
Ultrafast Backbone Protonation in Channelrhodopsin-1 Captured by Polarization Resolved Fs Vis-pump—IR-Probe Spectroscopy and Computational Methods


Abstract
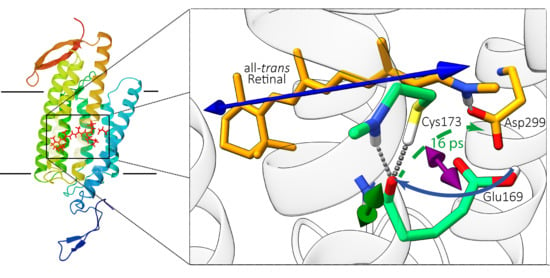
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
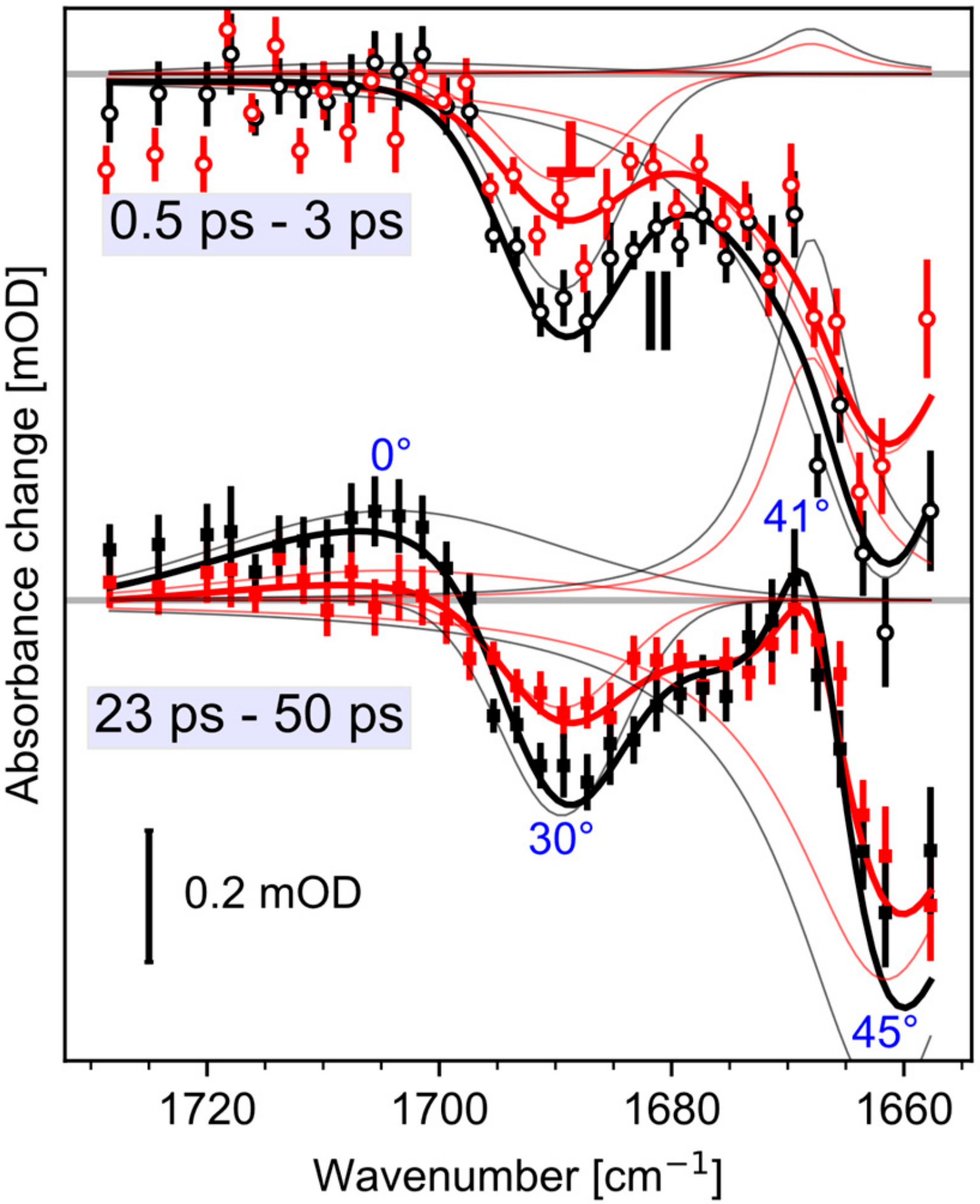{kind=link}
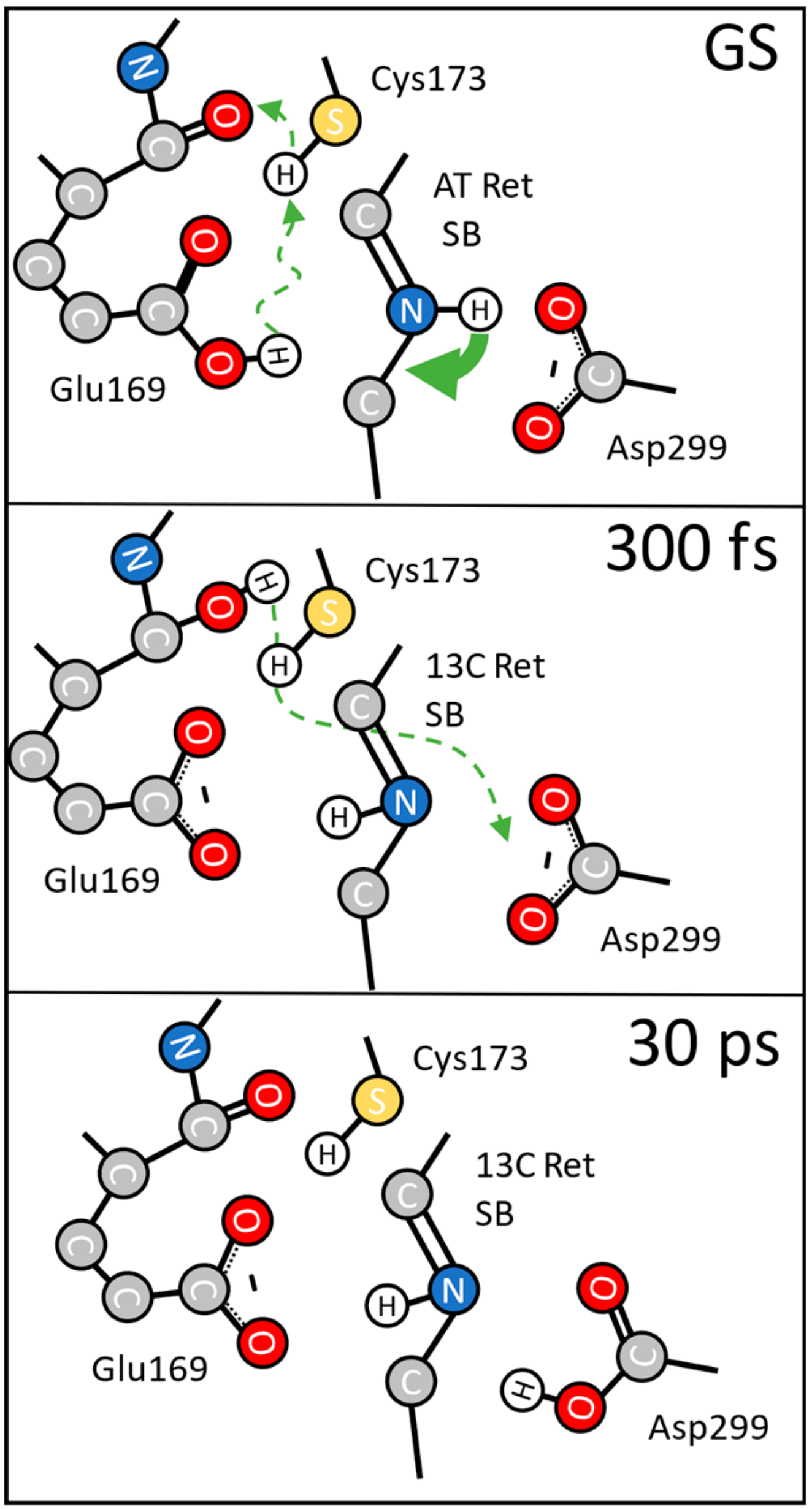{kind=link}
1. Introduction
2. Results
2.1. Computational Results
2.2. Experimental Results
2.3. Assignment of Protein Bands
2.4. Spectral Decomposition
3. Discussion
4. Materials and Methods
4.1. Experimental Methods
4.1.1. Sample Preparation
4.1.2. Spectroscopy
4.2. Theoretical Methods
4.2.1. Structural Modelling
4.2.2. Estimation of Vibrational tdm Orientation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deisseroth, K.; Hegemann, P. The form and function of channelrhodopsin. Science 2017, 357, eaan5544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegemann, P. Algal Sensory Photoreceptors. Annu. Rev. Plant Biol. 2008, 59, 167–189. [Google Scholar] [CrossRef] [PubMed]
- Tahara, S.; Kuramochi, H.; Takeuchi, S.; Tahara, T. Protein Dynamics Preceding Photoisomerization of the Retinal Chromophore in Bacteriorhodopsin Revealed by Deep-UV Femtosecond Stimulated Raman Spectroscopy. J. Phys. Chem. Lett. 2019, 10, 5422–5427. [Google Scholar] [CrossRef] [PubMed]
- Nogly, P.; Weinert, T.; James, D.; Carbajo, S.; Ozerov, D.; Furrer, A.; Gashi, D.; Borin, V.; Skopintsev, P.; Jaeger, K.; et al. Retinal isomerization in bacteriorhodopsin captured by a femtosecond x-ray laser. Science 2018, 361, eaat0094. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, G.N.; Colletier, J.-P.; Grünbein, M.L.; Yang, Y.; Stensitzki, T.; Batyuk, A.; Carbajo, S.; Doak, R.B.; Ehrenberg, D.; Foucar, L.; et al. Three-dimensional view of ultrafast dynamics in photoexcited bacteriorhodopsin. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Schenkl, S.; van Mourik, F.; van der Zwan, G.; Haacke, S.; Chergui, M. Probing the Ultrafast Charge Translocation of Photoexcited Retinal in Bacteriorhodopsin. Science 2005, 309, 917–920. [Google Scholar] [CrossRef]
- Neumann-Verhoefen, M.-K.; Neumann, K.; Bamann, C.; Radu, I.; Heberle, J.; Bamberg, E.; Wachtveitl, J. Ultrafast Infrared Spectroscopy on Channelrhodopsin-2 Reveals Efficient Energy Transfer from the Retinal Chromophore to the Protein. J. Am. Chem. Soc. 2013, 135, 6968–6976. [Google Scholar] [CrossRef]
- Bühl, E.; Eberhardt, P.; Bamann, C.; Bamberg, E.; Braun, M.; Wachtveitl, J. Ultrafast Protein Response in Channelrhodopsin-2 Studied by Time-Resolved Infrared Spectroscopy. J. Phys. Chem. Lett. 2018, 9, 7180–7184. [Google Scholar] [CrossRef]
- Gross, R.; Wolf, M.M.N.; Schumann, C.; Friedman, N.; Sheves, M.; Li, L.; Engelhard, M.; Trentmann, O.; Neuhaus, H.E.; Diller, R. Primary Photoinduced Protein Response in Bacteriorhodopsin and Sensory Rhodopsin II. J. Am. Chem. Soc. 2009, 131, 14868–14878. [Google Scholar] [CrossRef]
- Gross, R.; Schumann, C.; Wolf, M.M.N.; Herbst, J.; Diller, R.; Friedman, N.; Sheves, M. Ultrafast Protein Conformational Alterations in Bacteriorhodopsin and Its Locked Analogue BR5.12. J. Phys. Chem. B 2009, 113, 7851–7860. [Google Scholar] [CrossRef]
- Hou, S.-Y.; Govorunova, E.G.; Ntefidou, M.; Lane, C.E.; Spudich, E.N.; Sineshchekov, O.A.; Spudich, J.L. Diversity of Chlamydomonas Channelrhodopsins. Photochem. Photobiol. 2012, 88, 119–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogren, J.I.; Mamaev, S.; Russano, D.; Li, H.; Spudich, J.L.; Rothschild, K.J. Retinal Chromophore Structure and Schiff Base Interactions in Red-Shifted Channelrhodopsin-1 from Chlamydomonas augustae. Biochemistry 2014, 53, 3961–3970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogren, J.I.; Yi, A.; Mamaev, S.; Li, H.; Lugtenburg, J.; DeGrip, W.J.; Spudich, J.L.; Rothschild, K.J. Comparison of the Structural Changes Occurring during the Primary Phototransition of Two Different Channelrhodopsins from Chlamydomonas Algae. Biochemistry 2015, 54, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Muders, V.; Kerruth, S.; Lórenz-Fonfría, V.A.; Bamann, C.; Heberle, J.; Schlesinger, R. Resonance Raman and FTIR spectroscopic characterization of the closed and open states of channelrhodopsin-1. FEBS Lett. 2014, 588, 2301–2306. [Google Scholar] [CrossRef] [PubMed]
- Ogren, J.I.; Yi, A.; Mamaev, S.; Li, H.; Spudich, J.L.; Rothschild, K.J. Proton Transfers in a Channelrhodopsin-1 Studied by Fourier Transform Infrared (FTIR) Difference Spectroscopy and Site-directed Mutagenesis. J. Biol. Chem. 2015, 290, 12719–12730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stensitzki, T.; Muders, V.; Schlesinger, R.; Heberle, J.; Heyne, K. The primary photoreaction of channelrhodopsin-1: Wavelength dependent photoreactions induced by ground-state heterogeneity. Front. Mol. Biosci. 2015, 2, 41. [Google Scholar] [CrossRef] [Green Version]
- Stensitzki, T.; Yang, Y.; Muders, V.; Schlesinger, R.; Heberle, J.; Heyne, K. Femtosecond infrared spectroscopy of channelrhodopsin-1 chromophore isomerization. Struct. Dyn. 2016, 3, 043208. [Google Scholar] [CrossRef] [Green Version]
- Schnedermann, C.; Muders, V.; Ehrenberg, D.; Schlesinger, R.; Kukura, P.; Heberle, J. Vibronic Dynamics of the Ultrafast all- trans to 13- cis Photoisomerization of Retinal in Channelrhodopsin-1. J. Am. Chem. Soc. 2016, 138, 4757–4762. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Linke, M.; von Haimberger, T.; Hahn, J.; Matute, R.; González, L.; Schmieder, P.; Heyne, K. Real-Time Tracking of Phytochrome’s Orientational Changes During Pr Photoisomerization. J. Am. Chem. Soc. 2012, 134, 1408–1411. [Google Scholar] [CrossRef]
- Rubtsov, I.V.; Wang, J.; Hochstrasser, R.M. Vibrational Coupling between Amide-I and Amide-A Modes Revealed by Femtosecond Two Color Infrared Spectroscopy. J. Phys. Chem. A 2003, 107, 3384–3396. [Google Scholar] [CrossRef]
- Ogren, J.I. Vibrational Spectroscopy of an Optogenetic Rhodopsin: A Biophysical Study of Molecular Mechanisms. Ph.D. Thesis, Boston University, Boston, MA, USA, 2015. [Google Scholar]
- Nie, B.; Stutzman, J.; Xie, A. A Vibrational Spectral Maker for Probing the Hydrogen-Bonding Status of Protonated Asp and Glu Residues. Biophys. J. 2005, 88, 2833–2847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newville, M.; Stensitzki, T.; Allen, D.B.; Ingargiola, A. LMFIT: Non-Linear Least-Square Minimization and Curve-Fitting for Python (Version 0.8.0); Zenodo: Geneva, Switzerland, 2014. [Google Scholar] [CrossRef]
- Earnest, T.N.; Herzfeld, J.; Rothschild, K.J. Polarized Fourier transform infrared spectroscopy of bacteriorhodopsin. Transmembrane alpha helices are resistant to hydrogen/deuterium exchange. Biophys. J. 1990, 58, 1539–1546. [Google Scholar] [CrossRef] [Green Version]
- Kamiya, K.; Boero, M.; Shiraishi, K.; Oshiyama, A. Enol-to-keto Tautomerism of Peptide Groups. J. Phys. Chem. B 2006, 110, 4443–4450. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, S.; Muramoto, K.; Shinzawa-Itoh, K.; Aoyama, H.; Tsukihara, T.; Ogura, T.; Shimokata, K.; Katayama, Y.; Shimada, H. Reaction mechanism of bovine heart cytochrome c oxidase. Biochim. Biophys. Acta (BBA) Bioenerg. 2006, 1757, 395–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukihara, T.; Shimokata, K.; Katayama, Y.; Shimada, H.; Muramoto, K.; Aoyama, H.; Mochizuki, M.; Shinzawa-Itoh, K.; Yamashita, E.; Yao, M.; et al. The low-spin heme of cytochrome c oxidase as the driving element of the proton-pumping process. Proc. Natl. Acad. Sci. USA 2003, 100, 15304–15309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamiya, K.; Boero, M.; Tateno, M.; Shiraishi, K.; Oshiyama, A. Possible Mechanism of Proton Transfer through Peptide Groups in the H-Pathway of the Bovine Cytochrome c Oxidase. J. Am. Chem. Soc. 2007, 129, 9663–9673. [Google Scholar] [CrossRef]
- Kato, H.E.; Zhang, F.; Yizhar, O.; Ramakrishnan, C.; Nishizawa, T.; Hirata, K.; Ito, J.; Aita, Y.; Tsukazaki, T.; Hayashi, S.; et al. Crystal structure of the channelrhodopsin light-gated cation channel. Nature 2012, 482, 369–374. [Google Scholar] [CrossRef] [Green Version]
- Volkov, O.; Kovalev, K.; Polovinkin, V.; Borshchevskiy, V.; Bamann, C.; Astashkin, R.; Marin, E.; Popov, A.; Balandin, T.; Willbold, D.; et al. Structural insights into ion conduction by channelrhodopsin 2. Science 2017, 358, eaan8862. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Vierock, J.; Oishi, S.; Rodriguez-Rozada, S.; Taniguchi, R.; Yamashita, K.; Wiegert, J.S.; Nishizawa, T.; Hegemann, P.; Nureki, O. Crystal structure of the red light-activated channelrhodopsin Chrimson. Nat. Commun. 2018, 9, 3949. [Google Scholar] [CrossRef] [Green Version]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [Green Version]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, P.; de Vries, A.H.; Guest, M.F.; Schreckenbach, G.; Catlow, C.R.A.; French, S.A.; Sokol, A.A.; Bromley, S.T.; Thiel, W.; Turner, A.J.; et al. QUASI: A general purpose implementation of the QM/MM approach and its application to problems in catalysis. J. Mol. Struc. Theochem. 2003, 632, 1–28. [Google Scholar] [CrossRef]
- Kästner, J.; Carr, J.M.; Keal, T.W.; Thiel, W.; Wander, A.; Sherwood, P. DL-FIND: An Open-Source Geometry Optimizer for Atomistic Simulations. J. Phys. Chem. A 2009, 113, 11856–11865. [Google Scholar] [CrossRef] [PubMed]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins †. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Adam, S.; Bondar, A.-N. Mechanism by which water and protein electrostatic interactions control proton transfer at the active site of channelrhodopsin. PLoS ONE 2018, 13, e0201298. [Google Scholar] [CrossRef]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.; Darden, T.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Götz, A.W.; Homeyer, N.; et al. Amber 16; University of California: Oakland, CA, USA, 2016. [Google Scholar]
- Götz, A.W.; Clark, M.A.; Walker, R.C. An extensible interface for QM/MM Molecular Dynamics Simulations with AMBER. J. Comput. Chem. 2014, 35, 95–108. Available online: https://onlinelibrary.wiley.com/doi/full/10.1002/jcc.23444 (accessed on 1 October 2019). [CrossRef] [Green Version]
- Christiansen, O.; Koch, H.; Jørgensen, P. The second-order approximate coupled cluster singles and doubles model CC2. Chem. Phys. Lett. 1995, 243, 409–418. [Google Scholar] [CrossRef]
- Weigend, F.; Köhn, A.; Hättig, C. Efficient use of the correlation consistent basis sets in resolution of the identity MP2 calculations. J. Chem. Phys. 2002, 116, 3175–3183. [Google Scholar] [CrossRef]
- Furche, F.; Ahlrichs, R.; Hättig, C.; Klopper, W.; Sierka, M.; Weigend, F. Turbomole. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 91–100. [Google Scholar] [CrossRef]
- TURBOMOLE V7.3 2018, a Development of University of Karlsruhe and Forschungszentrum Karls-ruhe GmbH, 1989–2018, TURBOMOLE GmbH, Since 2007. Available online: http://www.turbomole.com (accessed on 14 February 2020).
Sample Availability: Samples of the compounds are available from the author R.S. |





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stensitzki, T.; Adam, S.; Schlesinger, R.; Schapiro, I.; Heyne, K. Ultrafast Backbone Protonation in Channelrhodopsin-1 Captured by Polarization Resolved Fs Vis-pump—IR-Probe Spectroscopy and Computational Methods. Molecules 2020, 25, 848. https://doi.org/10.3390/molecules25040848
Stensitzki T, Adam S, Schlesinger R, Schapiro I, Heyne K. Ultrafast Backbone Protonation in Channelrhodopsin-1 Captured by Polarization Resolved Fs Vis-pump—IR-Probe Spectroscopy and Computational Methods. Molecules. 2020; 25(4):848. https://doi.org/10.3390/molecules25040848
Chicago/Turabian StyleStensitzki, Till, Suliman Adam, Ramona Schlesinger, Igor Schapiro, and Karsten Heyne. 2020. "Ultrafast Backbone Protonation in Channelrhodopsin-1 Captured by Polarization Resolved Fs Vis-pump—IR-Probe Spectroscopy and Computational Methods" Molecules 25, no. 4: 848. https://doi.org/10.3390/molecules25040848