
Synthesis of Functionalized Diethyl(pyrrolidin-2-yl)phosphonate and Diethyl(5-oxopyrrolidin-2-yl)phosphonate


Bioorganic Chemistry Laboratory, Faculty of Pharmacy, Medical University of Lodz, 90-151 Lodz, Poland
*
Authors to whom correspondence should be addressed.
Molecules 2021, 26(11), 3160; https://doi.org/10.3390/molecules26113160
Submission received: 22 April 2021
/
Revised: 12 May 2021
/
Accepted: 22 May 2021
/
Published: 25 May 2021
(This article belongs to the Special Issue Design, Synthesis, and Analysis of Potential Drugs)
Abstract
:Short and efficient syntheses of functionalized (pyrrolidin-2-yl)phosphonate and (5-oxopyrrolidin-2-yl)phosphonate have been developed. The synthetic strategy involved the diastereospecific 1,3-dipolar cycloaddition of N-benzyl-C-(diethoxyphosphoryl)nitrone to cis-1,4-dihydroxybut-2-ene and dimethyl maleate, respectively. O,O-Diethyl 3-carbamoyl-4-hydroxy(5-oxopyrrolidin-2-yl)phosphonate was obtained from O,O-diethyl 2-benzyl-4,5-dimethoxycarbonyl(isoxazolidin-3-yl)phosphonate by hydrogenation and subsequent treatment with ammonia, whereas transformation of O,O-diethyl 2-benzyl-4,5-dihydroxymethyl(isoxazolidin-3-yl)phosphonate into O,O-diethyl 3-aminomethyl-4-hydroxy(pyrrolidin-2-yl)phosphonate was accomplished by mesylation followed by hydrogenolysis to undergo intramolecular cyclization and the introduction of amino group via ammonolysis. Stereochemistry of the isoxazolidine cycloadducts, as well as the final functionalized (pyrrolidin-2-yl)- and (5-oxopyrrolidin-2-yl)phosphonates were established based on conformational analyses using vicinal H–H, H–P, and C–P couplings and supported by the observed diagnostic NOESY correlation signals.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Pyrrolidine is an important fragment of many natural products [1,2,3,4] that can be exemplified by complex structures of swainsonin [5], monocotaline [6], lasiocarpine [7], and senecionine [8]. Pyrrolidine and pyrrolidinone moieties are also present in small biologically active molecules. For example, L-proline 1 and its hydoxylated analogue 2 (Figure 1) are the essential components of collagen, accounting for 30% of its composition and playing key roles in the stability of the collagen [9,10].
On the other hand, pyroglutamic acid 3 (Figure 1) is formed as a result of glutamate dehydration [11]. This is an intermediate substrate involved in the glutathione synthesis [12]. For decades, the basic structure of pyroglutamic acid has been modified and resulted in the syntheses of pharmacologically active compounds such as piracetam 4, oxiracetam 5, nebracetam 6, and its morpholine derivative 7 (Figure 1), which belong to “nootropic drugs” used in treatment of CNS diseases such as epilepsy and depression [13,14,15,16,17,18].
Hydroxylated pyrrolidine derivatives 8 and 9 (Figure 1) affected brain Glu levels and at the same time they did not exhibit brain and hepatic toxicity. The only disadvantage of these compounds is their inability to overcome the blood-brain interface [19]. Polyhydroxylated derivative of pyrrolidine 10, its enantiomer and 11 (Figure 1) have been obtained as inhibitors of α-glucosidases. Moreover, compound 11 demonstrates superior control of blood glucose levels [20]. On the other hand, antibiotic activity of several pyrrolidone-containing compounds has been observed, including compounds 12 and 13 [21] containing pyrrolidone ring incorporated in bicyclic system, as well as 14 (derivative of equisetin having additional methyl group at C3 in octahydronaphtalenyl moiety) acting on some multi-drug resistant bacteria [22] (Figure 1), and their resistance to β-lactamase have been recognized.
Over the decades, the importance of phosphonates in medicinal chemistry has been recognized [23,24,25]. Numerous phosphonates have been reported as analogues of biologically important compounds, including inhibitors of several enzymes, as well as antibacterial, antiviral, and fungicidal agents. Phosphonates have also been applied as mimetics of hydroxy- and amino acids in studies on their mode of action in biochemical transformations [26]. For this reason, phosphoproline 15 (Figure 2) [27] and its functionalized analogues received considerable attention and some of them have been successfully incorporated in biologically active systems such as analogues of dipeptides [28,29,30,31,32,33]. On the other hand, pyrrolidinone-containing phosphonate 16 (Figure 2), as a mixture of cyclic and non-cyclic form, has been recognized as inhibit NMCA β-lactamase. Moreover, a good activity of the cyclic form in the mixture of 16a and 16b tested against R39 D,D-peptidase has been proved [34].
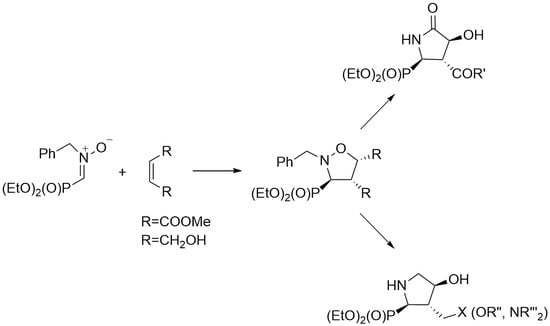
Several years ago, stereoisomers of analogues of proline as respective diethyl phosphonates 17 (Figure 2) hydroxylated at C4 in pyrrolidine ring have been synthesized in our research group [35]. Herein the syntheses of phosphonates 18 and 19 containing pyrrolidine framework functionalized at C3 and C4 are described (Scheme 1). Since N-substituted C-phosphorylated nitrones [36] have been successfully applied in the synthesis of various (isoxazolidin-3-yl)phosphonates [36,37] we found them suitable also for the preparation of isoxazolidines 20 and 21, which could be then transformed into the designed compounds 18 and 19 or their functionalized analogues. While isoxazolidine cycloadducts obtained from allyl alcohol and C-phosphorylated nitrone have already been successfully transformed into compound 17 (Figure 2) having hydroxy group at C4 in pyrrolidine skeleton [35], the application of 1,4-dihydroxybut-2-ene in 1,3-dipolar cycloaddition would allow to synthesis pyrrolidine 19 functionalized in both C3 and C4 positions. On the other hand, rearrangement of isoxazolidine 20 to pyrrolidinone 18 would be possible following the strategy demonstrated for several examples of differently functionalized systems [38,39,40,41].
2. Results and Discussion
The nitrone 22 was synthesized and fully characterized previously [36]. Cycloaddition of nitrone 22 with dimethyl maleate was then performed and led to the formation of (isoxazolidin-3-yl)phosphonate 20 as a single diastereoisomer in 84% yield after chromatographic purification. Similarly, reaction of nitrone 22 with cis-1,4-dihydroxybut-2-ene gave diastereoisomeric cycloadduct 21 in 70% yield after column chromatography (Scheme 2). In both cases, formation of single diastereoisomeric product (20 or 21) was proved by the analyses of the 31P and 1H NMR spectra of the crude product.
On the other hand, when maleic anhydride was used in the 1,3-dipolar cycloaddition with nitrone 22 isoxazolidine 23 was obtained exclusively in good yield (Scheme 3).
Since cis-alkenes were used for cycloadditions (Scheme 2 and Scheme 3), the cis relationship between HC4 and HC5 protons in 20 and 21, as well as in 23 can be arbitrarily assigned. To establish a relative configuration of (isoxazolidin-3-yl)phosphonate 18 the detailed conformational analysis was performed based on HCCP [42], HCCP [43,44], and CCCP [45,46] vicinal constants extracted from the 1H and 13C-NMR spectra. The vicinal couplings (J(H-C3C4-H) = 8.0 Hz, J(H-C4C5-H) = 8.3 Hz, J(H-C4C3-P) = 16.0 Hz, J(P-CC-C5) = 8.0 Hz, and J(P-CC-CO) = 5.5 Hz) indicate the 3E conformation of isoxazolidine ring. In this conformation the pseudoequatorially located diethoxyphosphoryl group at C3 is in trans relationship to both COOMe groups at C4 and C5 positions (Figure 3). On the other hand, to gather evidences for the spatial orientation of substituents in relation to the isoxazolidine moiety in (isoxazolidin-3-yl)phosphonate 21, NOESY experiment was performed. NOE diagnostic signals between HC4 and CH2OP as well as between HC3 and C4-CH2OH protons were noticed (Figure 3), which fully support the trans relationship between the HC3 and HC4 protons.
Next, transformation of (isoxazolidin-3-yl)phosphonate 20 into (5-oxopyrrolidin-2-yl)phosphonate 24 was performed (Scheme 4). Hydrogenation of the N–O bond together with the removal of benzyl group in isoxazolidine 20 released the free amino group, which subsequently became involved in spontaneous intramolecular cyclization to γ-lactam to produce phosphonate 18 in good yield (75%). When hydrogenation was carried out at a pressure of 15 bar, reaction time was significantly shortened (24 h vs. 5 h); moreover, the application of this procedure allowed to isolate compound 16 in higher yield (94%). Since configurations of all stereogenic centers in isoxazolidine ring (namely at C3, C4, and C5) remain unchanged during this transformation, relative configuration of γ-lactam ring (at C2, C3, and C4, respectively) in 18 can be established unambiguously. 3-Methoxycarbonyl-(5-oxopyrrolidin-2-yl)phosphonate 18 was then successfully transformed into 3-carbamoyl derivative 24 via ammonolysis (56%).
In order to support the already established relative stereochemistry of (5-oxopyrrolidin-2-yl)phosphonates 18 and 24, conformational analyses were undertaken. Based on the vicinal couplings found in 1H and 13C-NMR spectra of 18 (J(H-C3C4-H) = 8.8 Hz, J(H-C2C3-H) = 8.8 Hz, J(H-C3C2-P) = 17.6 Hz, J(P-CC-C4) = 7.8 Hz and J(P-CN-CO) = 7.8 Hz) and 24 (J(H-C3C4-H) = 9.1 Hz, J(H-C2C3-H) = 9.0 Hz, J(H-C3C2-P) = 18.0 Hz, J(P-CC-C4) = 8.7 Hz and J(P-CN-CO) = 7.9 Hz), the preferred 3E conformation of oxopyrrolidine ring was established in both phosphonates 18 and 24. In this conformation all substituents at C2, C3, and C4, namely OH, COR, and P(O)(OEt)2 groups are located equatorially, consequently hydrogen atoms occupy axial positions (Figure 4). Moreover, when NOESY experiments were performed for both phosphonates 18 and 24, NOE diagnostic signals between HC4 and HC2 protons were noticed, which fully support their cis orientations.
On the other hand, (isoxazolidin-3-yl)phosphonate 21 was found to be a good substrate for the synthesis of functionalized phosphoproline derivative (Scheme 5). Phosphonate 21 was first mesylated to produce O,O-dimesyl derivative 25, which was then subjected to hydrogenolysis to accomplish cleavage of N–O bond followed by removal of benzyl group together with intramolecular cyclization to form pyrrolidine ring in compound 26 [35]. The resulted crude ammonium mesylate 26 was then neutralized with potassium carbonate in chloroform to give functionalized phosphoproline analogue 27 (38% yield in two steps). Again, when hydrogenation was carried out at 15 bar pressure, the reaction time required for transformation of (isoxazolidin-3-yl)phosphonate 25 into ammonium mesylate 26 was significantly shortened (22 h vs. 6 h), and after neutralization with potassium carbonate phosphoproline analogue 27 was obtained efficiently (70% yield in two steps). Finally, compound 27 was reacted with morpholine to give 28 in 33% yield. Alternatively, mesyl group in 27 was changed to amino function by treatment with sodium azide followed by hydrogenolysis in the presence of Boc2O to produce phosphonate 30 (Scheme 5).
3. Materials and Methods
3.1. General Information
1H-NMR spectra were taken in CDCl3, C6D6 or CD3OD on a Bruker Avance III (600 MHz, Bruker Instruments, Karlsruhe, Germany). For spectra recorded in CDCl3 and C6D6 TMS was used as an internal standard; chemical shifts δ are given in ppm with respect to TMS and coupling constants J in Hz. 13C-NMR and 31P-NMR spectra were recorded in a 1H-decoupled mode for CDCl3, C6D6 or CD3OD solutions on the Bruker Avance III (600 MHz) spectrometer at 151 and 243 MHz, respectively. IR spectral data were measured on a Bruker Alpha-T FT-IR spectrometer (Bruker Optik GmbH, Ettlingen, Germany). Melting points were determined on a Boetius apparatus and are uncorrected. Elemental analyses were performed by the Microanalytical Laboratory of the Faculty of Pharmacy (Medical University of Lodz) on a Perkin Elmer PE 2400 CHNS analyzer (Perkin-Elmer Corp., Norwalk, CT, USA), and their results were found to be in good agreement (±0.3%) with the calculated values. Experiments at 15 bar pressure were carried out in a Büchi pressure reactor (Büchi AG, Uster, Switzerland).
The following adsorbents were used: column chromatography, Merck silica gel 60 (70–230 mesh), analytical TLC, Merck TLC plastic sheets silica gel 60 F254. TLC plates were developed in chloroform-methanol solvent systems. Visualization of spots was affected with iodine vapors. All solvents were purified by methods described in the literature.
1H-, 13C- and 31P-NMR spectra of all new synthesized compounds are provided in Supplementary Materials.
3.2. General Procedure for the Synthesis of Isoxazolidines 20, 21 and 23
A solution of nitrone 22 (2.0 mmol) and alkene (2.2 mmol) in toluene (4 mL) were stirred at 60 °C for 24 to 96 h (until the disappearance of the nitrone). The reaction mixture was concentrated in vacuo. The crude product was purified by silica gel column.
Dimethyl 2-benzyl-3-(diethoxyphosphoryl)isoxazolidine-4,5-dicarboxylate (20). Compound 20 was prepared from nitrone 22 (2.00 mmol, 0.542 g) and dimethyl maleate (2.20 mmol, 0.276 mL) and purified by column chromatography on a silica gel column with hexane-ethyl acetate (3:2, v/v) and crystallization (chloroform-hexane). Yield: 84% (0.697 g) as a white solid; m.p. = 75–78 °C. IR (KBr, cm−1): νmax= 2989, 2953, 2908, 1765, 1738, 1442, 1315, 1269, 1221, 1171, 1057, 1026, 984, 743, 707. 1H NMR (600 MHz, CDCl3): δ = 7.45–7.42 (m, 2H, HAr), 7.34–7.31 (m, 2H, HAr), 7.29–7.27 (m, 1H, HAr), 4.70 (d, 3JH5–H4 = 8.3 Hz, 1H, HC5), 4.52 (d, 2JHa–Hb=14.0 Hz, 1H, HaHbCPh), 4.28–4.16 (m, 5H, 2 × CH2OP, HaHbCPh), 3.95 (ddd, 3JH4–P = 16.3 Hz, 3JH4–H5 = 8.3 Hz,3JH4–H3 = 8,0 Hz, 1H, HC4), 3.80 (dd, 3JH3–H4 = 8.0 Hz, 2JH3–P = 2.8 Hz, 1H, HC3), 3.73 (s, 3H, CH3O), 3.71 (s, 3H, CH3O), 1.35 (t, 3J = 7.0 Hz, 3H, CH3CH2OP), 1.31 (t, 3J= 7.0 Hz, 3H, CH3CH2OP). 13C NMR (151 MHz, CDCl3): δ = 169.29 (d, 3JPCCC = 5.5 Hz, C(O)C4), 168.88 (C(O)C5), 137.16, 129.10, 128.26, 127.41, 77.39 (d, 3JPCCC = 8.0 Hz, C5), 63.96 (d, 1JPC = 169.6 Hz, C3) 63.93 (d, 3J = 6.5 Hz, CH2N), 62.90 (d, 2J = 5.7 Hz, COP), 62.88 (d, 2J = 5.7 Hz, COP), 53.45 (CH3O), 52.76 (CH3O), 52.47 (C4), 16.54 (d, 3J = 5.8 Hz, CCOP), 16.35 (d, 3J = 5.7 Hz, CCOP). 31P NMR (243 MHz, CDCl3): δ = 19.14. Analysis Calculated for C18H26NO8P: C, 52.05; H, 6.31; N, 3.37; Found: C, 52.05; H, 6.31; N, 3.36.
Diethyl 2-benzyl-4,5-dihydroxy-isoxazolidinyl-3-phosphonate (21). Compound 21 was prepared from nitrone 22 (2.00 mmol, 0.542 g) and cis-2-butene-1,4-diol (2.20 mmol, 0.194 mL) and purified by column chromatography on a silica gel column with chloroform-methanol (50:1, v/v). Yield: 70% (0.502 g) as a yellowish oil. IR (film, cm−1): νmax= 3385, 3064, 3032, 1497, 1229, 1050, 1025, 973, 740, 573. 1H NMR (600 MHz, CDCl3): δ = 7.40–7.38 (m, 2H, HAr), 7.35–7.30 (m, 2H, HAr), 7.30–7.26 (m, 1H, HAr), 4.55 (d, 2JHA-HB = 14.5 Hz, 1H, HAHBCPh), 4.32–4.20 (m, 4H, 2 × CH2OP), 4.13 (ddd, 3JH5–H4 = 9.7 Hz, 3JH5–Ha = 5.9 Hz, 3JH5–Hb = 3.5 Hz, 1H, HC5), 3.89 (d, 2JHA–HB = 14.5 Hz, 1H, HAHBCPh), 3.86 (dd, 2JHa–Hb = 12.5 Hz, 3JHa–H5 = 5.9 Hz, 1H, HaHbC-C5), 3.76 (dd, 2JHa–Hb = 12.5 Hz, 3JHb–H5 = 3.5 Hz, 1H, HaHbC-C5), 3.78–3.74 (m, 2H, CH2-C4), 3.10–3.00 (m, 2H, HC4, HC3), 1.38 (t, 3J = 7.2 Hz, 3H, CH3CH2OP), 1.36 (t, 3J= 7.2 Hz, 3H, CH3CH2OP). 1H NMR (600 MHz, C6D6): δ = 7.54–7.51 (m, 2H, HAr), 7.24–7.21 (m, 2H, HAr), 7.14–7.10 (m, 1H, HAr), 4.54 (d, 2JHA–HB = 14.4 Hz, 1H, HAHBCPh), 4.28 (dt, 3JH5–H4 = 6.8 Hz, 3JH5–Ha = 6.8 Hz, 3JH5–Hb = 4.2 Hz, 1H, HC5), 4.08–3.94 (m, 4H, 2 × CH2OP), 3.89 (d, 2JHA–HB = 14.4 Hz, 1H, HAHBCPh), 3.89 (dd, 2JHa–Hb = 12.0 Hz, 3JHa–H5 = 6.8 Hz, 1H, HaHbC-C5), 3.87 (dd, 2JHa–Hb = 11.4 Hz, 3JHa–H4 = 7.9 Hz, 1H, HaHbC-C4), 3.79 (dd, 2JHa–Hb = 12.0 Hz, 3JHb–H5 = 4.2 Hz, 1H, HaHbC-C5), 3.76 (dd, 2JHa–Hb = 11.4 Hz, 3JHb–H4 = 4.0 Hz, 1H, HaHbC-C4), 3.20 (ddddd, 3JH4–P = 18.7 Hz, 3JH4–Ha = 7.9 Hz, 3JH4–H5 = 6.8 Hz, 3JH4–H3 = 6.8 Hz, 3JH4–Hb = 4.0 Hz, 1H, HC4), 3.07 (dd, 3JH3–H4 = 6.8 Hz, 2JH3–P = 1.1 Hz, 1H, HC3), 1.06 (t, 3J = 7.0 Hz, 3H, CH3CH2OP), 1.04 (t, 3J = 7.0 Hz, 3H, CH3CH2OP). 13C NMR (151 MHz, C6D6): δ = 137.94, 129.02, 128.13, 127.99, 127.11, 79.53 (d, 3JPCCC = 7.4 Hz, C5), 63.38 (d, 1JPC = 169.5 Hz, C3), 63.13 (d, 2J = 6.6 Hz, COP), 62.75 (d, 2J = 6.6 Hz, COP), 62.66 (d, 3J = 3.4 Hz, CH2N), 60.59 (d, 3J = 7.5 Hz, CH2C4), 59.33 (CH2C5), 49.72 (C4), 16.19 (d, 3J = 5.7 Hz, CCOP), 16.13 (d, 3J = 5.7 Hz, CCOP). 31P NMR (243 MHz, CDCl3): δ = 22.05 ppm. 31P NMR (243 MHz, C6D6): δ = 22.94. Analysis Calculated for C16H26NO6P: C, 53.48; H, 7.29; N, 3.90. Found: C, 53.25; H, 7.16; N, 4.20.
2-Benzyl-3-(diethoxyphosphoryl)isoxazolidine-4,5-dicarboxylic acid (23). Compound 23 was prepared from nitrone 22 (2.00 mmol, 0.542 g) and maleic anhydride (2.20 mmol, 0.146 mL) and purified by column chromatography on a silica gel column with chloroform-methanol (10:1, v/v). Yield: 74% (0.573 g) as a yellowish oil. IR (film, cm−1): νmax= 3040, 2984, 2925, 1738, 1494 1206, 1045, 1020. 1H NMR (600 MHz, CDCl3): δ = 7.40–7.38 (m, 2H, HAr), 7.32–7.28 (m, 2H, HAr), 7.25–7.20 (m, 1H, HAr), 6.50–6.10 (br s, 2H, 2 × OH), 4.76 (d, 3JH5–H4 = 8.3 Hz, 1H, HC5), 4.41 (d, 2JHa–Hb=14.0 Hz, 1H, HaHbCPh), 4.23 (d, 2JHa–Hb=14.0 Hz, 1H, HaHbCPh), 4.25–4.15 (m, 4H, 2 × CH2OP), 4.02 (ddd, 3JH4–P = 16.3 Hz, 3JH4–H5 = 8.3 Hz,3JH4–H3 = 8,0 Hz, 1H, HC4), 3.89 (dd, 3JH3–H4 = 8.0 Hz, 2JH3–P = 3.1 Hz, 1H, HC3), 1.33 (t, 3J = 7.1 Hz, 3H, CH3CH2OP), 1.31 (t, 3J= 7.1 Hz, 3H, CH3CH2OP). 13C NMR (151 MHz, CDCl3): δ = 172.48 (C=O), 171.97 (C=O), 137.11, 129.10, 128.28, 127.42, 77.78 (br s, C5), 64.29 (d, 2J = 6.6 Hz, COP), 64.11 (d, 1JPC = 170.8 Hz, C3), 64.00 (d, 2J = 6.4 Hz, COP), 62.74 (br s, CH2N), 53.43 (C4), 16.42 (d, 3J = 5.6 Hz, CCOP), 16.31 (d, 3J = 5.6 Hz, CCOP). 31P NMR (243 MHz, CDCl3): δ = 19.85 ppm. Analysis Calculated for C16H22NO8P: C, 49.62; H, 5.73; N, 3.62. Found: C, 49.90; H, 5.52; N, 3.86.
3.3. Preparation of γ-Lactam 18
Procedure A: A solution of isoxazolidine 20 (0.045 g, 0.108 mmol) in methanol (1 mL) was kept under an atmospheric pressure of hydrogen over 20% Pd(OH)2–C (1.4 mg) at room temperature for 24 h. The suspension was filtered through a layer of Celite. The solution was concentrated, and the residue was chromatographed on silica gel column with chloroform-methanol (100:1, v/v) to give pure γ-lactam 18 (0.024 g, 0.081 mmol, 75%).
Procedure B: A solution of isoxazolidine 20 (0.208 g, 0.50 mmol) in methanol (5 mL) was kept in a pressure reactor under 15 bar pressure of hydrogen over 20% Pd(OH)2-C (6.5 mg) at room temperature for 5 h. The suspension was filtered through a layer of Celite. The solution was concentrated, and the residue was chromatographed on a silica gel column with chloroform-methanol (100:1, v/v) to give pure γ-lactam 18 (0.138 g, 94%).
Methyl 2-(diethoxyphosphoryl)-4-hydroxy-5-oxopyrrolidine-3-carboxylate (18). White solid; m.p. = 120–124 °C. IR (KBr, cm−1): νmax= 3298, 3189, 3126, 2989, 2957, 2925, 2796, 1741, 1713, 1441, 1374, 1248, 1170, 1049, 1010, 862, 753. 1H NMR (600 MHz, CDCl3): δ = 7.15 (brs, 1H, NH), 4.54 (d, 3JH3–OH = 7.6 Hz, 1H, OH), 4.46 (dd, 3JH3–H4 = 8.8 Hz, 3JH3–OH = 7.6 Hz, 1H, HC4), 4.25–4.14 (m, 4H, 2 × CH2OP), 4.07 (dd, 3JH2–H3 = 8.8 Hz, 2JH2–P = 3.2 Hz, 1H, HC2), 3.79 (s, 3H, CH3O), 3.40 (dt, 3JH3–P = 17.6 Hz, 3JH4–H3 = 8.8 Hz, 3JH3–H2 = 8.8 Hz, 1H, HC3), 1.35 (t, 3J = 7.0 Hz, 3H, CH3CH2OP), 1.32 (t, 3J= 7.0 Hz, 3H, CH3CH2OP). 13C NMR (151 MHz, CDCl3): δ = 174.94 (d, 3JPCNC = 7.8 Hz, C5), 171.35 (d, 3JPCCC = 2.3 Hz, C(O)CH3), 72.89 (d, 3JPCCC = 7.8 Hz, C4), 63.77 (d, 2J = 7.1 Hz, COP), 63.66 (d, 2J = 7.1 Hz, COP), 52.82 (CH3O), 49.72 (d, 2JCCP = 4.3 Hz, C3), 48.65 (d, 1JCP = 167.1 Hz, CP), 16.41 (d, J = 6.0 Hz, CCOP), 16.36 (d, J = 6.0 Hz, CCOP). 31P NMR (243 MHz, CDCl3) δ = 20.63. Analysis Calculated for C10H18NO7P: C, 40.68; H, 6.15; N, 4.74. Found: C, 40.88; H, 6.23; N, 4.91.
3.4. Ammonolysis of 18
To a solution of γ-lactam 18 (0.030 g, 0.10 mmol) in methanol (0.5 mL), aqueous NH3, (25%, 0.4 mL) was added. The homogenous mixture was stirred at room temperature for 17 h. Solvents were removed in vacuo, and the residue was evaporated with anhydrous methanol (3 × 5 mL), chloroform (3 × 5 mL) and chromatographed on silica gel with chloroform-methanol (10:1, v/v) to give pure 24 (0.016 g, 60%).
2-(diethoxyphosphoryl)-4-hydroxy-5-oxopyrrolidine-3-carboxamide (24). White solid; m.p. = 133–135 °C. IR (KBr, cm−1): νmax= 3300, 3199, 2986, 1711, 1679, 1231, 1045, 1020. 1H NMR (600 MHz, CD3OD): δ = 4.40 (d, 3JH3-H4 = 9.0 Hz, 1H, HC4), 4.25–4.20 (m, 4H, 2 × CH2OP), 4.08 (dd, 3JH2–H3 = 9.0 Hz, 2JH2–P = 2.0 Hz, 1H, HC2), 3.18 (dt, 3JH3–P = 18.0 Hz, 3JH4–H3 = 9.0 Hz, 3JH3–H2 = 9.0 Hz, 1H, HC3), 1.38 (t, 3J = 7.0 Hz, 6H, 2 × CH3CH2OP). 13C NMR (151 MHz, CD3OD): δ = 177.19 (d, 3JPCNC = 7.9 Hz, C5), 174.65 (C(O)NH2), 74.11 (d, 3JPCCC = 8.7 Hz, C4), 64.91 (d, 2J = 6.7 Hz, COP), 64.82 (d, 2J = 6.7 Hz, COP), 51.83 (d, 2JCCP = 4.3 Hz, C3), 49.51 (d, 1JCP = 167.4 Hz, CP), 16.74 (d, J = 5.6 Hz, CCOP), 16.67 (d, J = 5.4 Hz, CCOP). 31P NMR (243 MHz, CD3OD) δ = 21.25. Analysis Calculated for C9H17N2O6P: C, 38.58; H, 6.12; N, 10.00. Found: C, 38.29; H, 6.33; N, 9.92.
3.5. Mesylation of (Isoxazolidin-3-yl)Phosphonate 21
To a solution of isoxazolidine 21 (0.188 g, 0.523 mmol) in methylene chloride (6 mL) triethylamine (1.569 mmol, 0.219 mL) and mesyl chloride (1.569 mmol, 0.122 mL) were added at 0°C. The reaction mixture was stirred at this temperature for 2 h. The residue was washed with water (3 × 3 mL) and dried over MgSO4. The solution was concentrated, and the residue was chromatographed on silica gel column with chloroform to give pure dimesylate 25 (0.250 g, 96%).
Diethyl 2-benzyl-4,5-(dimesyloxymethyl)-3-phosphonate (25). Colourless oil. IR (film, cm−1): νmax= 3030, 2986, 2934, 1358, 1243, 1177, 1051, 1023, 964, 812, 754, 628. 1H NMR (600 MHz, CDCl3): δ = 7.39–7.33 (m, 4H, HAr), 7.31–7.28 (m, 1H, HAr), 4.59 (d, 2JHa–Hb= 14.3 Hz, 1H, HAHBCPh), 4.46 (dd, 2JHa–Hb = 11.6 Hz, 3JHa–H4 = 3.9 Hz, 1H, HaHbC-C4), 4.40 (dd, 2JHa–Hb = 11.6 Hz, 3JHa–H4 = 6.8 Hz, 1H, HaHbC-C4), 4.33–4.21 (m, 7H, 2 × CH2OP, HC5, HaHbC-C5), 3.88 (d, 2JHA–HB=14.3 Hz, 1H, HAHBCPh), 3.27 (ddddd, 3JH4–P = 17.3 Hz, 3JH4–Ha = 6.8 Hz, 3JH4–H5 = 8.9 Hz, 3JH4–H3 = 6.6 Hz, 3JH4–Hb = 3.9 Hz, 1H, HC4), 3.04 (s, 3H, CH3), 2.97 (dd, 3JH3–H4 = 6.6 Hz, 2JH3–P = 3.1 Hz, 1H, HC3), 2.89 (s, 3H, CH3), 1.41 (t, 3J = 7.1 Hz, 3H, CH3CH2OP), 1.39 (t, 3J= 7.1 Hz, 3H, CH3CH2OP). 13C NMR (151 MHz, CDCl3): δ = 136.22, 129.30, 128.29, 127.65, 75.93 (d, 3JPCCC = 7.6 Hz, C5), 66.55 (CH2-C4), (65.86 (d, 3J = 8.0 Hz, CH2-C4), 64.01 (d, 2J = 6.6 Hz, COP), 63.40 (d, 1JPC = 170.7 Hz, C3), 62.95 (d, 2J = 6.6 Hz, COP), 62.36 (d, 3J = 3.2 Hz, CH2N), 46.63 (d, 2J = 2.0 Hz, C4), 37.65 (CH3S), 37.57 (CH3S), 16.64 (d, 3J = 5.5 Hz, CCOP), 16.48 (d, 3J = 5.5 Hz, CCOP). 31P NMR (243 MHz, CDCl3): δ = 19.73. Analysis Calculated for C18H30NO10PS2: C, 41.94; H, 5.87; N, 2.72. Found C, 42.05; H, 5.93; N, 3.01.
3.6. The Synthesis of (Pyrrolidin-2-yl)Phosphonate 24 from Dimesylate 25
Procedure A: A solution of dimesylate 25 (0.20 g, 0.39 mmol) in methanol (2.5 mL) was kept under atmospheric pressure of hydrogen over 20% Pd(OH)2-C (3.9 mg) at room temperature for 22 h. The reaction progress was controlled by TLC. The suspension was filtered through a layer of Celite. The solution was concentrated to give ammonium mesylate (26), which was then dissolved in chloroform (4 mL) and anhydrous potassium carbonate (0.108 g, 0.78 mmol) was added. The reaction mixture was stirred at room temperature for 3 h. Then anhydrous MgSO4 was added, and the suspension was filtered through a layer of Celite. The solution was concentrated, and the residue was chromatographed on silica gel column with chloroform-methanol (100:1, v/v) to give pure 27 (0.049 g, 38%).
Procedure B: A solution of dimesylate 25 (0.236 g, 0.46 mmol) in methanol (5 mL) was kept in a pressure reactor under 15 bar pressure of hydrogen over 20% Pd(OH)2-C (10 mg) at room temperature for 6 h. The suspension was filtered through a layer of Celite. The solution was concentrated to give ammonium mesylate (26), which was then dissolved in chloroform (10 mL) and anhydrous potassium carbonate (0.127 g, 0.92 mmol) was added. The reaction mixture was stirred at room temperature for 3 h. Then anhydrous MgSO4 was added, and the suspension was filtered through a layer of Celite. The solution was concentrated, and the residue was chromatographed on silica gel column with chloroform-methanol (100:1, v/v) to give pure 27 (0.106 g, 70%).
Diethyl 4-hydroxy-3-mesyloxymethyl(pyrrolidin-2-yl)-phosphonate (27). Colorless oil. IR (film, cm−1): νmax= 3386, 2986, 2875, 1644, 1352, 1222, 1174, 1025, 970, 818. 1H NMR (600 MHz, CDCl3): δ = 4.31 (dd, J = 10.2 Hz, J = 5.6 Hz, 1H, HCH-C3), 4.25–4.15 (m, 6H, HC4, HCH-C3, 2 × CH2OP), 3.26 (dd, 2JH2–P = 7.9 Hz, 3JH2–H3 = 5.8 Hz, 1H, HC2), 3.17 (dd, 2JHa–Hb = 11.5 Hz, 3JH5–H = 5.3 Hz, 1H, HHC5), 3.10 (dd, 2JHa–Hb = 11.4 Hz, 3JH5–H = 3.0 Hz, 1H, HHC5), 3.06 (s, 3H, CH3S), 2.65 (ddddd, 3JH3–P = 18.5 Hz, 3JH3–H2 = 5.8 Hz, 3JH3–H4 = 5.8 Hz, 3JH3–H = 5.6 Hz, 3JH3–H = 2.7 Hz, 1H, HC3), 1.36 (t, 3J = 7.1 Hz, 3H, CH3CH2OP), 1.35 (t, 3J = 7.1 Hz, 3H, CH3CH2OP). 1H NMR (600 MHz, CD3OD): δ = 4.45 (dd, J = 10.3 Hz, J = 4.3 Hz, 1H, CH2-C3), 4.35 (dd, J = 10.3 Hz, J = 5.2 Hz, 1H, CH2-C3), 4.27–4.20 (m, 4H, 2 × CH2OP), 4.18 (ddd, J = 5.9 Hz, J = 5.8 Hz, J = 5.7 Hz, 1H, HC4), 3.30 (dd, 2JH2–P = 8.9 Hz, 3JH2–H3 = 8.8 Hz, 1H, HC2), 3.15 (s, 3H, CH3S), 3.06 (dd, 2JHa–Hb = 11.5 Hz, 3JH5–H = 8.9 Hz, 1H, HHC5), 2.86 (ddd, 2JHa–Hb = 11.5 Hz, 3JH5–H = 5.7 Hz, 3JH5–CN–H = 1.1 Hz, 1H, HHC5), 2.47 (ddddd, 3JH3–P = 18.0 Hz, 3JH3–H2 = 8.8 Hz, 3JH3–H4 = 5.8 Hz, 3JH3–H = 5.2 Hz, 3JH3–H = 4.3 Hz, 1H, HC3), 1.39 (t, 3J = 7.1 Hz, 3H, CH3CH2OP), 1.38 (t, 3J = 7.1 Hz, 3H, CH3CH2OP). 13C NMR (151 MHz, CD3OD): δ = 74.65 (d, 3JPCCC = 7.9 Hz, C4), 69.44 (d, 3JPCCC = 2.6 Hz, COMs), 64.48 (d, J = 7.1 Hz, COP), 64.21 (d, J = 7.2 Hz, COP), 55.35 (d, 1JPC = 167.5 Hz, C2), 55.09 (d, 3JPCNC = 8.9 Hz, C5), 50.19 (C3), 37.13 (CH3S), 16.80 (d, J = 5.6 Hz, POCC), 16.80 (d, J = 5.7 Hz, POCC). 31P NMR (243 MHz, CDCl3): δ = 26.33. Analysis Calculated for C10H22NO7PS: C, 36.25; H, 6.69; N, 4.23. Found: C, 36.46; H, 6.96; N, 4.20.
3.7. Synthesis of (Pyrrolidin-2-yl)Phosphonate 28
(Pyrrolidin-2-yl)-phosphonate 27 (0.040 g, 0.12 mmol) with morpholine (1.5 mL) was kept at room temperature for 39 h. After that chloroform (15 mL) was added. The solution was washed with water (2 × 10 mL), dried over MgSO4 and concentrated. The residue was chromatographed on silica gel column with chloroform-methanol (100:1, 50:1, v/v) to give pure 28 (0.014, 33%).
Diethyl (4-hydroxy-3-(piperidin-1-ylmeylyl)pyrrolidin-2-yl)phosphonate (28). Colourless oil. IR (film, cm−1): νmax= 3442, 2960, 2930, 2859, 2815, 1646, 1446, 1222, 1049, 1027. 1H NMR (600 MHz, CDCl3): δ = 4.30–4.15 (m, 4H, 2 × CH2OP), 4.08–4.05 (br m, 1H, HC4), 3.75–3.70 (br m, 4H), 3.37 (dd, 2JH2–P = 5.4 Hz, 3JH2–H3 = 5.4 Hz, 1H, HC2), 3.27 (dd, 2JHa–Hb = 10.9 Hz, 3JH5–H = 5.6 Hz, 1H, HHC5), 3.03 (dd, 2JHa–Hb = 10.9 Hz, 3JH5–H = 4.5 Hz, 1H, HHC5), 2.60–2.45 (m, 7H, HC3, 3 × CH2-C3), 1.38 (t, 3J = 7.1 Hz, 3H, CH3CH2OP), 1.36 (t, 3J = 7.1 Hz, 3H, CH3CH2OP). 13C NMR (151 MHz, CDCl3): δ = 77.16 (d, 3JPCCC = 5.4 Hz, C4), 66.69 (CH2-O-CH2), 61.11 (d, 3JPCCC = 8.8 Hz, CH2N), 63.12 (d, J = 7.4 Hz, COP), 62.37 (d, J = 7.5 Hz, COP), 55.64 (d, 1JPC = 166.1 Hz, C2), 53.75 (2 × CH2N), 53.60 (d, 3JPCNC = 6.6 Hz, C5), 45.40 (C3), 16.62 (d, J = 5.5 Hz, POCC), 16.54 (d, J = 5.9 Hz, POCC). 31P NMR (243 MHz, CDCl3): δ = 28.04. Analysis Calculated for C13H27N2O5P: C, 48.44; H, 8.44; N, 8.69. Found: C, 48.27; H, 8.48; N, 8.93.
3.8. Synthesis of azide 29
To a solution of mesylate 27 (0.100 g, 0.30 mmol) in methanol (2 mL) sodium azide was added (0.060 g, 0.90 mmol). The reaction mixture was stirred at 60°C for 96 h. The solvent was removed and the reside was chromatographed on silica gel column with chloroform-methanol (100:1, 50:1, v/v) to give pure azide 29 (0.036 g, 43%).
Diethyl (3-(azidomethyl)-4-hydroxypyrrolidin-2-yl)phosphonate (29). Colourless oil. IR (film, cm−1): νmax= 3355, 2985, 2933, 2872, 2103, 1648, 1445, 1354, 1225, 1175, 1026, 970. 1H NMR (600 MHz, CDCl3): δ = 4.26–4.13 (m, 4H, 2 × CH2OP), 4.11 (ddd, 3JH4–H5 = 5.2 Hz, 3JH4–H5 = 2.5 Hz, 3JH4–H3 = 2.5 Hz, 1H, HC4), 3.45 (ddd, 2JH–H = 12.2 Hz, 2JH–H3 = 6.7 Hz, 4JH–P = 0.9 Hz, 1H, HCHN3), 3.40 (dd, 2JH–H = 12.2 Hz, 2JH–H3 = 6.7 Hz, 1H, HCHN3), 3.23 (dd, 2JH2–P = 7.2 Hz, 3JH2–H3 = 4.8 Hz, 1H, HC2), 3.17 (dd, 2JHa–Hb = 11.3 Hz, 3JH5–H4 = 5.2 Hz, 1H, HHC5), 3.07 (dd, 2JHa–Hb = 11.3 Hz, 3JH5–H4 = 2.5 Hz, 1H, HHC5), 2.50 (ddddd, 3JH3–P = 14.8 Hz, 3JH3–H = 6.7 Hz, 3JH3–H = 6.7 Hz, 3JH3–H2 = 4.8 Hz, 3JH3–H4 = 2.5 Hz, 1H, HC3), 1.36 (t, 3J = 7.1 Hz, 6H, 2 × CH3CH2OP). 13C NMR (151 MHz, CDCl3): δ = 75.36 (d, 3JPCCC = 5.5 Hz, C4), 63.36 (d, J = 6.9 Hz, COP), 62.74 (d, J = 7.0 Hz, COP), 55.94 (d, 1JPC = 164.1 Hz, C2), 54.87 (d, 3JPCNC = 8.6 Hz, C5), 52.48 (d, 3JPCCC = 10.5 Hz, CH2N3), 49.35 (C3), 16.57 (d, J = 5.5 Hz, POCC), 16.53 (d, J = 5.5 Hz, POCC). 31P NMR (243 MHz, CDCl3): δ = 27.42. Analysis Calculated for C9H19N4O4P: C, 38.85; H, 6.88; N, 20.14. Found: C, 39.03; H, 6.94; N, 20.11.
3.9. Synthesis of (Pyrrolidin-2-yl)Phosphonate 30
A solution of azide 29 (0.036 g, 0.13 mmol) Boc2O (0.125 g, 0.572 mmol) in ethanol (0.5 mL) was kept under atmospheric pressure of hydrogen over 20% Pd(OH)2-C (1.7 mg) at room temperature for 35 h. The suspension was filtered through a layer of Celite. The solution was concentrated, and the residue was chromatographed on silica gel column with chloroform-methanol (100:1, 50:1, v/v) to give pure 27 (0.050 g, 69%).
tert-butyl 3-{[(tert-butoxycarbonyl)amino]methyl}-2-(diethoxyphosphoryl)-4-hydroxypyrrolidine-1-carboxylate (30). Colorless oil. IR (film, cm−1): νmax= 3312, 2980, 2933, 1743, 1703, 1519, 1279, 1254, 1165, 1028. 1H NMR (600 MHz, CDCl3): δ = 5.40–5.00 (br s, 1H, OH), 4.80–4.72 (br m, 1H, HC4), 4.25–4.15 (br m, 5H, 2 × CH2OP and HCHN), 4.10–4.00 (br m, 1H, HCHN), 3.48–3.40 (br m, 1H, HCH5), 3.28–3.20 (br m, 2H, HC2 and HCHC5), 2.85–2.70 (m, 1H, HC3), 1.50 (s, 9H, (CH3)3C), 1.48 (s, 9H, (CH3)3C), 1.46 (br s, 9H, (CH3)3C), 1.35 (t, J = 7.0 Hz, 6H, 2 × CH3CH2OP). 13C NMR (151 MHz, CDCl3): δ = 165.12 (C=O), 153.75 (C=O), 152.98 (C=O), 82.86 (C(CH3)3), 80.78 (C(CH3)3), 79.37 (C(CH3)3), 76.01 (very broad s, C4, 60%) and 75.13 (very broad s, C4, 40%), 62.83 (d, J = 7.2 Hz, CH2OP), 62.53 (br d, J ~ 7 Hz, CH2OP), 55.76 (very broad d, J ~ 167 Hz, C2, 40%) and 54.75 (very broad d, J ~ 164 Hz, C2, 60%), 50.47 (very broad s, C5, 60%) and 50.11 (very broad s, C5, 40%), 47.00 (very broad s, CH2N, 40%) and 45.45 (very broad s, CH2N, 60%), 42.46 (C3, 60%) and 41.93 (C3, 40%), 28.37, 28.26, 27.71, 16.48 (d, J = 5.7 Hz, POCC), 16.43 (very broad s, POCC). 31P NMR (243 MHz, CDCl3): δ = 24.06. Analysis Calculated for C24H45N2O10P: C, 52.16; H, 8.21; N, 5.07. Found: C, 52.32; H, 8.14; N, 5.02.
4. Conclusions
The 1,3-dipolar cycloadditions of N-benzyl-C-(diethoxyphosphoryl)nitrone 22 with dimethyl maleate and cis-1,4-dihydroxybut-2-ene, as well as maleic anhydride proceeded diastereospecifically to give cycloadducts 20, 21 and 23, respectively. Isoxazolidine 20 was smoothly hydrogenated to substituted (5-oxopyrrolidin-2-yl)phosphonate 18 and subsequently transformed into derivative 24 by exchanging of COOMe at C3 into amido function. For transformation of isoxazolidine 21 into functionalized derivative of (pyrrolidin-2-yl)phosphonate 28, reaction sequence consisted of a standard mesylation of both hydroxy groups, a hydrogenolytic cleavage of the N–O bond, removal of benzyl group followed by spontaneous formation of the pyrrolidine ring by intramolecular SN2 reaction and finally exchanging the other mesyloxy group to amino function. Since 3-methoxycarbonyl-(5-oxopyrrolidin-2-yl)phosphonate 18 and 3-mesyloxymethyl(pyrrolidin-2-yl)phosphonate 27 contain reactive groups, i.e., COOMe at C3 in 18 and MsO at C3 in 27, studies on their further functionalization based on their reactions with other nucleophiles are underway in our laboratory. The presented methodology could also be adopted for the synthesis of other stereoisomeric isoxazolidines via application of respective trans-alkenes in 1,3-dipolar cycloaddition to C-phosphorylated nitron 22. Moreover, the syntheses elaborated herein pave the way for new enantiomerically pure functionalized phosphonate analogues of prolines, substituted glutamic acid by application of the N-chiral C-phosphorylated nitrones.
Supplementary Materials
Figure S1: 1H NMR Spectrum for 20 in CDCl3, Figure S2: 13C NMR Spectrum for 20 in CDCl3, Figure S3: 31P NMR Spectrum for 20 in CDCl3, Figure S4: 1H NMR Spectrum for 21 in CDCl3, Figure S5: 1H NMR Spectrum for 21 in C6D6, Figure S6: 13C NMR Spectrum for 21 in C6D6, Figure S7: 31P NMR Spectrum for 21 in CDCl3, Figure S8: 31P NMR Spectrum for 21 in C6D6, Figure S9: 1H NMR Spectrum for 23 in CDCl3, Figure S10: 13C NMR Spectrum for 23 in CDCl3, Figure S11: 31P NMR Spectrum for 23 in CDCl3, Figure S12: 1H NMR Spectrum for 18 in CDCl3, Figure S13: 13C NMR Spectrum for 18 in CDCl3, Figure S14: 31P NMR Spectrum for 18 in CDCl3, Figure S15: 1H NMR Spectrum for 24 in CD3OD, Figure S16: 13C NMR Spectrum for 24 in CD3OD, Figure S17: 31P NMR Spectrum for 24 in CD3OD, Figure S18: 1H NMR Spectrum for 25 in CDCl3, Figure S19: 13C NMR Spectrum for 25 in CDCl3, Figure S20: 31P NMR Spectrum for 25 in CDCl3, Figure S21: 1H NMR Spectrum for 27 in CDCl3, Figure S22: 1H NMR Spectrum for 27 in CD3OD, Figure S23: 13C NMR Spectrum for 27 in CD3OD, Figure S24: 31P NMR Spectrum for 27 in CDCl3, Figure S25: 1H NMR Spectrum for 28 in CDCl3, Figure S26: 13C NMR Spectrum for 28 in CDCl3, Figure S27: 31P NMR Spectrum for 28 in CDCl3, Figure S28: 1H NMR Spectrum for 29 in CDCl3, Figure S29: 13C NMR Spectrum for 29 in CDCl3, Figure S30: 31P NMR Spectrum for 29 in CDCl3, Figure S31: 1H NMR Spectrum for 30 in CDCl3, Figure S32: 1H NMR Spectrum for 30 in CDCl3, Figure S33: 31P NMR Spectrum for 30 in CDCl3.
Author Contributions
Conceptualization, I.E.G. and D.G.P.; methodology, I.R. and A.H.; synthesis, I.R. and A.H.; investigation, D.G.P., I.R., A.H. and I.E.G.; resources and project administration, D.G.P., I.R. and I.E.G.; writing—original draft preparation, I.E.G., I.R. and D.G.P.; writing—review and editing, I.E.G., I.R. and D.G.P.; supervision, I.E.G. and D.G.P.; funding acquisition, I.E.G. and D.G.P. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Medical University of Lodz (internal fund 503/3-014-01/503-31-001).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples are not available.
References
- Massiot, G.; Delaude, C. Pyrrolidine Alkaloids. In The Alkaloids: Chemistry and Pharmacology; Brossi, A., Ed.; Harcourt Brace Jovanovich: Bethesda, MD, USA, 1986; Volume 27, pp. 269–322. [Google Scholar]
- Ali, M.S.; Ahmad, F.; Ahmad, V.U.; Azhar, I.; Usmanghani, K. Unusual Chemical Constituents of Lotus garcinii (Fabaceae). Turk. J. Chem. 2001, 25, 107–112. [Google Scholar]
- Zhu, Y.; Wang, Y.; Gu, B.-B.; Yang, F.; Jiao, W.-H.; Hu, G.-H.; Yu, H.-B.; Han, B.-N.; Zhang, W.; Shen, Y.; et al. Antifungal bromopyrrole alkaloids from the South China Sea sponge Agelas sp. Tetrahedron 2016, 72, 2964–2971. [Google Scholar] [CrossRef]
- Walsh, C.T. Nature loves nitrogen heterocycles. Tetrahedron Lett. 2015, 56, 3075–3081. [Google Scholar] [CrossRef]
- Colegate, S.M.; Dorling, P.R.; Huxtable, C.R. A Spectroscopic Investigation of Swainsonine: An α-Mannosidase Inhibitor Isolated from Swainsona canescens. Aust. J. Chem. 1979, 32, 2257–2264. [Google Scholar] [CrossRef]
- Culvenor, C.C.J.; Smith, L.W. The Alkaloids of Crotalaria Spectabilis roth. Aust. J. Chem. 1957, 10, 474–479. [Google Scholar] [CrossRef]
- Hay, D.G.; Mackay, M.F.; Culvenor, C.C.J. The structure of Lasiocarpine: Pyrrolizidine Alkaloid. Acta Crysallogr. 1982, 38, 155–159. [Google Scholar] [CrossRef]
- Hamid, H.K.; Kadhim, E.J. Extraction, isolation and characterization of Pyrrolizidine Alkaloids present in Senecio vulgaris Linn grown in Iraq. J. Pharmacogn. Phytochem. 2016, 5, 28–37. [Google Scholar]
- Szpak, P. Fish bone chemistry and ultrastructure: Implications for taphonomy and stable isotope analysis. J. Archaeol. Sci. 2011, 38, 3358–3372. [Google Scholar] [CrossRef]
- Nelson, D.L.; Cox, M.M. Lehninge’s Principles of Biochemistry, 5th ed.; W. H. Freeman and Company: New York, NY, USA, 2008; pp. 124–128. [Google Scholar]
- Abraham, G.N.; Podell, D.N. Pyroglutamic acid. Non-metabolic formation, function in proteins and peptides, and characteristics of the enzymes effecting its removal. Mol. Cell. Biochem. 1981, 38, 181–190. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [Green Version]
- Malykh, A.G.; Sadaie, M.R. Piracetam and Piracetam–Like Drugs from Basic Science to Novel Clinical Applications to CNS Disorders. Drugs 2010, 70, 287–312. [Google Scholar] [CrossRef]
- Gouliaev, A.H.; Senning, A. Piracetam and other structurally related nootropics. Brain Res. Rev. 1994, 19, 180–222. [Google Scholar] [CrossRef]
- Winblad, B. Piracetam: A Review of Pharmacological Properties and Clinical Uses. CNS Drug Rev. 2005, 11, 169–182. [Google Scholar] [CrossRef]
- Shorvon, S. Pyrrolidone derivatives. Lancet 2001, 358, 1885–1892. [Google Scholar] [CrossRef]
- Weber, K.-H.; Walther, G.; Schneide, O.; Hinzen, D.; Kuhn, F.J.; Lehr, E. Preparation and formulation of piperazinyl and morpholinyl substituted 1-(benzyl or pyridylmethyl)-4-or-5-aminomethyl-pyrrolidin-2-ones and their nootropic use. U.S. Patent Application No. 4,767,759, 30 August 1988. [Google Scholar]
- Lima, E.C.; Domingos, J.L.O.; Dias, A.G.; Costa, P.R.R. First stereoselective synthesis and assignment of the absolute configuration of the nebracetam eutomer and derivatives. Tetrahedron Asymmetry 2008, 19, 1161–1165. [Google Scholar] [CrossRef]
- Cecioni, S.; Aouadi, K.; Guiard, J.; Parrot, S.; Strazielle, N.; Blondel, S.; Ghersi–Egea, J.-F.; Chapelle, C.; Denoroy, L.; Praly, J.-P. Novel routes to either racemic or enantiopure α-amino-(4-hydroxypyrrolidin-3-yl)acetic acid derivatives and biological evaluation of a new promising pharmacological scaffold. Eur. J. Med. Chem. 2015, 98, 237–249. [Google Scholar] [CrossRef]
- Jenkinson, S.F.; Best, D.; Saville, A.W.; Mui, J.; Martinez, R.F.; Nakagawa, S.; Kunimatsu, T.; Alonzi, D.S.; Butters, T.D.; Norez, C.; et al. C-Branched Iminosugars: Alpha-Glucosidase Inhibition by Enantiomers of isoDMDP, isoDGDP, and isoDAB–L–isoDMDP Compared to Miglitol and Miglustat. J. Org. Chem. 2013, 78, 7380–7397. [Google Scholar] [CrossRef]
- Aszodi, J.; Rowlands, D.A.; Mauvais, P.; Collette, P.; Bonnefoy, A.; Lampilas, M. Design and synthesis of bridged γ-lactams as analogues of β-lactam antibiotics. Bioorg. Med. Chem. Lett. 2004, 14, 2489–2492. [Google Scholar]
- Sugie, Y.; Inagaki, S.; Kato, Y.; Nishida, H.; Pang, C.-H.; Saito, T.; Sakemi, S.; Dib–Hajj, F.; Mueller, J.P.; Sutcliffe, J.; et al. CJ-21,058, a new SecA Inhibitor isolated from a fungus. J. Antibiot. 2002, 55, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Hilderbrand, R.L. The Role of Phosphonates in Living Systems; CRC Press: Boca Raton, FL, USA, 1983. [Google Scholar]
- Engel, R. Synthesis of Carbon-Phosphorus Bond; CRC Press: Boca Raton, FL, USA, 1988. [Google Scholar]
- Kukhar, V.P.; Hudson, H.P. (Eds.) Aminophosphonic and Aminophosphinic Acids. Chemistry and Biological Activity; Wiley: New York, NY, USA, 1999. [Google Scholar]
- Yamamoto, H.; Inokawa, S. Sugar Analogs Having Phosphorus in the Hemiacetal Ring. Adv. Carbohydr. Chem. Biochem. 1984, 42, 135–191. [Google Scholar]
- Denhel, A.; Lavielle, G. α-Aminophosphonates. II. Alkylation: Synthesis of α-aminophosphonic acids. Bull. Soc. Chim. Fr. 1978, 9, 95–96. [Google Scholar]
- Moonen, K.; Laureyn, I.; Stevens, C.V. Synthetic methods for azaheterocyclic phosphonates and their biological activity. Chem. Rev. 2004, 104, 6177–6215. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.A.; Wu, Y.; Xu, H.; Zhang, J. Asymmetric Synthesis of Cis-5-Substituted Pyrrolidine 2-Phosphonates Using Metal Carbenoid NH Insertion and δ-Amino β-Ketophosphonates. Org. Lett. 2004, 6, 4523–4526. [Google Scholar] [CrossRef] [PubMed]
- Renaud, P.; Seebach, D. Enantiomerenreine Pyrrolidin-Derivate aus trans-4-Hydroxy-L-prolin durch elektrochemische oxidative Decarboxylierung und Titantetrachlorid-vermittelte Umsetzung mit Nukleophilen. Helv. Chem. Acta 1986, 69, 1704–1710. [Google Scholar] [CrossRef]
- El Khalabi, R.; El Hallaoui, A.; Ouazzani, F.; Elachqar, A.; Atmani, A.; Roumestant, M.L.; Viallefont, P.; Martinez, J. Syntheis of Phosphonic Analogues of 4-Hydroxyproline and 5-Hydroxypipecolic Acid. Prep. Biochem. Biotechnol. 2000, 30, 295–304. [Google Scholar] [CrossRef]
- Boto, A.; Gallardo, J.A.; Hernández, R.; Saavedra, C.J. One-pot synthesis of α-amino phosphonates from α-amino acids and β-amino alcohols. Tetrahedron Lett. 2005, 46, 7807–7811. [Google Scholar] [CrossRef]
- Chevrier, C.; Le Nouen, D.; Defoin, A.; Tarnus, C. Synthesis of Amino-L-Lyxose Phosphonates as Fucosyl-Phosphate Mimics. Eur. J. Org. Chem. 2006, 2384–2392. [Google Scholar] [CrossRef]
- Beck, J.; Gharbi, S.; Herteg–Fernea, A.; Vercheval, L.; Bebrone, C.; Lassaux, P.; Zervosen, A.; Marchand–Brynaert, J. Aminophosphonic Acids and Aminobis(phosphonic acids) as Potential Inhibitors of Penicillin–Binding Proteins. Eur. J. Org. Chem. 2009, 1, 85–97. [Google Scholar] [CrossRef]
- Piotrowska, D.G.; Głowacka, I.E. Enantiomerically pure phosphonate analogues of cis- and trans-4-hydroxyprolines. Tetrahedron Asymmetry 2007, 18, 1351–1363. [Google Scholar] [CrossRef]
- Piotrowska, D.G. N-Substituted C-diethoxyphosphorylated nitrones as useful synthons for the synthesis of α-aminophosphonates. Tetrahedron Lett. 2006, 47, 5363–5366. [Google Scholar] [CrossRef]
- Piotrowska, D.G. Stereochemistry of substituted isoxazolidines derived from N-methyl C-diethyoxyphosphorylated nitrone. Tetrahedron 2006, 62, 12306–12317. [Google Scholar] [CrossRef]
- Merino, P.; Mates, J.A.; Revuelta, J.; Tejero, T.; Chiachio, U.; Romeo, G.; Iannazzo, D.; Romeo, R. Experimental and theoretical study of the 1,3-dipolar cycloaddition between D-glyceraldehyde nitrones and acrylates. Diastereoselective approach to 4-hydroxy pyroglutamic acid derivatives. Tetrahedron Asymmetry 2002, 13, 173–190. [Google Scholar] [CrossRef]
- Merion, P.; Pádár, P.; Delso, I.; Thirumalaikumar, M.; Tejero, T.; Kovács, L. Stereoselective synthesis of pyrrolidinyl glycines from nitrones: Complementarity of nucleophilic addition and 1,3-dipolar cycloaddition. Tetrahedron Lett. 2006, 47, 5013–5016. [Google Scholar] [CrossRef]
- Argyropoulos, N.G.; Gkiziss, P.; Coutouli-Argyropoulou, E. A convenient synthesis of new enantiomerically pure rihydroxypyrrolizidines using L-erythrose glycosylhydroxylamine as a masked acyclic chiral nitrone. Tetrahedron 2008, 64, 8752–8758. [Google Scholar] [CrossRef]
- Piotrowska, D.G.; Głowacka, I.E.; Schols, D.; Snoeck, R.; Andrei, G.; Gotkowska, J. Novel Isoxazolidine and γ-Lactam Analogues of Homonucleosides. Molecules 2019, 24, 4014. [Google Scholar] [CrossRef] [Green Version]
- Karplus, M. Vicinal Proton Coupling in Nuclear Magnetic Resonance. J. Am. Chem. Soc. 1963, 85, 2870–2871. [Google Scholar] [CrossRef]
- Benezra, C. NMR of phosphonates. VI. Variation of vicinal phosphorus-31-carbon-carbon-proton couplings with dihedral angle in phosphonates. J. Am. Chem. Soc. 1973, 95, 6890–6894. [Google Scholar] [CrossRef]
- Neeser, J.-R.; Tronchet, J.M.J.; Charollais, E.J. Structural analysis of 3-C-phosphonates, -phosphinates, and -phosphine oxides of branched-chain sugars. Can. J. Chem. 1983, 61, 2112–2120. [Google Scholar] [CrossRef]
- Adiwidjaja, G.; Meyer, B.; Thiem, J. Darstellung and Kristallstruktur von endo-2-Dimethylphosphono-exo-2-hydroxy-(–)-camphan zur Bestimmung von 3J(CCCP)-Vicinalkopplungen. Z. Naturforsch. 1979, 34, 1547–1551. [Google Scholar] [CrossRef]
- Buchanan, G.W.; Bourque, K.; Seeley, A. 13C- and 31P-NMR spectra of 1-diethylphosphono-1-hydroxycycloalkanes. Magn. Res. Chem. 1986, 24, 360–367. [Google Scholar] [CrossRef]
Figure 1.
Examples of pyrrolidine- and pyrrolidone-containing biologically active compounds.

Figure 2.
Phosphoproline 15 and its functionalized analogues 16 and 17.

Scheme 1.
Retrosynthesis of 18 and 19.

Scheme 2.
Reaction and conditions: a. toluene, 60 °C (reaction time: 24 h for the synthesis of 20 and 96 h for the synthesis of 21).
Scheme 2.
Reaction and conditions: a. toluene, 60 °C (reaction time: 24 h for the synthesis of 20 and 96 h for the synthesis of 21).

Scheme 3.
Reaction and conditions: a. toluene, 24 h, 60 °C, 74%.

Figure 3.
The preferred conformation of 20 and the most important NOESY correlation for phosphonate 21 (blue arrows).
Figure 3.
The preferred conformation of 20 and the most important NOESY correlation for phosphonate 21 (blue arrows).

Scheme 4.
Synthesis of γ-lactam 24. Reaction and conditions: a. H2, Pd(OH)2–C, MeOH, rt, 1.01 bar, 24 h, 75% or H2, Pd(OH)2–C, MeOH, rt, 15 bar, 5 h, 94%; b. aq. NH3, MeOH, 17 h, 56%.
Scheme 4.
Synthesis of γ-lactam 24. Reaction and conditions: a. H2, Pd(OH)2–C, MeOH, rt, 1.01 bar, 24 h, 75% or H2, Pd(OH)2–C, MeOH, rt, 15 bar, 5 h, 94%; b. aq. NH3, MeOH, 17 h, 56%.

Figure 4.
The preferred conformations of 18 and 24 with the most important NOESY correlations (blue arrow).
Figure 4.
The preferred conformations of 18 and 24 with the most important NOESY correlations (blue arrow).

Scheme 5.
Synthesis of functionalized proline analogues 28 and 30. Reaction and conditions: a. MsCl, Et3N, CH2Cl2, 0 °C, 2h, 96%; b. H2, Pd(OH)2–C, MeOH, 1.01 bar, 22 h or H2, Pd(OH)2–C, MeOH, 15 bar, 6 h; c. K2CO3, CHCl3, rt, 3 h, (38% and 70% in two steps: b and c); d. morpholine, neat, rt, 39 h, 33%; e. NaN3, MeOH, 60 °C, 96 h, 43%; f. H2, Pd(OH)2–C, Boc2O, EtOH, rt, 35 h, 69%.
Scheme 5.
Synthesis of functionalized proline analogues 28 and 30. Reaction and conditions: a. MsCl, Et3N, CH2Cl2, 0 °C, 2h, 96%; b. H2, Pd(OH)2–C, MeOH, 1.01 bar, 22 h or H2, Pd(OH)2–C, MeOH, 15 bar, 6 h; c. K2CO3, CHCl3, rt, 3 h, (38% and 70% in two steps: b and c); d. morpholine, neat, rt, 39 h, 33%; e. NaN3, MeOH, 60 °C, 96 h, 43%; f. H2, Pd(OH)2–C, Boc2O, EtOH, rt, 35 h, 69%.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Głowacka, I.E.; Hartwich, A.; Rozpara, I.; Piotrowska, D.G. Synthesis of Functionalized Diethyl(pyrrolidin-2-yl)phosphonate and Diethyl(5-oxopyrrolidin-2-yl)phosphonate. Molecules 2021, 26, 3160. https://doi.org/10.3390/molecules26113160
AMA Style
Głowacka IE, Hartwich A, Rozpara I, Piotrowska DG. Synthesis of Functionalized Diethyl(pyrrolidin-2-yl)phosphonate and Diethyl(5-oxopyrrolidin-2-yl)phosphonate. Molecules. 2021; 26(11):3160. https://doi.org/10.3390/molecules26113160
Chicago/Turabian StyleGłowacka, Iwona E., Anna Hartwich, Iwona Rozpara, and Dorota G. Piotrowska. 2021. "Synthesis of Functionalized Diethyl(pyrrolidin-2-yl)phosphonate and Diethyl(5-oxopyrrolidin-2-yl)phosphonate" Molecules 26, no. 11: 3160. https://doi.org/10.3390/molecules26113160