3.3. Procedures and Spectroscopic Data for the Synthesis of the [18F]FLT-ProTide
Synthesis of
(2S)-ethyl-2-((chloro(phenyl)phosphoryl)amino)propanoate (
10). C
11H
15ClNO
4P; MW: 291.6. Compound 10 was synthesized according to standard procedure [
19]. Anhydrous triethylamine (2 eq; 0.662 mL; 0.480 g; 4.74 mmol) was added to the phenyldichlorophosphate
(9) (1 eq; 0.354 mL; 0.500 g; 2.37 mmol) and
l-alanine ethyl ester hydrochloride salt (1 eq; 0.364 g; 2.37 mmol) in anhydrous CH
2Cl
2 (5 mL) to obtain the final product 10 as a yellowish oil that was used without further purification. Yield: 92%.
1H NMR (500 MHz, CDCl
3): δ 7.35–7.41 (m, 2H, Ar-H), 7.21–7.30 (m, 3H, Ar-H), 4.53 (m, 1H, NH), 4.21 (m, 1H, CH), 3.95 (m, 2H, CH
2), 1.51 (m, 3H, CH
3), 1.23 (m, 3H, CH
3).
31P NMR (202 MHz, CDCl
3): δ 7.71, 8.05. Spectroscopic data in agreement with literature [
29,
30,
31].
Synthesis of
(2S)-ethyl2-(((((2R,3S,5R)-3-fluoro-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)propanoate (
11). C
21H
27FN
3O
8P, MW: 499.4. FLT (
8) (1 eq; 0.100 g; 0.41 mmol) in anhydrous THF, was reacted with
tBuMgCl (1.5 eq; 0.08 mL) under nitrogen atmosphere. The reaction mixture was stirred at rt for 30 min. A solution of the phosphorochloridate
10 (2 eq; 0.239 g; 0.82 mmol) in anhydrous THF was then added dropwise and the reaction mixture was left to stir overnight. The solvent was then evaporated under reduced pressure and the residue was purified by silica gel column chromatography (CH
2Cl
2/CH
3OH from 100% CH
2Cl
2 to 95% CH
2Cl
2) to give the FLT ProTide
(11) as a yellowish oil. Yield: 23.5%. Rf: 0.44 in 90% CH
2Cl
2/10% CH
3OH TLC system.
1H NMR (500 MHz, CDCl
3): δ 8.60 (br s, 1H, NH), 7.55 (s, 1H, H-6), 7.33–7.41 (m, 2H, Ar-H), 7.20–7.29 (m, 3H, Ar-H), 6.23–6.30 (m, 1H, H-1′), 5.33 (m, 1H, H-3′), 4.43–4.53 (m, 2H, NH, H-4′), 4.21 (m, 1H, CH), 3.95 (m, 2H, CH
2), 3.35-3.51 (m, 2H, H-5′, H-5”), 2.48–2.51 (m, 1H, H-2”), 2.55–2.33 (m, 1H, H-2′), 1.93–1.74 (m, 3H, CH
3, thy), 1.30–1.18 (d, J = 6.9, CH
3-ala).
19F NMR (479 MHz, CDCl
3): δ −173.70, −175.20.
31P NMR (202 MHz, CDCl
3): δ 4.34, 4.12. MS (ESI
+): 498.1 [M − H
+]. HPLC: Rt: 10.8 min; Purity > 96%; [Gradient: (0′) 95%H
2O/5% CH
3CN − (5′) 50% H
2O/50% CH
3CN − (15′) 50% H
2O/50% CH
3CN − (20′) 95% H
2O/5% CH
3CN]. Spectroscopic data in agreement with literature [
17].
Synthesis of
(3R,5R)-3-(hydroxymethyl)-8-methyl-2,3-dihydro-5H,9H-2,5-methanopyrimido[2,1-b][1,5,3]
dioxazepin-9-one (
12). MF: C
10H
12N
2O
4; MW: 224.22. Thymidine (
3) (1 eq, 0.250g, 1.32mmol) and triphenylphosphine (Ph
3P) (2 eq, 0.541 g, 2.64 mmol) were suspended in anhydrous acetonitrile (20 mL) and cooled down to −15 °C. Diisopropylazadicarboxylate (DIAD) (2 eq, 0.406 mL, 0.417 g, 2.64 mmol) was then added dropwise maintaining the temperature below −5 °C with vigorous stirring. The reaction was allowed to stir for 5h at 0 °C and then again cooled down to −20 °C. Cold ethyl acetate (20 mL) was added and the reaction was stirred for a further 15 min. A white precipitate was formed and was collected by Buchner filtration. The filtrate was washed with cold ethyl acetate and evaporated to dryness. The resulting crude compound was purified by silica gel column chromatography using 90% CH
2Cl
2/10% CH
3OH as eluent to obtain the product
12 as a white solid. Yield: 52%. Rf: 0.5.
1H NMR (500 MHz, DMSO-
d6): δ 7.55 (d,
J = 1.2, 1H, ArH), 5.80 (d,
J = 3.9, 1H, H-1′), 5.23 (brs, 1H, H-3′), 5.01 (t, 1H, 5′-OH), 4.20 (m, 1H, H-4′), 3.51 (m, 2H, H-5′, H-5”), 2.55 (d,
J = 1.2, H-2′,1H), 2.47 (ddd,
J1,8 = 19.0,
J1,4 = 6.7,
J1,2 = 3.0, 1H, H-2”), 1.76 (d,
J = 1.1, 3H, CH
3). Spectroscopic data in agreement with literature [
32].
Synthesis of
1-((2R,4R,5R)-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (
13). MF: C
10H
14N
2O
5; MW: 242. 3′-anhydrothymidine (
12) (0.200 g; 0.892 mmol) in aq. 1.5 M NaOH (3.33 mL) was stirred in methanol (30 mL) under reflux for 3 h. Upon heating, the solution changed colour from clear to golden-brown. The reaction was monitored by TLC chromatography. When the conversion into the final product was confirmed, the solvent was evaporated under reduced pressure. The resulting crude compound was purified by silica gel column chromatography (CH
2Cl
2/CH
3OH gradient from 100% to 90% of CH
2Cl
2) to obtain the final product (13) as a white powder. Yield: 64%. Rf: 0.4 in 90% CH
2Cl
2/10% CH
3OH TLC system.
1H NMR (500 MHz, DMSO-
d6): δ 11.24 (s, 1H, NH), 7.78 (s, 1H, H-6), 6.07 (dd,
J = 8.5, 2.44, 1H, H-1′), 5.25 (d,
J = 3.35, 1H, 3′-OH), 4.67 (t,
J = 5.49, 1H, 5′-OH), 4.23 (m, 1H, H-3′), 3.60–3.84 (m, 3H, H-4′, H-5′ and H-5”), 2.55–2.59 (m, 1H, H-2”), 1.84 (dd,
J = 14.95,
J = 2.14, 1H, H-2′), 1.76 (s, 3H, CH
3). Spectroscopic data in agreement with literature [
32].
Synthesis of
((2S)-ethyl-2-(((((2R,3R,5R)-3-hydroxy-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)propanoate (
14). MF: C
21H
28N
3O
9P; MW: 497.4. Compound
14 was synthesised according to standard procedure [
16]. 1-(2-deoxy-β-lyxofuranoxyl thymidine) (
13) (1 eq; 0.175 g; 0.721 mmol) was reacted with NMI (5 eq; 0.287 mL; 0.297 g, 3.62 mmol) and ethyl-(2-chloro(phenyl)phosphorylamino)propanoate (
10) (3 eq; 0.633 g; 2.17 mmol) to obtain a crude residue that was purified by silica gel column chromatography (97% CH
2Cl
2/3% CH
3OH) to afford the final compound
14 as a white solid. Yield: 18.4%. Rf: 0.4.
1H NMR (500 MHz, CDCl
3): δ 7.58 (d, J = 7.0, 1H, H-6), 7.35–7.43 (m, 2H, Ar-H), 7.20–7.29 (m, 3H, Ar-H), 6.25–6.32 (m, 1H, H-1′), 5.22 (m, 1H, 3′-OH), 4.94 (m, 1H, H-3′), 4.31–4.52 (m, 2H, NH, H-4′), 3.95–4.03 (m, 1H, CH), 3.68 (d, J = 7.0, 2H, CH
2-ester), 3.35 (d, J = 16.0, 2H, H-5′, H-5”), 2.48–2.51 (m, 1H, H-2”), 2.09–2.25 (m, 1H, H-2′), 1.85 (d, J = 10.1, 3H, CH
3-thy), 1.23 (m, 3H, CH
3-ala), 1.16 (d, J = 7.2, 3H, CH
3-ester).
31P NMR (200 MHz, CDCl
3): δ 5.34, 4.98.
Synthesis of (2S)-ethyl-2-(((((2R,3R,5R)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3-((methylsulfonyl)oxy)tetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)propanoate (4). MF: C22H30N3O11PS; MW: 575.5. Triethylamine (10 eq; 1.06 mL; 0.769 g; 7.6 mmol) and mesyl chloride (4 eq; 0.235 mL; 0.348 g; 3.04 mmol) were reacted with a solution of compound 14 (1 eq; 0.378 g; 0.76 mmol) in anhydrous CH2Cl2 (20 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 10 min and then warmed to rt and stirred for 1.5 h. The crude mixture was diluted with sat. NaHCO3 solution and extracted with CH2Cl2. After drying over Na2SO4, the solution was reduced under reduced pressure and the resulting crude compound was purified by silica gel column chromatography (CH2Cl2/CH3OH gradient from 100% CH2Cl2 to 95% CH2Cl2) to give the product 4 as a white solid. Yield: 29.5%. Rf: 0.45 in 90% CH2Cl2/10% CH3OH TLC system. 1H NMR (500 MHz, CDCl3): δ 8.93–8.96 (s, 1H, NH, thy), 7.33–7.32 (d, J = 7.0, 1H, H-6), 7.24–7.28 (m, 2H, Ar-H), 7.10–7.15 (m, 3H, Ar-H), 6.23–6.21 (m, 2H, H-1′), 5.19–5.15 (s, 1H, NH-ala), 3.84–4.37 (m, 7H, H-3′, H-4′, CH, H-5′, H-5”, CH2-ethyl), 2.96–3.01 (s, 3H, SO2CH3), 2.72–2.75 (m, 1H, H-2”), 2.38–2.42 (m, 1H, H-2′), 1.87 (d, J = 1.2, 3H, CH3-thy), 1.28–1.33 (m, 3H, CH3-ala), 1.19–1.16 (m, 3H, CH3-ethyl). 13C NMR (125 MHz, CDCl3) δ 173.7–173.4 (C=O, acetyl), 163.6 (C-2), 150.42 (C-1), 135.1 (C-4), 129.8 (C-2; C-6Ar), 125.2 (C-4Ar), 120.2 (C-3; C-5Ar), 111.6 (C-3), 83.5 (C-1′), 79.9 (C-3′), 77.3 (C-4′), 63.7 (C-5′), 61.7 (CH2-ethyl), 50.8 (CH-ala), 39.2 (C-2′), 38.8 (CH3-mesyl), 21.34 (CH3-ethyl), 14.04 (CH3-ala), 12.76 (CH3-thy). 31P NMR (202 MHz, CDCl3): δ 2.92, 2.63. MS(ESI+): 576.2 [M + H+]. HPLC: Rt: 13.2 min; Purity > 98%; [Gradient: (0′) 95% H2O/5% CH3CN − (5′) 50% H2O/50% CH3CN − (15′) 50% H2O/50% CH3CN − (20′) 95% H2O/5% CH3CN].
Synthesis of (2S)-ethyl-2-(((((2R,3R,5R)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3-(tosyloxy)tetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)propanoate (5). MF: C28H34N3O11PS; MW: 651.6. To a solution of compound 14 (1 eq; 0.181 g; 0.364 mmol) in pyridine (5 mL), tosyl chloride (2 eq; 0.138 g; 0.727 mmol) and silver trifluoromethanesulfonate (AgOTf) (2 eq; 0.186 g; 0.727 mmol) were added at 0 °C. The reaction was stirred for 1 h and then slowly allowed to warm to rt and stirred for another 2 h. The reaction mixture was then diluted with EtOAc, filtered, and the filtrate was washed with H2O and brine. The organic layer was dried over anhydrous Na2SO4 and the solvent was evaporated under reduced pressure. The crude residue was purified by silica gel column chromatography (95% CH2Cl2/5% CH3OH) to furnish the tosylated compound 5 as a yellowish solid. Yield: 35%. Rf: 0.55. 1H NMR (500 MHz, CDCl3) δ 7.76–7.75 (d, J = 1.9, 1H, H6 Ar), 7.34 (dd, J = 15.7, 8.6, 4H, Ar-H tosyl), 7.27–7.17 (m, 5H, Ar), 6.19–5.20 (td, J = 7.8, 2.9, 1H, H-1′), 4.45–3.74 (m, 10H), 2.72–2.61 (m, 1H, CH-ala), 2.46 (s, 3H, CH3, tosyl), 1.85 (s, 3H, CH3, thy), 1.39 (t, J = 7.2 Hz, 3H, CH3, ethyl), 1.31 (m, 3H, CH3-ala). 13C NMR (126 MHz, CDCl3) δ 173.65, 173.59 (C-ala), 163.52 (C1-thy), 150.58, 150.53 (C3-thy), 150.25, 150.19 (C1-tosyl), 145.99, 145.98 (C1-phenyl), 135.04, 134.95 (CH-thy), 133.05, 132.90 (C4-tosyl), 130.27, 130.21 (CH, C2, C6-tosyl), 129.75, 129.70 (CH, C2, C6-phenyl), 127.64, 127.58 (CH, C4-phenyl), 125.10, 120.35 (CH, C3, C5-phenyl), 120.31, 120.21 (CH, C3, C5-tosyl), 111.15, 110.98 (C3-thy), 84.22, 83.99 (CH, C1′), 80.96, 80.90 (CH, C3′), 80.72, 80.66 (CH, C4′), 63.89, 63.85 (CH2, C5′), 63.33, 63.29 (CH2, ethyl), 50.39, 50.38 (CH, ala), 39.03 (CH2, C3′), 21.69, 21.00 (CH3-ethyl), 20.96, 20.95 (CH3-tosyl), 14.12 (CH3, ala), 12.49, 12.44 (CH3-thy). 31P NMR (202 MHz, CDCl3) δ 2.78, 2.66. MS (ESI)+: 652.2 [M + H+]; 674.1 [M + Na+]. HPLC: Rt: 16.03 min; Purity > 98%; [Gradient: (0′) 95% H2O/5% CH3CN − (5′) 50% H2O/50% CH3CN − (15′) 50% H2O/50% CH3CN − (20′) 95% H2O/5% CH3CN].
Synthesis of (S)-ethyl-2-(((((2R,3R,5R)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3-(((4-nitrophenyl)sulfonyl)oxy)tetrahydrofuran-2yl)methoxy)(phenoxy)phosphoryl)amino)propanoate (6). MF: C27H31N4O13PS; MW: 682.5. The ProTide 14 (1 eq; 1.16 g; 2.34 mmol) was dissolved in pyridine (20 mL) at 0 °C. 4-nitrobenzenesulfonylchloride (nosyl chloride) (2 eq; 1.06 g; 4.79 mmol) and silver trifluoromethanesulfonate (AgOTf) (2 eq; 1.23 g; 4.79 mmol) were added and the reaction mixture was stirred at 0 °C. After 1h the reaction mixture was allowed to slowly warm to rt and stirred for another 2 h. The reaction mixture was then diluted with EtOAc, filtered, and the filtrate was washed with H2O and brine. The organic layer was dried over anhydrous Na2SO4 and the solvent was evaporated under reduced pressure. Purification of the crude residue was accomplished by silica gel column chromatography (95% CH2Cl2/5% CH3OH) to give the desired compound 6 as a yellowish solid. Yield: 60%. Rf: 0.6. 1H NMR (500 MHz, CDCl3) δ 8.77–8.67 (s, 1H, NH, thy), 8.41–8.39 (d, J = 2.2, 2H-Ar, nosyl), 8.14–8.08 (m, 2H-Ar, nosyl), 7.71 (ddd, J = 7.6, 4.7, 1.7, 1H, H6), 7.42–7.30 (m, 5H-Ar), 6.28–5.28 (m, 1H-H-1′), 4.51–3.75 (m, 7H), 2.79–2.46 (m, 1H, CH-ala), 1.96–1.86 (m, 2H, H-2′, H-2′’), 1.37 (t, J = 7.6, 3H, CH3 ester), 1.31–1.25 (m, 6H, CH3-thy, CH3-ala). 13C NMR (126 MHz, CDCl3) δ 173.66, 173.36 (C-ala), 163.48 (C2, thy), 151.13, 151.10 (C1, thy), 150.29, 150.25 (C1, nosyl), 141.45, 141.35 (C1, phenyl), 134.73, 134.67 (CH, thy), 129.86, 129.81 (CH, C2-6, nosyl), 129.15, 129.13 (CH, C2, C6, phenyl), 125.28 (CH, C4, phenyl), 124.84, 124.77 (CH, C3, C5, phenyl), 120.15, 120.11 (CH, C3, C5, nosyl), 120.06, 120.02, 111.36, 111.23 (C, C3, thy), 84.14, 84.00 (CH, C1′), 80.58, 80.52 (CH, C3′), 80.25, 80.18 (CH, C4′), 63.17, 63.14 (CH2, C5′), 62.82, 62.79 (CH2, ethyl), 50.41, 50.20 (CH, ala), 39.18, 39.16 (CH2, C2′), 20.94, 20.90 (CH3, ethyl), 14.12, 14.11 (CH3, ala), 12.58, 12.56 (CH3, thy). 31P NMR (202 MHz, CDCl3) δ 2.75, 2.48. MS (ESI)+: 705.1 [M + Na+]. HPLC: Rt: 15.88 min; Purity > 99%; [Gradient: (0′) 95% H2O/5% CH3CN − (5′) 50% H2O/50% CH3CN − (15′) 50% H2O/50% CH3CN − (20′) 95% H2O/5% CH3CN].
Synthesis of tert-butyl-3-((2R,4R,5R)-5-(((((tert-butoxycarbonyl)((S)-1-ethoxy-1-oxopropan-2-yl)amino)(phenoxy)phosphoryl)oxy)methyl)-4-(((4-nitrophenyl)sulfonyl)oxy)tetrahydrofuran-2-yl)-5-methyl-2,6-dioxo-3,6-dihydropyrimidine-1(2H)-carboxylate (7). MF: C37H47N4O17PS. MW: 882.83. The nosylated ProTide (6) (1 eq, 0.050 g, 0.073 mmol) was dissolved in pyridine (6 mL) at rt under nitrogen atmosphere. To the stirring solution, di(tert-butyl)dicarbonate (Boc2O) (1.3 eq, 0.021 mL, 0.020 g, 0.095 mmol) was added dropwise and the reaction was stirred for 16h. The crude mixture was evaporated under reduced pressure and was purified by silica gel column chromatography (CH2Cl2/CH3OH gradient from 100% CH2Cl2 to 95% CH2Cl2) to give the final di-protected nosylated derivate 7 as a yellowish oil. Yield: 56%. Rf: 0.67 in 90% CH2Cl2/10% CH3OH as TLC system. 1H NMR (500 MHz, CDCl3) δ 8.47–8.36 (d, 2H, J = 2.2, Ar, nosyl), 8.17–8.08 (m, 2H, Ar, nosyl), 7.38 (m, 1H, Ar), 7.32–7.24 (m, 4H), 7.17 (s, 1H, thy), 6.31–6.24 (m, 1H-H-1′), 4.51–3.75 (m, 7H), 2.78–2.43 (m, 1H, CH-Ala), 2.31–2.23 (m, 1H-H-2′), 2.01 (m, 3H, CH3, thy), 1.53–1.49 (m, 9H, CH3, tert-butyl), 1.44 (m, 9H, CH3, tert-butyl), 1.38 (t, J = 7.6, 3H, CH3, ester), 1.31 (m, 3H, CH3, ala). 13C NMR (126 MHz, CDCl3) δ 175.50, 174.29 (C, ala), 161.32 (C2, thy), 150.13, 150.08 (C1, thy), 150.02, 150.00 (C1, nosyl) 143.51, 142.21 (C1, phenyl), 132.71, 132.23 (CH, thy), 130.68, 129.99 (CH, C2–C6, nosyl), 129.34, 129.5 (CH, C2, C6, phenyl), 126.28 (CH, C4, phenyl), 125.79, 124.85 (CH, C3, C5, phenyl), 120.15, 120.13 (CH, C3,C5, nosyl), 120.06, 120.02, 111.36, 111.23 (C, C3, thy), 84.13, 84.10 (CH, C1′), 80.78–80.77 (C-tert-butyl), 80.58, 80.52 (CH, C3′), 80.25, 80.18 (CH, C4′), 63.21, 63.20 (CH2, C5′), 62.79, 62.77 (CH2, ethyl), 50.39, 50.30 (CH, ala), 39.18, 39.15 (CH2, C2′), 28.41–28.23 (CH3, tert-butyl), 20.94, 20.90 (CH3, ethyl), 14.12, 14.11 (CH3, ala), 12.58, 12.56 (CH3, thy). 31P NMR: (202 MHz, CDCl3): δ 2.61, 2.53. MS (ESI+): 905.83 [M + Na+]. HPLC: Rt: 17.1 min; Purity > 99%; [Gradient: (0′) 95% H2O/5% CH3CN − (5′) 50% H2O/50% CH3CN − (15′) 50% H2O/50% CH3CN − (20′) 95% H2O/5% CH3CN].
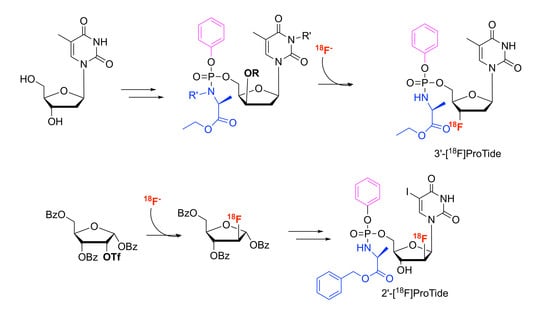
Synthesis of tert-butyl-3-((2R,4S,5R)-5-((((N-((S)-1-ethoxy-1-oxopropan-2-yl)-3,3-dimethylbutanamido)(phenoxy)phosphoryl)oxy)methyl)-4-[18F]fluoro-tetrahydrofuran-2-yl)-5-methyl-2,6-dioxo-3,6-dihydropyrimidine-1(2H)-carboxylate (15). MF: C32H4518FN3O11P; MW: 696.70. Aqueous [18F]fluoride (2–8 GBq) produced by the cyclotron was trapped in a QMA cartridge and was then eluted through the cartridge by an aqueous solution of KHCO3 and Kryptofix in CH3CN. The resulting [18F]F-/KHCO3/Kryptofix complex was dried by an azeotropic distillation with anhydrous CH3CN (2 × 1 mL) under reduced pressure and a stream of nitrogen. A solution of the precursor 7 (10 mg) in anhydrous CH3CN (1 mL) was added and the reaction was stirred for 30 min at 95 °C. The resulting reaction mixture was passed through an alumina cartridge to obtain the radiolabelled product 15. The reaction mixture was analysed by analytical radio HPLC: Rt: 15 min (analytical HPLC: (0′) 95% H2O/5% CH3CN − (5′) 50% H2O/50% CH3CN − (15′) 50% H2O/50% CH3CN − (20′) 95% H2O/5% CH3CN).
Synthesis of ethyl ((((2R,3S,5R)-3-[18F]fluoro-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)-l-alaninate (1). MF: C21H2718FN3O8P; MW: 498.43. To the Boc protected [18F]-radiolabelled ProTide (15), a solution of 2 N HCl (1 mL) was added and the reaction was stirred for 10 min. The solution was then neutralised with 2 N NaOH (1 mL). The crude mixture was then purified by semi-preparative HPLC and the desired compound was eluted after 35 min at a flow rate of 3.5 mL/min using 70% H2O/30% CH3CN as mobile phase. The compound was then dried under a stream of nitrogen, taken up in saline and flushed through a sterility filter to obtain the aqueous solution of [18F]FLT ProTide (1). RCY of 15–30% (n = 5, decay-corrected from end of bombardment (EoB)), with high radiochemical purities (97%) and molar activities of 56 GBq/μmol. The total synthesis time was 130 min after the end of bombardment (EoB). The reaction mixture was analyzed by analytical radio-HPLC. Analytical HPLC: (0′) 95% H2O/5% CH3CN − (5′) 50% H2O/50% CH3CN − (15′) 50% H2O/50% CH3CN − (20′) 95% H2O/5% CH3CN); Rt: 9.5 min.
3.4. Procedures and Analytical Data for the Synthesis of the [18F]FIAU ProTide
Synthesis of
2′-deoxy-2′-α-fluoro-5-iodouridine (
19). MW: 372.1; MF: C
9H
10FIN
2O
4. Iodine (1.2 eq; 1.24 g; 4.87 mmol) and ceric ammonium nitrate (CAN) (1 eq; 2.23 g; 4.062 mmol) were added to a stirring solution of 2′-β-fluoro-2′-deoxyuridine (
18) (2 eq; 2.0 g; 8.12 mmol) in anhydrous acetonitrile (50 mL). The mixture was stirred at 75 °C for 1 h and was then quenched with a saturated solution of Na
2S
2O
3 and concentrated under reduced pressure. The residue was then re-dissolved in ethyl acetate and washed twice with saturated NaCl. The organic layer was dried over MgSO
4, filtered and concentrated to give compound
19 as a pale yellow solid. Yield: 60%. HPLC: Rt: 2.3 min; Purity > 95% (98% H
2O/2% CH
3CN).
1H NMR (500 MHz, DMSO-
d6): δ 11.69 (s, 1H, NH), 8.53 (s, 1H, 6-CH), 5.86 (d,
J = 15.8, 1H, 1’-CH), 5.60 (d,
J = 6.4, 1H, 3’-OH), 5.39 (t,
J = 4.5, 1H, 3’-CH), 5.04 (dd,
J = 5.2, 4.1, 1H, 2’-CH), 4.18 (ddd,
J = 23.4, 11.4, 7.2, 1H, 4’-CH), 3.90 (d,
J = 8.2, 1H, 5’-OH), 3.85–3.79 (m, 1H, 5’-CH), 3.63–3.58 (m, 1H, 5’-CH).
13C NMR (125 MHz, DMSO-
d6): δ 167.88 (C=O), 165.01 (C=O), 145.02 (C-6), 125.19 (CH, C-2’), 121.26 (CH, C-1’), 115.81 (CH, C-4’), 61.11 (CH, C-5), 57.30 (CH, C-3’), 45.87 (CH, C-5’).
19F NMR (470 MHz, DMSO-
d6): δ −202.09. Spectroscopic data in agreement with literature [
10].
Synthesis of
benzyl(chloro(phenoxy)phosphoryl)-L-alaninate (
20). MW: 353.73; MF: C
16H
17ClNO
4P. Compound
20 was synthesised according to the standard procedure [
16]. Anhydrous triethylamine (2 eq; 1.26 mL; 0.918 g; 9.08 mmol), phenyl dichlorophosphate (1 eq; 0.678 mL; 0.958 g; 4.54 mmol) and L-alanine benzyl ester hydrochloride salt (1 eq; 1.50 g; 4.54 mmol) were reacted to give compound
20 as a yellowish oil. Yield: 88%.
1H NMR (500 MHz, CDCl
3): δ 7.54–7.47 (m, 7H, Ar-H), 7.46–7.40 (m, 3H, Ar-H), 5.27 (d,
J = 8.4, 2H, CH
2-ester), 4.69 (d,
J = 9.9, 1H, NH), 4.13 (dd,
J = 34.4, 29.8, 1H, CH-ala), 1.52 (m, 3H, CH
3-ala).
31P NMR (202 MHz, CDCl
3): δ 8.03, 7.75. Spectroscopic data in agreement with literature [
29,
30,
31].
Synthesis of
benzyl((((2R,3R,4S,5R)-4-fluoro-3-hydroxy-5-(5-iodo-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)-l-alaninate (
21). MF: C
25H
26FIN
3O
9P; MW: 689.3. Compound
21 was prepared according to the standard procedure [
16]. 1-(3-Fluoro-4-hydroxy-5-(hydromethyl)tetrahydrofuran-2-yl)-5-iodopyrimidine-2,4(1H,3H)-dione (
19) (1 eq; 0.400 g; 1.07 mmol) and NMI (5 eq; 0.424 mL; 0.439 g; 5.35 mmol) were reacted with benzyl 2-(chloro(phenoxy)phosphorylamino)propanoate (3 eq; 1.05 g; 3.22 mmol) (
20). The crude mixture was purified by silica gel column chromatography (CH
2Cl
2/CH
3OH from 100% CH
2Cl
2 to 95% CH
2Cl
2) to obtain the product
21 as a yellowish oil. Yield: 10%.
1H NMR (500 MHz, CDCl
3): δ 10.59 (s, 1H, 3-NH), 7.89 (s, 1H, 6-CH), 7.53–7.48 (m, 2H, CH-phenyl), 7.45 (t, J = 8.0, 2H, CH-phenyl), 7.41–7.38 (m, 2H, CH-benz), 7.17–7.15 (m, 2H, CH-benz), 7.13 (t, J = 8.0, 1H, CH-benzyl), 7.12 (t, J = 7.4, 1H, CH-phenyl), 5.99 (m, 1H, 1’-CH), 5.79 (dd, J = 47.6, 4.6, 1H, 2’-CH), 5.14 (m, 2H, CH
2-benz), 4.90 (m, 2H, 5’-CH
2), 4.39 (m, 1H, 4’-CH), 4.27 (m, 1H, 3’-CH), 4.19 (m, 1H, 3’-OH), 4.02 (m, 1H, NH-ala), 3.99 (m, 1H, CH-ala), 1.29 (d, J = 7.0, 3H, CH
3-ala).
13C NMR (125 MHz, CDCl
3): δ 171.9 (C=O, ala), 168.12 (C=O, C-4), 144.18 (C=O, C-2), 149.0 (C-phenyl), 147.91 (C-benzyl), 145.5, 145.1 (CH, C-6), 128.31–121.48 (CH, Ar-C), 94.3 (C-5), 92.10 (CH, C-4’), 89.13 (CH, C-2’), 83.6, 82.97 (CH, C-1’), 81.08, 80.97 (CH
2, C-ester), 69.61, 68.14 (CH, C-3’), 67.23 (CH, C-5’), 52.8 (CH-ala), 21.98 (CH
3-ala).
19F NMR (470 MHz, CDCl
3): δ -200.91, -201.15.
31P NMR (202 MHz, CDCl
3): δ 3.90, 3.86. MS (ESI)
+: 690.3 [M + H
+]. HPLC: Rt: 13.4 min; purity > 97%; [Gradient: (0′) 95% H
2O/5% CH
3CN − (5′) 50% H
2O/50% CH
3CN − (15′) 50% H
2O/50% CH
3CN − (20′) 95% H
2O/5% CH
3CN].
Synthesis of (2R,3S,4R,5R)-5-((benzoyloxy)methyl)-3-[18F]fluoro-tetrahydrofuran-2,4-diyl-dibenzoate (17). MF: C26H2118FO7 MW: 463.4 Aqueous [18F]fluoride (4.11 GBq), produced by the cyclotron was trapped in a QMA cartridge before it was eluted with an aqueous solution of KHCO3 and Kryptofix in anhydrous CH3CN. The [18F]F-/KHCO3/Kryptofix complex was dried by co-evaporation with anhydrous CH3CN (2 × 1 mL) under reduced pressure and a stream of nitrogen. A solution of the triflate precursor (16) (10 mg) in anhydrous CH3CN (1 mL) was added to the reaction vial and the reaction was stirred for 30 min at 95 °C. The mixture was passed through an alumina cartridge to obtain the radiolabelled product 17 that was used for next step without further purification. The reaction mixture was analysed by analytical HPLC. Rt: 8.3 min (100% H2O).
Synthesis of
5-iodo-2,4-bis((trimethylsilyl)oxy)pyrimidine (
23). MF: C
10H
19IN
2O
2Si
2; MW: 382.3. To a solution of 5-iodouracil (
22) (1 eq; 10 mg; 0.042 mmol) in dichloroethane (500 μL), hexamethyldisilazane (11.4 eq; 100 μL; 0.0774 mg; 0.479 mmol) and TMSOTf (13.1 eq; 100 μL, 0.123 mg; 0.549 mmol) were added. The mixture was stirred for 2 h at 85 °C and was used for next step without further purification. The purity of the compound was assessed by analytical HPLC (Rt = 6.9 min; 88% H
2O/12% CH
3CN) and LC-MS ([M + H
+]: 383.2). Spectroscopic data was in agreement with literature [
10].
Synthesis of
1-((2R,3S,4R,5R)-3-[18F]fluoro-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-5-iodopyrimidine-2,4(1H,3H)-dione ([18F]FIAU) (
24). MF: C
9H
1018FN
2O
5 MW: 371.0. Compound
17 was delivered to a vial containing 2-4-bis(trimethylsilyl)-5-iodouracil (
23). The mixture was then heated at 85 °C for 60 min. To this mixture, 0.5 M of NaOCH
3 in CH
3OH (1 mL) was then added and the reaction was stirred at 85 °C for another 5 min. The precipitate was then reconstituted in water (1 mL) and neutralised with 6 N HCl. The reaction mixture was analyzed by analytical HPLC showing the formation of the 2 anomers (α and β) of the 2′-deoxy-2′-fluoro-5-iodouridine. Analytical HPLC: 98% H
2O/2% CH
3CN; Rt: α anomer 2.1 min; β anomer 2.9 min). The anomeric mixture was then purified by semi-preparative HPLC and the target compound (
24) was eluted after 7.3 min at a flow rate of 3.5 mL/min using 20% CH
3CN/80% H
2O as mobile phase to obtain the final compound. HPLC: Rt: 2.1 min; 98% H
2O/2% CH
3CN. Data in agreement with literature [
10].
Synthesis of [18F]FIAU ProTide (2). MF: C25H2618FIN3O9P; MW: 688.3. To [18F]FIAU (24), NMI (0.1 mL) and a solution of benzyl-2-(chloro(benzyloxy)phosphorylamino)propanoate (20) (0.050 g) in anhydrous THF (0.5 mL) were added together under nitrogen atmosphere. The reaction mixture was stirred at 50 °C for 20 min and then dried under a flow of nitrogen, re-dissolved in CH3CN and purified via semi-preparative HPLC. The compound 2 was eluted after 23 min at a flow rate of 3.5 mL/min using 50% CH3CN/50% H2O as the mobile phase. The solvent was then removed from the mixture under a stream of nitrogen. The final product was then re-formulated in saline and flushed through a sterility filter to furnish a clean sterile aqueous solution of [18F]FIAU ProTide (2). Radiochemical reactions were carried out using starting activities between 4–15 GBq, leading to RCY’s of 1–5% (n = 7, decay-corrected from end of bombardment (EoB)), with high radiochemical purities (98%) and molar activities of 53 GBq/μmol. The total synthesis time was 240 min after the end of bombardment (EoB). Analytical HPLC: (0′) 95% H2O/5% CH3CN - (5′) 50% H2O/50% CH3CN- (15′) 50% H2O/50% CH3CN- (20′) 95% H2O/5% CH3CN); Rt: 12.3 min.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}