synthesis of AM1710, Suzuki cross-coupling reaction of aryl halides 44, 47, 36, 53, and 37 with either boronic acid 21 or 13, followed by one-step demethylation/lactonization afforded cannabilactones 46, 49, 51, 55, and 60, respectively, in satisfactory yields.
It is worthy to note that the treatment of biphenyl 50 (Scheme 4c) with boron tribromide induces multiple transformations and provides the desirable benzyl bromide 51 in a single operation and in 68% yield. These transformations involve the cleavage of all methyl ether groups, lactonization, and introduction of the C11 bromide. Additionally, the Suzuki cross-coupling reaction of methyl 4-bromo-2-iodobenzoate (53, Scheme 4d) proceeded with exclusive regioselectivity at the iodo carbon giving biphenyl 54 in 62% yield [38]. Cannabilactones 51, 55, and 60 were used as intermediates to prepare additional C9/C11 and C1 modified analogs via further functional group transformations as follows. The etherification of 51 (Scheme 4c) with methanol, ethanol, or 2-propanol in the presence of FeSO4·7H2O in a microwave reactor at 130 °C for 1 h led to the corresponding ethers 52a–52c in 97–99% yields [39]. The treatment of bromide 51 with tetrabutylammonium azide in dichloromethane at room temperature provided azide 52d in a 96% yield. The attempted sulfonylation of aryl bromide 55 (Scheme 4d) without masking the phenolic hydroxyl group did not lead to the desired product. Thus, 55 was treated with methoxymethyl chloride in the presence of N,N-diisopropylethylamine to produce 56 in 92% yield. The palladium-catalyzed sulfonylation of aryl bromide 56 using 1,4-diazabicyclo[2.2.2]octane bis(sulfur dioxide) adduct (DABSO) [40], followed by in situ treatment of the resulting sulfinate intermediate with N-fluorobenzene sulfonimide provided sulfonyl fluoride 57 in 71% yield [41]. This was followed by removal of the methoxymethyl protecting group with stoichiometric scandium(III) triflate in the presence of excess ethanol to give cannabilactone 58 in 91% yield [42]. Cannabilactone 60 (Scheme 4e) with a silyl ether protecting group at the C11 position can be used as an intermediate to synthesize analogs with further modifications at the C1 phenolic hydroxyl group. Such analogs would be useful to explore the role of this hydroxyl group in the cannabilactone template in terms of CB2 receptor recognition and activation. This is based on earlier observations that the C1 phenolic hydroxyl group in classical cannabinoids is important for activity, predominantly for CB1 [43,44,45,46,47]. On the other hand, compound 60 can be converted to C11-hydroxyl cannabilactone 62 in good yields via methoxymethyl ether protection and subsequent removal of the silyl ether protecting group. Compound 62 with a methoxymethyl ether protecting group at the C1 position can serve as an intermediate to prepare analogs with modifications on the C11 hydroxyl group. Thus, our approach provides a useful tool to prepare cannabilactone analogs with selective modifications on a hydroxyl group at either the C1 or C9/C11 position.
The new method for the synthesis of cannabilactones also provides a short approach to prepare analogs of cannabinol (CBN), which is another understudied bioactive constituent of the cannabis plant. For example, in earlier work, we synthesized the CBN analog AM4089, which is a potent CB1 agonist, in 11 steps in ca. 1.1% overall yield [30]. Key steps in this synthesis were a Friedel–Craft alkylation reaction and an oxidative aromatization with sulfur at 250 °C. Both reactions proceeded in low yields (32% and 34%, respectively). Following the approach reported here, AM4089 was synthesized in seven steps in 12.5% overall yield (Scheme 3f and Scheme 5a). Treatment of cannabilactone 39 (Scheme 5a) with excess methylmagnesium bromide in THF at reflux [16,30] followed by cyclization and simultaneous cleavage of the silyl ether group with p-toluenesulfonic acid [48] afforded AM4089 (63) in one-pot reaction and in 65% yield from 39.
Finally, thiocannabilactone 66 (Scheme 5b) with modifications at two potential pharmacophores, the C6 and C9, was prepared from the deuterated analog of AM1710 (49, Scheme 5b). Silyl ether protection of 49 gave 64 in 89% yield. Subsequent treatment of 64 with the Lawesson reagent [49,50] in toluene at reflux afforded thioester 65 in 76% yield. This was followed by fluorodesilylation using tetrabutylammonium fluoride to provide 66 in 82% yield.
3. Experimental Section
All reagents and solvents were purchased from Sigma-Aldrich Chemical Company, unless otherwise specified, and used without further purification. All anhydrous reactions were performed under a static argon atmosphere in flame-dried glassware using scrupulously dry solvents. Flash column chromatography employed silica gel 60 (230–400 mesh). All compounds were demonstrated to be homogeneous by analytical thin layer chromatography (TLC) on pre-coated silica gel TLC plates (Merck, 60 F245 on glass, layer thickness 250 mm), and chromatograms were visualized by phosphomolybdic acid staining. IR spectra were recorded on a PerkinElmer Spectrum One FT-IR spectrometer. NMR spectra were recorded in the indicated solvent on Varian 500 (1H-NMR at 500 MHz, 13C-NMR at 126 MHz), and Bruker 400 (1H-NMR at 400 MHz, 13C-NMR at 100 MHz) NMR spectrometers, and chemical shifts are reported in units of δ relative to internal tetramethylsilane (TMS). Multiplicities are indicated as br (broadened), s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), and coupling constants (J) and are reported in hertz (Hz). Mass spectral data are reported in the form of m/z (intensity relative to base = 100). Purities of compounds were determined by LC/MS analysis using a Waters MicroMass ZQ system (electrospray ionization (ESI) with a Waters-2525 binary gradient module coupled to a photodiode array detector (Waters-2996) and evaporative light scattering (ELS) detector (Waters-2424) using a XTerra MS C18 (5 µm, 4.6 mm × 50 mm column and acetonitrile/water) and were >95%.
Methyl 2′,5,6′-trimethoxy-4′-(2-methyloctan-2-yl)-[1,1′-biphenyl]-2-carboxylate (15). Argon was bubbled through a mixture of boronic acid 13 [27] (1.43 g, 4.65 mmol), methyl 2-bromo-4-methoxybenzoate (14, 950 mg, 3.88 mmol), and Cs2CO3 (5.06 g, 15.5 mmol) in DME/H2O (5:1, 10.5 mL of DME and 2.1 mL of H2O) for 10 min. The Pd(PPh3)4 (448 mg, 0.39 mmol) catalyst was added to the mixture while argon bubbling was maintained through the mixture, and degassing was continued for an additional 5 min. The reaction mixture was microwaved with stirring at 110 °C for 45 min in a Biotage apparatus [25]. Then, the mixture was cooled to room temperature and filtered through a short celite pad. Volatiles were removed under reduced pressure, and Et2O was added. The ethereal solution was washed with water and brine, dried over MgSO4, and concentrated under reduced pressure. Purification by flash column chromatography on silica gel (10–30% EtOAc/hexane) gave 15 (1.4 g, 84% yield) as a yellow oil. IR (thin film, cm−1) 2930, 1727 (CO), 1600, 1575, 1282, 1256, 1238, 1121, 1032, 831, 781, 671; 1H-NMR (500 MHz, CDCl3) δ 7.93 (d, J = 8.8 Hz, 1H, Ar-H), 6.86 (dd, J = 8.8, 1.5 Hz, 1H, Ar-H), 6.82 (d, J = 1.5 Hz, 1H, Ar-H), 6.56 (s, 2H, Ar-H), 3.82 (s, 3H, −OCH3), 3.68 (s, 6H, −OCH3), 3.54 (s, 3H, −COOCH3), 1.65–1.57 (m, 2H), 1.32 (s, 6H, >C(CH3)2), 1.24 (m, 6H), 1.11 (m, 2H), 0.84 (t, J = 6.7 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 167.9, 161.7, 156.6, 150.9, 137.5, 131.8, 124.1, 117.7, 116.7, 112.3, 102.2, 55.9, 55.3, 51.2, 44.7, 38.2, 31.8, 30.0, 28.9, 24.7, 22.6, 14.1. Mass spectrum (ESI) m/z (relative intensity) 451 (M+ + Na, 30), 397 (M+ + H − MeOH, 100). LC/MS analysis (Waters MicroMass ZQ system) showed retention time 5.9 min for the title compound.
1-Hydroxy-9-methoxy-3-(2-methyloctan-2-yl)-6H-benzo[c]chromen-6-one (1a). To a solution of 15 (100 mg, 0.23 mmol) in anhydrous CH2Cl2 (3.0 mL) at 0 °C was added dropwise 9-Iodo-9-borabicyclo[3.3.1]nonane (9-iodo-9-BBN, 0.78 mL, 1.0 M solution in hexane, 0.78 mmol). The reaction mixture was stirred at 0 °C for 1 h; then, it was warmed to room temperature over a 3-h period and kept stirring at room temperature overnight. Then, the mixture was treated with 0.5 mL of ethanolamine and stirred for an additional 30 min. Solvent evaporation and purification by flash column chromatography on silica gel (CH2Cl2/Et2O/hexane, 2:2:6) gave 1a (67 mg, 78%) as a white solid. m.p. 149–151 °C. Literature [16], m.p. 149–151 °C. IR (thin film, cm−1) 3210 (OH) 1676 (CO), 1606, 1401, 1302, 1229, 1110; 1H NMR (500 MHz, CDCl3) δ 8.49 (d, J = 2.5 Hz, 1H, Ar-H), δ 8.35 (d, J = 8.9 Hz, 1H, Ar-H), 7.07 (dd, J = 8.9, 2.5 Hz, 1H, Ar-H), 6.96 (d, J = 1.8 Hz, 1H, Ar-H), 6.67 (d, J = 1.8 Hz, 1H, Ar-H), 5.66 (brs, 1H, OH), 3.97 (s, 3H, OCH3), 1.60–1.57 (m, 2H, CH2), 1.29 (s, 6H, 2 × CH3), 1.25–1.18 (m, 6H, 3 × CH2), 1.08–1.03 (m, 2H, CH2), 0.84 (t, J = 6.7 Hz, 3H, CH3). HRMS (ESI) for C23H29O4: calculated 369.2066; found 369.2056. Mass spectrum (ESI) m/z (relative intensity) 369 (M+ + H, 100). LC/MS analysis (Waters MicroMass ZQ system) showed retention time 5.5 min for the title compound.
1,9-Dihydroxy-3-(2-methyloctan-2-yl)-6H-benzo[c]chromen-6-one (1b). To a solution of 15 (43 mg, 0.1 mmol) in CH2Cl2 (3.0 mL) under an argon atmosphere at room temperature, 1 M solution of BBr3 in CH2Cl2 (1.2 mL, 1.2 mmol) was added, and the reaction mixture was refluxed for 2 days. After the mixture was cooled to room temperature, water was added slowly, and the organic material was extracted with CH2Cl2. The combined organic extracts were washed with 5% aqueous NaHCO3, water, and brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography with EtOAc/hexane (3/7) as eluent to afford 1b (26 mg, 74%) as an off-white foam. m.p. 104–108 °C. Literature [16], m.p. 103–108 °C. IR (thin film, cm−1) 3319 (OH), 2929, 1687 (CO), 1622, 1600, 1410, 1277, 1217, 1108, 1057, 748; 1H-NMR (400 MHz, MeOD-d4) δ 8.58 (d, J = 2.4 Hz, 1H, Ar-H), 8.17 (d, J = 8.7 Hz, 1H, Ar-H), 8.94 (dd, J = 8.7, 2.4 Hz, 1H, Ar-H), 6.82 (d, J = 1.7 Hz, 1H, Ar-H), 6.77 (d, J = 1.7 Hz, 1H, Ar-H), 1.65–1.55 (m, 2H), 1.28 (s, 6H), 1.25–1.14 (m, 6H), 1.13–1.01 (m, 2H), 0.82 (t, J = 6.8 Hz, 3H); 13C-NMR (100 MHz, MeOD-d4) δ 165.1, 163.9, 157.7, 154.1, 154.0, 138.9, 133.2, 116.9, 113.8, 113.0, 110.7, 106.9, 105.6, 45.3, 38.8, 32.9, 31.0, 29.1, 25.8, 23.6, 14.4. HRMS (ESI) for C22H27O4: calculated 355.1919; found 355.1912. Mass spectrum (ESI) m/z (relative intensity) 355 (M+ + H, 100). LC/MS analysis (Waters MicroMass ZQ system) showed retention time 5.2 min for the title compound.
The experimental procedures for the synthesis and characterization data of novel compounds, the novel experimental procedures for the synthesis and characterization data of reported compounds, and the reproductions of NMR spectra for representative and final compounds can be found at the supplementary materials.
4. Conclusions
Following our earlier work including a preceding article of this special issue, an efficient synthesis of cannabilactones has been achieved via Suzuki cross-coupling reaction followed by one-step demethylation/cyclization. The two key cannabilactone prototypes AM1710 and AM1714 were obtained selectively in high overall yields and in a lesser number of synthetic steps when compared to our earlier synthesis. The new synthetic approach was used to prepare cannabilactone analogs and derivatives with modifications at four potential pharmacophoric regions including the C3 side chain, as well as the C9/C11, C1, and C6 positions.
Supplementary Materials
The following are available online, (i) experimental procedures for the synthesis and characterization data of novel compounds 17, 18, 19, 21, 22, 23, 25, 26, 27, 28, 30, 31, 32, 33, 35, 37, 38, 39, 43, 45, 46, 47, 48, 49, 50, 51, 52a–d, 54, 55, 56, 57, 58, 59, 60, 61, 62, 64, 65, and 66; (ii) novel experimental procedures for the synthesis and characterization data of reported compounds 34, 40, 42, and 63; (iii) reproductions of NMR spectra for representative and final compounds 1a (AM1710), 1b (AM1714), 19, 23, 27, 28, 32, 33, 39, 40, 46, 49, 51, 52a–d, 54, 55, 56, 57, 58, 60, 62, 63, and 66.
Author Contributions
Conceptualization, A.M. and S.P.N.; Methodology, Y.L., T.C.H., M.B., M.P.L.-A., C.I.-T. and S.P.N.; Validation, A.M., S.P.N., Y.L., T.C.H., M.B., M.P.L.-A., and C.I.-T.; Formal Analysis, A.M., S.P.N., Y.L., T.C.H., M.B., M.P.L.-A., and C.I.-T.; Writing—Original Draft Preparation, S.P.N, T.C.H., and Y.L.; Writing—Review and Editing, S.P.N. and T.C.H.; Supervision, A.M. and S.P.N.; Funding Acquisition, A.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the National Institutes of Health: grand numbers: DA009158, DA045020, and DA041307-01.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pacher, P.; Batkai, S.; Kunos, G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makriyannis, A. 2012 Division of medicinal chemistry award address. Trekking the cannabinoid road: A personal perspective. J. Med. Chem. 2014, 57, 3891–3911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodhams, S.G.; Chapman, V.; Finn, D.P.; Hohmann, A.G.; Neugebauer, V. The cannabinoid system and pain. Neuropharmacology 2017, 124, 105–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, J.; Hohmann, A.G. The endocannabinoid system and cancer: Therapeutic implication. Br. J. Pharmacol. 2011, 163, 1447–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Console-Bram, L.; Marcu, J.; Abood, M.E. Cannabinoid receptors: Nomenclature and pharmacological principles. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 38, 4–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howlett, A.C.; Barth, F.; Bonner, T.I.; Cabral, G.; Casellas, P.; Devane, W.A.; Felder, C.C.; Herkenham, M.; Mackie, K.; Martin, B.R.; et al. International union of pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 2002, 54, 161–202. [Google Scholar] [CrossRef]
- Mackie, K. Cannabinoid receptors: Where they are and what they do. J. Neuroendocrinol. 2008, 20, 10–14. [Google Scholar] [CrossRef]
- Han, S.; Thatte, J.; Buzard, D.J.; Jones, R.M. Therapeutic utility of cannabinoid receptor type 2 (CB2) selective agonists. J. Med. Chem. 2013, 56, 8224–8256. [Google Scholar] [CrossRef]
- Spinelli, F.; Capparelli, E.; Abate, C.; Colabufo, N.A.; Contino, M. Perspectives of cannabinoid type 2 receptor (CB2R) ligands in neurodegenerative disorders: Structure–affinity relationship (SAfiR) and structure–activity relationship (SAR) studies. J. Med. Chem. 2017, 60, 9913–9931. [Google Scholar] [CrossRef]
- Li, X.T.; Hua, T.; Vemuri, K.; Ho, J.H.; Wu, Y.R.; Wu, L.J.; Popov, P.; Benchama, O.; Zvonok, N.; Locke, K.; et al. Crystal structure of the human cannabinoid receptor CB2. Cell 2019, 176, 459–467. [Google Scholar] [CrossRef] [Green Version]
- Hua, T.; Li, X.; Wu, L.; Iliopoulos-Tsoutsouvas, C.; Wang, Y.; Wu, M.; Shen, L.; Johnston, C.A.; Nikas, S.P.; Song, F.; et al. Activation and signaling mechanism revealed by cannabinoid receptor-Gi complex structures. Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Xing, C.; Zhuang, Y.; Xu, T.-H.; Feng, Z.; Zhou, X.E.; Chen, M.; Wang, L.; Meng, X.; Xue, Y.; Wang, J.; et al. Cryo-EM structure of the human cannabinoid receptor CB2-Gi signaling complex. Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.H.; Yin, J.; Chapman, K.; Grzemska, M.; Clark, L.; Wang, J.M.; Rosenbaum, D.M. High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature 2016, 540, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Hua, T.; Vemuri, K.; Pu, M.C.; Qu, L.; Han, G.W.; Wu, Y.R.; Zhao, S.W.; Shui, W.Q.; Li, S.S.; Korde, A.; et al. Crystal structure of the human cannabinoid receptor CB1. Cell 2016, 167, 750–762. [Google Scholar] [CrossRef] [Green Version]
- Hua, T.; Vemuri, K.; Nikas, S.P.; Laprairie, R.B.; Wu, Y.; Qu, L.; Pu, M.; Korde, A.; Jiang, S.; Ho, J.H.; et al. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 2017, 547, 468–471. [Google Scholar] [CrossRef]
- Khanolkar, A.D.; Lu, D.; Ibrahim, M.; Duclos, R.I.; Thakur, G.A.; Malan, T.P.; Porreca, F.; Veerappan, V.; Tian, X.; George, C.; et al. Cannabilactones: A novel class of CB2 selective agonists with peripheral analgesic activity. J. Med. Chem. 2007, 50, 6493–6500. [Google Scholar] [CrossRef]
- Alapafuja, S.O.; Nikas, S.P.; Ho, T.C.; Tong, F.; Benchama, O.; Makriyannis, A. Chain substituted cannabilactones with selectivity for the CB2 cannabinoid receptor. Molecules 2019, 24, 3559. [Google Scholar] [CrossRef] [Green Version]
- Rahn, E.J.; Thakur, G.A.; Wood, J.A.T.; Zvonok, A.M.; Makriyannis, A.; Hohmann, A.G. Pharmacological characterization of AM1710, a putative cannabinoid CB2 agonist from the cannabilactone class: Antinociception without central nervous system side-effects. Pharmacol. Biochem. Behav. 2011, 98, 493–502. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.T.; Guindon, J.; Cornett, B.L.; Makriyannis, A.; Mackie, K.; Hohmann, A.G. Chronic cannabinoid receptor 2 activation reverses paclitaxel neuropathy without tolerance or cannabinoid receptor 1-dependent withdrawal. Biol. Psychiatry 2015, 77, 475–487. [Google Scholar] [CrossRef] [Green Version]
- Li, A.L.; Lin, X.; Dhopeshwarkar, A.S.; Thomaz, A.C.; Carey, L.M.; Liu, Y.; Nikas, S.P.; Makriyannis, A.; Mackie, K.; Hohmann, A.G. Cannabinoid CB2 agonist AM1710 differentially suppresses distinct pathological pain states and attenuates morphine tolerance and withdrawal. Mol. Pharmacol. 2019, 95, 155–168. [Google Scholar] [CrossRef] [Green Version]
- Nikas, S.P.; Alapafuja, S.O.; Papanastasiou, I.; Paronis, C.A.; Shukla, V.G.; Papahatjis, D.P.; Bowman, A.L.; Halikhedkar, A.; Han, X.W.; Makriyannis, A. Novel 1′,1′-chain substituted hexahydrocannabinols: 9/beta-1-hydroxy-3-(1-hexyl-cyclobut-l-yl)hexahydrocannabinol (AM2389) a highly potent cannabinoid receptor 1 (CB1) agonist. J. Med. Chem. 2010, 53, 6996–7010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, G.A.; Nikas, S.P.; Li, C.; Makriyannis, A. Structural requirements for cannabinoid receptor probes. Handb. Exp. Pharmacol. 2005, 168, 209–246. [Google Scholar]
- Papahatjis, D.P.; Nahmias, V.R.; Nikas, S.P.; Andreou, T.; Alapafuja, S.O.; Tsotinis, A.; Guo, J.; Fan, P.; Makriyannis, A. C1′-cycloalkyl side chain pharmacophore in tetrahydrocannabinols. J. Med. Chem. 2007, 50, 4048–4060. [Google Scholar] [CrossRef] [PubMed]
- Papahatjis, D.P.; Nikas, S.P.; Kourouli, T.; Chari, R.; Xu, W.; Pertwee, R.G.; Makriyannis, A. Pharmacophoric requirements for the cannabinoid side chain. Probing the cannabinoid receptor subsite at C1′. J. Med. Chem. 2003, 46, 3221–3229. [Google Scholar] [CrossRef] [PubMed]
- Miyaura, N.; Suzuki, A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef] [Green Version]
- Alonso, E.; Ramón, D.J.; Yus, M. Naphthalene-catalysed lithiation of N,N-diisopropylbenzamide and its methoxy derivatives. Tetrahedron 1998, 54, 13629–13638. [Google Scholar] [CrossRef]
- Makriyannis, A.; Nikas, S.P.; Alapafuja, S.O.; Shukla, V.G. Monoacylglycerol Lipase Inhibitors for Modulation of Cannabinoid Activity. Patent WO2009052319A1, 23 April 2009. [Google Scholar]
- Busch-Petersen, J.; Hill, W.A.; Fan, P.S.; Khanolkar, A.; Xie, X.Q.; Tius, M.A.; Makriyannis, A. Unsaturated side chain beta-11-hydroxyhexahydrocannabinol analogs. J. Med. Chem. 1996, 39, 3790–3796. [Google Scholar] [CrossRef]
- Lu, D.; Meng, Z.; Thakur, G.A.; Fan, P.; Steed, J.; Tartal, C.L.; Hurst, D.P.; Reggio, P.H.; Deschamps, J.R.; Parrish, D.A.; et al. Adamantyl cannabinoids: a novel class of cannabinergic ligands. J. Med. Chem. 2005, 48, 4576–4585. [Google Scholar] [CrossRef]
- Thakur, G.A.; Bajaj, S.; Paronis, C.; Peng, Y.; Bowman, A.L.; Barak, L.S.; Caron, M.G.; Parrish, D.; Deschamps, J.R.; Makriyannis, A. Novel adamantyl cannabinoids as CB1 receptor probes. J. Med. Chem. 2013, 56, 3904–3921. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, G.; Tius, M.A.; Zhou, H.; Nikas, S.P.; Halikhedkar, A.; Mallipeddi, S.; Makriyannis, A. 3′-functionalized adamantyl cannabinoid receptor probes. J. Med. Chem. 2015, 58, 3104–3116. [Google Scholar] [CrossRef] [Green Version]
- Nikas, S.P.; Sharma, R.; Paronis, C.A.; Kulkarni, S.; Thakur, G.A.; Hurst, D.; Wood, J.T.; Gifford, R.S.; Rajarshi, G.; Liu, Y.; et al. Probing the carboxyester side chain in controlled deactivation (-)-delta(8)-tetrahydrocannabinols. J. Med. Chem. 2015, 58, 665–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makriyannis, A.; D’Souza, M.R.; Bajaj, S.; Nikas, S.P.; Thakur, G.A. 2-Cycloalkyl Resorcinol Cannabinergic Ligands. Patent WO2014062965A1, 24 April 2014. [Google Scholar]
- Kulkarni, S.; Nikas, S.P.; Sharma, R.; Jiang, S.; Paronis, C.A.; Leonard, M.Z.; Zhang, B.; Honrao, C.; Mallipeddi, S.; Raghav, J.G.; et al. Novel C-ring-hydroxy-substituted controlled deactivation cannabinergic analogues. J. Med. Chem. 2016, 59, 6903–6919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gant, T.G. Using deuterium in drug discovery: Leaving the label in the drug. J. Med. Chem. 2014, 57, 3595–3611. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, A.; Jones, L.H. Sulfonyl fluorides as privileged warheads in chemical biology. Chem. Sci. 2015, 6, 2650–2659. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Peng, L. Photoactivatable lipid probes for studying biomembranes by photoaffinity labeling. Chem. Rev. 2013, 113, 7880–7929. [Google Scholar] [CrossRef]
- Rodríguez, G.; Albrecht, M.; Schoenmaker, J.; Ford, A.; Lutz, M.; Spek, A.L.; van Koten, G. Bifunctional pincer-type organometallics as substrates for organic transformations and as novel building blocks for polymetallic materials. J. Am. Chem. Soc. 2002, 124, 5127–5138. [Google Scholar] [CrossRef] [Green Version]
- Joshi, G.; Adimurthy, S. New method for the synthesis of benzyl alkyl ethers mediated by FeSO4. Synth. Commun. 2011, 41, 720–728. [Google Scholar] [CrossRef]
- Emmett, E.J.; Hayter, B.R.; Willis, M.C. Palladium-catalyzed synthesis of ammonium sulfinates from aryl halides and a sulfur dioxide surrogate: A gas- and reductant-free process. Angew. Chem. Int. Ed. 2014, 53, 10204–10208. [Google Scholar] [CrossRef] [Green Version]
- Davies, A.T.; Curto, J.M.; Bagley, S.W.; Willis, M.C. One-pot palladium-catalyzed synthesis of sulfonyl fluorides from aryl bromides. Chem. Sci. 2017, 8, 1233–1237. [Google Scholar] [CrossRef] [Green Version]
- Ho, T.C.; Shimada, N.; Tius, M.A.; Nikas, S.P.; Zhang, W.; Makriyannis, A. C1′-azacycloalkyl hexahydrocannabinols. J. Org. Chem. 2017, 82, 7839–7849. [Google Scholar] [CrossRef]
- Crocker, P.J.; Mahadevan, A.; Wiley, J.L.; Martin, B.R.; Razdan, R.K. The role of fluorine substitution in the structure-activity relationships (SAR) of classical cannabinoids. Bioorg. Med. Chem. Lett. 2007, 17, 1504–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gareau, Y.; Dufresne, C.; Gallant, M.; Rochette, C.; Sawyer, N.; Slipetz, D.M.; Tremblay, N.; Weech, P.K.; Metters, K.M.; Labelle, M. Structure activity relationships of tetrahydrocannabinol analogues on human cannabinoid receptors. Bioorg. Med. Chem. Lett. 1996, 6, 189–194. [Google Scholar] [CrossRef]
- Huffman, J.W.; Hepburn, S.A.; Lyutenko, N.; Thompson, A.L.S.; Wiley, J.L.; Selley, D.E.; Martin, B.R. 1-Bromo-3-(1′,1′-dimethylalkyl)-1-deoxy-Δ(8)-tetrahydrocannabinols: New selective ligands for the cannabinoid CB(2) receptor. Bioorg. Med. Chem. 2010, 18, 7809–7815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huffman, J.W.; Yu, S.; Showalter, V.; Abood, M.E.; Wiley, J.L.; Compton, D.R.; Martin, B.R.; Bramblett, R.D.; Reggio, P.H. Synthesis and pharmacology of a very potent cannabinoid lacking a phenolic hydroxyl with high affinity for the CB2 receptor. J. Med. Chem. 1996, 39, 3875–3877. [Google Scholar] [CrossRef]
- Burdick, D.; DeOrazio, R.; Guzzo, P.; Habershaw, A.; Helle, M.; Paul, B.; Wolf, M. Synthesis and structure–activity relationship of substitutions at the C-1 position of Δ9-tetrahydrocannabinol. Bioorg. Med. Chem. Lett. 2010, 20, 1424–1426. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Yanagita, R.C.; Hamada, N.; Murakami, A.; Takahashi, H.; Saito, N.; Nagai, H.; Irie, K. A simple analogue of tumor-promoting aplysiatoxin is an antineoplastic agent rather than a tumor promoter: Development of a synthetically accessible protein kinase C activator with bryostatin-like activity. J. Am. Chem. Soc. 2009, 131, 7573–7579. [Google Scholar] [CrossRef]
- Bunnelle, W.H.; Mckinnis, B.R.; Narayanan, B.A. Difluorination of esters—Preparation of α,α-difluoro ethers. J. Org. Chem. 1990, 55, 768–770. [Google Scholar] [CrossRef]
- Cava, M.P.; Levinson, M.I. Thionation reactions of Lawesson reagents. Tetrahedron 1985, 41, 5061–5087. [Google Scholar] [CrossRef]
Sample Availability: Not available. |
Figure 1.
(A), structures of AM1710 and AM1714 and potential pharmacophores of the cannabilactone template. (B), representative tetrahydrocannabinol (THC) and hexahydrocannabinol (HHC) analogs with the well-defined pharmacophoric regions.
Figure 1.
(A), structures of AM1710 and AM1714 and potential pharmacophores of the cannabilactone template. (B), representative tetrahydrocannabinol (THC) and hexahydrocannabinol (HHC) analogs with the well-defined pharmacophoric regions.
Scheme 1.
Earlier synthesis of AM1710 and AM1714 (6–7 steps, 27–38% overall yields) a. a Reagents and conditions: (a) Diisopropylamine, Et3N, CH2Cl2, rt, 2 h, 75% [26]; (b) i. sec-BuLi, N,N,N′,N′-tetramethylethylenediamine (TMEDA), THF, −78 °C, 1 h; ii. B(OCH3)3, −78 °C to rt, 12 h; iii. 5% aq. HCl, 96%; (c) Br2, CCl4, 0 °C, 20 min, 85%; (d) Pd(Ph3P)4, Ba(OH)2, DME/H2O, 110 °C, microwave irradiation, 15 min, 83%; (e) 9-iodo-9-BBN, CH2Cl2, 0 °C, 4 h; (f) AcOH, reflux, 5 h, 76% from 8; (g) BBr3, CH2Cl2, reflux, 12 h, 72%.
Scheme 1.
Earlier synthesis of AM1710 and AM1714 (6–7 steps, 27–38% overall yields) a. a Reagents and conditions: (a) Diisopropylamine, Et3N, CH2Cl2, rt, 2 h, 75% [26]; (b) i. sec-BuLi, N,N,N′,N′-tetramethylethylenediamine (TMEDA), THF, −78 °C, 1 h; ii. B(OCH3)3, −78 °C to rt, 12 h; iii. 5% aq. HCl, 96%; (c) Br2, CCl4, 0 °C, 20 min, 85%; (d) Pd(Ph3P)4, Ba(OH)2, DME/H2O, 110 °C, microwave irradiation, 15 min, 83%; (e) 9-iodo-9-BBN, CH2Cl2, 0 °C, 4 h; (f) AcOH, reflux, 5 h, 76% from 8; (g) BBr3, CH2Cl2, reflux, 12 h, 72%.
Scheme 2.
New synthesis of functionalized cannabilactones.
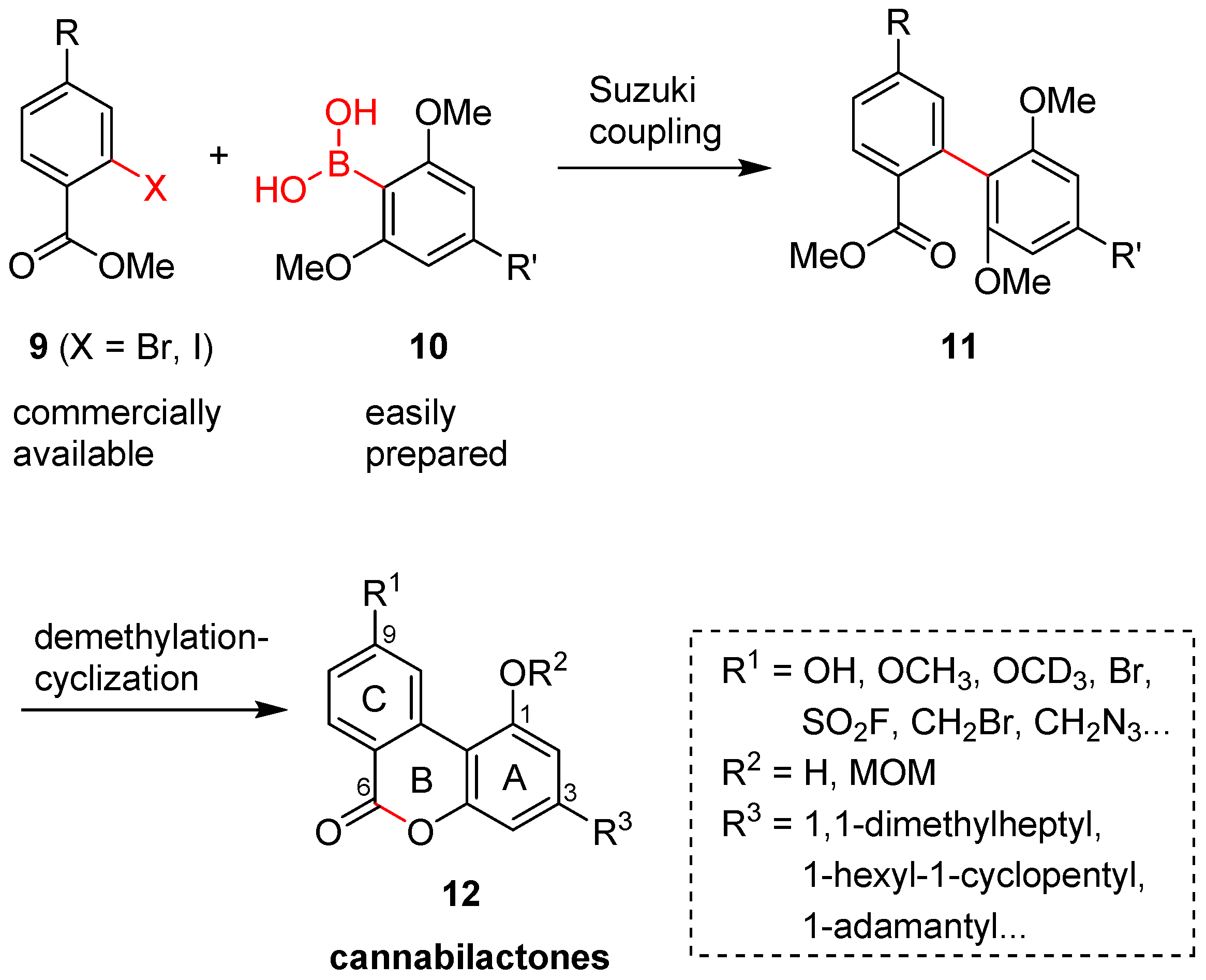
Scheme 3.
Synthesis of AM1710, AM1714, and C3 side chain modified cannabilactones (a–g) a. a Reagents and conditions: (a) i. n-BuLi, THF, −78 °C, 45 min, 10 °C, 1.5 h, −78 °C 30 min; ii. B(OMe)3, −78 °C to rt, overnight; iii. 5% aq. HCl, 88% for 13, 65% for 17, 80% for 21, 73% for 25, 61% for 30, and 61% for 35; (b) Cs2CO3, DME/H2O, Pd(Ph3P)4, 110 °C, microwave irradiation, 45 min, 84% for 15, 69% for 18, 81% for 22, 76% for 26, 84% for 31, and 83% for 38; (c) 9-iodo-9-BBN, CH2Cl2, 0 °C to rt, overnight, 78% for AM1710, 75% for 19, 70% for 23, 67% for 27, 68% for 32, and 73% for 39; (d) BBr3, CH2Cl2, 2 days, reflux, 74% for AM1714; (e) NaCN, DMSO, rt, overnight, 90%; (f) AgNO3, CH3CN, rt, 5 h, 91%; (g) TIPSOTf, 2,6-lutidine, DCM, 0 °C to rt, 2 h, 89% (h) TBAF, THF, rt, 2 h, 90%; (i) 1-adamantanol, MeSO3H, 50 °C, 3 h, 58%; (j) Tf2NPh, Et3N, 4-(dimethylamino)pyridine (DMAP), CH2Cl2, reflux, 18 h, 95%; (k) PdCl2(Ph3P)2, dppp, n-Bu3N, HCOOH, polymethylhydrosiloxan (PMHS), DMF, reflux, 19 h, 92%.
Scheme 3.
Synthesis of AM1710, AM1714, and C3 side chain modified cannabilactones (a–g) a. a Reagents and conditions: (a) i. n-BuLi, THF, −78 °C, 45 min, 10 °C, 1.5 h, −78 °C 30 min; ii. B(OMe)3, −78 °C to rt, overnight; iii. 5% aq. HCl, 88% for 13, 65% for 17, 80% for 21, 73% for 25, 61% for 30, and 61% for 35; (b) Cs2CO3, DME/H2O, Pd(Ph3P)4, 110 °C, microwave irradiation, 45 min, 84% for 15, 69% for 18, 81% for 22, 76% for 26, 84% for 31, and 83% for 38; (c) 9-iodo-9-BBN, CH2Cl2, 0 °C to rt, overnight, 78% for AM1710, 75% for 19, 70% for 23, 67% for 27, 68% for 32, and 73% for 39; (d) BBr3, CH2Cl2, 2 days, reflux, 74% for AM1714; (e) NaCN, DMSO, rt, overnight, 90%; (f) AgNO3, CH3CN, rt, 5 h, 91%; (g) TIPSOTf, 2,6-lutidine, DCM, 0 °C to rt, 2 h, 89% (h) TBAF, THF, rt, 2 h, 90%; (i) 1-adamantanol, MeSO3H, 50 °C, 3 h, 58%; (j) Tf2NPh, Et3N, 4-(dimethylamino)pyridine (DMAP), CH2Cl2, reflux, 18 h, 95%; (k) PdCl2(Ph3P)2, dppp, n-Bu3N, HCOOH, polymethylhydrosiloxan (PMHS), DMF, reflux, 19 h, 92%.
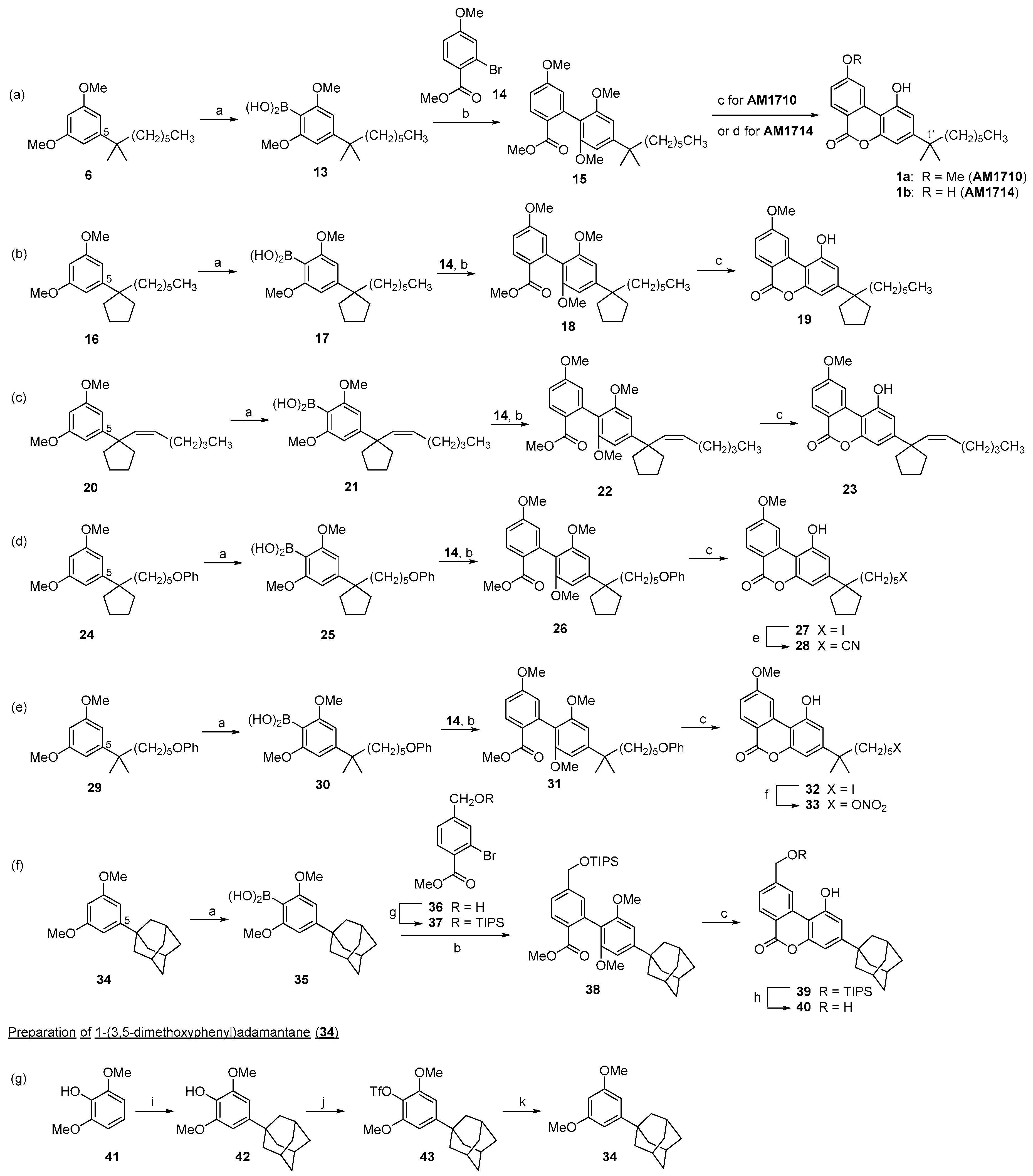
Scheme 4.
Synthesis of C9/C11 and C1 modified cannabilactones (a–e) a. a Reagents and conditions: (a) Cs2CO3, DME/H2O, Pd(Ph3P)4, 110 °C, microwave irradiation, 45 min, 82% for 45, 81% for 48, 80% for 50, 62% for 54, 86% for 59; (b) 9-iodo-9-BBN, DCM, 0°C to rt, overnight, 65% for 46, 75% for 49, 73% for 55, 60% for 60; (c) K2CO3, CD3I, DMF, 0 °C to rt, 3 h, 86%; (d) BBr3, CH2Cl2, −78 °C to rt, overnight, 68%; (e) FeSO4·7H2O, CH2Cl2, MeOH, EtOH, or propan-2-ol, 130 °C, microwave, 1 h, 97–99% for 52a–52c; (f) n-Bu4N+N3−, CH2Cl2, rt, 2 days, 96% for 52d; (g) MOMCl, DIPEA, CH2Cl2, 0 °C to rt, 1 h, 92%; (h) DABSO, PdCl2(Amphos)2, Et3N, propan-2-ol, 75 °C, 24 h, then NFSI, rt, 3 h, 71%; (i) Sc(OTf)3, EtOH/CH3CN, rt, 8 h, 91%; (j) MOMCl, DIPEA, CH2Cl2, 0 °C to rt, overnight, 98%; (k) TBAF, THF, 0 °C to rt, 30 min, 97%.
Scheme 4.
Synthesis of C9/C11 and C1 modified cannabilactones (a–e) a. a Reagents and conditions: (a) Cs2CO3, DME/H2O, Pd(Ph3P)4, 110 °C, microwave irradiation, 45 min, 82% for 45, 81% for 48, 80% for 50, 62% for 54, 86% for 59; (b) 9-iodo-9-BBN, DCM, 0°C to rt, overnight, 65% for 46, 75% for 49, 73% for 55, 60% for 60; (c) K2CO3, CD3I, DMF, 0 °C to rt, 3 h, 86%; (d) BBr3, CH2Cl2, −78 °C to rt, overnight, 68%; (e) FeSO4·7H2O, CH2Cl2, MeOH, EtOH, or propan-2-ol, 130 °C, microwave, 1 h, 97–99% for 52a–52c; (f) n-Bu4N+N3−, CH2Cl2, rt, 2 days, 96% for 52d; (g) MOMCl, DIPEA, CH2Cl2, 0 °C to rt, 1 h, 92%; (h) DABSO, PdCl2(Amphos)2, Et3N, propan-2-ol, 75 °C, 24 h, then NFSI, rt, 3 h, 71%; (i) Sc(OTf)3, EtOH/CH3CN, rt, 8 h, 91%; (j) MOMCl, DIPEA, CH2Cl2, 0 °C to rt, overnight, 98%; (k) TBAF, THF, 0 °C to rt, 30 min, 97%.
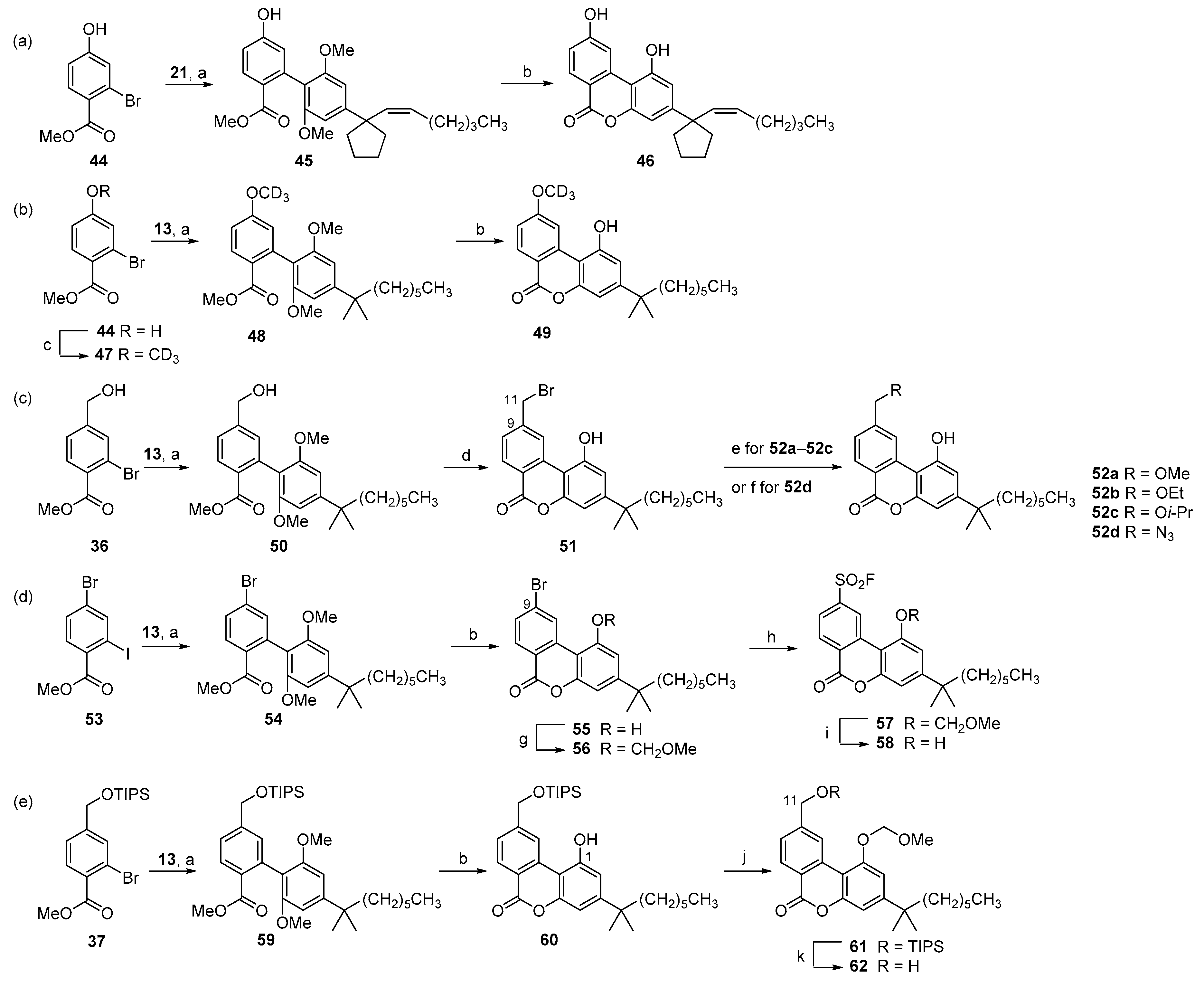
Scheme 5.
Synthesis of AM4089 and thiocannabilactone (a,b) a. a Reagents and conditions: (a) CH3MgBr, Et2O/THF, reflux, 2 h; p-TsOH·H2O, CHCl3, rt, 12 h, 65% from 39; (b) TIPSOTf, 2,6-lutidine, CH2Cl2, 0 °C to rt, 2 h, 89%; (c) Lawesson reagent, toluene, reflux, 24 h, 76%; (d) TBAF, THF, −40 °C, 30 min, 82%.
Scheme 5.
Synthesis of AM4089 and thiocannabilactone (a,b) a. a Reagents and conditions: (a) CH3MgBr, Et2O/THF, reflux, 2 h; p-TsOH·H2O, CHCl3, rt, 12 h, 65% from 39; (b) TIPSOTf, 2,6-lutidine, CH2Cl2, 0 °C to rt, 2 h, 89%; (c) Lawesson reagent, toluene, reflux, 24 h, 76%; (d) TBAF, THF, −40 °C, 30 min, 82%.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Liu, Y.; Ho, T.C.; Baradwan, M.; Pascual Lopez-Alberca, M.; Iliopoulos-Tsoutsouvas, C.; Nikas, S.P.; Makriyannis, A. Synthesis of Functionalized Cannabilactones. Molecules 2020, 25, 684. https://doi.org/10.3390/molecules25030684
AMA Style
Liu Y, Ho TC, Baradwan M, Pascual Lopez-Alberca M, Iliopoulos-Tsoutsouvas C, Nikas SP, Makriyannis A. Synthesis of Functionalized Cannabilactones. Molecules. 2020; 25(3):684. https://doi.org/10.3390/molecules25030684
Chicago/Turabian StyleLiu, Yingpeng, Thanh C. Ho, Mohammed Baradwan, Maria Pascual Lopez-Alberca, Christos Iliopoulos-Tsoutsouvas, Spyros P. Nikas, and Alexandros Makriyannis. 2020. "Synthesis of Functionalized Cannabilactones" Molecules 25, no. 3: 684. https://doi.org/10.3390/molecules25030684