
Multistep Photochemical Reactions of Polypyridine-Based Ruthenium Nitrosyl Complexes in Dimethylsulfoxide

 and
and
Abstract
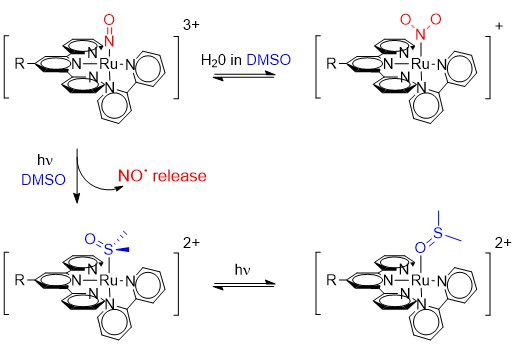
:
1. Introduction
2. Results and Discussion
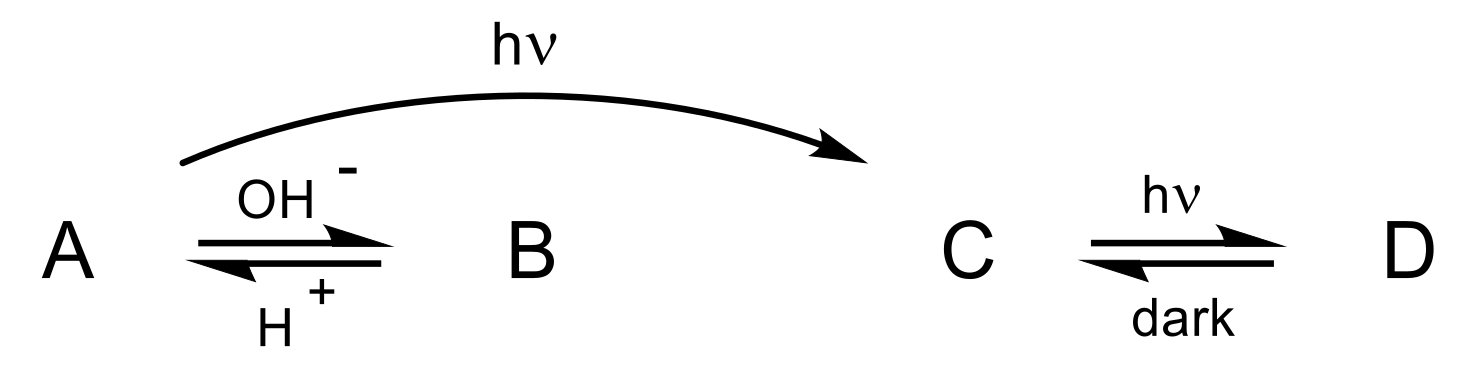
2.1. Photo-Chemical Behavior of [Ru(R-tpy)(bpy)(NO)]3+ in DMSO: the First Step
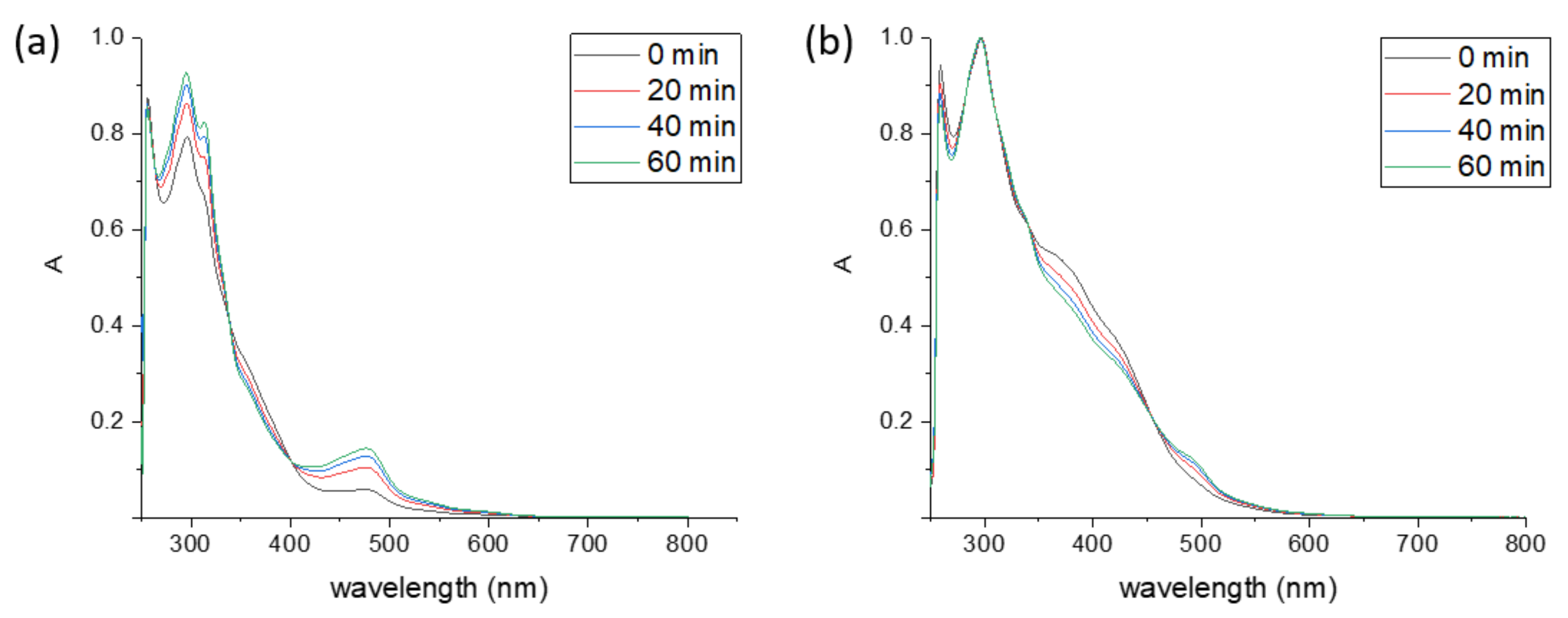
2.1.1. Nature of Form C
2.1.2. Nature of Form B
2.1.3. Origin of Form C
2.1.4. Origin of Form B
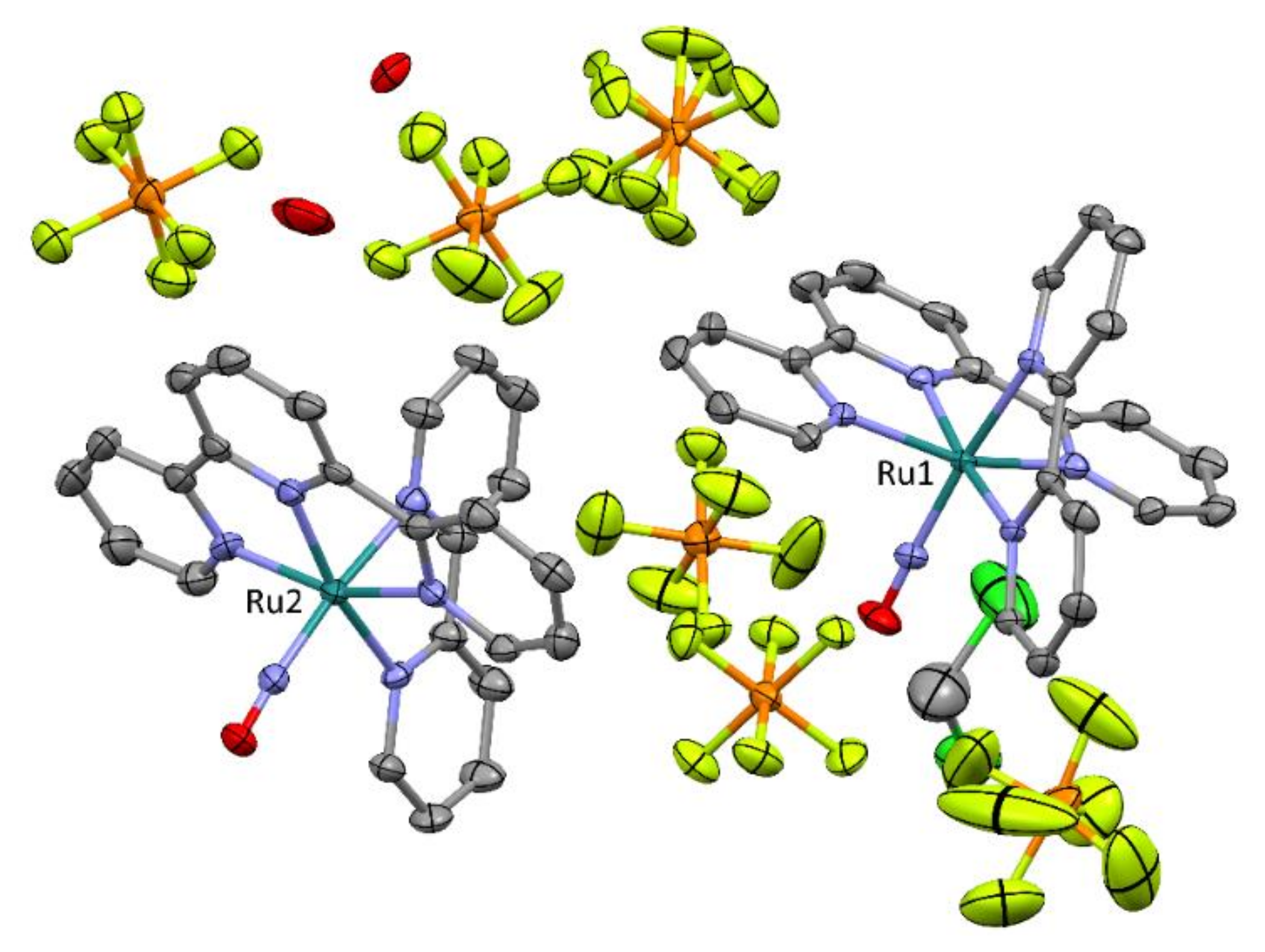
2.2. Crystal Structures Description
2.3. Photochemical Behavior of [Ru(R-tpy)(bpy)(NO)]3+ in DMSO: the Second Step
3. Materials and Methods
3.1. Starting Materials and Equipment
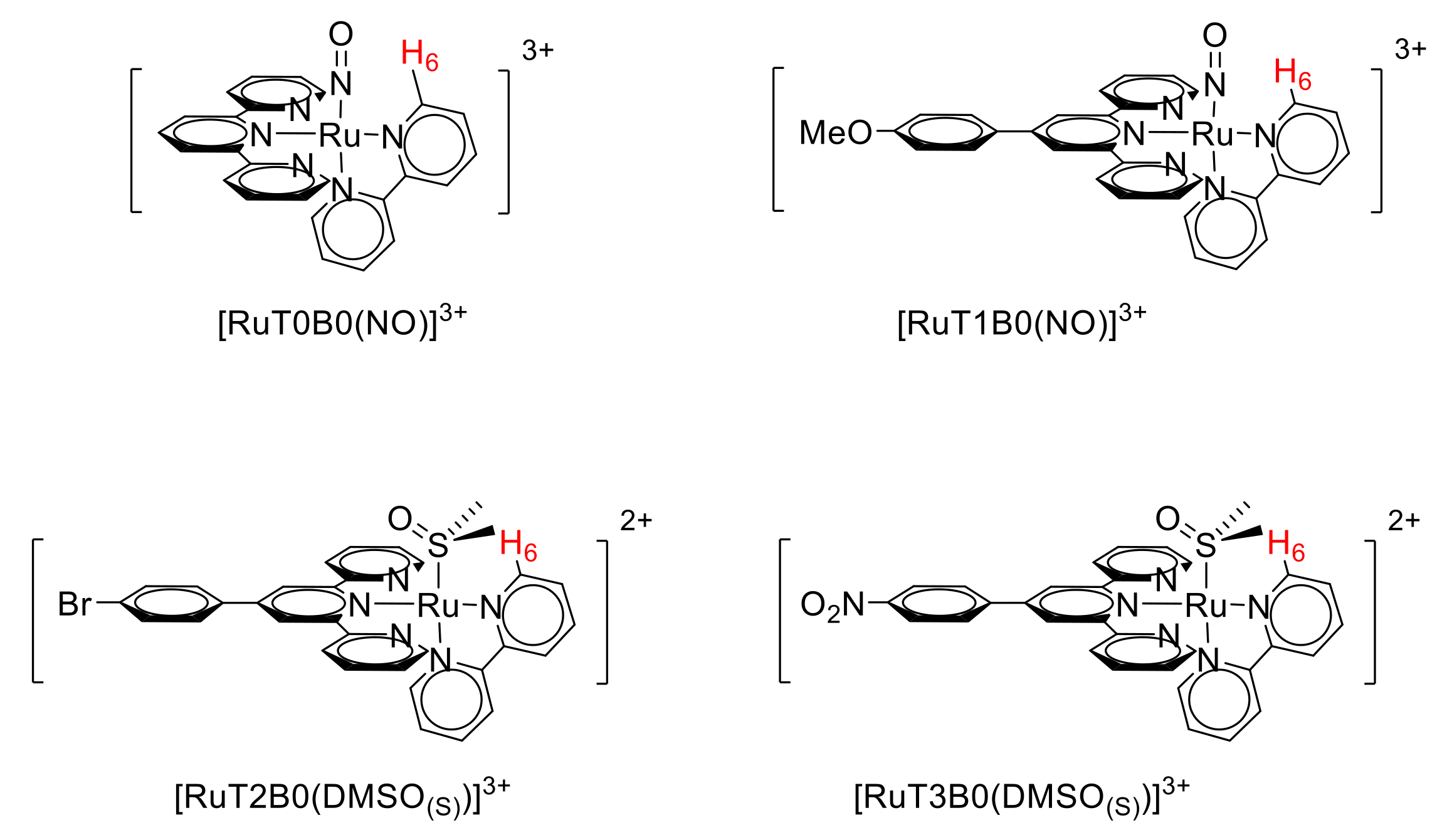
3.2. Synthesis of Ru-DMSO Complexes (Form C)
3.2.1. [RuT0B0(DMSO)(S)](PF6)2
3.2.2. [RuT1B0(DMSO)(S)](PF6)2
3.2.3. [RuT2B0(DMSO)(S)](PF6)2
3.2.4. [RuT3B0(DMSO)(S)](PF6)2
3.3. Synthesis of Form D, by Photo-Isomerization of Form C
3.4. X-ray Studies
3.5. Computational Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ignarro, L.; Freeman, B. Nitric Oxide, Biology and Pathobiology, 3rd ed.; Academic Press: Cambridge, MA, USA, 2017. [Google Scholar]
- Xiang, H.J.; Guo, M.; Liu, J.G. Transition-Metal Nitrosyls for Photocontrolled Nitric Oxide Delivery. Eur. J. Inorg. Chem. 2017, 2017, 1586–1595. [Google Scholar] [CrossRef] [Green Version]
- Mingos, D.M.P. Nitrosyl Complexes in Inorganic Chemistry, Biochemistry and Medicine; Springer: Berlin, Germany, 2014; Volume 154. [Google Scholar]
- Fry, N.L.; Mascharak, P.K. Photoactive Ruthenium Nitrosyls as NO Donors: How to Sensitize Them toward Visible Light. Acc. Chem. Res. 2011, 44, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.J.; Mascharak, P.K. Photoactive ruthenium nitrosyls: Effects of light and potential application as NO donors. Coord. Chem. Rev. 2008, 252, 2093–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikhailov, A.A.; Vorobyev, V.A.; Nadolinny, V.A.; Patrushev, Y.V.; Yudina, Y.S.; Kostin, G.A. Primary and secondary photochemical transformations of biologically active precursor-Nitro-Nitrosyl ruthenium complex. J. Photochem. Photobiol. A Chem. 2019, 373, 37–44. [Google Scholar] [CrossRef]
- Orlowska, E.; Babak, M.V.; Doemoetoer, O.; Enyedy, E.A.; Rapta, P.; Zalibera, M.; Bucinsky, L.; Malcek, M.; Govind, C.; Karunakaran, V.; et al. NO Releasing and Anticancer Properties of Octahedral Ruthenium–Nitrosyl Complexes with Equatorial 1H-Indazole Ligands. Inorg. Chem. 2018, 57, 10702–10717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crisalli, M.A.; Franco, L.P.; Silva, B.R.; Holanda, A.K.M.; Bendhack, L.M.; Da Silva, R.S.; Ford, P.C. Nitric oxide release from a photoactive water-soluble ruthenium nitrosyl. Biological effects. J. Coord. Chem. 2018, 71, 1690–1703. [Google Scholar] [CrossRef]
- Kumar, R.; Kumar, S.; Bala, M.; Ratnam, A.; Singh, U.P.; Ghosh, K. Unprecedented oxidation of aldimine to carboxamido function during reactivity studies on ruthenium complex with acidified nitrite solution: Synthesis of ruthenium nitrosyl complex having {RuNO}6 moiety and photorelease of coordinated NO. J. Organomet. Chem. 2018, 863, 77–83. [Google Scholar] [CrossRef]
- Guo, M.; Xiang, H.J.; Wang, Y.; Zhang, Q.L.; An, L.; Yang, S.P.; Ma, Y.; Wang, Y.; Liu, J.G. Ruthenium nitrosyl functionalized graphene quantum dots as an efficient nanoplatform for NIR-light-controlled and mitochondria-targeted delivery of nitric oxide combined with photothermal therapy. Chem. Commun. 2017, 53, 3253–3256. [Google Scholar] [CrossRef]
- Rodrigues, G.L.S.; Rocha, W.R. Formation and Release of NO from Ruthenium Nitrosyl Ammine Complexes [Ru(NH3)5(NO)]2+/3+ in Aqueous Solution: A Theoretical Investigation. J. Phys. Chem. B 2016, 120, 11821–11833. [Google Scholar] [CrossRef]
- Cacita, N.; Possato, B.; da Silva, C.F.N.; Paulo, C.M.; Formiga, A.L.B.; Bendhack, L.M.; Nikolaou, S. Investigation of a novel trinuclear μ-oxo ruthenium complex as a potential nitric oxide releaser for biological purposes. Inorg. Chim. Acta. 2015, 429, 114–121. [Google Scholar] [CrossRef]
- Pereira, J.C.M.; Souza, M.L.; Franco, D.W. Nitric Oxide and Nitroxyl Products from the Reaction of L-Cysteine with trans-[RuNO(NH3)4P(OEt)3](PF6)3. Eur. J. Inorg. Chem. 2015, 2015, 1005–1011. [Google Scholar] [CrossRef]
- Reynolds, W.R. Dimethyl Sulfoxide in Inorganic Chemistry. In Progress in Inorganic Chemistry; Lippard, S.J., Ed.; Interscience: New York, NY, USA, 1970; Volume 12. [Google Scholar]
- Cordones, A.A.; Lee, J.H.; Hong, K.; Cho, H.; Garg, K.; Boggio-Pasqua, M.; Rack, J.J.; Huse, N.; Schoenlein, R.W.; Kim, T.K. Transient metal-centered states mediate isomerization of a photochromic ruthenium-sulfoxide complex. Nat. Commun. 2018, 9, 1989. [Google Scholar] [CrossRef] [PubMed]
- Rack, J.J.; Mockus, N.V. Room-Temperature Photochromism in cis- and trans-[Ru(bpy)2(dmso)2]2+. Inorg. Chem. 2003, 42, 5792–5794. [Google Scholar] [CrossRef] [PubMed]
- Rachford, A.A.; Rack, J.J. Picosecond isomerization in photochromic ruthenium–dimethyl sulfoxide complexes. J. Am. Chem. Soc. 2006, 128, 14318–14324. [Google Scholar] [CrossRef]
- Göttle, A.J.; Dixon, I.M.; Alary, F.; Heully, J.-L.; Boggio-Pasqua, M. Adiabatic versus nonadiabatic photoisomerization in photochromic ruthenium sulfoxide complexes: A mechanistic picture from density functional theory calculations. J. Am. Chem. Soc. 2011, 133, 9172–9174. [Google Scholar] [CrossRef]
- Ciofini, I.; Daul, C.; Adamo, C. Phototriggered linkage isomerization in ruthenium–dimethylsulfoxyde complexes: Insights from theory. J. Phys. Chem. A 2003, 107, 11182–11190. [Google Scholar] [CrossRef]
- McClure, B.A.; Rack, J.J. Isomerization in photochromic ruthenium sulfoxide complexes. Eur. J. Inorg. Chem. 2010, 2010, 3895–3904. [Google Scholar] [CrossRef]
- Pawlicki, M.; Collins, H.A.; Denning, R.G.; Anderson, H.L. Two-photon absorption and the design of two-photon dyes. Angew. Chem. Int. Ed. 2009, 48, 3244–3266. [Google Scholar] [CrossRef]
- Strehmel, B.; Strehmel, V. Two-photon physical organic, and polymer chemistry: Theory, techniques, chromophore design, and applications. Adv. Photochem. 2007, 29, 111–341. [Google Scholar] [CrossRef]
- Bukhanko, V.; Lacroix, P.G.; Sasaki, I.; Tassé, M.; Mallet-Ladeira, S.; Voitenko, Z.; Malfant, I. Mechanism and oxidation state involved in the nitric oxide (NO) photorelease in a terpyridine-bipyridine-based ruthenium nitrosyl complex. Inorg. Chim. Acta 2018, 482, 195–205. [Google Scholar] [CrossRef]
- Roose, M.; Tassé, M.; Lacroix, P.G.; Malfant, I. Nitric oxide (NO) photo-release in a series of ruthenium–nitrosyl complexes: New experimental insights in the search for a comprehensive mechanism. New J. Chem. 2019, 43, 755–767. [Google Scholar] [CrossRef]
- Garcia, J.S.; Alary, F.; Boggio-Pasqua, M.; Dixon, I.M.; Heully, J.-L. Is photoisomerization required for NO photorelease in ruthenium nitrosyl complexes? J. Mol. Model. 2016, 22, 1–10. [Google Scholar] [CrossRef] [PubMed]
- García, J.S.; Alary, F.; Boggio-Pasqua, M.; Dixon, I.M.; Malfant, I.; Heully, J.-L. Establishing the Two-Photon Linkage Isomerization Mechanism in the Nitrosyl Complex trans-[RuCl(NO)(py)4]2+ by DFT and TDDFT. Inorg. Chem. 2015, 54, 8310–8318. [Google Scholar] [CrossRef] [PubMed]
- Tfouni, E.; Krieger, M.; McGarvey, B.R.; Franco, D.W. Structure, chemical and photochemical reactivity and biological activity of some ruthenium amine nitrosyl complexes. Coord. Chem. Rev. 2003, 236, 57–69. [Google Scholar] [CrossRef]
- Cormary, B.; Malfant, I.; Buron-Le Cointre, M.; Toupet, L.; Delley, B.; Schaniel, D.; Morckus, N.; Woike, T.; Fejfarova, K.; Petricek, V.; et al. [Ru(py)4Cl NO)](PF6)2· 0.5 H2O: A model system for structural determination and ab initio calculations of photo-induced linkage NO isomers. Acta Crystallogr. Sect. B 2009, 65, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Schaniel, D.; Cormary, B.; Malfant, I.; Valade, L.; Woike, T.; Delley, B.; Kraemer, K.W.; Guedel, H.U. Photogeneration of two metastable NO linkage isomers with high populations of up to 76% in trans-[RuCl(py)4(NO)][PF6]2· ½H2O. Phys. Chem. Chem. Phys. 2007, 9, 3717–3724. [Google Scholar] [CrossRef]
- Mikhailov, A.A.; Wenger, E.; Kostin, G.A.; Schaniel, D. Room-Temperature Photogeneration of Nitrosyl Linkage Isomers in Ruthenium Nitrosyl Complexes. Chem. Eur. J. 2019, 25, 7569–7574. [Google Scholar] [CrossRef] [Green Version]
- Karidi, K.; Garoufis, A.; Hadjiliadis, N.; Lutz, M.; Spek, L.; Reedijk, J. Synthesis, Characterization, and DNA-Binding Studies of Nitro(oligopyridine)ruthenium(II) Complexes. Inorg. Chem. 2006, 45, 10282–10292. [Google Scholar] [CrossRef] [Green Version]
- Xiang, J.C.; Gao, Q.H.; Wu, A.X. Solvents as Reagents in Organic Synthesis: Reactions and Applications; Wiley-VCH, Wiley: Hoboken, NJ, USA, 2017; pp. 315–353. [Google Scholar]
- Liang, Y.F.; Wu, K.; Song, S.; Li, X.; Huang, X.; Jiao, N. I2- or NBS-Catalyzed Highly Efficient α-Hydroxylation of Ketones with Dimethyl Sulfoxide. Org. Lett. 2015, 17, 876–8792. [Google Scholar] [CrossRef]
- Cao, Z.; Lv, H.; Liu, Y.; Nie, Z.; Liu, H.; Yang, T.; Luo, W.; Liu, Q.; Guo, C. Dimethyl Sulfoxide Oxygen Donor-Based Annulation of Ketones and Ammonium Persulfate: Regioselective Synthesis of 2, 4-disubstituted Oxazoles. Adv. Synth. Catal. 2019, 361, 1632–1640. [Google Scholar] [CrossRef]
- Zhang, X.; Li, S.S.; Wang, L.; Xu, L.; Xiao, J.; Liu, Z.J. 2-Methylquinoline promoted oxidative ring-opening of N-sulfonyl aziridines with DMSO: Facile synthesis of α-amino aryl ketones. Tetrahedron 2016, 72, 8073–8077. [Google Scholar] [CrossRef]
- Sultana, F.; Shaik, S.P.; Alarifi, A.; Srivastava, A.K.; Kamal, A. Transition-Metal-Free Oxidative Cross-Coupling of Methylhetarenes with Imidazoheterocycles towards Efficient C(sp2)−H Carbonylation. Asian J. Org. Chem. 2017, 6, 890–897. [Google Scholar] [CrossRef]
- Enemark, J.H.; Feltham, R.R. Principles of structure, bonding, and reactivity for metal nitrosyl complexes. Coord. Chem. Rev. 1974, 13, 339–406. [Google Scholar]
- Ogasawara, M.; Huang, D.; Streib, W.E.; Huffman, J.C.; Gallego-Planas, N.; Maseras, F.; Eisenstein, O.; Caulton, K.G. RuX(CO)(NO)L2 and Ru(CO)(NO)L2+: Ru(0) or Ru(II) or In Between? J. Am. Chem. Soc. 1997, 119, 8642–8651. [Google Scholar] [CrossRef]
- Rack, J.J.; Winkler, J.R.; Gray, H.B. Phototriggered Ru(II)-dimethylsulfoxide linkage isomerization in crystals and films. J. Am. Chem. Soc. 2001, 123, 2432–2433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enriquez-Cabrera, A.; Sasaki, I.; Bukhanko, V.; Tassé, M.; Mallet-Ladeira, S.; Lacroix, P.G.; Barba-Barba, R.M.; Ramos-Ortiz, G.; Farfan, N.; Voitenko, Z.; et al. Replacing Two Chlorido Ligands by a Bipyridine Ligand in Ruthenium Nitrosyl Complexes with NO-Release Capabilities: A Comparative Study. Eur. J. Inorg. Chem. 2017, 1446–1456. [Google Scholar] [CrossRef]
- Enriquez-Cabrera, A.; Lacroix, P.G.; Sasaki, I.; Mallet-Ladeira, S.; Farfan, N.; Barba-Barba, R.M.; Ramos-Ortiz, G.; Malfant, I. Comparison of Carbazole and Fluorene Donating Effects on the Two-Photon Absorption and Nitric Oxide Photorelease Capabilities of a Ruthenium–Nitrosyl Complex. Eur. J. Inorg. Chem. 2018, 2018, 531–543. [Google Scholar] [CrossRef] [Green Version]
- King, A.W.; Wang, L.; Rack, J.J. Excited state dynamics and isomerization in ruthenium sulfoxide complexes. Acc. Chem. Res. 2015, 48, 1115–1122. [Google Scholar] [CrossRef]
- Rack, J.J. Electron transfer triggered sulfoxide isomerization in ruthenium and osmium complexes. Coord. Chem. Rev. 2009, 253, 78–85. [Google Scholar] [CrossRef]
- Roeker, L.; Dobson, J.C.; Vining, W.J.; Meyer, T.J. Oxygen atom transfer in the oxidations of dimethyl sulfide and dimethyl sulfoxide by bis(bipyridine)oxo(pyridine)ruthenium(2+). Inorg. Chem. 1987, 26, 779–781. [Google Scholar] [CrossRef]
- Root, M.J.; Deutsch, E. Synthesis and characterization of (bipyridine)(terpyridine)(chalcogenoether) ruthenium (II) complexes. Kinetics and mechanism of the hydrogen peroxide oxidation of [(bpy)(tpy)RuS(CH3)2]2+ to [(bpy)(tpy)RuS(O)(CH3)2]2+. Kinetics of the aquation of [(bpy)(tpy)RuS(O)(CH3)2]2+. Inorg. Chem. 1985, 24, 1464–1471. [Google Scholar]
- Göttle, A.J.; Alary, F.; Dixon, I.M.; Heully, J.-L.; Boggio-Pasqua, M. Unravelling the S → O Linkage Photoisomerization Mechanisms in cis- and trans-[Ru(bpy)2(DMSO)2]2+ Using Density Functional Theory. Inorg. Chem. 2014, 53, 6752–6760. [Google Scholar] [CrossRef] [PubMed]
- Murphy, W.R., Jr.; Takeuchi, K.; Barley, M.H.; Meyer, T.J. Mechanism of reduction of bound nitrite to ammonia. Inorg. Chem. 1986, 25, 1041–1053. [Google Scholar] [CrossRef]
- Pipes, D.W.; Meyer, T.J. Comparisons between polypyridyl nitrosyl complexes of osmium (II) and ruthenium (II). Inorg. Chem. 1984, 23, 2466–2472. [Google Scholar] [CrossRef]
- Patel, M.N.; Joshi, H.N.; Patel, C.R. Interactions with herring sperm DNA and biological studies of sparfloxacin drug-based copper (II) compounds. Appl. Organometal. Chem. 2012, 26, 641–649. [Google Scholar] [CrossRef]
- Koohmareh, G.A.; Sharifi, M. Synthesis, characterization, and coordination behavior of copoly(styrene-maleimide) functionalized with terpyridine. J. Appl. Polym. Sci. 2010, 116, 179–183. [Google Scholar] [CrossRef]
- Takeuchi, K.J.; Thompson, M.S.; Pipes, D.W.; Meyer, T.J. Redox and spectral properties of monooxo polypyridyl complexes of ruthenium and osmium in aqueous media. Inorg. Chem. 1984, 23, 1845–1851. [Google Scholar] [CrossRef]
- Palatinus, L.; Chapuis, G. SUPERFLIP–a computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Cryst. 2007, 40, 786–790. [Google Scholar] [CrossRef] [Green Version]
- Betteridge, P.W.; Carruthers, J.R.; Cooper, R.I.; Prout, K.; Watkin, D.J. CRYSTALS version 12: Software for guided crystal structure analysis. J. Appl. Cryst. 2003, 36, 1487. [Google Scholar] [CrossRef]
- Blessing, R.H. Blessing R H. An empirical correction for absorption anisotropy. Acta Crystallogr. Sect. A 1995, 51, 33–38. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comp. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- London, F. The quantic theory of inter-atomic currents in aromatic combinations. J. Phys. Radium 1937, 8, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. 1. Gauge-invariant LCAO method for N.M.R. chemical shifts. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient Implementation of the Gauge-Independent Atomic Orbital Method for NMR Chemical Shift Calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand X | [RuT0B0(X)](PF6)3 | [RuT1B0(X)](PF6)3 |
|---|---|---|
| Cl− | 10.20 ppm | 10.26 ppm |
| NO2− (form B) | 9.755 ppm | 9.77 ppm a |
| NO (form A) | 9.47 ppm | 9.50 ppm a |
| Structures | Δ(δH-6) |
|---|---|
| [RuT1B0(η2-NO)]3+ | −0.33 ppm |
| [RuT1B0(ON)]3+ | −0.14 ppm |
| [RuT1B0(NO2)]+ | +0.93 ppm |
| [RuT1B0(DMSO(S))]2+ | +1.34 ppm |
| [RuT1B0(DMSO(O))]2+ | +0.74 ppm |
| [RuT0B0(NO)](PF6)3 ½ CH2Cl2–H2O | [RuT1B0(DMSO(S))](PF6)2 CH3CN | |
|---|---|---|
| Chemical formula | C25.5H22ClF18N6O2P3Ru | C36H34F12N6O2P2RuS1 |
| Molecular weight (g/mol) | 1015.90 | 1005.76 |
| Crystal system | monoclinic | triclinic |
| Space group | P21/n | P-1 |
| a (Å) | 19.2309(9) | 12.6477(6) |
| b (Å) | 19.2739(10) | 13.2076(5) |
| c (Å) | 19.7628(10) | 13.2282(7) |
| a (deg) | 90 | 97.4744(14) |
| b (deg) | 106.5407(17) | 109.0108(14) |
| g (deg) | 90 | 106.0250(14) |
| V (Å3) | 7022.0(6) | 1948.27(16) |
| Z | 8 | 2 |
| T (K) | 110 | 113 |
| μ (MoKα) | 0.793 | 0.639 |
| Dcalc (g/cm3) | 1.92 | 1.71 |
| T (K) | 110 | 113 |
| Reflect. measured | 173754 | 99152 |
| 2θ range | 1.30° ≤ 2θ ≤ 28.34° | 1.65° ≤ 2θ ≤ 30.61° |
| Reflect. unique | 17439 | 11796 |
| Rint | 0.0777 | 0.0432 |
| Refinement on | F | F |
| Nb Parameters | 1048 | 541 |
| R1 (I > nσ(I)) | 0.0767, n = 2.5 | 0.0294, n = 3 |
| wR2 (I > nσ(I)) | 0.0964, n = 2.5 | 0.0305, n = 3 |
| Residual electron density (ē.Å−3) | 4.41/−1.77 | 1.02/−0.91 |
| [RuT0B0(NO)]3+ | [RuT1B0(DMSO(S))]2+ | ||
|---|---|---|---|
| Complex 1 (Ru1) | Complex 2 (Ru2) | ||
| Ru-NO | |||
| Ru-N | 1.766(6) | 1.770(7) | |
| N-O | 1.119(8) | 1.135(9) | |
| Ru-N-O | 177.7(6) | 175.3(6) | |
| Ru-DMSO | |||
| Ru-S | 2.2666(4) | ||
| S-O | 1.4826(12) | ||
| Ru-tpy | |||
| Ru-Ncentral | 1.998(6) | 1.996(6) | 1.9735(12) |
| Ru-Nlateral | 2.093(6) | 2.064(7) | 2.0738(12) |
| 2.101(6) | 2.084(7) | 2.0842(12) | |
| Ru-bpy | |||
| Ru-NtransNO | 2.082(6) | 2.092(7) | |
| Ru-NtransDMSO | 2.0912(12) | ||
| Ru-N | 2.089(6) | 2.077(6) | 2.1037(12) |
| Complex | Substituent on Terpyridine | Ru-DMSO(O)/Ru-DMSO(S) | Computed ε365 (L mol−1 cm−1) | |
|---|---|---|---|---|
| Experimental 1 | Computed 2 | |||
| [RuT0B0(DMSO(S))]2+ | H | 42% | 53% | 6021 |
| [RuT0B0(DMSO(O))]2+ | H | 5317 | ||
| [RuT1B0(DMSO(S))]2+ | MeO-phenyl | 25% | 32% | 9806 |
| [RuT1B0(DMSO(O))]2+ | MeO-phenyl | 21,112 | ||
| [RuT2B0(DMSO(S))]2+ | Br-phenyl | 31% | 47% | 12,365 |
| [RuT2B0(DMSO(S))]2+ | Br-phenyl | 14,186 | ||
| [RuT3B0(DMSO(S))]2+ | NO2-phenyl | 46% | 64% | 14,034 |
| [RuT3B0(DMSO(S))]2+ | NO2-phenyl | 7794 | ||
| Compound | UV-vis | DFT Computed Data | ||||||
|---|---|---|---|---|---|---|---|---|
| λmax | ε | Transition a | λ | f | λmaxb | Composition c | Character d | |
| [RuT0B0(DMSO(S))]2+ | 419 | 6680 | S0 → S6 | 395 | 0.104 | 395 | 0.53 χ129 → 132 + 0.41 χ129 → 133 | Ru(d) → π(bpy/tpy) |
| [RuT0B0(DMSO(O))]2+ | S0 → S6 | 462 | 0.103 | 456 | 0.38 χ128 → 133 – 0.32 χ129 → 1313 | Ru(d) → π(tpy) | ||
| [RuT1B0(DMSO(S))]2+ | 436 | 6680 | S0 → S3 | 440 | 0.311 | 430 | 0.61 χ158 → 159 | π(PhOMe)Ru(d) →π(tpy) |
| [RuT1B0(DMSO(O))]2+ | S0 → S6 | 474 | 0.134 | 472 | 0.40 χ158 → 161 + 0.35 χ158 → 160 | π(PhOMe)Ru(d) → π(tpy) | ||
| [RuT2B0(DMSO(S))]2+ | 433 | 11150 | S0 → S4 | 427 | 0.159 | 409 | 0.53 χ167 → 170 – 0.42 χ165 → 168 | Ru(d) → π(bpy) |
| [RuT2B0(DMSO(O))]2+ | S0 → S6 | 470 | 0.154 | 466 | 0.35 χ167 → 169 – 0.34 χ166 → 168 | Ru(d) → π(tpy) | ||
| [RuT3B0(DMSO(S))]2+ | 438 | 14500 | S0 → S4 | 432 | 0.210 | 412 | 0.56 χ159 → 162 | Ru(d) → π(tpyPhNO2) |
| [RuT3B0(DMSO(O))]2+ | S0 → S5 | 477 | 0.181 | 478 | 0.34 χ161 → 164 + 0.32 χ161 → 163 | Ru(d) → π(tpyPhNO2) | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchenko, N.; Lacroix, P.G.; Bukhanko, V.; Tassé, M.; Duhayon, C.; Boggio-Pasqua, M.; Malfant, I. Multistep Photochemical Reactions of Polypyridine-Based Ruthenium Nitrosyl Complexes in Dimethylsulfoxide. Molecules 2020, 25, 2205. https://doi.org/10.3390/molecules25092205
Marchenko N, Lacroix PG, Bukhanko V, Tassé M, Duhayon C, Boggio-Pasqua M, Malfant I. Multistep Photochemical Reactions of Polypyridine-Based Ruthenium Nitrosyl Complexes in Dimethylsulfoxide. Molecules. 2020; 25(9):2205. https://doi.org/10.3390/molecules25092205
Chicago/Turabian StyleMarchenko, Nataliia, Pascal G. Lacroix, Valerii Bukhanko, Marine Tassé, Carine Duhayon, Martial Boggio-Pasqua, and Isabelle Malfant. 2020. "Multistep Photochemical Reactions of Polypyridine-Based Ruthenium Nitrosyl Complexes in Dimethylsulfoxide" Molecules 25, no. 9: 2205. https://doi.org/10.3390/molecules25092205