
The Nrf2 Pathway in Ischemic Stroke: A Review
by
 , and
, and
Marcelo Farina
1,*, 
Leonardo Eugênio Vieira
1,
Brigitta Buttari
2 ,
,
Elisabetta Profumo
2 and
Luciano Saso
3,*
1
Department of Biochemistry, Federal University of Santa Catarina, 88040-900 Florianópolis, Brazil
2
Department of Cardiovascular, Endocrine-Metabolic Diseases, and Aging, Italian National Institute of Health, 00161 Rome, Italy
3
Department of Physiology and Pharmacology “Vittorio Erspamer”, Sapienza University of Rome, 00185 Rome, Italy
*
Authors to whom correspondence should be addressed.
Molecules 2021, 26(16), 5001; https://doi.org/10.3390/molecules26165001
Submission received: 23 July 2021
/
Revised: 13 August 2021
/
Accepted: 14 August 2021
/
Published: 18 August 2021
(This article belongs to the Special Issue Molecules Medicinal Chemistry Reviews)
Abstract
:Ischemic stroke, characterized by the sudden loss of blood flow in specific area(s) of the brain, is the leading cause of permanent disability and is among the leading causes of death worldwide. The only approved pharmacological treatment for acute ischemic stroke (intravenous thrombolysis with recombinant tissue plasminogen activator) has significant clinical limitations and does not consider the complex set of events taking place after the onset of ischemic stroke (ischemic cascade), which is characterized by significant pro-oxidative events. The transcription factor Nuclear factor erythroid 2-related factor 2 (Nrf2), which regulates the expression of a great number of antioxidant and/or defense proteins, has been pointed as a potential pharmacological target involved in the mitigation of deleterious oxidative events taking place at the ischemic cascade. This review summarizes studies concerning the protective role of Nrf2 in experimental models of ischemic stroke, emphasizing molecular events resulting from ischemic stroke that are, in parallel, modulated by Nrf2. Considering the acute nature of ischemic stroke, we discuss the challenges in using a putative pharmacological strategy (Nrf2 activator) that relies upon transcription, translation and metabolically active cells in treating ischemic stroke patients.
1. Introduction
Ischemic stroke, the most common one, accounts for approximately 87% of all stroke cases. It is characterized by the sudden loss of blood flow caused by thrombosis or embolism that occludes cerebral vessel(s) supplying specific area(s) of the brain [1]. Ischemic stroke is the leading cause of permanent disability and is among the leading causes of death worldwide. Globally, one in six people will have a stroke in their lifetime and around 14 million have a stroke each year. Even though stroke was initially classified as a condition affecting blood vessels, it was recently reclassified and is currently considered a neurological disorder; this led to improvements in acute healthcare and acquisition of research funding for stroke [2].
There is a great number of modifiable and non-modifiable risk factors for ischemic stroke. Among the non-modifiable risk factors, the most important ones are (i) age (the incidence of stroke increases with age [3]), (ii) sex (incidence is greater at younger ages in women, but increases with older age in men [4]), (iii) ethnicity (Hispanic and black populations are at higher risk of stroke than white populations [5]) and (iv) genetics (parental stroke by 65 years of age was associated with a 3-fold increase in risk of offspring stroke [6]. Among the modifiable risk factors, the most important ones are (i) hypertension (high blood pressure is one of the predominant risk factors and a 10 mm Hg increase in systolic blood pressure has been associated with a 38% increased stroke risk [7]), (ii) hyperglycemia (impaired glucose tolerance is an independent risk factor for future stroke [8]), (iii) atrial fibrillation (contributes to 15% of all strokes [9]), (iv) hyperlipidemia (total plasma cholesterol is positively associated with risk of stroke, while plasmatic levels of high-density lipoprotein are negatively associated with risk of stroke [10]), (v) smoking (tobacco smoking is directly linked to increased risk of stroke [11]) and (vi) insufficient physical inactivity and poor diet (lack of exercise increases the chances of a stroke episode and poor diet influences the risk of stroke, contributing to hypertension, hyperlipidemia and diabetes [12,13]. In addition to the aforementioned factors, socioeconomic variation also affects the occurrence and/or outcomes of stroke; for broader information regarding risk factors for ischemic stroke, see reference [14].
Regarding treatments, intravenous thrombolysis with recombinant tissue plasminogen activator is the only approved pharmacological treatment for acute ischemic stroke. It has significant beneficial effects in acute ischemic stroke when administered between 3 and 4.5 h after the onset of symptoms [15], although an extension of this time interval is currently under debate [16]. This thrombolytic treatment aims at stimulating fibrinolysis to allow for clot removal, but does not consider the complex set of events taking place after the onset of ischemic stroke, called ischemic cascade (discussed below).
A great number of experimental studies has been performed in order to discover drugs able to mitigate the neurodegeneration following ischemic episodes. Of particular importance, oxidative stress has been highlighted as a potential pharmacological target in ischemic stroke, which is in line with the increased generation and decreased detoxification of oxidant molecules leading to stroke-mediated neurodegeneration [17]. Among the potential oxidative stress-related molecular targets, the transcription factor Nrf2 (Nuclear factor erythroid 2-related factor 2), which regulates the expression of a great number of antioxidant and/or defense proteins (discussed below), has been pointed as a potential pharmacological target involved in the mitigation of deleterious oxidative events taking place at the ischemic cascade. In this review, we summarized studies concerning the protective role of Nrf2 in experimental models of ischemic stroke, emphasizing molecular events resulting from ischemic stroke that are, in parallel, modulated by Nrf2. We also reviewed the available experimental literature concerning the effects of Nrf2 activators in ischemic stroke models, discussing the potential pharmacological use of Nrf2 activators in ischemic stroke patients.
2. The Ischemic Cascade and Oxidative Consequences
Ischemic stroke is characterized by the interruption or sudden restriction of cerebral blood flow in specific area(s) of the brain. Considering that brain metabolism is greatly dependent upon blood-derived glucose and oxygen, which allow for the proper functioning of glycolysis, tricarboxylic acid cycle and mitochondrial electron transport chain [18], ischemic stroke leads to major changes in cellular bioenergetics. The lack of proper adenosine triphosphate (ATP) levels represents a primary metabolic change resulting from ischemia; it is a main trigger for the initiation of a series of molecular deleterious events known as ischemic cascade, which is particularly detrimental to neurons due to its highly oxidative and glucose-dependent metabolism.
Because of the crucial role of ATP in maintaining cellular (especially, neuronal) ionic homeostasis, a significant ionic imbalance occurs few minutes after ischemia, with the abnormal influx of Na+ and efflux of K+, contributing to extensive depolarization and water transport into cells [19], which leads to cytotoxic edema. The low ATP synthesis, followed by Na+/K+ imbalance (due to Na+/K+ ATPase), also decreases the uptake of glutamate, the main excitatory neurotransmitter. This phenomenon is related to the fact that the action potential induced by glutamate on postsynaptic receptors is terminated by its clearance from the synaptic cleft by transporters located in neurons and, remarkably, in glial cells. Several glutamate transporters are dependent on extracellular Na+, thus on the activity of Na+/K+ ATPase. The impaired removal of glutamate, or even its release through the reverse operation of their transporters, represents an important event in the ischemic cascade, leading to neuronal toxicity due to excessive excitatory neurotransmission (excitotoxicity) [20]. In fact, increased glutamate levels in the synaptic cleft cause overstimulation of neuronal post-synaptic glutamate receptors, stimulating sodium and calcium influx. This may induce cytoplasmic calcium overload and activation of diverse enzymes, such as phospholipases, proteases and nucleases, which drive the breakdown of phospholipids, proteins and nucleases [20]. Of note, the depolarization of adjacent neurons produces a further calcium influx and additional glutamate release, leading to local amplification of the ischemic damage [21].
Cytoplasmic calcium overload is also detrimental to the mitochondrial function, being linked to the mitochondrial production of oxidant molecules [22]. Some mechanisms related to the pro-oxidative role of calcium in the ischemic cascade include a calcium-stimulated increase in metabolic rate, nitric oxide production and cardiolipin peroxidation. Thus, the excitatory overstimulation resulting from low ATP levels may culminate in oxidative damage, which represents a critical event leading to neuronal damage in this hypoxic phase of stroke [23]. In addition, it is important to mention that the synthesis of glutathione (GSH), a main low-molecular weight intracellular antioxidant, is dependent upon ATP. Consequently, decreased GSH synthesis subsequent to low ATP levels may also contribute to the redox imbalance and oxidative damage resulting from ischemia.
Even though fast reoxygenation, via reperfusion, is a desired step required to mitigate the metabolic stress that takes place in ischemic stroke, reoxygenation may also contribute to the generation of reactive oxidants [24], thus exacerbating the ischemia-reperfusion oxidative injury. Approximately 4 decades ago, considering the low levels of molecular oxygen in ischemic tissues, there was no reason to suppose that ischemia involved elevated production of oxygen-derived reactive species. However, evidence showed that a significant part of the damage resulting from ischemia may be more accurately called reperfusion injury or post-ischemic injury. Indeed, much of the injury was shown to occur not during the period of hypoxia but rather during the period when molecular oxygen is reintroduced to the tissue [25]. In this scenario, experimental evidence showing the protective effects of superoxide dismutase (SOD) indicated that superoxide was a critical molecule in ischemic (or post-ischemic) events [26,27]. Concerning the mechanisms mediating the generation of oxygen radicals in the ischemic cascade, a main source of superoxide in post-ischemic tissues is the enzyme xanthine oxidase, which is usually synthesized as a dehydrogenase (type D) and able to catalyze the conversion of xanthine into uric acid with no production of superoxide. However, under certain conditions (pro-oxidative environment and increased intracellular calcium levels), xanthine dehydrogenase is converted into xanthine oxidase in vivo in ischemic tissues, catalyzing the conversion of xanthine into uric acid with production of superoxide [28]. In addition, the ischemia-related depletion of ATP is paralleled by an increase in the levels of AMP, adenosine, inosine and hypoxanthine and xanthine. These two last products of purine catabolism represent the substrate for xanthine oxidase. The “new enzyme” with oxidase activity and the availability of molecular oxygen during reperfusion represent two important factors that contribute to the oxidative stress taking place during the post-ischemic (reperfusion/reoxygenation) period [25]. Currently, since most of the studies concerning ischemia-reoxygenation do not dissociate ischemic- from post-ischemic-related injuries, the term ischemic-reperfusion injury (IRI) has been used to correctly refer to the damage resulting from ischemia following reoxygenation and/or reperfusion.
During the last decades, evidence has highlighted additional molecules that stimulate oxidative damage toward biomolecules in IRI. One of these molecules is phospholipase A2 (PLA2), whose activation represents a critical metabolic event in ischemic stroke, leading to the hydrolysis of membrane phospholipids and consequent release of lysophopholipids and free fatty acids (FFAs), including arachidonic acid, a metabolic precursor for pro-inflammatory eicosanoids [29]. PLA2 (mitochondrial secretory isoform) also catalyzes the hydrolysis of cardiolipin, leading to disruption of the mitochondrial respiratory chain and increased production of reactive oxygen species (ROS) [30]. In addition, the oxidative metabolism of arachidonic acid also generates ROS. Both events contribute to the occurrence of lipid peroxidation, whose end products (i.e., malondialdehyde and 4-hydroxynonenal) covalently bind to proteins/nucleic acids, altering their function and causing cellular damage [29]. FFAs released in PLA2-catalyzed reactions can accumulate following ischemic stroke, undergoing oxidative metabolism by non-enzymatic and enzymatic processes catalyzed mainly by cyclooxygenases (COXs) and lipoxygenases (LOXs), resulting in the formation of lipid oxoderivatives [31], which modulate inflammatory and pro-oxidative processes. In line with this, experimental evidence indicates that both COXs [32,33] and LOXs [34,35] represent potential pharmacological targets for stroke therapy. This experimental (nonclinical) evidence has provided new insights into the regulation of inflammatory and pro-oxidative events in the ischemic brain. However, the potential translation of such experimental data to clinic scenarios remains a matter of debate [36].
In addition to COXs and LOXs, nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs), a family of enzymes that catalyze the production of superoxide by transferring one electron from NADPH to molecular oxygen, have also been reported to exert detrimental effects on ischemic brain tissue. Evidence shows that NOX-knockout mice are resistant to damage due to experimental stroke and the infarct size and blood–brain barrier breakdown are enhanced in mice with pericyte-specific overexpression of NOX4 [37]. A recent experimental study has elegantly identified type 5 NADPH oxidase (NOX5) as a major player of IRI. Using in vitro organotypic cultures, the authors found that reoxygenation or calcium overload increased brain ROS levels in a NOX5-dependent manner. Based on in vivo approaches, the authors also showed that postischemic ROS formation, infarct volume and functional outcomes were worsened in NOX5-KI mice [38].
Based on the aforementioned evidence, it becomes clear that several pro-oxidative events display central roles for the occurrence of IRI. Some of these events include (i) calcium-mediated oxidative events (through increased metabolic rate, nitric oxide production and cardiolipin peroxidation), (ii) impaired GSH synthesis, (iii) increased superoxide generation (in dysfunctional mitochondria, as well as in xanthine oxidase and NOX-catalyzed reactions) and (iv) formation of lipid oxoderivatives (including in LOX- and COX-catalyzed reactions). The knowledge concerning the involvement of oxidative events in the ischemic cascade and consequently in IRI, was mostly derived from experimental studies (in vitro approaches and in vivo animal studies). Of note, such experimental studies have also elucidated several ischemia/reoxygenation-mediated biochemical and histological oxidative changes toward biomolecules, such as lipid peroxidation [39] and nucleic acid oxidation [40], as well as protein carbonylation [41] and nitrosylation [42]. Of note, increases in the levels of some of these oxidative-stress-related biomarkers have also been observed in patients with ischemic stroke [43], highlighting the significance of oxidative events in human IRI. Such findings suggest that drugs able to mitigate such oxidative events represent potential pharmacological strategies to treat ischemic stroke patients. Among the variety of molecules and/or targets that might be useful to mitigate IRI-mediated oxidative stress, there is the transcription factor Nrf2, which is the main topic of this review (Section 3).
3. Nrf2 Signaling Pathway and Ischemic Stroke
3.1. Overview of the Nrf2 Signaling Pathway
Nrf2 is a member of the cap’n’collar family of transcription factors and is present in various cell types. It consists of 605 amino acids with 7 highly conserved Nrf2-ECH domains (Neh1-7), which serve as a different functional region [44,45,46,47,48]. The Neh1 regulates DNA binding through the CNC–bZIP [49] and a nuclear localization signal (NLS) is responsible for the nuclear translocation of Nrf2 [50]. The Neh2, an N terminal regulatory domain, consists of DLG (low affinity) and ETGE (high affinity) motifs for the interaction with the Nrf2 negative regulator Kelch-like ECH-associated protein 1 (Keap1), which influences the stability and ubiquitination of Nrf2 [51]. The Neh3, Neh4 and Neh5 are transactivation domains mediating the interaction of Nrf2 with other coactivators [52,53], while the Neh5 domain is responsible for its cytoplasmic localization [54]. The Neh6 domain is a negative regulatory domain which binds to a β-transducin repeat-containing protein (β-TrCP), leading to Nrf2 ubiquitination, or regulates the Nrf2 stability by phosphorylation of serine residues [55]. The Neh7 domain inhibits the Nrf2-antioxidant response element (ARE) signaling pathway by promoting the binding of Nrf2 to the retinoic X receptor α (RXRα) [56].
In homeostatic conditions, Nrf2 stays in its inactive form within cells via Keap1. Keap1 is a cysteine-rich (27 cysteines), cytoplasmic, actin cytoskeleton-associated adapter zinc-metalloprotein of the Cul3/Rbx1 complex. It consists of five domains and Keap1/Cul3 homodimerization is regulated by the N-terminal portion of the intervening region with the BTB (Broad complex, Tramtrack and Bric-a-Brac) domain. The BTB domain of Keap1 plays a key role in sensing environmental electrophiles and is believed to be the target for several small molecule covalent activators of the Nrf2 pathway [57,58,59]. In the cytoplasm, Keap1 homodimerizes and binds to the cullin-based (Cul3) E3 ligase, forming Keap1-Cul3-RBX1 (Ring box protein-1) E3 ligase complex, that interacts with the Neh2 domain, forms the Keap1-Nrf2 complex and initiates degradation of Nrf2 by ubiquitination and proteasomal degradation [60,61,62].
In stress conditions (excessive accumulation of ROS, electrophilic molecules and proteotoxic stress), Nrf2 is released from the Keap1-Cul3-RBX1 complex and translocates into the nucleus, wherein it heterodimerizes with small Maf proteins (sMaf) and binds to the AREs on DNA, leading to the transcription of Nrf2 target genes [63]. The Nrf2/Keap1 pathway regulates a coordinated activation of a battery of cytoprotective genes that include biotransformation enzymes, antioxidant proteins, drug transporters, anti-apoptotic proteins and proteasome proteins. There are over 250 currently identified NRF2 target genes involved with redox regulation [44,58,64,65,66,67,68]. For example, target genes of Nrf2 are glutamate-cysteine ligase, NAD(P)H qui-none oxidoreductase 1 (NQO1), heme-oxygenase (HO-1), sulfiredoxin1 (SRXN1), heme-oxygenase (HO-1), glutathione S-transferase (GST), multidrug resistance-associated proteins (MRPs) and UDP-glucuronosyltransferase (UGT) [57]. Figure 1 depicts major molecules and events modulating Nrf2 stability and activation, as well as main downstream protein targets and their functions.
In addition, Nrf2 signaling takes part in the regulation of the cellular response to inflammation cooperating with NF-κB signaling pathways via suppression of pro-inflammatory genes, redox homeostasis and controls fundamental cellular processes, such as apoptosis, autophagy, angiogenesis, proliferation and cell migration [69,70]. Of note, Nrf2 can indirectly control the transcription of a host of non-ARE-containing genes. Indeed, functional AREs have been identified in the promoters of a number of transcription factors involved in DNA damage repair and apoptosis prevention [71,72,73].
Regulation of Nrf2 mainly occurs through the controlled maintenance of Nrf2 protein levels at the post-transcriptional and post-translational levels, as well as via epigenetic factors and interaction with other signaling pathways. It is important to note that its regulation mainly depends on the physiological and pathological context.
3.2. Molecular Events Linking Ischemic Stroke and Nrf2 Pathway
In a search using the terms “Nrf2” AND “ischemic stroke” in the PubMed database (https://pubmed.ncbi.nlm.nih.gov, accessed on 25 June 2021), it was possible to detect molecular players that are closely related to both topics. Among these players, there are several redox-active molecules (discussed below). Before discussing their potential (patho)physiological roles in the interplay between ischemic stroke and Nrf2 pathway, it is important to mention that most of the knowledge concerning molecular mechanisms involved in ischemic stroke and Nrf2 pathway comes primarily from experimental studies, including a great number of in vivo studies with rodents (mouse and rats). Of note, different experimental protocols have been developed to simulate different conditions, such as focal or global, as well as transient or permanent cerebral ischemia. Even though each of these protocols has specific features, the increased levels of reactive oxygen species and markers of oxidative damage are commonly observed in either focal or global, in addition to transient or permanent models [75,76,77,78]. Of note, data on the relationship between the Nrf2 pathway and ischemic stroke have been derived from these different experimental approaches (for a detailed review concerning such models, see reference [79]). Nonetheless, experimental ischemic models based on reperfusion/reoxygenation are expected to provide increased rates of oxidative stress considering the critical role of reoxygenation for the production of ROS [25].
As previously discussed (Section 2), increased levels of ROS are observed after either ischemia or ischemia/reoxygenation events. Hydrogen peroxide, a ROS whose levels are increased after ischemia/reoxygenation [27], is able to up regulate Nrf2 [80,81]. In agreement with this observation, experimental studies have reported the endogenous activation of Nrf2 following ischemic stroke, suggesting that this event represents a physiological response to the stress to which cells are subjected in ischemia/reperfusion (IR). Based on an IR protocol with a luciferase mouse model (a Keap1-dependent oxidative stress detector to visualize the Nrf2 expression from brain ischemia), Takagi and collaborators [82] showed increased levels of Nrf2 in the cerebral cortex and striatum of mice subjected to transient middle cerebral artery occlusion. The increased levels of Nrf2 were observed in both neurons and astrocytes and, notably, mainly in the penumbra zone. In another study based on an experimental model of transient cerebral ischemia [83], the authors reported the temporal and spatial distribution of Nrf2 in the nuclear and cytoplasmic compartments in cells in the ischemic core and peri-infarct regions and contralateral hemisphere of rat. Based on a quantitative immunohistochemical technique, these authors observed increased Nrf2 expression in brain sections in core and peri-infarct regions after 24 h reperfusion, with levels remaining elevated only in peri-infarct regions after 72 h. These two studies [82,83] provide evidence of Nrf2 activation following IR, suggesting that it represents an endogenous response resultant, at least partially, from ROS produced during transient ischemia.
On the other hand, several Nrf2-downstream proteins can mediate redox balance by neutralizing IR-derived ROS, such as superoxide anion and hydrogen peroxide. Particularly, superoxide dismutase and catalase, which metabolize superoxide anion and hydrogen peroxide, respectively, represent well-known Nrf2-downstream proteins [84,85,86,87]. Additional proteins, such as glutamate-cysteine ligase, glutathione peroxidase, glutathione reductase, thioredoxin reductase, heme oxygenase-1 and NADPH:quinone oxidoreductase, among others, are also known Nrf2-downstream molecules mediating redox balance and mitigating oxidative stress [74]. Table 1 presents experimental studies reporting the endogenous modulation of Nrf2 and/or its main Nrf2-downstream proteins after cerebral ischemia or ischemia-reperfusion. Although several studies presented in Table 1 were aimed at investigating protective effects of exogenously administrated Nrf2 activators against cerebral ischemic stroke, we have initially focused only on the potential endogenous modulation of Nrf2, evaluating the differences between control/sham and ischemic animals. It is noteworthy that most of these studies, which were based on either transient or permanent models of cerebral ischemia, reported significant increases in mRNA and/or protein expression of Nrf2 and Nrf2-downstream targets, indicating that the endogenous upregulation of Nrf2 represents an event resulting from cerebral ischemia or ischemia-reperfusion. Of note, some of these studies [88,89] indicated that Nrf2 knockout animals (Nrf2−/−) are more susceptible to cerebral ischemic stroke, highlighting the significance of this transcription factor in protecting the ischemic cerebral tissue. On the other hand, there are some studies (less frequent) reporting decreased gene and/or protein expression of Nrf2 and/or downstream proteins in the brain of ischemic animals, compared to controls. Although these apparent contradictory results may result from several causes, it is likely that the decreased gene and/or protein expression of Nrf2 and downstream proteins observed in ischemic animals in some studies can be related, at least partially, to an extreme rate of tissue damage, resulting in improper capability of performing transcription and translation.
As already discussed, ROS whose levels are increased after ischemia/reoxygenation, such as hydrogen peroxide [27], are able to upregulate Nrf2 [80,81]. On the other hand, there are specific Nrf2 downstream proteins capable of counteracting oxidants; this highlights an interesting interplay between ischemic stroke and Nrf2. From a Cartesian point of view, IR leads to increased levels of oxidants that, in turn, can activate Nrf2. On the other hand, Nrf2-downstream proteins can mitigate the deleterious effects of oxidants produced in excess during IR (see References from Table 1). Figure 2 depicts a schematic view of this interplay between ischemic stroke and Nrf2 pathway.
3.3. Effects of Nrf2 Modulators in Ischemic Stroke: Evidence from Experimental Studies
Given the pivotal role of Nrf2 in redox balance, several studies have reported its involvement in modulating cellular homeostasis in physiological and/or pathological conditions [64,147]. Particularly, promising neuroprotective effects of Nrf2 have been reported in diverse experimental models [148,149,150]. In the context of ischemic stroke, in addition to the protective effects exhibited by the endogenous activation of Nrf2 following ischemia or IR (discussed in Section 3.1, see Table 1), protective effects resulting from exogenously induced Nrf2 activation have also been reported, predominantly in rodent models (see examples discussed below). Of note, some Nrf2-activating compounds have displayed superior neuroprotective effects against IRI in wild-type compared to Nrf2−/− animals [89,151], confirming the involvement of Nrf2 in mediating the beneficial effects of these molecules. Although there is a large number of molecules capable of activating Nrf2 and exhibiting protective effects in ischemic stroke models (described in the last paragraph of this section), we provide a more detailed discussion on the most frequently reported agents, as follows.
3.3.1. Curcumin
Curcumin {1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione} (diferuloyl methane), a phytochemical compound extracted from Curcuma longa rhizomes, has been extensively studied for its multiple biological activities, including anti-inflammatory, antioxidant and anti-infective properties [152]. This polyphenol having a long history of use in traditional medicines of China and India, has a favorable safe profile. It is able to cross the BBB [153,154] with no toxicity, even at a high dose [155]. Curcumin has a Michael acceptor in the form of a α,β-unsaturated carbonyl group; thus, the main mechanism, by which it activates Nrf2, is by alkylating a protein thiol on the Keap-1-Nrf2 binding complex, which allows Nrf2 to translocate to the nucleus to initiate antioxidant gene expression changes [156,157]. Recently, mass spectrometric analysis revealed that curcumin binds to Keap1 Cys151 in the BTB domain, supporting that this amino acid is a critical target for curcumin modification of Keap1, which facilitates the liberation of Nrf2 [158]. Curcumin has been proven to exert neuroprotective effects and to prevent ischemic stroke through the attenuation of neurological dysfunction, infarction size, brain edema and BBB disruption [159,160,161,162,163,164,165,166,167,168,169] via anti-oxidant, anti-inflammatory and anti-apoptotic effects [170,171]. In vivo and in vitro [172,173], evidence demonstrates curcumin is an effective activator of Nrf2 in cerebral IR injury. The Nrf2/ARE signal pathway plays an important role at a very early time in rat brains subjected to middle cerebral artery occlusion (MCAO), a classic animal model of stroke. Indeed, both Nrf2 and HO-1 raise significantly in the first 3 h and maximize at 24 h after MCAO [174]. After systemic administration of curcumin, Nrf2 and HO-1 are further enhanced, the infarct size decreases and the brain edema improves [174]. Wu et al. [111] have demonstrated that, after stroke, curcumin administered by intraperitoneal injection (300 mg/kg) in rats inhibits oxidative stress, induces the expression of NQO1 and enhances the binding activity of Nrf2 to ARE. However, the PI3K/Akt pathway is necessary for curcumin effects, because blocking the PI3K/Akt signaling pathway abolishes the neuroprotective effects. To reduce the toxicity typically observed when curcumin is dissolved with DMSO or NaOH, Li et al. [169] administered curcumin dissolved in corn oil at 30 min after MCAO. The administration pre-reperfusion with curcumin reduced the subsequent IRI in the MCAO rat model as indicated by the reduction of brain edema, BBB disruption and neurological dysfunction at 24 h post reperfusion. The authors indicate that curcumin has significant neuroprotective effects after cerebral IRI by activating the Nrf2 pathway and by down-regulating NF-κB and MDA levels [169].
In hemorrhagic strokes, the lysis of red blood cells produces the release of hemin, a degradation product of hemoglobin. Hemin is a highly reactive compound and a dangerous molecule that is quickly accumulated and slowly degraded by HO, which causes damage in rat astrocytes and neurons [175]. The in vitro study by González-Reyes et al. [172] identifies curcumin as a neuroprotectant against hemin-induced damage in primary cultures of cerebellar granule neurons of rats. These in vitro data confirm that Nrf2 activation and antioxidant response (HO-1 and GSH) play a major role in the neuroprotective effect of curcumin. Although many experimental in vivo and in vitro studies have showed the protective effects of curcumin via the Nrf2 pathway, currently, no high-quality evidence showing that curcumin administration activates Nrf2 in humans is reported. The poor bioavailability of curcumin and its fast metabolism in humans are important factors to consider. Several approaches have been considered, including the adjuvant, the liposomal curcumin, curcumin nanoparticles and phospholipid complexes. The main structural modification of curcumin is to prepare the analogues without the β-diketone moiety, responsible for the instability and weak pharmacokinetic profiles of curcumin [176]. Furthermore, there is growing evidence that the addition of piperine may improve curcumin bioavailability [177,178]. Future well-controlled human intervention trials are needed to corroborate the neuroprotective effects of curcumin via the Nrf2 pathway observed in vitro and in animal studies of IRI and to advance our current understanding in humans.
3.3.2. Fumarate
Dimethyl fumarate (DMF) is derived from the simple organic acid fumaric acid, which is named after the earth smoke plant (Fumaria officinalis). While free fumaric acid is poorly absorbed, DMF is rapidly metabolized to monomethyl fumarate (MMF) [179]. With a broad efficacy, good safety and satisfying tolerability, the compound is the first-line oral drug for multiple sclerosis disease [180] and its immunomodulatory potential is also explored in other immune-mediated diseases [181,182,183,184]. Pleiotropic biological effects characterize DMF, including anti-oxidative stress and anti-apoptotic and immunomodulatory properties, as well as providing protection from microvascular dysfunction in a variety of tissues [185]. In vitro experiments have shown that MMF prevents detrimental pro-inflammatory response promoting, in a dose-dependent manner, the polarization of T lymphocytes toward the T-helper cell type 2 (Th2) phenotype, a T cell subset characterized by the production of interleukin-(IL)4, IL-5 [186] and IL-10 [187]. This Th2 shift was later linked to direct effects of fumaric acid esters on dendritic cells (DCs), thus inducing functional type II DCs with in vivo relevant suppression of the proinflammatory cytokines IL-12 and IL-23 [188,189]. At high dosages, fumaric acid esters were also shown to induce apoptosis in vitro [190]. Beyond its effects on T-cells and dendritic cells, DMF may also target several other immunologically active cell types [187,191,192,193,194,195,196].
In addition to its modulatory effects on immune cells, DMF may also possess neuroprotective capacity. DMF inhibits the production of nitric oxide (NO), IL-1β, TNF and IL-6 in astrocytes and microglia, increases plasma levels of IL-10 and suppresses macrophage infiltration into the brain during autoimmune encephalomyelitis (EAE) [194]. Abundant evidence indicates that fumaric acid esters activate the Nrf2-Keap1 pathway and increase the natural antioxidant responses in vivo and in vitro [197,198]. DMF leads to direct modification of Keap1 [188,197] and can suppress NF-κB transcription, induces detoxification enzymes (i.e., GSH reductase, c-glutamylcysteine synthetase and GSH synthetase) in astrocytes and microglial cells and modulates glutathione levels in cells [199]. Recent data indicate that systemic DMF treatment is involved in maintaining BBB integrity and improving neurological outcomes in a short-term model of hemorrhagic strokes [121,200,201] and ischemic stroke [202,203]. In all of these cases, abundant evidence indicates that DMF/MMF act via activation of the Keap1-Nrf2-ARE signaling pathway [121,200,202]. Indeed, the beneficial effect of DMF was lost in the Nrf2-KO animals, suggesting that its therapeutic effect is mainly through activating Nrf2. However, the long-term neuroprotective effects observed after DMF treatment are also related to its immunomodulatory ability via an Nrf2-independent mechanism [125].
Liu et al. [204,205] have also provided evidence on the protection derived by the pretreatment with DMF against ischemic damage in initial, acute and extended phases after hypoxia-ischemia (HI). By using a cerebral HI mouse model and transgenic loss-of-function of Nrf2 mice, the authors have observed that pre-treatment with DMF for 7 days prior to hypoxia-ischemia confers robust and prolonged Nrf2-dependent neuroprotection by involving anti-oxidative and anti-inflammatory response and the attenuation of reactive gliosis in astrocyte and microglia. Overall, these findings support the unique protective role of Nrf2 in the stroke field and may open a new window to utilize these endogenous neuroprotection mechanisms as preventive approach in the development and progression of cerebral ischemia pathology.
3.3.3. Resveratrol
Resveratrol (3,4,5-trihydroxystilbene) is a polyphenolic compound abundantly present in grapes and red wine [206]. It is well known for its antioxidant, anti-inflammatory and antiapoptotic properties [207,208,209,210,211] that it exerts by influencing multiple pathways [212]. Studies performed both in vitro and in vivo have provided evidence that resveratrol has neuroprotective effects. Resveratrol treatment of neuronal cell lines and hippocampal slice cultures exposed to oxygen and glucose deprivation (OGD)—a model of hypoxia/ischemia—promoted cell survival [213,214]. Recent studies demonstrated that resveratrol can protect hippocampal neurons from damage caused by transient cerebral ischemia [209]. In rodent models of ischemia, pre- and post-treatment with resveratrol determined a reduction of the infarct volume and brain edema [107,158], thus confirming the neuroprotective effects of this natural compound observed in in vitro models. Different mechanisms have been identified as responsible for the neuroprotective effects of resveratrol. Evidence exists demonstrating that it down-modulates the activity of the pro-apoptotic factors caspase-3 and Bax, promotes Bcl-2 expression and contrasts alteration of mitochondrial function [215,216], thus exerting anti-apoptotic effects. In a rat model of brain ischemia, it has been observed that resveratrol can induce neuroprotection by activating the PI3K/AKT signaling pathway, that has a key role in mediating cell survival, thus preventing neuronal death [211]. More recently, Hou et al. [217] deepened the understanding of the mechanisms involved in resveratrol-mediated neuroprotection using rats subjected to middle cerebral artery occlusion followed by reperfusion. They observed that pre-treatment with resveratrol for 7 days was able to reduce cerebral infarct area, neuronal damage and apoptosis and this was associated with increased expression of p-JAK2, p-STAT3, p-AKT and p-mTOR. The authors concluded that resveratrol is able to exert neuroprotective activity on cerebral IR by promoting the phosphorylation of key proteins of the JAK2/STAT3/PI3K/AKT/mTOR pathway. In vitro and in vivo studies also demonstrated an anti-inflammatory activity of resveratrol on activated microglia, as it effectively inhibits IL-1β, TNFα and nitric oxide production, together with NF-κB signaling and p38 phosphorylation [218,219,220,221], thus contrasting the deleterious effects of inflammation, an important factor involved in ischemic stroke. Another important property of resveratrol responsible for its neuroprotective effects is its antioxidant activity. It directly attenuates oxidative stress by scavenging ROS, thus inhibiting lipid peroxidation and DNA damage. In vitro and in vivo evidence exists demonstrating that the neuroprotective effects of resveratrol are due, at least in part, to its ability to activate the Keap1−Nrf2 pathway, which, in turn, modulates the expression of inflammatory mediators and of antioxidant enzymes [222,223]. Through the up-regulation of Nrf2 activity, resveratrol promotes the expression of ARE-regulated genes involved in the control of free radical levels [212]. In vitro experiments performed with neuronal cell lines and primary neuronal cells demonstrated that the activation of the Nrf2/ARE pathway by resveratrol promotes HO1 activity and the increase in glutathione and SOD levels [224,225]. The use of small interfering RNA in an in vitro oxidative stress model of endothelial cells showed that the antioxidant activity of resveratrol was inhibited if Nrf2 was knocked down [226]. In a recent study, Yang and colleagues [227] observed that in vitro resveratrol treatment of rat cortical neurons at different times reduced neuronal injury, decreasing lactate dehydrogenase and increasing SOD activity in a concentration-dependent manner. Cells treated with resveratrol showed increased cell viability and reduced apoptosis. The authors also observed that this treatment promoted the upregulation of Nrf2 and its translocation into the nucleus and the expression of NAD(P)H, NADPH quinone oxidoreductase 1 (NQO-1) and HO1, all of which are involved in contrasting oxidative stress. Of note, NQO-1 is able to reduce ROS levels, thus preventing cellular injury in brain ischemia and in neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease and multiple sclerosis [228].
Studies conducted in rats demonstrated that pre-treatment with resveratrol up-regulated Nrf2 expression and increased HO1 levels after cerebral IRI [107]. Moreover, in a mouse model of cerebral ischemia, Narayanan et al. [151] showed that resveratrol-mediated neuroprotection was reduced in Nrf2−/− mice, compared to wild type mice, thus demonstrating that resveratrol activity was Nrf2-dependent. These observations also confirm, in vivo, that the neuroprotective antioxidant activity of resveratrol is mediated, at least in part, by the activation of the Nrf2/ARE pathway. All these observations obtained using in vitro and in vivo models strongly sustain the therapeutic potential of resveratrol in ischemic cerebral damage. However, due to its rapid clearance from the circulation, further studies are needed to improve its efficacy in vivo.
3.3.4. Sulforaphane
Sulforaphane (4-methylsulfinylbutyl isothiocyanate) is a natural isothiocyanate derived from the hydrolysis of glucoraphanin, widely present in cabbage, broccoli and other vegetables belonging to the family Brassicaceae [229,230]. It is a pleiotropic compound with anti-tumor and anti-microbial activities, as widely demonstrated in experimental models [231,232,233]. Data obtained in animal models showed a protective activity of sulforaphane in IRI affecting different tissues, including kidney [234], retina [235] and intestine [236,237].
Evidence exists demonstrating that it has also neuroprotective effects. Studies in rodents have shown that sulforaphane is able to reduce the cerebral infarct volume following focal ischemia and cerebral edema in injured brain [238,239]. Ma and colleagues [240] demonstrated that in vivo treatment with sulforaphane inhibited the NF-κB signaling pathway, thus reducing the expression of pro-inflammatory cytokines, nitric oxide and cyclooxygenase-2 in rats subjected to middle cerebral artery occlusion. Data obtained using in vitro and in vivo models evidenced that the protective effects of this compound are mainly due to its ability to activate the Nrf2/ARE pathway [241,242]. In a neonatal hypoxia-ischemia model, Ping and colleagues demonstrated that treatment with sulforaphane upregulated Nrf2 and HO1 expression and reduced neurons apoptosis and brain tissue loss [243]. Furthermore, in vitro administration of this compound in cultures of astrocytes, before or after exposure to oxygen–glucose deprivation conditions (OGD), improved cell survival by activating the Nrf2 pathway [244]. Sulforaphane promoted Nrf2 expression in cardiac cells and epidermal cells by the methylation of the Nrf2 promoter [245,246]. It also interacts with thiol groups of Keap1 cysteines, thus affecting the Nrf2/Keap1 complex stability and contrasting Nrf2 degradation [246,247,248]. Sulforaphane-mediated activation of the Nrf2 pathway induces antioxidative and detoxifying enzymes, such as glutathione S-transferase (GST), HO1 and NQO-1, that, in turn, play a crucial role in neuroprotection. Recently, in a rat model of vascular cognitive impairment, which involves the permanent occlusion of carotid arteries, it was demonstrated that administration of sulforaphane reduced ischemic injuries and improved cognitive abilities [249]. The observed neuroprotection was associated with increased Nrf2 activation and HO1 expression. To confirm the role of Nrf2 in sulforaphane-mediated protective effects, the same authors set up in vitro experiments with endothelial cells subjected to OGD conditions. They observed that, if Nrf2 was knocked down, sulforaphane was no longer able to protect endothelial cells from OGD-mediated damage, thus concluding that sulforaphane preserves the integrity of the blood–brain barrier via Nrf2 activation [249].
Evidence obtained both in humans and animal models has shown that this natural compound is rapidly absorbed and accumulated in tissues and that it is able to pass the blood–brain barrier and to accumulate in the brain [250,251,252,253,254]. All these observations suggest that sulforaphane could be a potential therapeutic molecule to treat cerebral ischemia injury.
3.3.5. Tert-Butylhydroquinone
Tert-Butylhydroquinone (tBHQ) derives from the metabolism of the natural antioxidant compound butylated hydroxyanisole [88]. Several years ago, it obtained the approval for its use in humans [255,256] and it is widely used as a food additive. Studies conducted in different models of cerebral injury, including brain trauma and ischemic stroke, have demonstrated that it has neuroprotective effects [88,257]. In a rat model of IR, Shih et al. [88] observed that pretreatment with tBHQ reduced cerebral damage 24 h after stroke and it was associated with the increase of cortical GSH levels. The reduction of ischemic damage was observed even 1 month after and with both intracerebroventricular and intraperitoneal administration of tBHQ. Of note, tBHQ administration failed to induce cortical GSH increase and to reduce infarct size in Nrf2−/− mice, thus suggesting that the neuroprotective activity of tBHQ is Nrf2-dependent [88]. These results have been confirmed in more recent papers. In a rat model of subarachnoid hemorrhage (SAH), Wang and collaborators [258] observed that tBHQ administration after SAH preserved blood–brain barrier integrity, as demonstrated by its ability to inhibit the increase of blood–brain barrier permeability evaluated by Evans blue extravasation. It also reduced cortical apoptosis and oxidative stress levels, neuronal degeneration and clinical behavior deficits. Moreover, significantly higher protein and mRNA expression levels of Nrf2, Keap1, HO1 and NQO1 were observed in animals treated with tBHQ, compared to those treated with vehicle [258], thus indicating the role of Nrf2 activation in tBHQ-mediated neuroprotection. Similar results were obtained in another recent study conducted using a rat model of neonatal hypoxic-ischemic encephalopathy [143]. The authors observed that post-treatment of animals with tBHQ reduced neuronal apoptosis in the cerebral cortex, infarct size and neuronal damage. The administration of this compound also improved neurological reflex, motor coordination and memory deficits. Furthermore, animals subjected to tBHQ administration showed higher levels of Nrf2 into the nucleus and increased expression of Nrf2-regulated antioxidative genes. All these data strongly support the neuroprotective effects of tBHQ and that these effects are mediated, at least in part, by the activation of the Nrf2 pathway. Due to its 1,4 diphenolic structure, tBHQ is able to dissociate the Nrf2/Keap1 complex, thus promoting the translocation of Nrf2 into the nucleus and the expression of antioxidant genes [259,260,261]. However, evidence exists showing that tBHQ also has detrimental effects [262], likely due to reactions mediated by GSH-conjugates [263]. Sun and colleagues [264] conducted a research study using a murine permanent middle cerebral artery occlusion model and observed that tBHQ treatment was associated with a significant increase in mortality, compared to control. They also observed that tBHQ significantly increased brain volume and impaired mitochondrial function of cerebrovascular endothelial cells, suggesting that tBHQ, by altering the blood–brain barrier integrity, can exacerbate stroke damage. Therefore, further studies are needed to determine whether tBHQ is able to promote long-term neuroprotection without severe side effects [143] to clarify its potential as therapeutic agent for stroke.
3.3.6. Carbon Monoxide
The gaseous molecule carbon monoxide (CO) is associated with central nervous system toxicity. However, evidence also indicates that CO can be protective, depending on its concentration. CO is endogenously produced upon degradation of heme by HO. Heme oxygenase-1 (HO-1) participates in the cell defense against oxidative stress and is known to be induced by Nrf2 [265]. Zeynalov and Doré provided evidence in mice that CO can be therapeutic in IR brain injury [266] and its beneficial effect is mediated by activation of the Nrf2/Keap l/ARE/HO-1 pathway. Indeed, 250 ppm CO exposure promoted dissociation of Nrf2 from Keap1, increased the nuclear Nrf2 occupancy of AREs within the HO-1 promoter and induced time-dependent increases in HO-1 expression. Although the neuroprotection is completely lost in Nrf2−/− mice, the beneficial effects of CO were also likely caused by the activation of other protective mechanisms. CO may also act through activation of anti-inflammatory, anti-apoptotic and vasodilatory mechanisms [267,268]. In addition, CO has been reported to have early thrombolytic effects after ischemia [269]. The ability to activate the Nrf2 signaling pathway and to pass through the blood–brain barrier (BBB), in concert with other anti-inflammatory mechanisms, render the low concentrations of inhaled CO an emerging good candidate for neurologic protection after stroke.
In addition to the aforementioned compounds, which represent the most studied Nrf2 activators with neuroprotective effects in experimental models of ischemic stroke, additional drugs (less frequently investigated compounds) have also shown beneficial effects, such as tanshinol borneol ester [270], Apelin 13 [141], ezetimibe [271], rosmarinic acid [100], biochanin A [272], isoquercetin [273], trilabatin [137], forsythiaside A [274], octreotide [275], Korean Red Ginseng [89], Schizandrin A [276], leonurine [277], sinomenine [101], myricetin [139], diterpene ginkgolides [278], totarol [279], paeonol [91], alpha-lipoic acid [280], omega-3 fatty acids [281], nicotinamide mononucleotide [282], chlorogenic acid [144], eriocitrin [283], bicyclol [114] andrographolide [284], phyllanthin [285], neohesperidin [286], protocatechualdehyde [287], osthole [275], salidroside [288], palmatine [289], pelargonidin [290] and britanin [291].
4. Challenges/Perspectives on the Use of Nrf2 Activators in Ischemic Stroke Patients
The scientific literature reviewed herein provides compelling evidence that Nrf2 activation is neuroprotective in ischemic stroke models. By using in vivo experimental approaches based mainly on the induction of permanent or transient cerebral ischemia in rodents, researchers have shown that both the endogenous and exogenously induced Nrf2 activations display neuroprotective effects. Concerning the endogenous activation, most of the studies presented in Table 1 (Section 3.2) clearly show the upregulation of Nrf2 and downstream targets following ischemia or IR. Of note, some studies show that Nrf2 knockout animals were more susceptible to cerebral ischemic stroke (higher infarct sizes and more severe neurological deficits), indicating that such endogenous activation has a major role in mitigating IR-related damage. Concerning the exogenously-induced activation of Nrf2, several well-known Nrf2 activators (described in Section 3.3) have displayed neuroprotective effects in ischemic stroke models and, notably, some Nrf2-activating compounds had superior neuroprotective effects against IRI in wild-type compared to Nrf2 knockout animals [89,151]. Thus, the reviewed literature provides an optimistic scenery and indicates that Nrf2 modulators may represent promising pharmacological strategies to treat ischemic stroke patients in a near future.
As already discussed in Section 2, the pro-oxidative events mediating IRI are diverse and may result from either increased production or decreased neutralization of oxidants, which are also diverse with respect to their chemical characteristics, including reactivity. Moreover, inflammation, which is closely related to oxidative stress, also represents a key event resulting from IR. In this scenery, it is reasonable to suppose that the treatment of acute stroke with Nrf2 activators could have advantages, compared to strategies based on a unique mechanism of action, such as (i) free radical scavengers aimed to neutralize a specific radical specie, or (ii) inhibitors of specific radical-generating enzymes (i.e., NOX). This is based on the fact that Nrf2 activation may lead to the upregulation of diverse players that counterbalance impairments in proteostasis, redox and inflammatory control [74]; their combined action might simultaneously mitigate distinct deleterious events resulting from IR.
On the other hand, there are significant drawbacks and challenges that work against the successful translation of the preclinical efficacy of Nrf2 activators into the clinical conditions of ischemic stroke patients. Initially, it is important to take into account the acute nature of ischemic stroke and the relative fast cascade of events resulting from the sudden loss of blood flow. As a consequence of the impaired delivery of oxygen and nutrients to cells, the brain’s electrical activity and signs of awareness have been reported to disappear within seconds after severe ischemic stroke, while energy stores seem to be depleted within minutes [292]. In such kind of situation, the hypothetical neuroprotective pharmacological treatment should be performed as soon as possible and, in addition, it is essential that it has a relatively quick pharmacological result in order to minimize neurodegeneration and related sequelae. In addition to the good pharmacokinetic properties, it is desired that the drug has a mechanism of action that allows the occurrence of neuroprotection even in metabolically compromised cells. As already discussed at the beginning of Section 3.1, Nrf2 is a transcription factor that controls the expression of cytoprotective genes [44,58,64,65,66,67,68,258,293,294] and is involved in different cellular processes [69,70]. mRNA and protein syntheses represent events that depend on the proper cellular metabolic homeostasis, which is compromised in ischemic cells [19]. In this context, although Nrf2 has been pointed as a therapeutic target for human chronic diseases [74], it seems that the translation of the beneficial effects of Nrf2 activators, observed in preclinical models, into clinical sceneries of ischemic stroke (an acute condition) is less probable. There are two important separate areas of the ischemic brain, the ischemic core and the ischemic penumbra. During a vessel occlusion, the core area is the first to be damaged, while cells from the ischemic penumbra are predominantly damaged during the reperfusion/reoxygenation period, thus representing a target for neuroprotection shortly after ischemic stroke episodes [295]. Taking into account that the beneficial effects of Nrf2 activators commonly depend on transcription and translation, it is likely that their potential neuroprotective effects in clinical stroke (if any) are restricted to cells located far from the ischemic center, whose metabolic compromise is not sufficient to impair transcription and translation. In this regard, it is important to recapitulate the experimental study by Takagi and collaborators, which showed increased levels of Nrf2 in the cerebral cortex and striatum of mice subjected to transient middle cerebral artery occlusion; notably, such increase was observed particularly in the penumbra zone [82], which is less metabolically compromised compared to the ischemic core. This experimental evidence reinforces the idea that metabolically impaired cells located near the ischemic core are unable to properly upregulate Nrf2 and related downstream proteins. This represents a conceivable disadvantage in using Nrf2 activators to treat extreme acute metabolic impairments, such as severe ischemic stroke.
This supposed disadvantage of using Nrf2 activators to treat acute conditions is in line with the idea that timing is crucial in modulating Nrf2 in disease [64]. Within this panorama, the literature reviewed herein indicates a significant number of studies using pretreatments when evaluating neuroprotective effects of Nrf2 activators in models of experimental ischemic stroke. Considering the acute nature of ischemic stroke, translating experimental results on Nrf2 activators into real clinical conditions seems to be less likely when data are derived from protocols based on pretreatments. This seems to be particular important considering that (i) Nrf2-derived biological effects are greatly dependent on transcription and translation and (ii) acute ischemic stroke leads to quick cell metabolic impairment. The design of protocols that properly mimic the real conditions of ischemic stroke patients (i.e., post-treatment with drugs after the diagnosis of stroke) will certainly maximize the possibility of progression of Nrf2 activators from bench to clinical conditions if the aim is to treat (not prevent) ischemic stroke.
In summary, the literature reviewed herein has unequivocally shown neuroprotective effects of the exogenously induced Nrf2 activation in experimental models of ischemic stroke, providing a positive panorama and indicating that Nrf2 modulators may represent promising pharmacological strategies to treat ischemic stroke patients in a near future. However, the acute nature of ischemic stroke represents a challenge when using a putative pharmacological strategy (Nrf2 activator) that relies upon transcription, translation and metabolically active cells. In this context, the execution of experimental protocols able to mimic real conditions of ischemic stroke patients in order to progress Nrf2 activators from preclinical studies to clinical practices seems crucial.
Funding
This research was funded by CNPq-Brazil, grant numbers 404666/2018-3 and 302952/2018-7.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Taoufik, E.; Probert, L. Ischemic Neuronal Damage. CPD 2008, 14, 3565–3573. [Google Scholar] [CrossRef]
- Shakir, R.; Norrving, B. Stroke in ICD-11: The end of a long exile. Lancet 2017, 389, 2373. [Google Scholar] [CrossRef] [Green Version]
- George, M.G.; Tong, X.; Kuklina, E.V.; Labarthe, D.R. Trends in stroke hospitalizations and associated risk factors among children and young adults, 1995–2008. Ann. Neurol. 2011, 70, 713–721. [Google Scholar] [CrossRef]
- Appelros, P.; Stegmayr, B.; Terént, A. Sex Differences in Stroke Epidemiology: A Systematic Review. Stroke 2009, 40, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M. The sixth report of the JoInt. National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure, and 1999 World Health Organization-International Society of Hypertension Guidelines for the Management of Hypertension. Nihon Rinsho 2000, 58 (Suppl. 1), 267–275. [Google Scholar]
- Seshadri, S.; Beiser, A.; Pikula, A.; Himali, J.J.; Kelly-Hayes, M.; Debette, S.; DeStefano, A.L.; Romero, J.R.; Kase, C.S.; Wolf, P.A. Parental Occurrence of Stroke and Risk of Stroke in Their Children: The Framingham Study. Circulation 2010, 121, 1304–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, M.J.; Xavier, D.; Liu, L.; Zhang, H.; Chin, S.L.; Rao-Melacini, P.; Rangarajan, S.; Islam, S.; Pais, P.; McQueen, M.J.; et al. Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): A case-control study. Lancet 2010, 376, 112–123. [Google Scholar] [CrossRef]
- Vermeer, S.E.; Sandee, W.; Algra, A.; Koudstaal, P.J.; Kappelle, L.J.; Dippel, D.W.J. Impaired Glucose Tolerance Increases Stroke Risk in Nondiabetic Patients With Transient Ischemic Attack or Minor Ischemic Stroke. Stroke 2006, 37, 1413–1417. [Google Scholar] [CrossRef] [Green Version]
- Romero, J.R.; Morris, J.; Pikula, A. Stroke prevention: Modifying risk factors. Ther. Adv. Cardiovasc. Dis. 2008, 2, 287–303. [Google Scholar] [CrossRef] [Green Version]
- Denti, L.; Cecchetti, A.; Annoni, V.; Merli, M.F.; Ablondi, F.; Valenti, G. The role of lipid profile in determining the risk of ischemic stroke in the elderly: A case–control study. Arch. Gerontol. Geriatr. 2003, 37, 51–62. [Google Scholar] [CrossRef]
- Bhat, V.M.; Cole, J.W.; Sorkin, J.D.; Wozniak, M.A.; Malarcher, A.M.; Giles, W.H.; Stern, B.J.; Kittner, S.J. Dose-Response Relationship Between Cigarette Smoking and Risk of Ischemic Stroke in Young Women. Stroke 2008, 39, 2439–2443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonnell, M.N.; Hillier, S.L.; Hooker, S.P.; Le, A.; Judd, S.E.; Howard, V.J. Physical Activity Frequency and Risk of Incident Stroke in a National US Study of Blacks and Whites. Stroke 2013, 44, 2519–2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estruch, R.; Ros, E.; Martínez-González, M.A. Mediterranean Diet for Primary Prevention of Cardiovascular Disease. N. Engl. J. Med. 2013, 369, 672–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuriakose, D.; Xiao, Z. Pathophysiology and Treatment of Stroke: Present Status and Future Perspectives. Int. J. Mol. Sci. 2020, 21, 7609. [Google Scholar] [CrossRef] [PubMed]
- Hacke, W.; Kaste, M.; Bluhmki, E.; Brozman, M.; Dávalos, A.; Guidetti, D.; Larrue, V.; Lees, K.R.; Medeghri, Z.; Machnig, T.; et al. Thrombolysis with Alteplase 3 to 4.5 Hours after Acute Ischemic Stroke. N. Engl. J. Med. 2008, 359, 1317–1329. [Google Scholar] [CrossRef] [Green Version]
- Rabinstein, A.A. Update on Treatment of Acute Ischemic Stroke. Continuum 2020, 26, 268–286. [Google Scholar] [CrossRef] [PubMed]
- Orellana-Urzúa, S.; Rojas, I.; Líbano, L.; Rodrigo, R. Pathophysiology of Ischemic Stroke: Role of Oxidative Stress. CPD 2020, 26, 4246–4260. [Google Scholar] [CrossRef]
- Dienel, G.A. Brain Glucose Metabolism: Integration of Energetics with Function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef]
- Kaplan, J.H. Biochemistry of Na,K-ATPase. Annu. Rev. Biochem. 2002, 71, 511–535. [Google Scholar] [CrossRef]
- Choi, D.W. Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Front. Neurosci. 2020, 14, 579953. [Google Scholar] [CrossRef] [PubMed]
- Sattler, R.; Tymianski, M. Molecular mechanisms of calcium-dependent excitotoxicity. J. Mol. Med. 2000, 78, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Depp, C.; Bas-Orth, C.; Schroeder, L.; Hellwig, A.; Bading, H. Synaptic Activity Protects Neurons Against Calcium-Mediated Oxidation and Contraction of Mitochondria During Excitotoxicity. Antioxid. Redox Signal. 2018, 29, 1109–1124. [Google Scholar] [CrossRef] [PubMed]
- Quillinan, N.; Herson, P.S.; Traystman, R.J. Neuropathophysiology of Brain Injury. Anesthesiol. Clin. 2016, 34, 453–464. [Google Scholar] [CrossRef] [Green Version]
- Saito, A.; Maier, C.M.; Narasimhan, P.; Nishi, T.; Song, Y.S.; Yu, F.; Liu, J.; Lee, Y.-S.; Nito, C.; Kamada, H.; et al. Oxidative Stress and Neuronal Death/Survival Signaling in Cerebral Ischemia. Mol. Neurobiol. 2005, 31, 105–116. [Google Scholar] [CrossRef]
- Epstein, F.H.; McCord, J.M. Oxygen-Derived Free Radicals in Postischemic Tissue Injury. N. Engl. J. Med. 1985, 312, 159–163. [Google Scholar] [CrossRef]
- Uyama, O.; Matsuyama, T.; Michishita, H.; Nakamura, H.; Sugita, M. Protective effects of human recombinant superoxide dismutase on transient ischemic injury of CA1 neurons in gerbils. Stroke 1992, 23, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Morooka, H.; Hirotsune, N.; Wani, T.; Ohmoto, T. Histochemical Demonstration of Free Radicals (H22O2) in Ischemic Brain Edema and Protective Effects of Human Recombinant Superoxide Dismutase on Ischemic Neuronal Damage. In Brain Edema IX; Ito, U., Baethmann, A., Hossmann, K.-A., Kuroiwa, T., Marmarou, A., Reulen, H.-J., Takakura, K., Eds.; Springer: Vienna, Austria, 1994; pp. 307–309. ISBN 978-3-7091-9336-5. [Google Scholar]
- Kinuta, Y.; Kikuchi, H.; Ishikawa, M. Ischaemic Brain Oedema and Xanthine-Xanthine Oxidase System. In Brain Edema VIII; Reulen, H.-J., Baethmann, A., Fenstermacher, J., Marmarou, A., Spatz, M., Eds.; Springer: Vienna, Austria, 1990; pp. 192–194. ISBN 978-3-7091-9117-0. [Google Scholar]
- Muralikrishna Adibhatla, R.; Hatcher, J.F. Phospholipase A2, reactive oxygen species, and lipid peroxidation in cerebral ischemia. Free Radic. Biol. Med. 2006, 40, 376–387. [Google Scholar] [CrossRef]
- Nakahara, I.; Kikuchi, H.; Taki, W.; Nishi, S.; Kito, M.; Yonekawa, Y.; Goto, Y.; Ogata, N. Changes in major phospholipids of mitochondria during postischemic reperfusion in rat brain. J. Neurosurg. 1992, 76, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Chomova, M.; Zitnanova, I. Look into brain energy crisis and membrane pathophysiology in ischemia and reperfusion. Stress 2016, 19, 341–348. [Google Scholar] [CrossRef]
- Lake, E.M.R.; Mester, J.; Thomason, L.A.; Adams, C.; Bazzigaluppi, P.; Koletar, M.; Janik, R.; Carlen, P.; McLaurin, J.; Stanisz, G.J.; et al. Modulation of the peri-infarct neurogliovascular function by delayed COX-1 inhibition: Delayed COX-1 Inhibition Stroke Treatment. J. Magn. Reson. Imaging 2017, 46, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Yang, Y.; DeMars, K.M.; Rosenberg, G.A.; Candelario-Jalil, E. Genetic Deletion or Pharmacological Inhibition of Cyclooxygenase-2 Reduces Blood-Brain Barrier Damage in Experimental Ischemic Stroke. Front. Neurol. 2020, 11, 887. [Google Scholar] [CrossRef] [PubMed]
- Leyen, K.V. Lipoxygenase: An Emerging Target for Stroke Therapy. CNSNDDT 2013, 12, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zuo, F.; Wu, H. Blockage of cytosolic phospholipase A2 alpha by monoclonal antibody attenuates focal ischemic brain damage in mice. Biosci Trends 2017, 11, 439–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Yu, Y. Small molecules targeting cyclooxygenase/prostanoid cascade in experimental brain ischemia: Do they translate? Med. Res. Rev. 2021, 41, 828–857. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Ago, T.; Kitazono, T.; Nabika, T. NADPH Oxidase-Related Pathophysiology in Experimental Models of Stroke. Int. J. Mol. Sci. 2017, 18, 2123. [Google Scholar] [CrossRef] [Green Version]
- Casas, A.I.; Kleikers, P.W.M.; Geuss, E.; Langhauser, F.; Adler, T.; Busch, D.H.; Gailus-Durner, V.; de Angelis, M.H.; Egea, J.; Lopez, M.G.; et al. Calcium-dependent blood-brain barrier breakdown by NOX5 limits postreperfusion benefit in stroke. J. Clin. Investig. 2019, 129, 1772–1778. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhang, Y.; Zhao, X.; Shao, L.; Liu, G.; Sun, C.; Xu, R.; Zhang, Z. ACSL4 exacerbates ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain Behav. Immun. 2021, 93, 312–321. [Google Scholar] [CrossRef]
- Kimura-Ohba, S.; Yang, Y. Oxidative DNA Damage Mediated by Intranuclear MMP Activity Is Associated with Neuronal Apoptosis in Ischemic Stroke. Oxid. Med. Cell. Longev. 2016, 2016, 6927328. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, H.; Wang, L.; Konishi, T. Prevention of Cerebral Oxidative Injury by Post-ischemic Intravenous Administration of Shengmai San. Am. J. Chin. Med. 2006, 34, 591–600. [Google Scholar] [CrossRef]
- Narne, P.; Pandey, V.; Phanithi, P.B. Role of Nitric Oxide and Hydrogen Sulfide in Ischemic Stroke and the Emergent Epigenetic Underpinnings. Mol. Neurobiol. 2019, 56, 1749–1769. [Google Scholar] [CrossRef]
- Dogan, O.; Kisa, U.; Erdemoglu, A.K.; Kacmaz, M.; Caglayan, O.; Kurku, H. Oxidative and nitrosative stress in patients with ischemic stroke. LaboratoriumsMedizin 2018, 42, 195–200. [Google Scholar] [CrossRef]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 2009, 47, 1304–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connell, M.A.; Hayes, J.D. The Keap1/Nrf2 pathway in health and disease: From the bench to the clinic. Biochem. Soc. Trans. 2015, 43, 687–689. [Google Scholar] [CrossRef] [Green Version]
- Sova, M.; Saso, L. Design and development of Nrf2 modulators for cancer chemoprevention and therapy: A review. Drug Des. Dev. Ther. 2018, 12, 3181–3197. [Google Scholar] [CrossRef] [Green Version]
- Zheng, F.; Gonçalves, F.M.; Abiko, Y.; Li, H.; Kumagai, Y.; Aschner, M. Redox toxicology of environmental chemicals causing oxidative stress. Redox Biol. 2020, 34, 101475. [Google Scholar] [CrossRef]
- Cores, Á.; Piquero, M.; Villacampa, M.; León, R.; Menéndez, J.C. NRF2 Regulation Processes as a Source of Potential Drug Targets against Neurodegenerative Diseases. Biomolecules 2020, 10, 904. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP Augments Promoter-Specific DNA Binding of Nrf2 during the Antioxidant Response. Mol. Cell. Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef] [Green Version]
- Theodore, M.; Kawai, Y.; Yang, J.; Kleshchenko, Y.; Reddy, S.P.; Villalta, F.; Arinze, I.J. Multiple Nuclear Localization Signals Function in the Nuclear Import of the Transcription Factor Nrf2. J. Biol. Chem. 2008, 283, 8984–8994. [Google Scholar] [CrossRef] [Green Version]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [Green Version]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The Carboxy-Terminal Neh3 Domain of Nrf2 Is Required for Transcriptional Activation. Mol. Cell. Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef] [Green Version]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription: Synergistic activation of Nrf2 by CBP. Genes Cells 2001, 6, 857–868. [Google Scholar] [CrossRef]
- Krajka-Kuźniak, V.; Paluszczak, J.; Baer-Dubowska, W. The Nrf2-ARE signaling pathway: An update on its regulation and possible role in cancer prevention and treatment. Pharmacol. Rep. 2017, 69, 393–402. [Google Scholar] [CrossRef]
- Rada, P.; Rojo, A.I.; Evrard-Todeschi, N.; Innamorato, N.G.; Cotte, A.; Jaworski, T.; Tobón-Velasco, J.C.; Devijver, H.; García-Mayoral, M.F.; Van Leuven, F.; et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/β-TrCP axis. Mol. Cell. Biol. 2012, 32, 3486–3499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRα Inhibits the NRF2-ARE Signaling Pathway through a Direct Interaction with the Neh7 Domain of NRF2. Cancer Res. 2013, 73, 3097–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Mittal, R. Nrf2: A potential therapeutic target for diabetic neuropathy. Inflammopharmacology 2017, 25, 393–402. [Google Scholar] [CrossRef] [PubMed]
- David, J.A.; Rifkin, W.J.; Rabbani, P.S.; Ceradini, D.J. The Nrf2/Keap1/ARE Pathway and Oxidative Stress as a Therapeutic Target in Type II Diabetes Mellitus. J. Diabetes Res. 2017, 2017, 4826724. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.-L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB Protein Is an Adaptor That Bridges Nrf2 to a Cul3-Based E3 Ligase: Oxidative Stress Sensing by a Cul3-Keap1 Ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D.; Lo, S.-C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 Is a Redox-Regulated Substrate Adaptor Protein for a Cul3-Dependent Ubiquitin Ligase Complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; McMahon, M.; Chowdhry, S.; Dinkova-Kostova, A.T. Cancer Chemoprevention Mechanisms Mediated through the Keap1–Nrf2 Pathway. Antioxid. Redox Signal. 2010, 13, 1713–1748. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575. [Google Scholar] [CrossRef]
- Sykiotis, G.P.; Bohmann, D. Stress-Activated Cap’n’collar Transcription Factors in Aging and Human Disease. Sci. Signal. 2010, 3, re3. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe, Á.J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, S.; Pal, D.; Sandur, S.K. Nrf2 facilitates repair of radiation induced DNA damage through homologous recombination repair pathway in a ROS independent manner in cancer cells. Mutat. Res. 2015, 779, 33–45. [Google Scholar] [CrossRef]
- Niture, S.K.; Jaiswal, A.K. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J. Biol. Chem. 2012, 287, 9873–9886. [Google Scholar] [CrossRef] [Green Version]
- Niture, S.K.; Jaiswal, A.K. Nrf2-induced antiapoptotic Bcl-xL protein enhances cell survival and drug resistance. Free Radic. Biol. Med. 2013, 57, 119–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; León, R.; López, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, Y.-Z.; Miao, H.; Cheng, J.-J.; Qi, J.-M. Effects of Amelioration of Total Flavonoids from Stems and Leaves of Scutellaria baicalensis Georgi on Cognitive Deficits, Neuronal Damage and Free Radicals Disorder Induced by Cerebral Ischemia in Rats. Biol. Pharma. Bull. 2006, 29, 805–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.-H.; Kuo, H.-C.; Lee, K.-F.; Tsai, T.-H. Magnolol protects neurons against ischemia injury via the downregulation of p38/MAPK, CHOP and nitrotyrosine. Toxicol. Appl. Pharmacol. 2014, 279, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.-J.; Xie, G.-N.; Liu, L.; Fu, Z.-J.; Zhang, Z.-W.; Teng, L.-Z. Sesamol attenuates oxidative stress, apoptosis and inflammation in focal cerebral ischemia/reperfusion injury. Exp. Ther. Med. 2017, 14, 841–847. [Google Scholar] [CrossRef]
- Ahmari, M.; Sharafi, A.; Mahmoudi, J.; Jafari-Anarkoli, I.; Gharbavi, M.; Hosseini, M.-J. Selegiline (l-Deprenyl) Mitigated Oxidative Stress, Cognitive Abnormalities, and Histopathological Change in Rats: Alternative Therapy in Transient Global Ischemia. J. Mol. Neurosci. 2020, 70, 1639–1648. [Google Scholar] [CrossRef]
- Liu, L.; Locascio, L.M.; Doré, S. Critical Role of Nrf2 in Experimental Ischemic Stroke. Front. Pharmacol. 2019, 10, 153. [Google Scholar] [CrossRef] [Green Version]
- Purdom-Dickinson, S.E.; Sheveleva, E.V.; Sun, H.; Chen, Q.M. Translational Control of Nrf2 Protein in Activation of Antioxidant Response by Oxidants. Mol. Pharmacol. 2007, 72, 1074–1081. [Google Scholar] [CrossRef]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [Green Version]
- Takagi, T.; Kitashoji, A.; Iwawaki, T.; Tsuruma, K.; Shimazawa, M.; Yoshimura, S.; Iwama, T.; Hara, H. Temporal activation of Nrf2 in the penumbra and Nrf2 activator-mediated neuroprotection in ischemia-reperfusion injury. Free Radic. Biol. Med. 2014, 72, 124–133. [Google Scholar] [CrossRef]
- Srivastava, S.; Alfieri, A.; Siow, R.C.M.; Mann, G.E.; Fraser, P.A. Temporal and spatial distribution of Nrf2 in rat brain following stroke: Quantification of nuclear to cytoplasmic Nrf2 content using a novel immunohistochemical technique: Quantification of cerebral Nrf2 expression in stroke. J. Physiol. 2013, 591, 3525–3538. [Google Scholar] [CrossRef]
- Dong, J.; Sulik, K.K.; Chen, S. Nrf2-Mediated Transcriptional Induction of Antioxidant Response in Mouse Embryos Exposed to Ethanol in vivo: Implications for the Prevention of Fetal Alcohol Spectrum Disorders. Antioxid. Redox Signal. 2008, 10, 2023–2033. [Google Scholar] [CrossRef] [Green Version]
- Kubo, E.; Chhunchha, B.; Singh, P.; Sasaki, H.; Singh, D.P. Sulforaphane reactivates cellular antioxidant defense by inducing Nrf2/ARE/Prdx6 activity during aging and oxidative stress. Sci. Rep. 2017, 7, 14130. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.-F.; Lu, J.-J.; Cao, Y.; Wang, W.; Li, H.-H.; Chen, J.-G.; Wang, F.; Wu, P.-F. Sulforaphane alleviates ethanol-mediated central inhibition and reverses chronic stress-induced aggravation of acute alcoholism via targeting Nrf2-regulated catalase expression. Neuropharmacology 2020, 176, 108235. [Google Scholar] [CrossRef]
- Dhaliwal, N.; Dhaliwal, J.; Singh, A.; Chopra, K. Dimethyl fumarate attenuates 2-VO-induced vascular dementia via activating the Nrf2 signaling pathway in rats. Inflammopharmacology 2021, 29, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.Y. A Small-Molecule-Inducible Nrf2-Mediated Antioxidant Response Provides Effective Prophylaxis against Cerebral Ischemia In Vivo. J. Neurosci. 2005, 25, 10321–10335. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Vollmer, M.K.; Fernandez, V.M.; Dweik, Y.; Kim, H.; Doré, S. Korean Red Ginseng Pretreatment Protects Against Long-Term Sensorimotor Deficits After Ischemic Stroke Likely Through Nrf2. Front. Cell. Neurosci. 2018, 12, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, N.; Ikeda, Y.; Ohta, Y.; Deguchi, K.; Tian, F.; Shang, J.; Matsuura, T.; Abe, K. Expression of Keap1-Nrf2 system and antioxidative proteins in mouse brain after transient middle cerebral artery occlusion. Brain Res. 2011, 1370, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Fu, B.; Zhang, X.; Zhao, T.; Chen, L.; Zhang, J.; Wang, X. Paeonol pretreatment attenuates cerebral ischemic injury via upregulating expression of pAkt, Nrf2, HO-1 and ameliorating BBB permeability in mice. Brain Res. Bull. 2014, 109, 61–67. [Google Scholar] [CrossRef]
- Li, L.; Zhang, X.; Cui, L.; Wang, L.; Liu, H.; Ji, H.; Du, Y. Ursolic acid promotes the neuroprotection by activating Nrf2 pathway after cerebral ischemia in mice. Brain Res. 2013, 1497, 32–39. [Google Scholar] [CrossRef]
- Nakano, Y.; Yamashita, T.; Li, Q.; Sato, K.; Ohta, Y.; Morihara, R.; Hishikawa, N.; Abe, K. Time-dependent change of in vivo optical imaging of oxidative stress in a mouse stroke model. J. Neurosci. Res. 2017, 95, 2030–2039. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Jing, X.; Wei, X.; Perez, R.G.; Ren, M.; Zhang, X.; Lou, H. S-allyl cysteine activates the Nrf2-dependent antioxidant response and protects neurons against ischemic injury in vitro and in vivo. J. Neurochem. 2015, 133, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Ya, B.-L.; Li, H.-F.; Wang, H.-Y.; Wu, F.; Xin, Q.; Cheng, H.-J.; Li, W.-J.; Lin, N.; Ba, Z.-H.; Zhang, R.-J.; et al. 5-HMF attenuates striatum oxidative damage via Nrf2/ARE signaling pathway following transient global cerebral ischemia. Cell Stress Chaperones 2017, 22, 55–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Wei, R.; Zhang, L.; Tan, Y.; Qian, C. Sirtuin 6 protects the brain from cerebral ischemia/reperfusion injury through NRF2 activation. Neuroscience 2017, 366, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Yan, H.; Jiao, Y.; Ohta, Y.; Liu, X.; Li, X.; Morihara, R.; Nakano, Y.; Fukui, Y.; Shi, X.; et al. Therapeutic Effects of Pretreatment with Tocovid on Oxidative Stress in Postischemic Mice Brain. J. Stroke Cerebrovasc. Dis. 2018, 27, 2096–2105. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.; Lyu, Z.; Yang, X.; Pan, Z.; Lou, J.; Dong, T. Epigallocatechin-3-gallate promotes angiogenesis via up-regulation of Nfr2 signaling pathway in a mouse model of ischemic stroke. Behav. Brain Res. 2017, 321, 79–86. [Google Scholar] [CrossRef]
- Yamauchi, K.; Nakano, Y.; Imai, T.; Takagi, T.; Tsuruma, K.; Shimazawa, M.; Iwama, T.; Hara, H. A novel nuclear factor erythroid 2-related factor 2 (Nrf2) activator RS9 attenuates brain injury after ischemia reperfusion in mice. Neuroscience 2016, 333, 302–310. [Google Scholar] [CrossRef]
- Cui, H.-Y.; Zhang, X.-J.; Yang, Y.; Zhang, C.; Zhu, C.-H.; Miao, J.-Y.; Chen, R. Rosmarinic acid elicits neuroprotection in ischemic stroke via Nrf2 and heme oxygenase 1 signaling. Neural Regen. Res. 2018, 13, 2119. [Google Scholar] [CrossRef]
- Bi, F.; Zhang, Y.; Liu, W.; Xie, K. Sinomenine activation of Nrf2 signaling prevents inflammation and cerebral injury in a mouse model of ischemic stroke. Exp. Ther. Med. 2021, 21, 647. [Google Scholar] [CrossRef]
- Wang, H.; Wei, W.; Lan, X.; Liu, N.; Li, Y.; Ma, H.; Sun, T.; Peng, X.; Zhuang, C.; Yu, J. Neuroprotective Effect of Swertiamain on Cerebral Ischemia/Reperfusion Injury by Inducing the Nrf2 Protective Pathway. ACS Chem. Neurosci. 2019, 10, 2276–2286. [Google Scholar] [CrossRef]
- Yang, M.-Y.; Yu, Q.-L.; Huang, Y.-S.; Yang, G. Neuroprotective effects of andrographolide derivative CX-10 in transient focal ischemia in rat: Involvement of Nrf2/AE and TLR/NF-κB signaling. Pharmacol. Res. 2019, 144, 227–234. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, X.; Yang, Y.; Zhang, L.; Cui, L.; Zhang, C.; Chen, R.; Xie, Y.; He, J.; He, W. Tert-butylhydroquinone enhanced angiogenesis and astrocyte activation by activating nuclear factor-E2-related factor 2/heme oxygenase-1 after focal cerebral ischemia in mice. Microvasc. Res. 2019, 126, 103891. [Google Scholar] [CrossRef] [PubMed]
- Jianrong, S.; Yanjun, Z.; Chen, Y.; Jianwen, X. DUSP14 rescues cerebral ischemia/reperfusion (IR) injury by reducing inflammation and apoptosis via the activation of Nrf-2. Biochem. Biophys. Res. Commun. 2019, 509, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, W.; Lv, C.; Wang, Y.; Ma, B.; Zhang, H.; Fan, Z.; Li, M.; Li, X. A novel biscoumarin compound ameliorates cerebral ischemia reperfusion-induced mitochondrial oxidative injury via Nrf2/Keap1/ARE signaling. Neuropharmacology 2020, 167, 107918. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Fan, C.; Chen, N.; Huang, J.; Yang, Q. Resveratrol pretreatment attenuates cerebral ischemic injury by upregulating expression of transcription factor Nrf2 and HO-1 in rats. Neurochem. Res. 2011, 36, 2352–2362. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-Y.; Kuan, Y.-H.; Li, J.-R.; Chen, W.-Y.; Ou, Y.-C.; Pan, H.-C.; Liao, S.-L.; Raung, S.-L.; Chang, C.-J.; Chen, C.-J. Docosahexaenoic acid reduces cellular inflammatory response following permanent focal cerebral ischemia in rats. J. Nutr. Biochem. 2013, 24, 2127–2137. [Google Scholar] [CrossRef] [PubMed]
- Kao, T.-K.; Chang, C.-Y.; Ou, Y.-C.; Chen, W.-Y.; Kuan, Y.-H.; Pan, H.-C.; Liao, S.-L.; Li, G.-Z.; Chen, C.-J. Tetramethylpyrazine reduces cellular inflammatory response following permanent focal cerebral ischemia in rats. Exp. Neurol. 2013, 247, 188–201. [Google Scholar] [CrossRef]
- Alfieri, A.; Srivastava, S.; Siow, R.C.M.; Cash, D.; Modo, M.; Duchen, M.R.; Fraser, P.A.; Williams, S.C.R.; Mann, G.E. Sulforaphane preconditioning of the Nrf2/HO-1 defense pathway protects the cerebral vasculature against blood–brain barrier disruption and neurological deficits in stroke. Free Radic. Biol. Med. 2013, 65, 1012–1022. [Google Scholar] [CrossRef]
- Wu, J.; Li, Q.; Wang, X.; Yu, S.; Li, L.; Wu, X.; Chen, Y.; Zhao, J.; Zhao, Y. Neuroprotection by Curcumin in Ischemic Brain Injury Involves the Akt/Nrf2 Pathway. PLoS ONE 2013, 8, e59843. [Google Scholar] [CrossRef] [Green Version]
- Peng, B.; Zhao, P.; Lu, Y.-P.; Chen, M.-M.; Sun, H.; Wu, X.-M.; Zhu, L. Z-ligustilide activates the Nrf2/HO-1 pathway and protects against cerebral ischemia-reperfusion injury in vivo and in vitro. Brain Res. 2013, 1520, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Wang, M.; Jing, X.; Shi, H.; Ren, M.; Lou, H. (−)-Epigallocatechin Gallate Protects Against Cerebral Ischemia-Induced Oxidative Stress via Nrf2/ARE Signaling. Neurochem. Res. 2014, 39, 1292–1299. [Google Scholar] [CrossRef]
- Zhang, J.; Fu, B.; Zhang, X.; Zhang, L.; Bai, X.; Zhao, X.; Chen, L.; Cui, L.; Zhu, C.; Wang, L.; et al. Bicyclol upregulates transcription factor Nrf2, HO-1 expression and protects rat brains against focal ischemia. Brain Res. Bull. 2014, 100, 38–43. [Google Scholar] [CrossRef]
- Meng, H.; Guo, J.; Wang, H.; Yan, P.; Niu, X.; Zhang, J. Erythropoietin activates Keap1-Nrf2/ARE pathway in rat brain after ischemia. Int. J. Neurosci. 2014, 124, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Sun, J.; Lv, G.; Yu, Y.; Wang, G.; Xie, K.; Jiao, Y.; Yu, Y. Sevoflurane postconditioning attenuates cerebral ischemia-reperfusion injury via protein kinase B/nuclear factor-erythroid 2-related factor 2 pathway activation. Int. J. Dev. Neurosci. 2014, 38, 79–86. [Google Scholar] [CrossRef]
- Guo, H.; Li, M.; Liu, Q.; Guo, L.; Ma, M.; Wang, S.; Yu, B.; Hu, L.-M. Danhong Injection Attenuates Ischemia/Reperfusion-Induced Brain Damage Which is Associating with Nrf2 Levels In Vivo and In Vitro. Neurochem. Res. 2014, 39, 1817–1824. [Google Scholar] [CrossRef] [PubMed]
- Ashabi, G.; Khalaj, L.; Khodagholi, F.; Goudarzvand, M.; Sarkaki, A. Pre-treatment with metformin activates Nrf2 antioxidant pathways and inhibits inflammatory responses through induction of AMPK after transient global cerebral ischemia. Metab. Brain Dis. 2015, 30, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-Y.; Kao, T.-K.; Chen, W.-Y.; Ou, Y.-C.; Li, J.-R.; Liao, S.-L.; Raung, S.-L.; Chen, C.-J. Tetramethylpyrazine inhibits neutrophil activation following permanent cerebral ischemia in rats. Biochem. Biophys. Res. Commun. 2015, 463, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhu, K.; Liu, Y.; Wu, X.; Wu, J.; Zhao, Y.; Zhao, J. Targeting thioredoxin-1 with siRNA exacerbates oxidative stress injury after cerebral ischemia/reperfusion in rats. Neuroscience 2015, 284, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qiu, J.; Wang, Z.; You, W.; Wu, L.; Ji, C.; Chen, G. Dimethylfumarate alleviates early brain injury and secondary cognitive deficits after experimental subarachnoid hemorrhage via activation of Keap1-Nrf2-ARE system. J. Neurosurg. 2015, 123, 915–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Park, Y.H.; Jeon, Y.T.; Hwang, J.W.; Lim, Y.J.; Kim, E.; Park, S.Y.; Park, H.P. Sevoflurane post-conditioning increases nuclear factor erythroid 2-related factor and haemoxygenase-1 expression via protein kinase C pathway in a rat model of transient global cerebral ischaemia. Br. J. Anaesth. 2015, 114, 307–318. [Google Scholar] [CrossRef] [Green Version]
- Lou, J.; Cao, G.; Li, R.; Liu, J.; Dong, Z.; Xu, L. β-Caryophyllene Attenuates Focal Cerebral Ischemia-Reperfusion Injury by Nrf2/HO-1 Pathway in Rats. Neurochem. Res. 2016, 41, 1291–1304. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, X.; Zhang, C.; Bai, X.; Zhang, J.; Zhao, X.; Chen, L.; Wang, L.; Zhu, C.; Cui, L.; et al. Nobiletin promotes antioxidant and anti-inflammatory responses and elicits protection against ischemic stroke in vivo. Brain Res. 2016, 1636, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Cai, J.; Kostuk, E.W.; Rosenwasser, R.; Iacovitti, L. Fumarate modulates the immune/inflammatory response and rescues nerve cells and neurological function after stroke in rats. J. Neuroinflamm. 2016, 13, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xian, J.W.; Choi, A.Y.-T.; Lau, C.B.-S.; Leung, W.N.; Ng, C.F.; Chan, C.W. Gastrodia and Uncaria (tianma gouteng) water extract exerts antioxidative and antiapoptotic effects against cerebral ischemia in vitro and in vivo. Chin. Med. 2016, 11, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Chen, Y.; Yu, S.; Li, L.; Zhao, X.; Li, Q.; Zhao, J.; Zhao, Y. Neuroprotective effects of sulfiredoxin-1 during cerebral ischemia/reperfusion oxidative stress injury in rats. Brain Res. Bull. 2017, 132, 99–108. [Google Scholar] [CrossRef]
- Mršić-Pelčić, J.; Pilipović, K.; Pelčić, G.; Vitezić, D.; Župan, G. Decrease in Oxidative Stress Parameters after Post-Ischaemic Recombinant Human Erythropoietin Administration in the Hippocampus of Rats Exposed to Focal Cerebral Ischaemia. Basic Clin. Pharmacol. Toxicol. 2017, 121, 453–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atef, R.M.; Agha, A.M.; Abdel-Rhaman, A.-R.A.; Nassar, N.N. The Ying and Yang of Adenosine A1 and A2A Receptors on ERK1/2 Activation in a Rat Model of Global Cerebral Ischemia Reperfusion Injury. Mol. Neurobiol. 2018, 55, 1284–1298. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, Y.; He, Q.; Li, L.; Xie, H.; Zhao, Y.; Zhao, J. Nrf2 inhibits NLRP3 inflammasome activation through regulating Trx1/TXNIP complex in cerebral ischemia reperfusion injury. Behav. Brain Res. 2018, 336, 32–39. [Google Scholar] [CrossRef]
- Miao, Z.-Y.; Xia, X.; Che, L.; Song, Y.-T. Genistein attenuates brain damage induced by transient cerebral ischemia through up-regulation of Nrf2 expression in ovariectomized rats. Neurol. Res. 2018, 40, 689–695. [Google Scholar] [CrossRef]
- Guo, H.; Adah, D.; James, P.B.; Liu, Q.; Li, G.; Ahmadu, P.; Chai, L.; Wang, S.; Liu, Y.; Hu, L. Xueshuantong Injection (Lyophilized) Attenuates Cerebral Ischemia/Reperfusion Injury by the Activation of Nrf2–VEGF Pathway. Neurochem. Res. 2018, 43, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhao, Y.; Li, Y.; Wu, J.; Yu, S.; Zhu, J.; Li, L.; Zhao, Y. Sestrin2 overexpression attenuates focal cerebral ischemic injury in rat by increasing Nrf2/HO-1 pathway-mediated angiogenesis. Neuroscience 2019, 410, 140–149. [Google Scholar] [CrossRef]
- Li, Y.; Wu, J.; Yu, S.; Zhu, J.; Zhou, Y.; Wang, P.; Li, L.; Zhao, Y. Sestrin2 promotes angiogenesis to alleviate brain injury by activating Nrf2 through regulating the interaction between p62 and Keap1 following photothrombotic stroke in rats. Brain Res. 2020, 1745, 146948. [Google Scholar] [CrossRef] [PubMed]
- Malik, I.; Shah, F.A.; Ali, T.; Tan, Z.; Alattar, A.; Ullah, N.; Khan, A.; Alshaman, R.; Li, S. Potent Natural Antioxidant Carveol Attenuates MCAO-Stress Induced Oxidative, Neurodegeneration by Regulating the Nrf-2 Pathway. Front. Neurosci. 2020, 14, 659. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, H.; Zhang, L.; Xu, Z.; Song, Y.; Wang, X.; Chu, R.; Xiao, Y.; Sun, M.; Ma, Y.; et al. Cottonseed Oil Alleviates Ischemic Stroke-Induced Oxidative Stress Injury Via Activating the Nrf2 Signaling Pathway. Mol. Neurobiol. 2021, 58, 2494–2507. [Google Scholar] [CrossRef]
- Gao, J.; Chen, N.; Li, N.; Xu, F.; Wang, W.; Lei, Y.; Shi, J.; Gong, Q. Neuroprotective Effects of Trilobatin, a Novel Naturally Occurring Sirt3 Agonist from Lithocarpus polystachyus Rehd., Mitigate Cerebral Ischemia/Reperfusion Injury: Involvement of TLR4/NF-κB and Nrf2/Keap-1 Signaling. Antioxid. Redox Signal. 2020, 33, 117–143. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Yue, Y.; Li, J.; Li, Z.; Li, X.; Niu, Y.; Xiang, J.; Ding, H. Procyanidin B2 attenuates neurological deficits and blood-brain barrier disruption in a rat model of cerebral ischemia. Mol. Nutr. Food Res. 2015, 59, 1930–1941. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Yue, Y.; Peng, A.; Zhang, L.; Xiang, J.; Cao, X.; Ding, H.; Yin, S. Myricetin ameliorates brain injury and neurological deficits via Nrf2 activation after experimental stroke in middle-aged rats. Food Funct. 2016, 7, 2624–2634. [Google Scholar] [CrossRef]
- Janyou, A.; Wicha, P.; Jittiwat, J.; Suksamrarn, A.; Tocharus, C.; Tocharus, J. Dihydrocapsaicin Attenuates Blood Brain Barrier and Cerebral Damage in Focal Cerebral Ischemia/Reperfusion via Oxidative Stress and Inflammatory. Sci. Rep. 2017, 7, 10556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, J.; Cui, J.; Yang, Z.; Guo, C.; Cao, J.; Xi, M.; Weng, Y.; Yin, Y.; Wang, Y.; Wei, G.; et al. Neuroprotective effect of Apelin 13 on ischemic stroke by activating AMPK/GSK-3β/Nrf2 signaling. J. Neuroinflamm. 2019, 16, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Li, X.; Wu, H.; Yang, Z.; Fei, L.; Zhu, J. Theaflavin attenuates cerebral ischemia/reperfusion injury by abolishing miRNA-128-3p-mediated Nrf2 inhibition and reducing oxidative stress. Mol. Med. Rep. 2019, 20, 4893–4904. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Tucker, L.D.; Yan, D.; Lu, Y.; Yang, L.; Wu, C.; Li, Y.; Zhang, Q. Tert-butylhydroquinone post-treatment attenuates neonatal hypoxic-ischemic brain damage in rats. Neurochem. Int. 2018, 116, 1–12. [Google Scholar] [CrossRef]
- Liu, D.; Wang, H.; Zhang, Y.; Zhang, Z. Protective Effects of Chlorogenic Acid on Cerebral Ischemia/Reperfusion Injury Rats by Regulating Oxidative Stress-Related Nrf2 Pathway. Drug Des. Dev. Ther. 2020, 14, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kam, K.-Y.; Yu, S.J.; Jeong, N.; Hong, J.H.; Jalin, A.M.A.A.; Lee, S.; Choi, Y.W.; Lee, C.K.; Kang, S.G. p-Hydroxybenzyl alcohol prevents brain injury and behavioral impairment by activating Nrf2, PDI, and neurotrophic factor genes in a rat model of brain ischemia. Mol. Cells 2011, 31, 209–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Li, R.; Tu, P.; Chen, J.; Zeng, K.; Jiang, Y. Total Glycosides of Cistanche deserticola Promote Neurological Function Recovery by Inducing Neurovascular Regeneration via Nrf-2/Keap-1 Pathway in MCAO/R Rats. Front. Pharmacol. 2020, 11, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, T.-C. Nuclear Factor-Erythroid 2-Related Factor 2 (Nrf2) and Mitochondrial Dynamics/Mitophagy in Neurological Diseases. Antioxidants 2020, 9, 617. [Google Scholar] [CrossRef]
- Jiang, S.; Deng, C.; Lv, J.; Fan, C.; Hu, W.; Di, S.; Yan, X.; Ma, Z.; Liang, Z.; Yang, Y. Nrf2 Weaves an Elaborate Network of Neuroprotection Against Stroke. Mol. Neurobiol. 2017, 54, 1440–1455. [Google Scholar] [CrossRef]
- Li, H.; Tang, Z.; Chu, P.; Song, Y.; Yang, Y.; Sun, B.; Niu, M.; Qaed, E.; Shopit, A.; Han, G.; et al. Neuroprotective effect of phosphocreatine on oxidative stress and mitochondrial dysfunction induced apoptosis in vitro and in vivo: Involvement of dual PI3K/Akt and Nrf2/HO-1 pathways. Free Radic. Biol. Med. 2018, 120, 228–238. [Google Scholar] [CrossRef]
- Zolnourian, A.; Galea, I.; Bulters, D. Neuroprotective Role of the Nrf2 Pathway in Subarachnoid Haemorrhage and Its Therapeutic Potential. Oxid. Med. Cell. Longev. 2019, 2019, 1–21. [Google Scholar] [CrossRef]
- Narayanan, S.V.; Dave, K.R.; Saul, I.; Perez-Pinzon, M.A. Resveratrol Preconditioning Protects Against Cerebral Ischemic Injury via Nuclear Erythroid 2–Related Factor 2. Stroke 2015, 46, 1626–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, N.B.; Jain, S.; Agarwal, N.K.; Mediratta, P.K.; Sharma, K.K. Modulation of pentylenetetrazole-induced kindling and oxidative stress by curcumin in mice. Phytomedicine 2011, 18, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin Inhibits Formation of Amyloid β Oligomers and Fibrils, Binds Plaques, and Reduces Amyloid in Vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Bu, Q.; Liu, X.; Hu, W.; Wang, Y. Neuroprotective effect of TAT-14-3-3ε fusion protein against cerebral ischemia/reperfusion injury in rats. PLoS ONE 2014, 9, e93334. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.; Sanderson, I.R.; MacDonald, T.T. Curcumin as a therapeutic agent: The evidence from in vitro, animal and human studies. Br. J. Nutr. 2010, 103, 1545–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balogun, E.; Hoque, M.; Gong, P.; Killeen, E.; Green, C.J.; Foresti, R.; Alam, J.; Motterlini, R. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem. J. 2003, 371, 887–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turpaev, K.T. Keap1-Nrf2 signaling pathway: Mechanisms of regulation and role in protection of cells against toxicity caused by xenobiotics and electrophiles. Biochem. Mosc. 2013, 78, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.W.; Chun, K.-S.; Kim, D.-H.; Kim, S.-J.; Kim, S.H.; Cho, N.-C.; Na, H.-K.; Surh, Y.-J. Curcumin induces stabilization of Nrf2 protein through Keap1 cysteine modification. Biochem. Pharmacol. 2020, 173, 113820. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.; Umar, S.; Ashafaq, M.; Akhtar, M.; Iqbal, Z.; Samim, M.; Ahmad, F.J. A comparative study of PNIPAM nanoparticles of curcumin, demethoxycurcumin, and bisdemethoxycurcumin and their effects on oxidative stress markers in experimental stroke. Protoplasma 2013, 250, 1327–1338. [Google Scholar] [CrossRef]
- Dohare, P.; Garg, P.; Jain, V.; Nath, C.; Ray, M. Dose dependence and therapeutic window for the neuroprotective effects of curcumin in thromboembolic model of rat. Behav. Brain Res. 2008, 193, 289–297. [Google Scholar] [CrossRef]
- Funk, J.L.; Frye, J.B.; Davis-Gorman, G.; Spera, A.L.; Bernas, M.J.; Witte, M.H.; Weinand, M.E.; Timmermann, B.N.; McDonagh, P.F.; Ritter, L. Curcuminoids Limit Neutrophil-Mediated Reperfusion Injury in Experimental Stroke by Targeting the Endothelium. Microcirculation 2013, 20, 544–554. [Google Scholar] [CrossRef]
- Ghoneim, A.I.; Abdel-Naim, A.B.; Khalifa, A.E.; El-Denshary, E.S. Protective effects of curcumin against ischaemia/reperfusion insult in rat forebrain. Pharmacol. Res. 2002, 46, 273–279. [Google Scholar] [CrossRef]
- Lapchak, P.A.; Schubert, D.R.; Maher, P.A. Delayed treatment with a novel neurotrophic compound reduces behavioral deficits in rabbit ischemic stroke: Neuroprotection from ischemic stroke. J. Neurochem. 2011, 116, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Rathore, P.; Dohare, P.; Varma, S.; Ray, A.; Sharma, U.; Jaganathanan, N.R.; Ray, M. Curcuma Oil: Reduces Early Accumulation of Oxidative Product and is Anti-apoptogenic in Transient Focal Ischemia in Rat Brain. Neurochem. Res. 2008, 33, 1672–1682. [Google Scholar] [CrossRef]
- Shukla, P.K.; Khanna, V.K.; Ali, M.M.; Khan, M.Y.; Srimal, R.C. Anti-ischemic effect of curcumin in rat brain. Neurochem. Res. 2008, 33, 1036–1043. [Google Scholar] [CrossRef]
- Thiyagarajan, M.; Sharma, S.S. Neuroprotective effect of curcumin in middle cerebral artery occlusion induced focal cerebral ischemia in rats. Life Sci. 2004, 74, 969–985. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yu, S.; Zheng, W.; Feng, G.; Luo, G.; Wang, L.; Zhao, Y. Curcumin improves outcomes and attenuates focal cerebral ischemic injury via antiapoptotic mechanisms in rats. Neurochem. Res. 2010, 35, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wang, W.; Sun, Y.J.; Hu, M.; Li, F.; Zhu, D.Y. Neuroprotective effect of curcumin on focal cerebral ischemic rats by preventing blood-brain barrier damage. Eur. J. Pharmacol. 2007, 561, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Suwanwela, N.C.; Patumraj, S. Curcumin by down-regulating NF-kB and elevating Nrf2, reduces brain edema and neurological dysfunction after cerebral I/R. Microvasc. Res. 2016, 106, 117–127. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, Y.; Zheng, W.; Lu, Y.; Feng, G.; Yu, S. Neuroprotective effect of curcumin on transient focal cerebral ischemia in rats. Brain Res. 2008, 1229, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, M.A.; Waeber, C. Remote ischemic preconditioning: Making the brain more tolerant, safely and inexpensively. Circulation 2011, 123, 709–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Reyes, S.; Guzmán-Beltrán, S.; Medina-Campos, O.N.; Pedraza-Chaverri, J. Curcumin Pretreatment Induces Nrf2 and an Antioxidant Response and Prevents Hemin-Induced Toxicity in Primary Cultures of Cerebellar Granule Neurons of Rats. Oxid. Med. Cell. Longev. 2013, 2013, 1–14. [Google Scholar] [CrossRef]
- Zhao, R.; Yang, B.; Wang, L.; Xue, P.; Deng, B.; Zhang, G.; Jiang, S.; Zhang, M.; Liu, M.; Pi, J.; et al. Curcumin protects human keratinocytes against inorganic arsenite-induced acute cytotoxicity through an NRF2-dependent mechanism. Oxid. Med. Cell. Longev. 2013, 2013, 412576. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhang, X.; Fan, H.; Liu, Y. Curcumin upregulates transcription factor Nrf2, HO-1 expression and protects rat brains against focal ischemia. Brain Res. 2009, 1282, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.N.; Robinson, S.R.; Dringen, R.; Bishop, G.M. Uptake, metabolism and toxicity of hemin in cultured neurons. Neurochem. Int. 2011, 58, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Liu, Z.; Liang, G. Promising curcumin-based drug design: Mono-carbonyl analogues of curcumin (MACs). Curr. Pharm. Des. 2013, 19, 2114–2135. [Google Scholar] [PubMed]
- Jangra, A.; Kwatra, M.; Singh, T.; Pant, R.; Kushwah, P.; Sharma, Y.; Saroha, B.; Datusalia, A.K.; Bezbaruah, B.K. Piperine Augments the Protective Effect of Curcumin Against Lipopolysaccharide-Induced Neurobehavioral and Neurochemical Deficits in Mice. Inflammation 2016, 39, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Antony, B.; Merina, B.; Iyer, V.; Judy, N.; Lennertz, K.; Joyal, S. A pilot cross-over study to evaluate human oral bioavailability of BCM-95® CG (BiocurcumaxTM), a novel bioenhanced preparation of curcumin. Indian J. Pharm. Sci. 2008, 70, 445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werdenberg, D.; Joshi, R.; Wolffram, S.; Merkle, H.P.; Langguth, P. Presystemic metabolism and intestinal absorption of antipsoriatic fumaric acid esters. Biopharm. Drug Dispos. 2003, 24, 259–273. [Google Scholar] [CrossRef]
- Linker, R.A.; Haghikia, A. Dimethyl fumarate in multiple sclerosis: Latest developments, evidence and place in therapy. Ther. Adv. Chronic Dis. 2016, 7, 198–207. [Google Scholar] [CrossRef] [Green Version]
- Reich, K.; Thaci, D.; Mrowietz, U.; Kamps, A.; Neureither, M.; Luger, T. Efficacy and safety of fumaric acid esters in the long-term treatment of psoriasis—A retrospective study (FUTURE). J. Dtsch. Dermatol. Ges. 2009, 7, 603–611. [Google Scholar] [CrossRef]
- Meissner, M.; Valesky, E.M.; Kippenberger, S.; Kaufmann, R. Dimethyl fumarate—Only an anti-psoriatic medication? J. Dtsch. Dermatol. Ges. 2012, 10, 793–801. [Google Scholar] [CrossRef]
- Fox, R.J.; Miller, D.H.; Phillips, J.T.; Hutchinson, M.; Havrdova, E.; Kita, M.; Yang, M.; Raghupathi, K.; Novas, M.; Sweetser, M.T.; et al. Placebo-Controlled Phase 3 Study of Oral BG-12 or Glatiramer in Multiple Sclerosis. N. Engl. J. Med. 2012, 367, 1087–1097. [Google Scholar] [CrossRef] [Green Version]
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; Sheikh, S.I.; et al. Placebo-Controlled Phase 3 Study of Oral BG-12 for Relapsing Multiple Sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [CrossRef] [Green Version]
- Salmen, A.; Gold, R. Mode of action and clinical studies with fumarates in multiple sclerosis. Exp. Neurol. 2014, 262 Pt A, 52–56. [Google Scholar] [CrossRef]
- De Jong, R.; Bezemer, A.C.; Zomerdijk, T.P.L.; van de Pouw-Kraan, T.; Ottenhoff, T.H.M.; Nibbering, P.H. Selective stimulation of T helper 2 cytokine responses by the anti-psoriasis agent monomethylfumarate. Eur. J. Immunol. 1996, 26, 2067–2074. [Google Scholar] [CrossRef]
- Asadullah, K.; Schmid, H.; Friedrich, M.; Randow, F.; Volk, H.-D.; Sterry, W.; Döcke, W.-D. Influence of monomethylfumarate on monocytic cytokine formation—Explanation for adverse and therapeutic effects in psoriasis? Arch. Dermatol. Res. 1997, 289, 623–630. [Google Scholar] [CrossRef]
- Litjens, N.H.R.; Rademaker, M.; Ravensbergen, B.; Rea, D.; van der Plas, M.J.A.; Thio, B.; Walding, A.; van Dissel, J.T.; Nibbering, P.H. Monomethylfumarate affects polarization of monocyte-derived dendritic cells resulting in down-regulated Th1 lymphocyte responses. Eur. J. Immunol. 2004, 34, 565–575. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Brück, J.; Kellerer, C.; Deng, C.; Peng, H.; Rothfuss, O.; Hussain, R.Z.; Gocke, A.R.; Respa, A.; Glocova, I.; et al. Fumarates improve psoriasis and multiple sclerosis by inducing type II dendritic cells. J. Exp. Med. 2011, 208, 2291–2303. [Google Scholar] [CrossRef]
- Treumer, F.; Zhu, K.; Gläser, R.; Mrowietz, U. Dimethylfumarate is a potent inducer of apoptosis in human T cells. J. Investig. Dermatol. 2003, 121, 1383–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loewe, R.; Holnthoner, W.; Gröger, M.; Pillinger, M.; Gruber, F.; Mechtcheriakova, D.; Hofer, E.; Wolff, K.; Petzelbauer, P. Dimethylfumarate inhibits TNF-induced nuclear entry of NF-kappa B/p65 in human endothelial cells. J. Immunol. 2002, 168, 4781–4787. [Google Scholar] [CrossRef] [PubMed]
- Nibbering, P.H.; Thio, B.; Zomerdijk, T.P.; Bezemer, A.C.; Beijersbergen, R.L.; van Furth, R. Effects of monomethylfumarate on human granulocytes. J. Investig. Dermatol. 1993, 101, 37–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ockenfels, H.M.; Schultewolter, T.; Ockenfels, G.; Funk, R.; Goos, M. The antipsoriatic agent dimethylfumarate immunomodulates T-cell cytokine secretion and inhibits cytokines of the psoriatic cytokine network. Br. J. Dermatol. 1998, 139, 390–395. [Google Scholar] [CrossRef]
- Schilling, S.; Goelz, S.; Linker, R.; Luehder, F.; Gold, R. Fumaric acid esters are effective in chronic experimental autoimmune encephalomyelitis and suppress macrophage infiltration. Clin. Exp. Immunol. 2006, 145, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Sebok, B.; Bonnekoh, B.; Vetter, R.; Schneider, I.; Gollnick, H.; Mahrle, G. The antipsoriatic dimethyl-fumarate suppresses interferon-gamma -induced ICAM-1 and HLA-DR expression on hyperproliferative keratinocytes. Quantification by a culture plate-directed APAAP-ELISA technique. Eur. J. Dermatol. 1998, 8, 29–32. [Google Scholar] [PubMed]
- Stoof, T.J.; Flier, J.; Sampat, S.; Nieboer, C.; Tensen, C.P.; Boorsma, D.M. The antipsoriatic drug dimethylfumarate strongly suppresses chemokine production in human keratinocytes and peripheral blood mononuclear cells. Br. J. Dermatol. 2001, 144, 1114–1120. [Google Scholar] [CrossRef]
- Linker, R.A.; Lee, D.-H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef] [Green Version]
- Scannevin, R.H.; Chollate, S.; Jung, M.; Shackett, M.; Patel, H.; Bista, P.; Zeng, W.; Ryan, S.; Yamamoto, M.; Lukashev, M.; et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J. Pharmacol. Exp. Ther. 2012, 341, 274–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.X.; Lisi, L.; Dello Russo, C.; Polak, P.E.; Sharp, A.; Weinberg, G.; Kalinin, S.; Feinstein, D.L. The anti-inflammatory effects of dimethyl fumarate in astrocytes involve glutathione and haem oxygenase-1. ASN Neuro 2011, 3, e00055. [Google Scholar] [CrossRef] [PubMed]
- Iniaghe, L.O.; Krafft, P.R.; Klebe, D.W.; Omogbai, E.K.I.; Zhang, J.H.; Tang, J. Dimethyl fumarate confers neuroprotection by casein kinase 2 phosphorylation of Nrf2 in murine intracerebral hemorrhage. Neurobiol. Dis. 2015, 82, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Sun, G.; Zhang, J.; Ting, S.-M.; Gonzales, N.; Aronowski, J. Dimethyl Fumarate Protects Brain From Damage Produced by Intracerebral Hemorrhage by Mechanism Involving Nrf2. Stroke 2015, 46, 1923–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunze, R.; Urrutia, A.; Hoffmann, A.; Liu, H.; Helluy, X.; Pham, M.; Reischl, S.; Korff, T.; Marti, H.H. Dimethyl fumarate attenuates cerebral edema formation by protecting the blood-brain barrier integrity. Exp. Neurol. 2015, 266, 99–111. [Google Scholar] [CrossRef]
- Lin-Holderer, J.; Li, L.; Gruneberg, D.; Marti, H.H.; Kunze, R. Fumaric acid esters promote neuronal survival upon ischemic stress through activation of the Nrf2 but not HIF-1 signaling pathway. Neuropharmacology 2016, 105, 228–240. [Google Scholar] [CrossRef]
- Liu, L.; Vollmer, M.K.; Ahmad, A.S.; Fernandez, V.M.; Kim, H.; Doré, S. Pretreatment with Korean red ginseng or dimethyl fumarate attenuates reactive gliosis and confers sustained neuroprotection against cerebral hypoxic-ischemic damage by an Nrf2-dependent mechanism. Free Radic. Biol. Med. 2019, 131, 98–114. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Vollmer, M.K.; Kelly, M.G.; Fernandez, V.M.; Fernandez, T.G.; Kim, H.; Doré, S. Reactive Gliosis Contributes to Nrf2-Dependent Neuroprotection by Pretreatment with Dimethyl Fumarate or Korean Red Ginseng Against Hypoxic-Ischemia: Focus on Hippocampal Injury. Mol. Neurobiol. 2020, 57, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, T.; Liu, H.; Wang, X.; Bo, S.; Xie, Y.; Bai, X.; Wu, L.; Wang, Z.; Liu, D. Resveratrol exerts antidepressant properties in the chronic unpredictable mild stress model through the regulation of oxidative stress and mTOR pathway in the rat hippocampus and prefrontal cortex. Behav. Brain Res. 2016, 302, 191–199. [Google Scholar] [CrossRef]
- Wang, L.; Wang, C.; Jia, Y.; Liu, Z.; Shu, X.; Liu, K. Resveratrol Increases Anti-Proliferative Activity of Bestatin Through Downregulating P-Glycoprotein Expression Via Inhibiting PI3K/Akt/mTOR Pathway in K562/ADR Cells. J. Cell. Biochem. 2016, 117, 1233–1239. [Google Scholar] [CrossRef]
- Sui, T.; Ma, L.; Bai, X.; Li, Q.; Xu, X. Resveratrol inhibits the phosphatidylinositide 3-kinase/protein kinase B/mammalian target of rapamycin signaling pathway in the human chronic myeloid leukemia K562 cell line. Oncol. Lett. 2014, 7, 2093–2098. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.-H.; Lee, H.; Lee, S.-R. Protective effect of resveratrol against neuronal damage following transient global cerebral ischemia in mice. J. Nutr. Biochem. 2016, 27, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Li, Y.-H.; Wang, J.-J.; Pan, J.; Lu, H. Endoplasmic reticulum stress could induce autophagy and apoptosis and enhance chemotherapy sensitivity in human esophageal cancer EC9706 cells by mediating PI3K/Akt/mTOR signaling pathway. Tumour Biol. 2017, 39, 101042831770574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simão, F.; Matté, A.; Pagnussat, A.S.; Netto, C.A.; Salbego, C.G. Resveratrol prevents CA1 neurons against ischemic injury by parallel modulation of both GSK-3β and CREB through PI3-K/Akt pathways: Resveratrol prevents ischemic injury through PI3-K. Eur. J. Neurosci. 2012, 36, 2899–2905. [Google Scholar] [CrossRef]
- Singh, N.; Agrawal, M.; Doré, S. Neuroprotective properties and mechanisms of resveratrol in in vitro and in vivo experimental cerebral stroke models. ACS Chem. Neurosci. 2013, 4, 1151–1162. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, M.; Kumar, V.; Singh, A.K.; Kashyap, M.P.; Khanna, V.K.; Siddiqui, M.A.; Pant, A.B. trans -Resveratrol Protects Ischemic PC12 Cells by Inhibiting the Hypoxia Associated Transcription Factors and Increasing the Levels of Antioxidant Defense Enzymes. ACS Chem. Neurosci. 2013, 4, 285–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamin, L.L.; Dillenburg-Pilla, P.; Argenta-Comiran, R.; Horn, A.P.; Simão, F.; Nassif, M.; Gerhardt, D.; Frozza, R.L.; Salbego, C. Protective effect of resveratrol against oxygen–glucose deprivation in organotypic hippocampal slice cultures: Involvement of PI3-K pathway. Neurobiol. Dis. 2006, 24, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Bournival, J.; Quessy, P.; Martinoli, M.-G. Protective Effects of Resveratrol and Quercetin Against MPP+ -Induced Oxidative Stress Act by Modulating Markers of Apoptotic Death in Dopaminergic Neurons. Cell Mol. Neurobiol. 2009, 29, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Zini, R.; Morin, C.; Bertelli, A.; Bertelli, A.A.; Tillement, J.P. Effects of resveratrol on the rat brain respiratory chain. Drugs Exp. Clin. Res. 1999, 25, 87–97. [Google Scholar]
- Hou, Y.; Wang, K.; Wan, W.; Cheng, Y.; Pu, X.; Ye, X. Resveratrol provides neuroprotection by regulating the JAK2/STAT3/PI3K/AKT/mTOR pathway after stroke in rats. Genes Dis. 2018, 5, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Candelario-Jalil, E.; de Oliveira, A.; Gräf, S.; Bhatia, H.S.; Hüll, M.; Muñoz, E.; Fiebich, B.L. Resveratrol potently reduces prostaglandin E2 production and free radical formation in lipopolysaccharide-activated primary rat microglia. J. Neuroinflamm. 2007, 4, 25. [Google Scholar] [CrossRef] [Green Version]
- Bi, X.L.; Yang, J.Y.; Dong, Y.X.; Wang, J.M.; Cui, Y.H.; Ikeshima, T.; Zhao, Y.Q.; Wu, C.F. Resveratrol inhibits nitric oxide and TNF-α production by lipopolysaccharide-activated microglia. Int. Immunopharmacol. 2005, 5, 185–193. [Google Scholar] [CrossRef]
- Kim, Y.A.; Kim, G.-Y.; Park, K.-Y.; Choi, Y.H. Resveratrol Inhibits Nitric Oxide and Prostaglandin E2 Production by Lipopolysaccharide-Activated C6 Microglia. J. Med. Food 2007, 10, 218–224. [Google Scholar] [CrossRef]
- Bureau, G.; Longpré, F.; Martinoli, M.-G. Resveratrol and quercetin, two natural polyphenols, reduce apoptotic neuronal cell death induced by neuroinflammation. J. Neurosci. Res. 2008, 86, 403–410. [Google Scholar] [CrossRef]
- Innamorato, N.G.; Rojo, A.I.; García-Yagüe, Á.J.; Yamamoto, M.; de Ceballos, M.L.; Cuadrado, A. The Transcription Factor Nrf2 Is a Therapeutic Target against Brain Inflammation. J. Immunol. 2008, 181, 680–689. [Google Scholar] [CrossRef] [Green Version]
- Shah, Z.A.; Li, R.-C.; Thimmulappa, R.K.; Kensler, T.W.; Yamamoto, M.; Biswal, S.; Doré, S. Role of reactive oxygen species in modulation of Nrf2 following ischemic reperfusion injury. Neuroscience 2007, 147, 53–59. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-Y.; Jang, J.-H.; Li, M.-H.; Surh, Y.-J. Resveratrol upregulates heme oxygenase-1 expression via activation of NF-E2-related factor 2 in PC12 cells. Biochem. Biophys. Res. Commun. 2005, 331, 993–1000. [Google Scholar] [CrossRef]
- Agrawal, M.; Kumar, V.; Kashyap, M.P.; Khanna, V.K.; Randhawa, G.S.; Pant, A.B. Ischemic insult induced apoptotic changes in PC12 cells: Protection by trans resveratrol. Eur. J. Pharmacol. 2011, 666, 5–11. [Google Scholar] [CrossRef]
- Ungvari, Z.; Bagi, Z.; Feher, A.; Recchia, F.A.; Sonntag, W.E.; Pearson, K.; de Cabo, R.; Csiszar, A. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H18–H24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Huang, J.; Shen, C.; Cheng, W.; Yu, P.; Wang, L.; Tang, F.; Guo, S.; Yang, Q.; Zhang, J. Resveratrol Treatment in Different Time-Attenuated Neuronal Apoptosis After Oxygen and Glucose Deprivation/Reoxygenation via Enhancing the Activation of Nrf-2 Signaling Pathway In Vitro. Cell Transplant. 2018, 27, 1789–1797. [Google Scholar] [CrossRef]
- Van Horssen, J.; Schreibelt, G.; Bö, L.; Montagne, L.; Drukarch, B.; van Muiswinkel, F.L.; de Vries, H.E. NAD(P)H:quinone oxidoreductase 1 expression in multiple sclerosis lesions. Free Radic. Biol. Med. 2006, 41, 311–317. [Google Scholar] [CrossRef]
- Ghawi, S.K.; Methven, L.; Niranjan, K. The potential to intensify sulforaphane formation in cooked broccoli (Brassica oleracea var. italica) using mustard seeds (Sinapis alba). Food Chem. 2013, 138, 1734–1741. [Google Scholar] [CrossRef] [PubMed]
- Hwang, E.-S.; Jeffery, E.H. Induction of Quinone Reductase by Sulforaphane and Sulforaphane N -Acetylcysteine Conjugate in Murine Hepatoma Cells. J. Med. Food 2005, 8, 198–203. [Google Scholar] [CrossRef]
- Clarke, J.D.; Dashwood, R.H.; Ho, E. Multi-targeted prevention of cancer by sulforaphane. Cancer Lett. 2008, 269, 291–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.; Chen, G.-D.; Zhou, X.; McGinnis, J.F.; Li, F.; Cao, W. Molecular mechanisms underlying cochlear degeneration in the tubby mouse and the therapeutic effect of sulforaphane. Neurochem. Int. 2009, 54, 172–179. [Google Scholar] [CrossRef] [Green Version]
- Talalay, P.; Fahey, J.W.; Healy, Z.R.; Wehage, S.L.; Benedict, A.L.; Min, C.; Dinkova-Kostova, A.T. Sulforaphane mobilizes cellular defenses that protect skin against damage by UV radiation. Proc. Natl. Acad. Sci. USA 2007, 104, 17500–17505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shokeir, A.A.; Barakat, N.; Hussein, A.M.; Awadalla, A.; Harraz, A.M.; Khater, S.; Hemmaid, K.; Kamal, A.I. Activation of Nrf2 by Ischemic Preconditioning and Sulforaphane in Renal Ischemia/Reperfusion Injury: A Comparative Experimental Study. Physiol Res. 2015, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; He, M.; Liu, R.; Brecha, N.C.; Yu, A.C.H.; Pu, M. Sulforaphane Protects Rodent Retinas against Ischemia-Reperfusion Injury through the Activation of the Nrf2/HO-1 Antioxidant Pathway. PLoS ONE 2014, 9, e114186. [Google Scholar] [CrossRef]
- Chen, Z.; Mohr, A.; Heitplatz, B.; Hansen, U.; Pascher, A.; Brockmann, J.G.; Becker, F. Sulforaphane Elicits Protective Effects in Intestinal Ischemia Reperfusion Injury. Int. J. Mol. Sci. 2020, 21, 5189. [Google Scholar] [CrossRef]
- Zhao, H.-D. Sulforaphane protects liver injury induced by intestinal ischemia reperfusion through Nrf2-ARE pathway. WJG 2010, 16, 3002. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Kobori, N.; Aronowski, J.; Dash, P.K. Sulforaphane reduces infarct volume following focal cerebral ischemia in rodents. Neurosci. Lett. 2006, 393, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Moore, A.N.; Clifton, G.L.; Dash, P.K. Sulforaphane enhances aquaporin-4 expression and decreases cerebral edema following traumatic brain injury. J. Neurosci. Res. 2005, 82, 499–506. [Google Scholar] [CrossRef]
- Ma, L.-L.; Xing, G.-P.; Yu, Y.; Liang, H.; Yu, T.-X.; Zheng, W.-H.; Lai, T.-B. Sulforaphane exerts neuroprotective effects via suppression of the inflammatory response in a rat model of focal cerebral ischemia. Int. J. Clin. Exp. Med. 2015, 8, 17811–17817. [Google Scholar]
- Tarozzi, A.; Angeloni, C.; Malaguti, M.; Morroni, F.; Hrelia, S.; Hrelia, P. Sulforaphane as a Potential Protective Phytochemical against Neurodegenerative Diseases. Oxid. Med. Cell. Longev. 2013, 2013, 1–10. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, T.; Mao, L.; Zhang, F. Sulforaphane Protects against Brain Diseases: Roles of Cytoprotective Enzymes. Austin J. Cerebrovasc. Dis. Stroke 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Ping, Z.; Liu, W.; Kang, Z.; Cai, J.; Wang, Q.; Cheng, N.; Wang, S.; Wang, S.; Zhang, J.H.; Sun, X. Sulforaphane protects brains against hypoxic–ischemic injury through induction of Nrf2-dependent phase 2 enzyme. Brain Res. 2010, 1343, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Danilov, C.A.; Chandrasekaran, K.; Racz, J.; Soane, L.; Zielke, C.; Fiskum, G. Sulforaphane protects astrocytes against oxidative stress and delayed death caused by oxygen and glucose deprivation. Glia 2009, 57, 645–656. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Cui, W.; Xin, Y.; Miao, X.; Barati, M.T.; Zhang, C.; Chen, Q.; Tan, Y.; Cui, T.; Zheng, Y.; et al. Prevention by sulforaphane of diabetic cardiomyopathy is associated with up-regulation of Nrf2 expression and transcription activation. J. Mol. Cell. Cardiol. 2013, 57, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.-Y.; Zhang, C.; Lee, J.H.; Shu, L.; Wu, T.-Y.; Khor, T.O.; Conney, A.H.; Lu, Y.-P.; Kong, A.-N.T. Requirement and Epigenetics Reprogramming of Nrf2 in Suppression of Tumor Promoter TPA-Induced Mouse Skin Cell Transformation by Sulforaphane. Cancer Prev. Res. 2014, 7, 319–329. [Google Scholar] [CrossRef] [Green Version]
- McMahon, M.; Lamont, D.J.; Beattie, K.A.; Hayes, J.D. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc. Natl. Acad. Sci. USA 2010, 107, 18838–18843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent Proteasomal Degradation of Transcription Factor Nrf2 Contributes to the Negative Regulation of Antioxidant Response Element-driven Gene Expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef] [Green Version]
- Mao, L.; Yang, T.; Li, X.; Lei, X.; Sun, Y.; Zhao, Y.; Zhang, W.; Gao, Y.; Sun, B.; Zhang, F. Protective effects of sulforaphane in experimental vascular cognitive impairment: Contribution of the Nrf2 pathway. J. Cereb. Blood Flow Metab. 2019, 39, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Egner, P.A.; Chen, J.G.; Wang, J.B.; Wu, Y.; Sun, Y.; Lu, J.H.; Zhu, J.; Zhang, Y.H.; Chen, Y.S.; Friesen, M.D.; et al. Bioavailability of Sulforaphane from Two Broccoli Sprout Beverages: Results of a Short-term, Cross-over Clinical Trial in Qidong, China. Cancer Prev. Res. 2011, 4, 384–395. [Google Scholar] [CrossRef] [Green Version]
- Hu, R.; Hebbar, V.; Kim, B.-R.; Chen, C.; Winnik, B.; Buckley, B.; Soteropoulos, P.; Tolias, P.; Hart, R.P.; Kong, A.-N.T. In Vivo Pharmacokinetics and Regulation of Gene Expression Profiles by Isothiocyanate Sulforaphane in the Rat. J. Pharmacol. Exp. Ther. 2004, 310, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Dinkova-Kostova, A.T.; Wade, K.L.; Zhang, Y.; Shapiro, T.A.; Talalay, P. Quantitative determination of dithiocarbamates in human plasma, serum, erythrocytes and urine: Pharmacokinetics of broccoli sprout isothiocyanates in humans. Clin. Chim. Acta 2002, 316, 43–53. [Google Scholar] [CrossRef]
- Jazwa, A.; Rojo, A.I.; Innamorato, N.G.; Hesse, M.; Fernández-Ruiz, J.; Cuadrado, A. Pharmacological Targeting of the Transcription Factor Nrf2 at the Basal Ganglia Provides Disease Modifying Therapy for Experimental Parkinsonism. Antioxid. Redox Signal. 2011, 14, 2347–2360. [Google Scholar] [CrossRef] [Green Version]
- Clarke, J.D.; Hsu, A.; Williams, D.E.; Dashwood, R.H.; Stevens, J.F.; Yamamoto, M.; Ho, E. Metabolism and Tissue Distribution of Sulforaphane in Nrf2 Knockout and Wild-Type Mice. Pharm. Res. 2011, 28, 3171–3179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Toxicology Program. NTP Toxicology and Carcinogenesis Studies of t-Butylhydroquinone (CAS No. 1948-33-0) in F344/N Rats and B6C3F(1) Mice (Feed Studies). Natl. Toxicol. Program. Tech. Rep. Ser. 1997, 459, 1–326. [Google Scholar]
- Joint FAO/WHO Expert Committee on Food Additives (Ed.) Evaluation of Certain Food Additives and Contaminants: Forty-Ninth Report of the Joint FAO/WHO Expert Committee on Food Additives; WHO Technical Report Series; World Health Organization: Geneva, Switzerland, 1999; ISBN 978-92-4-120884-0. [Google Scholar]
- Saykally, J.N.; Rachmany, L.; Hatic, H.; Shaer, A.; Rubovitch, V.; Pick, C.G.; Citron, B.A. The nuclear factor erythroid 2-like 2 activator, tert-butylhydroquinone, improves cognitive performance in mice after mild traumatic brain injury. Neuroscience 2012, 223, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ji, C.; Wu, L.; Qiu, J.; Li, Q.; Shao, Z.; Chen, G. Tert-Butylhydroquinone Alleviates Early Brain Injury and Cognitive Dysfunction after Experimental Subarachnoid Hemorrhage: Role of Keap1/Nrf2/ARE Pathway. PLoS ONE 2014, 9, e97685. [Google Scholar] [CrossRef] [Green Version]
- De Long, M.J.; Santamaria, A.B.; Talalay, P. Role of cytochrome P 1 in the induction of NAD(P)H:quinone reductase in a murine hepatoma cell line and its mutants. Carcinogenesis 1987, 8, 1549–1553. [Google Scholar] [CrossRef]
- Talalay, P. Mechanisms of induction of enzymes that protect against chemical carcinogenesis. Adv. Enzym. Regul. 1989, 28, 237–250. [Google Scholar] [CrossRef]
- Van Ommen, B.; Koster, A.; Verhagen, H.; van Bladeren, P.J. The glutathione conjugates of tert-butyl hydroquinone as potent redox cycling agents and possible reactive agents underlying the toxicity of butylated hydroxyanisole. Biochem. Biophys. Res. Commun. 1992, 189, 309–314. [Google Scholar] [CrossRef]
- Gharavi, N.; Haggarty, S.; El-Kadi, A.O.S. Chemoprotective and Carcinogenic Effects of tert-Butylhydroquinone and Its Metabolites. CDM 2007, 8, 1–7. [Google Scholar] [CrossRef]
- Peters, M.M.; Rivera, M.I.; Jones, T.W.; Monks, T.J.; Lau, S.S. Glutathione conjugates of tert-butyl-hydroquinone, a metabolite of the urinary tract tumor promoter 3-tert-butyl-hydroxyanisole, are toxic to kidney and bladder. Cancer Res. 1996, 56, 1006–1011. [Google Scholar]
- Sun, J.; Hu, H.; Ren, X.; Simpkins, J.W. Tert-butylhydroquinone compromises survival in murine experimental stroke. Neurotoxicol. Teratol. 2016, 54, 15–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev. Pharmacol. Toxicol 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Zeynalov, E.; Doré, S. Low doses of carbon monoxide protect against experimental focal brain ischemia. Neurotox Res. 2009, 15, 133–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otterbein, L.E. Carbon monoxide: Innovative anti-inflammatory properties of an age-old gas molecule. Antioxid. Redox Signal. 2002, 4, 309–319. [Google Scholar] [CrossRef]
- Leffler, C.W.; Parfenova, H.; Jaggar, J.H.; Wang, R. Carbon monoxide and hydrogen sulfide: Gaseous messengers in cerebrovascular circulation. J. Appl. Physiol. 2006, 100, 1065–1076. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-H.; Tsai, H.-L.; Chiang, M.-T.; Chau, L.-Y. Carbon Monoxide-Induced Early Thrombolysis Contributes to Heme Oxygenase-1-Mediated Inhibition of Neointimal Growth after Vascular Injury in Hypercholesterolemic Mice. J. Biomed. Sci. 2006, 13, 721–730. [Google Scholar] [CrossRef]
- Liao, S.; Wu, J.; Liu, R.; Wang, S.; Luo, J.; Yang, Y.; Qin, Y.; Li, T.; Zheng, X.; Song, J.; et al. A novel compound DBZ ameliorates neuroinflammation in LPS-stimulated microglia and ischemic stroke rats: Role of Akt(Ser473)/GSK3β(Ser9)-mediated Nrf2 activation. Redox Biol. 2020, 36, 101644. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wang, W.; Matei, N.; Li, X.; Pang, J.; Mo, J.; Chen, S.; Tang, J.; Yan, M.; Zhang, J.H. Ezetimibe Attenuates Oxidative Stress and Neuroinflammation via the AMPK/Nrf2/TXNIP Pathway after MCAO in Rats. Oxid. Med. Cell. Longev. 2020, 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Lu, H.; Qin, J.; Qu, S.; Wang, W.; Guo, Y.; Liao, W.; Song, M.; Chen, J.; Wang, Y. Biochanin A Provides Neuroprotection Against Cerebral Ischemia/Reperfusion Injury by Nrf2-Mediated Inhibition of Oxidative Stress and Inflammation Signaling Pathway in Rats. Med. Sci. Monit. 2019, 25, 8975–8983. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Zhang, H.; Zhang, J.; Yan, M. Isoquercetin attenuates oxidative stress and neuronal apoptosis after ischemia/reperfusion injury via Nrf2-mediated inhibition of the NOX4/ROS/NF-κB pathway. Chem. Biol. Interact. 2018, 284, 32–40. [Google Scholar] [CrossRef]
- Ma, T.; Shi, Y.; Wang, Y. Forsythiaside A protects against focal cerebral ischemic injury by mediating the activation of the Nrf2 and endoplasmic reticulum stress pathways. Mol. Med. Rep. 2019, 20, 1313–1320. [Google Scholar] [CrossRef]
- Chen, L.; Wang, L.; Zhang, X.; Cui, L.; Xing, Y.; Dong, L.; Liu, Z.; Li, Y.; Zhang, X.; Wang, C.; et al. The protection by Octreotide against experimental ischemic stroke: Up-regulated transcription factor Nrf2, HO-1 and down-regulated NF-κB expression. Brain Res. 2012, 1475, 80–87. [Google Scholar] [CrossRef]
- Zhou, F.; Wang, M.; Ju, J.; Wang, Y.; Liu, Z.; Zhao, X.; Yan, Y.; Yan, S.; Luo, X.; Fang, Y. Schizandrin A protects against cerebral ischemia-reperfusion injury by suppressing inflammation and oxidative stress and regulating the AMPK/Nrf2 pathway regulation. Am. J. Transl. Res. 2019, 11, 199–209. [Google Scholar] [PubMed]
- Xie, Y.; Zhang, X.; Zhang, C.; Yang, Y.; He, J.; Chen, Y. Protective effects of leonurine against ischemic stroke in mice by activating nuclear factor erythroid 2-related factor 2 pathway. CNS Neurosci. Ther. 2019, 25, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Song, J.; Yan, R.; Li, L.; Xiao, Z.; Zhou, W.; Wang, Z.; Xiao, W.; Du, G. Diterpene ginkgolides protect against cerebral ischemia/reperfusion damage in rats by activating Nrf2 and CREB through PI3K/Akt signaling. Acta Pharmacol. Sin. 2018, 39, 1259–1272. [Google Scholar] [CrossRef]
- Gao, Y.; Xu, X.; Chang, S.; Wang, Y.; Xu, Y.; Ran, S.; Huang, Z.; Li, P.; Li, J.; Zhang, L.; et al. Totarol prevents neuronal injury in vitro and ameliorates brain ischemic stroke: Potential roles of Akt activation and HO-1 induction. Toxicol. Appl. Pharmacol. 2015, 289, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Maharjan, S.; Wang, Q.; Sun, Y.; Han, X.; Wang, S.; Mao, Z.; Xin, Y.; Zhang, B. α-Lipoic Acid Promotes Neurological Recovery After Ischemic Stroke by Activating the Nrf2/HO-1 Pathway to Attenuate Oxidative Damage. Cell. Physiol. Biochem. 2017, 43, 1273–1287. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, S.; Mao, L.; Leak, R.K.; Shi, Y.; Zhang, W.; Hu, X.; Sun, B.; Cao, G.; Gao, Y.; et al. Omega-3 Fatty Acids Protect the Brain against Ischemic Injury by Activating Nrf2 and Upregulating Heme Oxygenase 1. J. Neurosci. 2014, 34, 1903–1915. [Google Scholar] [CrossRef]
- Wei, C.-C.; Kong, Y.-Y.; Li, G.-Q.; Guan, Y.-F.; Wang, P.; Miao, C.-Y. Nicotinamide mononucleotide attenuates brain injury after intracerebral hemorrhage by activating Nrf2/HO-1 signaling pathway. Sci. Rep. 2017, 7, 717. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Zhou, D.; Yan, B. Eriocitrin alleviates oxidative stress and inflammatory response in cerebral ischemia reperfusion rats by regulating phosphorylation levels of Nrf2/NQO-1/HO-1/NF-κB p65 proteins. Ann. Transl. Med. 2020, 8, 757. [Google Scholar] [CrossRef] [PubMed]
- Yen, T.-L.; Chen, R.-J.; Jayakumar, T.; Lu, W.-J.; Hsieh, C.-Y.; Hsu, M.-J.; Yang, C.-H.; Chang, C.-C.; Lin, Y.-K.; Lin, K.-H.; et al. Andrographolide stimulates p38 mitogen-activated protein kinase–nuclear factor erythroid-2-related factor 2–heme oxygenase 1 signaling in primary cerebral endothelial cells for definite protection against ischemic stroke in rats. Transl. Res. 2016, 170, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Yang, Q.; Yang, B.; Xu, H.; Nasif, O.; Muruganantham, S.; Chen, J. Phyllanthin Averts Oxidative Stress and Neuroinflammation in Cerebral Ischemic-Reperfusion Injury through Modulation of the NF-κB and AMPK/Nrf2 Pathways. J. Environ. Pathol. Toxicol. Oncol. 2021, 40, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-J.; Cui, P. Neohesperidin attenuates cerebral ischemia–reperfusion injury via inhibiting the apoptotic pathway and activating the Akt/Nrf2/HO-1 pathway. J. Asian Nat. Prod. Res. 2013, 15, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Wang, S.; Duan, J.; Jia, N.; Zhu, Y.; Ding, Y.; Guan, Y.; Wei, G.; Yin, Y.; Xi, M.; et al. Protocatechualdehyde Protects Against Cerebral Ischemia-Reperfusion-Induced Oxidative Injury Via Protein Kinase Cε/Nrf2/HO-1 Pathway. Mol. Neurobiol. 2017, 54, 833–845. [Google Scholar] [CrossRef]
- Han, J.; Xiao, Q.; Lin, Y.; Zheng, Z.; He, Z.; Hu, J.; Chen, L. Neuroprotective effects of salidroside on focal cerebral ischemia/reperfusion injury involves the nuclear erythroid 2-related factor 2 pathway. Neural Regen. Res. 2015, 10, 1989. [Google Scholar] [CrossRef]
- Tang, C.; Hong, J.; Hu, C.; Huang, C.; Gao, J.; Huang, J.; Wang, D.; Geng, Q.; Dong, Y. Palmatine Protects against Cerebral Ischemia/Reperfusion Injury by Activation of the AMPK/Nrf2 Pathway. Oxid. Med. Cell. Longev. 2021, 2021, 1–12. [Google Scholar] [CrossRef]
- Fu, K.; Chen, M.; Zheng, H.; Li, C.; Yang, F.; Niu, Q. Pelargonidin ameliorates MCAO-induced cerebral ischemia/reperfusion injury in rats by the action on the Nrf2/HO-1 pathway. Transl. Neurosci. 2021, 12, 20–31. [Google Scholar] [CrossRef]
- Wu, G.; Zhu, L.; Yuan, X.; Chen, H.; Xiong, R.; Zhang, S.; Cheng, H.; Shen, Y.; An, H.; Li, T.; et al. Britanin Ameliorates Cerebral Ischemia–Reperfusion Injury by Inducing the Nrf2 Protective Pathway. Antioxid. Redox Signal. 2017, 27, 754–768. [Google Scholar] [CrossRef]
- Xing, C.; Arai, K.; Lo, E.H.; Hommel, M. Pathophysiologic Cascades in Ischemic Stroke. Int. J. Stroke 2012, 7, 378–385. [Google Scholar] [CrossRef]
- Shaw, P.; Chattopadhyay, A. Nrf2–ARE signaling in cellular protection: Mechanism of action and the regulatory mechanisms. J. Cell. Physiol. 2020, 235, 3119–3130. [Google Scholar] [CrossRef]
- Stefanson, A.; Bakovic, M. Dietary Regulation of Keap1/Nrf2/ARE Pathway: Focus on Plant-Derived Compounds and Trace Minerals. Nutrients 2014, 6, 3777–3801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phipps, M.S.; Cronin, C.A. Management of acute ischemic stroke. BMJ 2020, 368, l6983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
General overview of the Nrf2 pathway. Major molecules and events modulating Nrf2 stability and activation, as well as main downstream protein targets and their functions are shown. (A) Nrf2 structure is composed by domains (i.e., Neh2 and Neh6) that, after redox or signaling regulation, affect Nrf2 stability. Under homeostatic conditions, Keap1 binds to Nrf2, directing this transcription factor to ubiquitination and subsequent degradation by the proteasome. Keap1 is a redox sensor that, upon oxidative thiol modification, loses its capability to repress Nrf2. Glycogen synthase kinase 3 (GSK-3)-mediated phosphorylation of Nrf2 represents an alternative mechanism, facilitating its ubiquitination and consequent degradation by the proteasome. (B) Under stress conditions (excessive accumulation of ROS, electrophilic molecules and proteotoxic stress), Nrf2-Keap1 interaction is disrupted and Nrf2 translocates into the nucleus, wherein it heterodimerizes with small Maf proteins (sMaf) and binds to an enhancer sequence termed ARE that is present in the regulatory regions of over 250 genes (ARE genes). (C) These ARE genes, whose encoded proteins participate in diverse cellular/metabolic events, display significant roles in counteracting imbalances in proteostasis, redox and inflammatory control. For a detailed review on the mechanisms mediating Nrf2 stability and activation, see [74]. CALCOCO2, calcium binding and coiled-coil domain 2; CD36, CD36 scavenger receptor; GPx, glutathione peroxidase; Gpx8, glutathione peroxidase 8; HO-1, heme oxygenase-1; IL 17D, interleukin-17D; NQO1, NADPH Quinone oxidoreductase enzyme; PSMB7, proteasome subunit b type-7; TrxR, thioredoxin reductase; ULK1, unc-51 like autophagy activating kinase 1.
Figure 1.
General overview of the Nrf2 pathway. Major molecules and events modulating Nrf2 stability and activation, as well as main downstream protein targets and their functions are shown. (A) Nrf2 structure is composed by domains (i.e., Neh2 and Neh6) that, after redox or signaling regulation, affect Nrf2 stability. Under homeostatic conditions, Keap1 binds to Nrf2, directing this transcription factor to ubiquitination and subsequent degradation by the proteasome. Keap1 is a redox sensor that, upon oxidative thiol modification, loses its capability to repress Nrf2. Glycogen synthase kinase 3 (GSK-3)-mediated phosphorylation of Nrf2 represents an alternative mechanism, facilitating its ubiquitination and consequent degradation by the proteasome. (B) Under stress conditions (excessive accumulation of ROS, electrophilic molecules and proteotoxic stress), Nrf2-Keap1 interaction is disrupted and Nrf2 translocates into the nucleus, wherein it heterodimerizes with small Maf proteins (sMaf) and binds to an enhancer sequence termed ARE that is present in the regulatory regions of over 250 genes (ARE genes). (C) These ARE genes, whose encoded proteins participate in diverse cellular/metabolic events, display significant roles in counteracting imbalances in proteostasis, redox and inflammatory control. For a detailed review on the mechanisms mediating Nrf2 stability and activation, see [74]. CALCOCO2, calcium binding and coiled-coil domain 2; CD36, CD36 scavenger receptor; GPx, glutathione peroxidase; Gpx8, glutathione peroxidase 8; HO-1, heme oxygenase-1; IL 17D, interleukin-17D; NQO1, NADPH Quinone oxidoreductase enzyme; PSMB7, proteasome subunit b type-7; TrxR, thioredoxin reductase; ULK1, unc-51 like autophagy activating kinase 1.

Figure 2.
Interplay between ischemic stroke and Nrf2. Ischemia-reperfusion leads to increased levels of oxidants (i.e., hydrogen peroxide (H2O2), superoxide anion (O2•−)) that, in turn, can activate Nrf2. Nrf2-downstream proteins (i.e., heme oxygenase-1 (HO-1), superoxide dismutase (SOD)) can mitigate the deleterious effects of oxidants produced in excess during IR, preventing oxidative stress and cell death.
Figure 2.
Interplay between ischemic stroke and Nrf2. Ischemia-reperfusion leads to increased levels of oxidants (i.e., hydrogen peroxide (H2O2), superoxide anion (O2•−)) that, in turn, can activate Nrf2. Nrf2-downstream proteins (i.e., heme oxygenase-1 (HO-1), superoxide dismutase (SOD)) can mitigate the deleterious effects of oxidants produced in excess during IR, preventing oxidative stress and cell death.


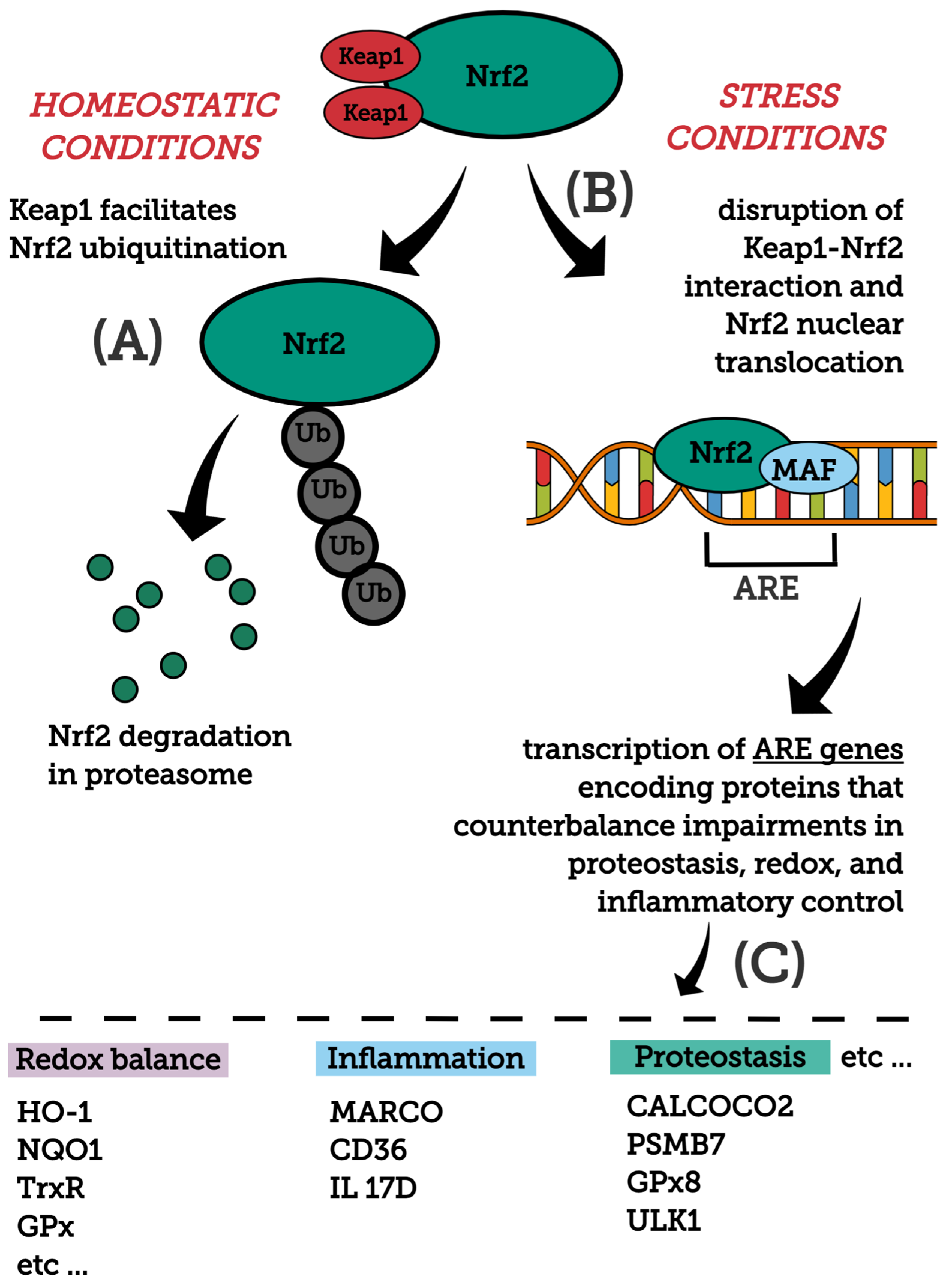{kind=link}
{kind=link}
Table 1.
In vivo experimental studies reporting the endogenous modulation of Nrf2 and/or Nrf2-downstream proteins after cerebral ischemia or ischemia-reperfusion.
Table 1.
In vivo experimental studies reporting the endogenous modulation of Nrf2 and/or Nrf2-downstream proteins after cerebral ischemia or ischemia-reperfusion.
| Species (Sex; Age) | Experimental Model | #Findings Indicating Endogenous Modulation of Nrf2 after Ischemic Stroke | Tissue | Ref. | ||
|---|---|---|---|---|---|---|
| Specific Findings | General Effect | |||||
| MICE | ICR mice (M; 8 weeks) | MCAO/R (1 h/2, 8, 24, 72 h) | ↑ Nrf2, HO-1 and Trx protein expression ↓ Keap1 protein expression | ↑ | ischemic brain tissue | [90] |
| ddY mice (M; 8–12 weeks) | MCAO/R (1 h/6, 24, 48 h) | ↑ Nrf2 and HO-1 protein expression | ↑ | ischemic cortex and striatum | [82] | |
| CD-1 mice (M; NI) | pMCAO (24 h) | ↑ nuclear Nrf2 translocation ↑ HO-1 protein expression ↑ SOD activity | ↑ | ischemic cerebral cortex | [91] | |
| ICR mice (M; 8–10 weeks) | MCAO/R (1 h/24 h) | ↑ Nrf2 and HO-1 mRNA levels ↑ nuclear Nrf2 and HO-1 protein expression | ↑ | ischemic cerebral cortex | [92] | |
| OKD48 mice (M, F; 11–13 weeks) | MCAO/R (45 min/ 12, 24, 72 h, 7 d) | ↑ Nrf2 and HO-1 protein expression | ↑ | peri-ischemic brain tissue | [93] | |
| Mice (NI) (M; 12 weeks) | MCAO/R (2 h/24 h) | ↑ Nrf2, HO-1, GCL protein expression | ↑ | ischemic brain tissue | [94] | |
| C57BL/6 mice (M; 8 weeks) | BCCAO/R (20 min/24 h) | ↑ Nrf2 DNA binding activity ↑ nuclear Nrf2, HO-1 and NQO1 protein expression | ↑ | striatum | [95] | |
| C57BL/6J mice (M; 8–10 weeks) | MCAO/R (1 h/24 h) | ↑ Nrf2 and HO-1 protein expression | ↑ | ischemic brain tissue | [96] | |
| ICR mice (M; 6 weeks) | MCAO/R (1 h/24–72 h, 7 d) | ↑ Nrf2 protein expression ↓ Keap1 protein expression | ↑ | ischemic brain tissue | [97] | |
| C57BL/6 mice (M; 10–18 weeks) | pMCAO (1 d–3 d) | ↑ NQO1, HO-1, SOD2 and GPX-1 protein expression | ↑ | ischemic brain tissue | [89] | |
| C57BL/6 mice (M; 10–12 weeks) | MCAO/R (1.5 h/7 d) | ↑ Nrf2 and SOD1 protein expression | ↑ | ischemic brain tissue | [98] | |
| ddY mice (M; 5–8 weeks) | MCAO/R (2 h/2, 6, 22 h) | ↑ Nrf2 and HO-1 protein expression | ↑ | ischemic brain tissue | [99] | |
| CD-1 mice (M; 4 weeks) | MCAO/R (1 h/24 h) | ↑ Nrf2 and HO-1 mRNA ↑ Nrf2 and HO-1 protein ↑ HO-1 activity | ↑ | ischemic brain tissue | [100] | |
| C57BL/6 mice (M; 12 weeks) | MCAO/R (1 h/3 d) | ↑ nuclear Nrf2, HO-1 and NQO1 protein expression | ↑ | cerebral tissue | [101] | |
| ICR mice (M; NI) | MCAO/R (2 h/24 h) | ↑ cytosolic Nrf2, HO-1 and NQO1 protein expression | ↑ | hippocampus | [102] | |
| BALB/c mice (M; 7 weeks) | MCAO/R (1.5 h/24–72 h) | ↑ Nrf2, HO-1 and iNOS protein expression ↓ SOD activity | ↑ | ischemic brain tissue | [103] | |
| ICR mice (M; 8 weeks) | p dMCAO (7 d) | ↑ Nrf2 protein expression | ↑ | ischemic cerebral cortex | [104] | |
| C57BL/6J mice (M; NI) | MCAO/R (1.5 h/24 h) | ↓ SOD 2, HO-1, NQO1 and Nrf2 protein expression ↓ SOD activity ↑ NADPH oxidase protein expression | ↓ | ischemic brain tissue | [105] | |
| C57BL/6 mice (M; NI) | MCAO/R (1 h/72 h) | ↓ Nrf2 protein expression | ↓ | hippocampus and cerebral cortex | [106] | |
| RATS | SD rats (M; adult) | MCAO/R (2 h/2, 6, 24 h) | ↑ Nrf2 and HO-1 protein expression ↑ Nrf2 and HO-1 mRNA levels | ↑ | ischemic cerebral cortex | [107] |
| SD rats (M; NI) | pMCAO (72 h) | ↑ Nrf2 and HO-1 protein expression | ↑ | ischemic cerebral cortex | [108] | |
| SD rats (M; adult) | pMCAO (72 h) | ↑ Nrf2 and HO-1 protein expression | ↑ | ischemic brain tissue | [109] | |
| SD rats (M; NI) | MCAO/R (70min/ 4, 24, 72 h) | ↑ Nrf2 and HO-1 protein expression | ↑ | ischemic brain tissue | [110] | |
| SD rats (M; 9 weeks) | MCAO/R (1 h/24 h) | ↑ Nrf2 and NQO1 protein expression ↑ Nrf2 binding activity to ARE | ↑ | ischemic brain tissue | [111] | |
| SD rats (M; adult) | MCAO/R (2 h/22 h) | ↑ Nrf2 (nuclear) and HO-1 protein expression | ↑ | cerebral cortex | [112] | |
| SD rats (M; adult) | MCAO/R (2 h/24 h) | ↑ Nrf2 protein expression ↑ HO-1 protein expression | ↑ | ischemic brain tissue | [113] | |
| SD rats (M; NI) | pMCAO (24 h) | ↑ nuclear Nrf2 translocation ↑ HO-1 protein expression ↑ Nrf2 and HO-1 mRNA levels | ↑ | ischemic brain tissue | [114] | |
| SD rats (M; NI) | pMCAO (24 h) | ↑ Nrf2 and HO-1 protein expression | ↑ | ischemic brain tissue | [115] | |
| SD rats (M; adult) | MCAO/R (1 h/ 72 h) | ↑ Nrf2 and NQO1 protein expression | ↑ | ischemic cerebral cortex | [116] | |
| Wistar rats (M; NI) | MCAO/R (1 h/72 h) | ↑ Nrf2, HO-1 and NQO1 mRNA levels | ↑ | ischemic brain tissue | [117] | |
| Wistar rats (M; 6 months) | BCCAO/R (30 min/72 h) | ↑ Nrf2 and HO-1 protein expression | ↑ | hippocampus | [118] | |
| SD rats (M; NI) | pMCAO (72 h) | ↑ Nrf2 and HO-1 protein expression | ↑ | ischemic brain tissue | [119] | |
| SD rats (M; 10–12 weeks) | MCAO/R (1 h/24 h) | ↑ Nrf2, Txr-1, Prdx1, Prdx2, Prdx3 and Prdx4 protein expression ↑ Nrf2, Txr-1, Prdx1, Prdx2, Prdx3 and Prdx4 mRNA levels | ↑ | ischemic brain tissue | [120] | |
| SD rats (M; NI) | MCAO/R (2 h/24 h) | ↑ Nrf2 protein expression ↑ Nrf2 mRNA levels ↓ SOD activity | ↑ | ischemic brain tissue | [121] | |
| SD rats (NI; NI) | BCCAO/R (10 min/1–7 d) | ↑ Nrf2 and HO-1 protein expression | ↑ | ischemic brain tissue | [122] | |
| SD rats (M; adult) | MCAO/R (1.5 h/24 h) | ↑ Nrf2 and HO-1 protein expression ↑ Nrf2 and HO-1 mRNA levels | ↑ | hippocampus | [123] | |
| SD rats (M; adult) | pMCAO (24 h) | ↑ nuclear Nrf2 translocation ↑ HO-1 and SOD1 protein expression ↑ HO-1 mRNA levels ↑ SOD1 activity | ↑ | ischemic cerebral cortex | [124] | |
| SD rats (M; adult) | MCAO/R (2 h/ 72 h) | ↑ Nrf2 protein expression ↑ HO-1 protein expression | ↑ | ischemic brain tissue | [125] | |
| SD rats (M; NI) | MCAO/R (2 h/7 d) | ↑ Nrf2 protein expression | ↑ | ischemic brain tissue | [126] | |
| SD rats (M; adult) | MCAO/R (1 h/24 h) | ↑ Nrf2, NQO1 and Srnx1 protein expression ↑ Prdx 1, Prdx 2, Prdx 3, Prdx 4 protein expression | ↑ | ischemic brain tissue | [127] | |
| Hannover-Wistar rats (M; NI) | MCAO/R (1 h/24 h) | ↑ Nrf2 protein expression ↑ SOD and GPx activity | ↑ | hippocampus | [128] | |
| Wistar rats (M; adult) | BCCAO/R (45 min/24 h) | ↑ iNOS and Nrf2 protein expression | ↑ | hippocampus | [129] | |
| SD rats (M; 60–80 days) | MCAO/R (1 h/24 h) | ↑ Nrf2 and Trx1 mRNA levels ↑ Trx1 protein expression | ↑ | ischemic brain tissue | [130] | |
| SD rats (F; adult) | MCAO/R (1.5 h/72 h) | ↑ Nrf2 and NQO1 protein expression | ↑ | ischemic brain tissue | [131] | |
| Wistar rats (M; adult) | MCAO/R (1.5 h/72 h) | ↑ Nrf2, HO-1 and NQO1 mRNA levels | ↑ | ischemic brain tissue | [132] | |
| SD rats (M; adult) | PCI (20 min) | ↑ Nrf2 and HO-1 protein expression | ↑ | cerebral cortex ischemic penumbra | [133] | |
| SD rats (M; NI) | Focal PTI | ↑ Nrf2 and HO-1 protein expression | ↑ | penumbra of cerebral infarction | [134] | |
| SD rats (M; 7–10 weeks) | MCAO/R (2 h/72 h) | ↑ Nrf2 and HO-1 protein expression | ↑ | cerebral cortex and striatum | [135] | |
| SD rats (M; NI) | MCAO/R (1 h/6 or 24 h) | ↑ Nrf2 protein expression ↓ SOD activity | ↑ | ischemic penumbra | [136] | |
| SD rats (M; Adult) | MCAO/R (2 h/72 h) | ↑ nuclear Nrf2 protein expression ↓ cytosolic Nrf2, NQO1 and HO-1 protein expression ↓ SOD activity | ↑ | ischemic brain tissue | [137] | |
| SD rats (M; 8 weeks) | MCAO/R (2 h/24 h) | ↑ HO-1 protein expression ↓ Nrf2 and Trx protein expression | ↑↓ | cerebral cortex | [113] | |
| SD rats (M; adult) | MCAO/R (2 h/72 h) | ↓ SOD activity ↓ Nrf2, HO-1 and NQO1 protein expression | ↓ | ipsilateral ischemic tissue | [138] | |
| SD rats (M; 10 months) | MCAO/R (2 h/48 h) | ↓ HO-1 protein expression ↓ SOD activity | ↓ | ipsilateral ischemic tissue | [139] | |
| Wistar rats (M; NI) | MCAO/R (2 h/24 h) | ↓ GPx and SOD activity ↓ Nrf2 and NQO1 protein expression | ↓ | ischemic brain tissue | [140] | |
| SD rats (M; 3 months) | MCAO/R (1.5 h/72 h) | ↓ NQO1, HO-1 and cytoplasmic Nrf2 protein expression | ↓ | ischemic brain tissue | [141] | |
| SD rats (M; 7–8 weeks) | MCAO/R (1.5 h/7 d) | ↓ Nrf2 mRNA levels | ↓ | ischemic brain tissue | [142] | |
| SD rats (M; NI) | MCAO/R (2 h/7 d) | ↓ SOD activity ↓ nuclear Nrf2, HO-1 and NQO1 protein expression ↓ Nrf2, HO-1 and NQO1 mRNA levels | ↓ | cerebral cortex | [143] | |
| SD rats (NI; NI) | BCCAO/R (10 min occlusion + 10 min reperfusion + 10 min occlusion) | ↓ Nrf2, NQO1 and HO-1 protein expression ↓ SOD activity | ↓ | ischemic brain tissue | [144] | |
| SD rats (F; NI) | MCAO/R (1 h/24 h) | ↓ Nrf2 protein expression | ↓ | hippocampus | [145] | |
| SD rats (M; NI) | MCAO/R (1.5 h/14 d) | ↓ Nrf2 protein expression ↑ Keap-1 protein expression ↓ SOD and catalase activities | ↓ | ischemic brain tissue | [146] | |
Abbreviations and symbols: ↑, activation. ↓, inhibition. M, male; F, female. SD rats, Sprague-Dawley rats. NI, not informed. BCCAO, bilateral common carotid arteries occlusion. MCAO, middle cerebral artery occlusion. MCAO/R, middle cerebral artery occlusion followed by reperfusion. PCI, photochemical cerebral ischemia. pdMCAO, permanent distal middle cerebral artery occlusion. pMCAO, permanent middle cerebral artery occlusion. PTI, photothrombotic ischemia model. ARE, antioxidant response element. HO-1, heme oxygenase 1. NQO1, NAD(P)H:quinone oxidoreductase 1. Prdx, peroxiredoxin. SOD, superoxide dismutase. Srnx1, sulfiredoxin-1. Trx1, thioredoxin. Notes: # Only significant differences between control/sham and ischemic animals were evaluated. HO-1, Heme oxygenase 1: Nrf2-downstream protein catalyzing the degradation of heme, producing biliverdin, ferrous iron and carbon monoxide. NQO1, NAD(P)H:quinone oxidoreductase 1: Nrf2-downstream protein catalyzing the two-electron reduction of quinones and a wide range of other organic compounds. Its physiological role is partly related to the reduction of free radical load in cells and the detoxification of xenobiotics. Prdxs, peroxiredoxins: ubiquitous family of Nrf2-downstream antioxidant enzymes involved in the reduction of peroxides (specifically hydrogen peroxide). SODs, superoxide dismutases: enzymes catalyzing the dismutation of the superoxide radical into molecular oxygen and hydrogen peroxide. Srnx, Sulfiredoxin: Nrf2-downstream protein member of the oxidoreductases family catalyzing reduction of oxidative modifications (i.e., sulfinic, disulfides, etc.). Trx, Thioredoxin: class of small redox (Nrf2-downstream) proteins playing important roles in redox signaling.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Farina, M.; Vieira, L.E.; Buttari, B.; Profumo, E.; Saso, L. The Nrf2 Pathway in Ischemic Stroke: A Review. Molecules 2021, 26, 5001. https://doi.org/10.3390/molecules26165001
AMA Style
Farina M, Vieira LE, Buttari B, Profumo E, Saso L. The Nrf2 Pathway in Ischemic Stroke: A Review. Molecules. 2021; 26(16):5001. https://doi.org/10.3390/molecules26165001
Chicago/Turabian StyleFarina, Marcelo, Leonardo Eugênio Vieira, Brigitta Buttari, Elisabetta Profumo, and Luciano Saso. 2021. "The Nrf2 Pathway in Ischemic Stroke: A Review" Molecules 26, no. 16: 5001. https://doi.org/10.3390/molecules26165001