
Review of Fluorescence Spectroscopy in Environmental Quality Applications


Abstract
:1. Introduction
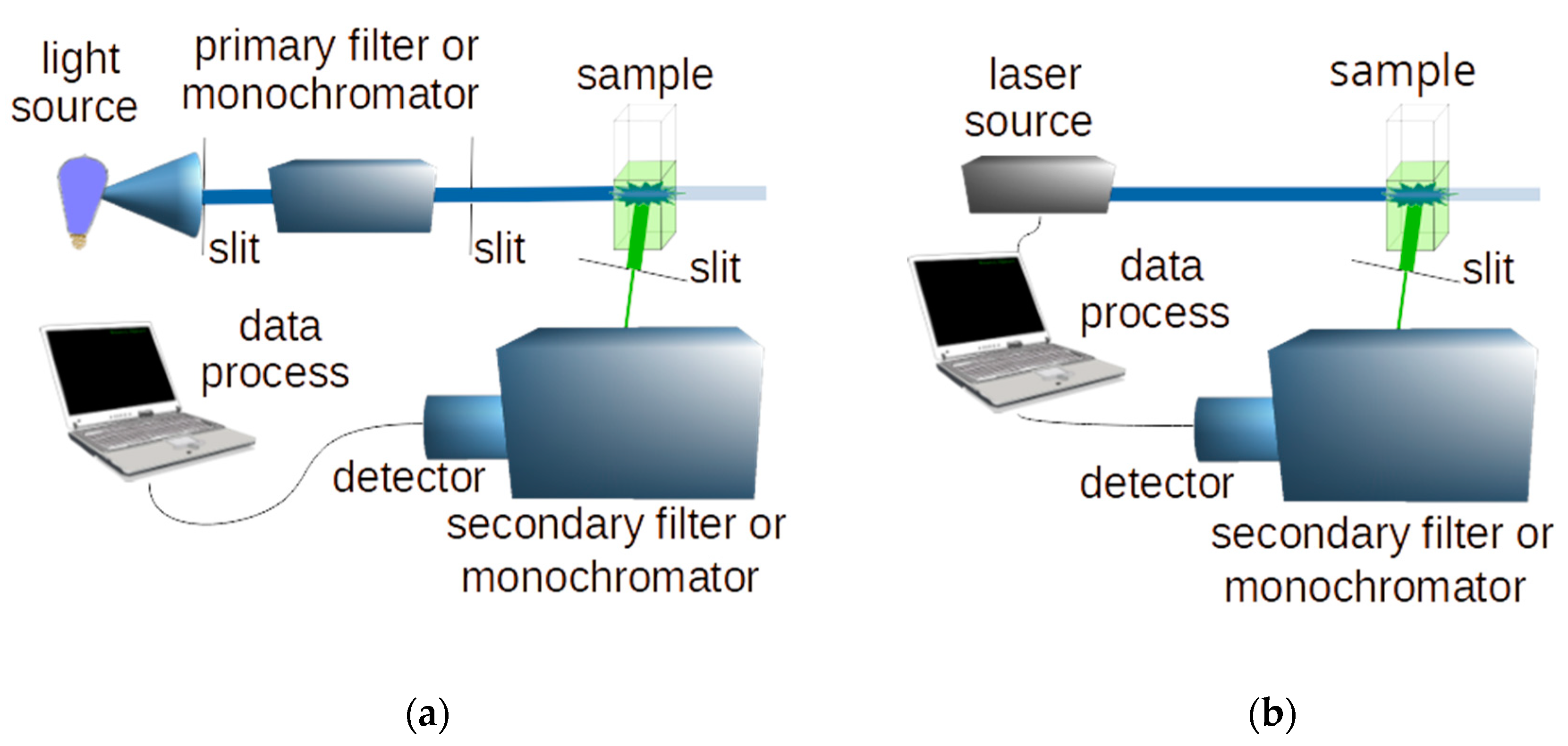
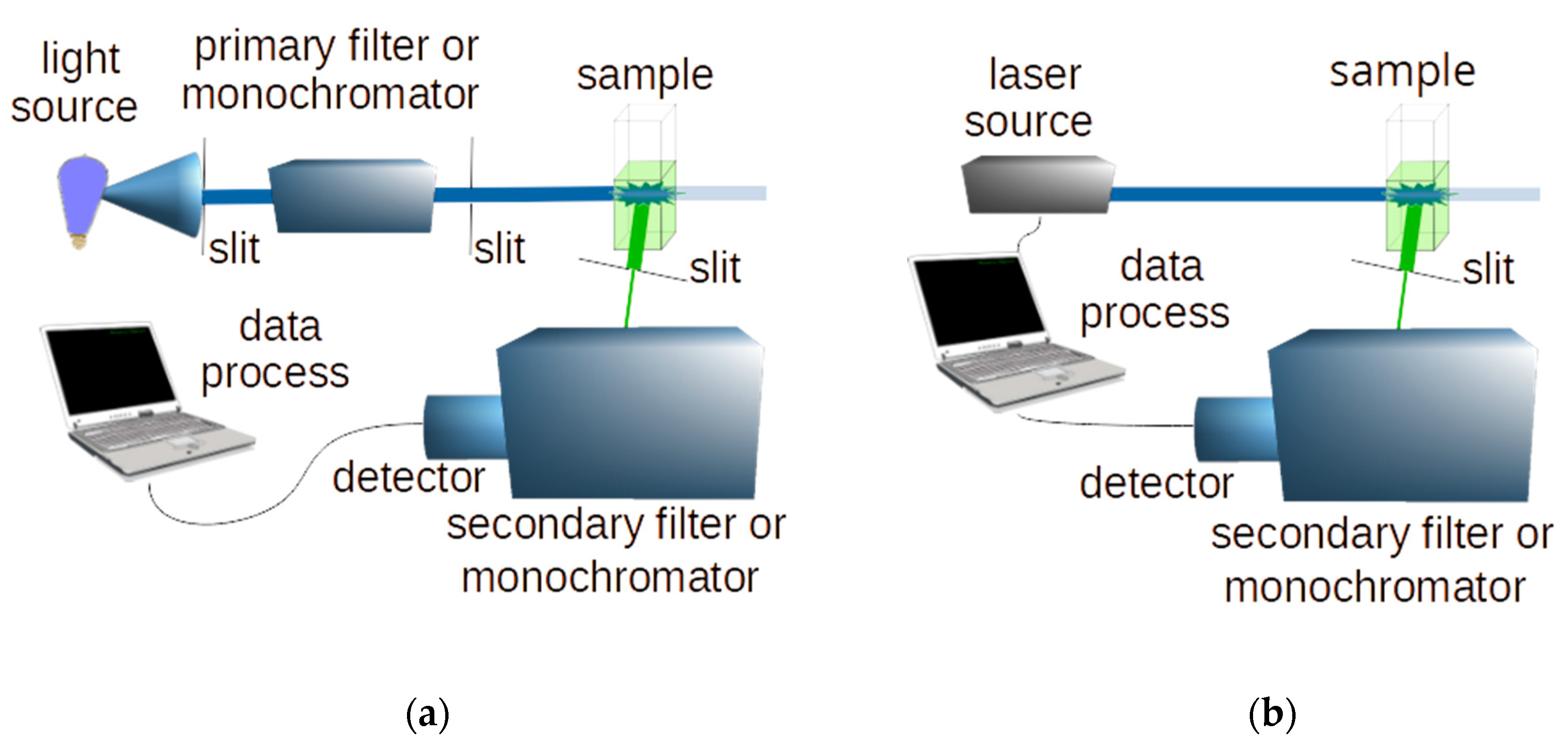
1.1. Light Sources
1.2. Detectors
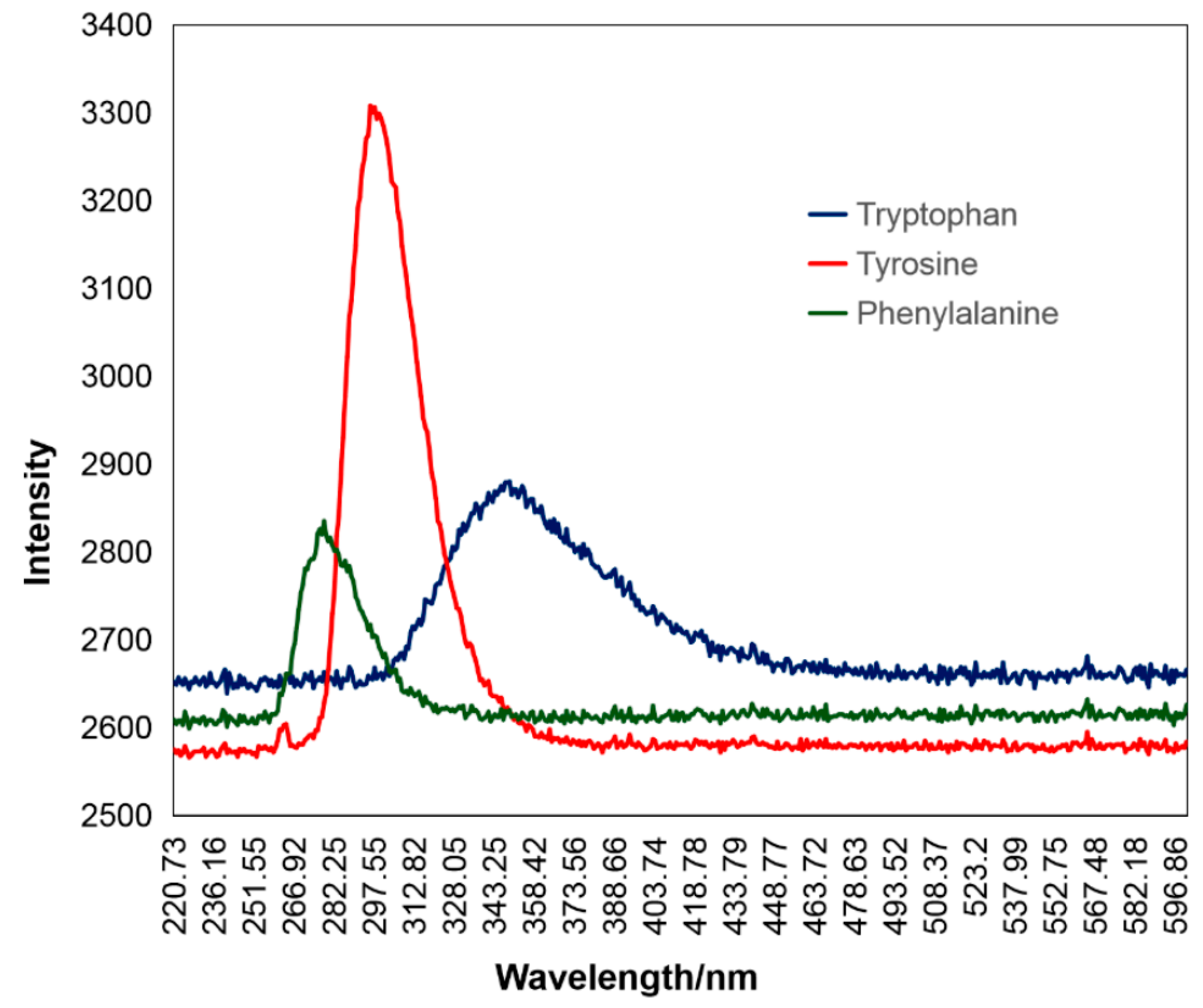
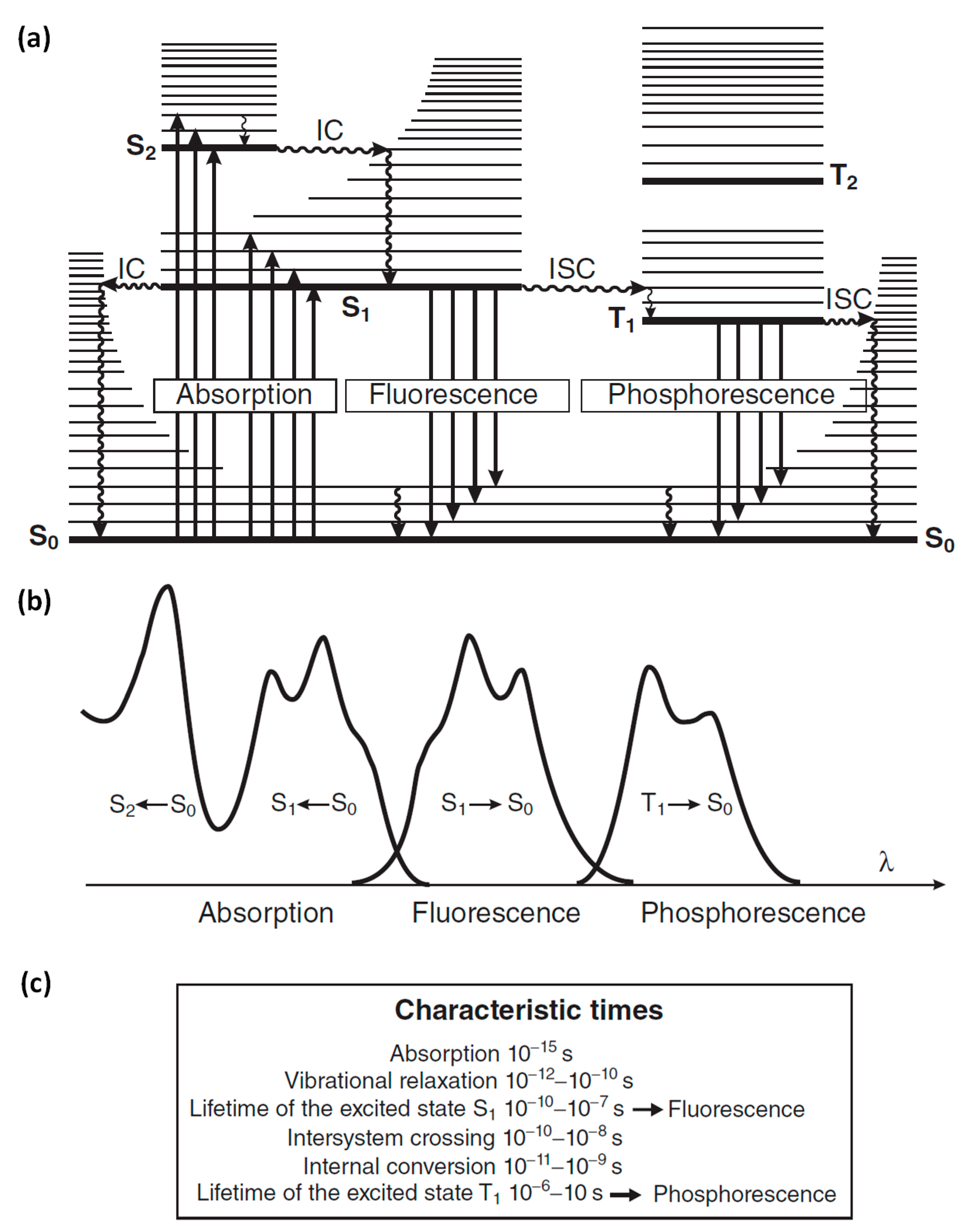
1.3. Fluorescent Compounds
1.4. Factors That Affect Fluorescence

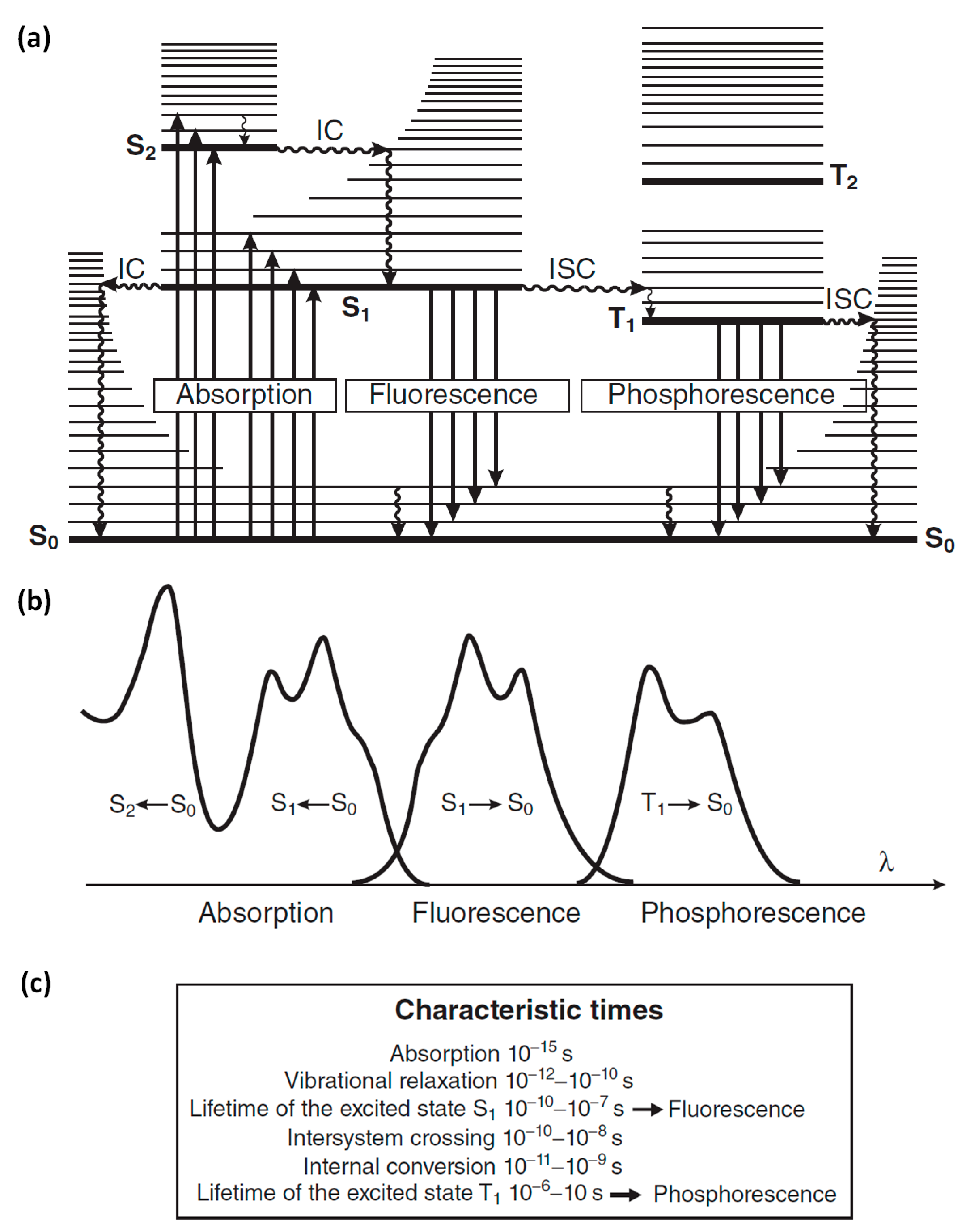{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Typical Fluorophores | Quenchers |
|---|---|
| Polycyclic aromatic hydrocarbons (PAHs) | Nitrocompounds, nitromethane [14,15] |
| Anthracene | Diethylaniline [16] |
| Tyrosine | Disulfides [17], phosphates [18] |
| Tryptophan, indole | Acrylamide [19,20], cations [21], anions [22] |
| Aromatic hydrocarbons | Aromatic and aliphatic amines, pyridinium salts [23] |
| Majority of known fluorophores | Oxygen [24,25] |
1.5. Types of Laser Induced Fluorescence
1.6. Fluorescence Recording
1.7. Interferences in Fluorescence Measurement
2. Applications of Fluorescence in Environmental Samples
2.1. Applications of Conventional Fluorescence in Various Types of Water
2.2. Applications of LIF in Various Types of Water
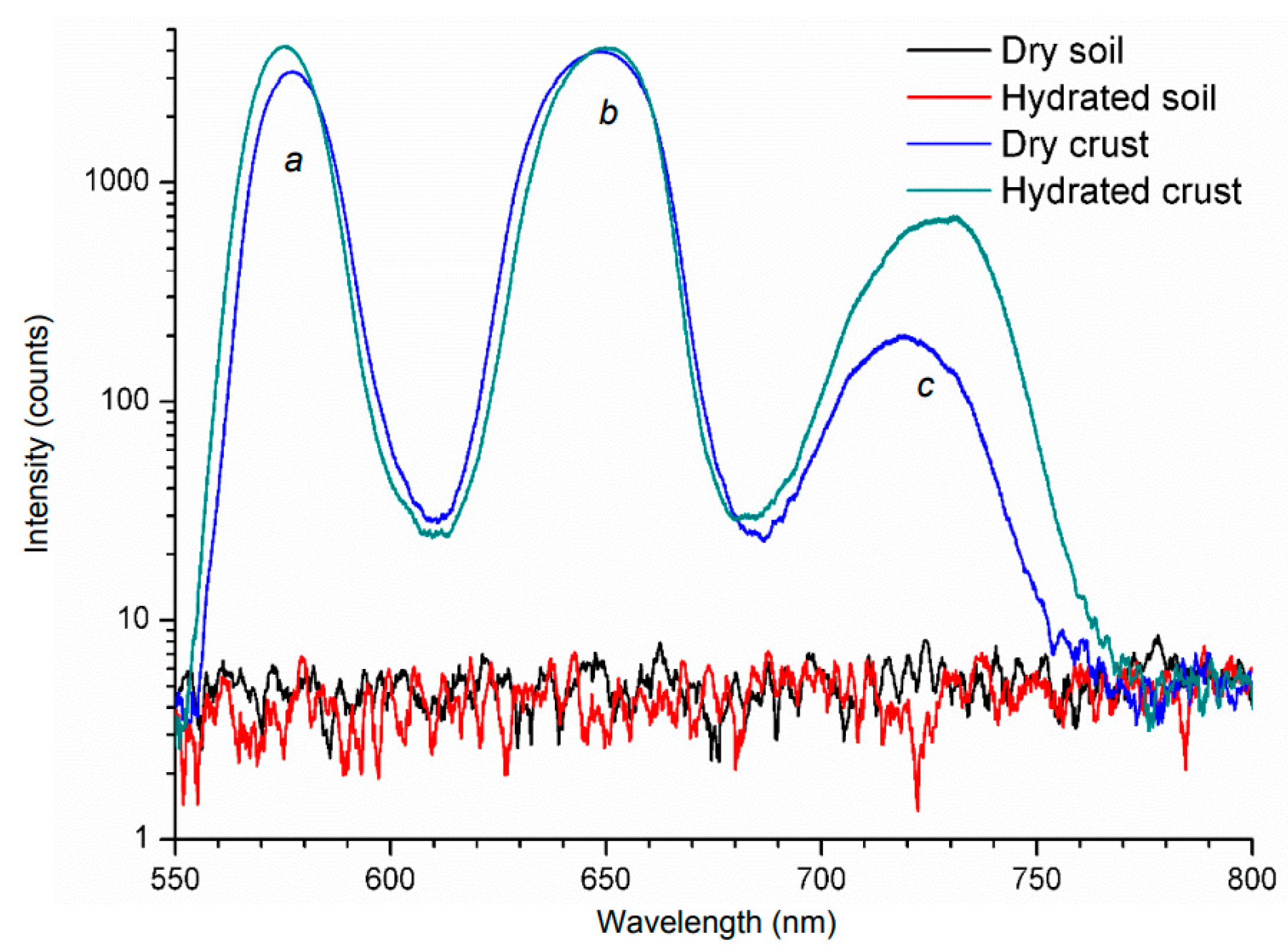
2.3. Applications of Conventional Fluorescence in Soils and Sediments
2.4. Applications of LIF in Soils and Sediments
3. Remote Sensing
4. Conclusions
Funding
Conflicts of Interest
References
- Valeur, B.; Berberan-Santos, M.N. Molecular Fluorescence: Principles and Applications, 2nd ed.; John Wiley and Sons: Weinheim, Germany, 2012. [Google Scholar]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: Baltimore, MD, USA, 2006. [Google Scholar]
- Amano, H.; Collazo, R.; De Santi, C.; Einfeldt, S.; Funato, M.; Glaab, J.; Zhang, Y. The 2020 UV emitter roadmap. J. Phys. D Appl. Phys. 2020, 53, 503001. [Google Scholar] [CrossRef]
- Abramczyk, H. Chapter 4: Lasers in Introduction to Laser Spectroscopy; Elsevier: Amsterdam, The Netherlands, 2005; pp. 59–106. [Google Scholar]
- Telle, H.H.; Urena, A.G. Laser Spectroscopy and Laser Imaging: An Introduction; CRC: Boca Raton, FL, USA, 2017. [Google Scholar] [CrossRef]
- Eichler, H.J.; Eichler, J.; Lux, O. Lasers; Springer International Publishing: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Pavlopoulos, T.G. Scaling of dye lasers with improved laser dyes. Prog. Quantum Electron. 2002, 26, 193–224. [Google Scholar] [CrossRef]
- Murray, J.R. Lasers for Spectroscopy. Laser Spectroscopy and Its Applications; CRC Press: Boca Raton, FL, USA, 2017. [Google Scholar] [CrossRef]
- Iwaya, M.; Tanaka, S.; Omori, T.; Yamada, K.; Hasegawa, R.; Shimokawa, M.; Miyake, H. Recent development of UV-B laser diodes. Jpn. J. Appl. Phys. 2022, 61, 040501. [Google Scholar] [CrossRef]
- Hitz, C.B.; Ewing, J.; Hecht, J. Semiconductor Lasers. In Introduction to Laser Technology; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2012. [Google Scholar] [CrossRef]
- Donati, S. Photodetectors: Devices, Circuits and Applications, 2nd ed.; Wiley: Hoboken, NJ, USA, 2021. [Google Scholar]
- Chen, R.F. Fluorescence Quantum Yields of Tryptophan and Tyrosine. Anal. Lett. 1967, 1, 35–42. [Google Scholar] [CrossRef]
- Joung, J.F.; Han, M.; Jeong, M.; Park, S. Experimental database of optical properties of organic compounds. Sci. Data 2020, 7, 1–6. [Google Scholar] [CrossRef]
- Dreeskamp, H.; Koch, E.; Zander, M. On the fluorescence quenching of polycyclic aromatic hydrocarbons by nitromethane. Z. Nat. 1975, 30, 1311–1314. [Google Scholar] [CrossRef]
- Pandey, A.; Yadav, A.; Bhawna; Pandey, S. Fluorescence quenching of polycyclic aromatic hydrocarbons within deep eutectic solvents and their aqueous mixtures. J. Lumin. 2017, 183, 494–506. [Google Scholar] [CrossRef]
- Guo, X.; Xu, H.; Guo, R. Fluorescence Quenching of Anthracene by N,N-Diethylaniline in the Sodium Dodecyl Sulfate/Benzyl Alcohol/Water System. J. Colloid Interface Sci. 2001, 240, 559–565. [Google Scholar] [CrossRef]
- Arian, S.; Benjamini, M.; Feitelson, J.; Stein, G. Interaction between tyrosine and divalent sulfur in fluorescence quenching and in the photochemistry of ribonuclease. Photochem. Photobiol. 1970, 12, 481–487. [Google Scholar] [CrossRef]
- Chen, R.F.; Cohen, P.F. Quenching of tyrosine fluorescence in proteins by phosphate. Arch. Biochem. Biophys. 1966, 114, 514–522. [Google Scholar] [CrossRef]
- Tallmadge, D.H.; Huebner, J.S.; Borkman, R.F. Acrylamide Quenching of Tryptophan Photochemistry and Photophysics. Photochem. Photobiol. 1989, 49, 81–386. [Google Scholar] [CrossRef] [PubMed]
- Froehlich, P.M.; Nelson, K. Fluorescence quenching of indoles by amides. J. Phys. Chem. 1978, 82, 2401–2403. [Google Scholar] [CrossRef]
- Wang, G.; Wang, A.-J.; Hu, K.-S. Tryptophan fluorescence quenching by alkaline earth metal cations in deionized bacteriorhodopsin. J. Photochem. Photobiol. B Biol. 2000, 59, 38–41. [Google Scholar] [CrossRef]
- Idress, M.; Ayaz, M.; Bibi, R.; Khan, M.N. Fluorescence Quenching of the Probes L-Tryptophan and Indole by Anions in Aqueous System. Anal. Sci. 2020, 36, 183–185. [Google Scholar] [CrossRef]
- Davis, G.A. Quenching of aromatic hydrocarbons by alkylpyridinium halides. J. Chem. Soc. Chem. Commun. 1973, 19, 728–729. [Google Scholar] [CrossRef]
- Willkinson, F. Quenching of electronically excited states by molecular oxygen in fluid solution. Pure Appl. Chem. 1997, 69, 851–858. [Google Scholar] [CrossRef]
- Ware, W.R. Oxygen Quenching of Fluorescence in Solution: An Experimental Study of the Diffusion Process. J. Phys. Chem. 1962, 66, 455–458. [Google Scholar] [CrossRef]
- Gemeda, F.T. A Review on Effect of Solvents on Fluorescent Spectra. Chem. Sci. Int. J. 2017, 18, 1–12. [Google Scholar] [CrossRef]
- Ghosh, K.; Schnitzer, M. Macromolecular structures of humic substances. Soil Sci. 1980, 129, 266–276. [Google Scholar] [CrossRef]
- Patel-Sorrentino, N.; Mounier, S.; Lukas, Y.; Benaim, J.Y. Effects of UV-visible irradiation on natural organic matter from the Amazon basin. Sci. Total Environ. 2004, 321, 231–239. [Google Scholar] [CrossRef]
- Baker, A. Thermal fluorescence quenching properties of dissolved organic matter. Water Res. 2005, 39, 4405–4412. [Google Scholar] [CrossRef] [PubMed]
- Mikalauskaite, K.; Ziaunys, M.; Sneideris, T.; Smirnovas, V. Effect of Ionic Strength on Thioflavin-T Affinity to Amyloid Fibrils and Its Fluorescence Intensity. Int. J. Mol. Sci. 2020, 21, 8916. [Google Scholar] [CrossRef] [PubMed]
- Donges, A.; Noll, R. Laser-Induced Fluorescence. In Laser Measurement Technology; Springer Series in Optical Sciences; Springer: Berlin/Heidelberg, Germany, 2015; Volume 188. [Google Scholar] [CrossRef]
- He, G.; Tan, L.; Zheng, Q.; Prasad, P. Multiphoton Absorbing Materials: Molecular Designs, Characterizations, and Applications. Chem. Rev. 2008, 108, 1245–1330. [Google Scholar] [CrossRef]
- Fitilis, I.; Fakis, M.; Polyzos, I.; Giannetas, V.; Persephonis, P.; Mikroyannidis, J. Strong two photon absorption and photophysical properties of symmetrical chromophores with electron accepting edge substituents. J. Phys. Chem. A 2008, 112, 4742–4748. [Google Scholar] [CrossRef]
- Noll, R. Combination of LIBS and LIF. In Laser-Induced Breakdown Spectroscopy; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar] [CrossRef]
- Anabitarte, F.; Cobo, A.; Lopez-Higuera, J.M. Laser-induced breakdown spectroscopy: Fundamentals, applications, and challenges. ISRN Spectrosc. 2012, 2012, 12. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, J.B.F. Multicomponent analysis by synchronous luminescence spectrometry. Nature 1971, 231, 64–67. [Google Scholar]
- Johnson, D.W.; Callis, J.B.; Christian, G.D. Rapid scanning fluorescence spectroscopy. Anal. Chem. 1977, 49, 747–757. [Google Scholar] [CrossRef]
- Warner, I.M.; Christian, G.D.; Davidson, E.R.; Callis, J.B. Analysis of multicomponent fluorescence data. Anal. Chem. 1977, 49, 2155–2159. [Google Scholar] [CrossRef]
- Gabor, R.; Baker, A.; McKnight, D.M.; Miller, M. Fluorescence indices and their interpretation. In Aquatic Organic Matter Fluorescence; Coble, P., Lead, J., Baker, A., Reynolds, D., Spencer, R., Eds.; Cambridge Environmental Chemistry Series; Cambridge University Press: Cambridge, UK, 2014; pp. 303–338. [Google Scholar] [CrossRef]
- Ohno, T. Fluorescence inner-filtering correction for determining the humification index of dissolved organic matter. Environ. Sci. Technol. 2002, 36, 742–746. [Google Scholar] [CrossRef]
- Kubista, M.; Sjoback, R.; Eriksson, S.; Albinsson, B. Experimental correction for the inner-filter effect in fluorescence spectra. Analyst 1994, 119, 417–419. [Google Scholar] [CrossRef]
- Brouwer, E.R.; Hermans, A.N.; Lingeman, H.; Brinkman, U.A.T. Determination of polycyclic aromatic hydrocarbons in surface water by column liquid chromatography with fluorescence detection, using on-line micelle-mediated sample preparation. J. Chromatogr. A 1994, 669, 45–57. [Google Scholar] [CrossRef]
- Gasperi, J.; Garnaud, S.; Rocher, V.; Moilleron, R. Priority pollutants in surface waters and settleable particles within a densely urbanised area: Case study of Paris (France). Sci. Total Environ. 2009, 407, 2900–2908. [Google Scholar] [CrossRef]
- Kotti, M.; Zacharioudaki, D.-E.; Kokinou, E.; Stavroulakis, G. Characterization of water quality of Almiros river (Northeastern Crete, Greece): Physicochemical parameters, polycyclic aromatic hydrocarbons and anionic detergents. Modeling Earth Syst. Environ. 2018, 4, 1285–1296. [Google Scholar] [CrossRef]
- Song, Y.; Wilke, B.; Song, X.; Gong, P.; Zhou, Q.; Yang, G. Polycyclic aromatic hydrocarbons (PAHs), polychlorinated biphenyls (PCBs) and heavy metals (HMs) as well as their genotoxicity in soil after long-term wastewater irrigation. Chemosphere 2006, 65, 1859–1868. [Google Scholar] [CrossRef] [PubMed]
- Chrysikou, L.; Gemenetzis, P.; Kouras, A.; Manoli, E.; Terzi, E.; Samara, C. Distribution of persistent organic pollutants, polycyclic aromatic hydrocarbons and trace elements in soil and vegetation following a large scale landfill fire in northern Greece. Environ. Intern. 2008, 34, 210–225. [Google Scholar] [CrossRef] [PubMed]
- Vane, C.; Harrison, I.; Kim, A. Polycyclic aromatic hydrocarbons (PAHs) and polychlorinated biphenyls (PCBs) in sediments from the Mersey Estuary, U.K. Sci. Total Environ. 2007, 374, 112–126. [Google Scholar] [CrossRef] [Green Version]
- Bihari, N.; Fafandel, M.; Hamer, B.; Kralj-Bilen, B. PAH content, toxicity and genotoxicity of coastal marine sediments from the Rovinj area, Northern Adriatic, Croatia. Sci. Total Environ. 2006, 366, 602–611. [Google Scholar] [CrossRef]
- Fua, L.; Liub, X.; Hub, J.; Zhaob, X.; Wangc, H.; Wanga, X. Application of dispersive liquid–liquid microextraction for the analysis of triazophos and carbaryl pesticides in water and fruit juice samples. Anal. Chim. Acta 2009, 632, 289–295. [Google Scholar] [CrossRef]
- Le Bot, B.; Colliaux, K.; Pelle, D.; Briens, C.; Seux, R.; Clement, M. Optimization and performance evaluation of the analysis of glyphosate and AMPA in water by HPLC with fluorescence detection. Chromatographia 2005, 56, 161–164. [Google Scholar] [CrossRef]
- Murillo Pulgarin, J.A.; Garcia Bermejo, L.F. Determination of the pesticide napropamide in soil, pepper, and tomato by micelle-stabilized room-temperature phosphorescence. J. Agric. Food Chem. 2002, 50, 1002–1008. [Google Scholar] [CrossRef]
- Salinas-Castillo, A.; Fernandez-Sanchez, J.F.; Segura-Carretero, A.; Fernandez-Gutierrez, A. Simple determination of herbicide napropamide in water and soil samples by room-temperature phosphorescence. Pest Manag. Sci. 2005, 61, 816–820. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Tan, J.; Tang, C.; Yu, Y.; Wang, Z. Multiresidue determination of fluoroquinolone, sulfonamide, trimethoprim, and chloramphenicol antibiotics in urban waters in China. Environ. Toxicol. Chem. 2008, 27, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Seifrtova, M.; Pena, A.; Lino, C.M.; Solich, P. Determination of fluoroquinolone antibiotics in hospital and municipal wastewaters in Coimbra by high performance liquid chromatography using a monolithic column and fluorescence detection. Anal. Bioanal. Chem. 2008, 391, 799–805. [Google Scholar] [CrossRef] [Green Version]
- Blackwell, P.A.; Holten, H.C.; Mab, H.P.; Halling-Sorensen, B.; Boxall, A.B.A.; Kaya, P. Ultrasonic extraction of veterinary antibiotics from soils and pig slurry with SPE clean-up and LC–UV and fluorescence detection. Talanta 2004, 64, 1058–1064. [Google Scholar] [CrossRef]
- Uslu, M.O.; Yediler, A.; Balcıoğlu, I.A.; Schulte-Hostede, S. Analysis and Sorption Behavior of Fluoroquinolones in Solid Matrices. Water Air Soil Pollut. 2008, 190, 55–63. [Google Scholar] [CrossRef]
- Robinson, B.J.; Hellou, J. Biodegradation of endocrine disrupting compounds in harbour seawater and sediments. Sci. Total Environ. 2009, 407, 5713–5718. [Google Scholar] [CrossRef]
- Sorensen, L.K.; Hansen, H. Determination of oxolinic acid in marine sediments by HPLC with fluorescence detection. J. Liq. Chromatogr. Relat. Technol. 2001, 24, 2469–2476. [Google Scholar] [CrossRef]
- Wie, Q.; Yang, J.; Zhang, Y.; Chemg, G.; Du, B. Determination of antimony (III) in environmental water samples in microemulsion system by the fluorescence quenching method. Talanta 2002, 58, 419–426. [Google Scholar]
- San Vicente, B.; Costa-Fernandez, J.M.; Jin, W.L.; Pereiro, R.; Sanz-Medel, A. Determination of trace levels of mercury in water samples based on room temperature phosphorescence energy transfer. Anal. Chim. Acta 2002, 455, 179–186. [Google Scholar]
- Coble, P.G.; Green, S.A.; Blough, N.V.; Gagosian, R.B. Characterization of disove organic matter in the Black Sea by fluorescence spectroscopy. Nature 1990, 348, 432–435. [Google Scholar] [CrossRef]
- Coble, P.G. Characterization of marine and terrestial DOM in seawater using excitation-emission matrix spectroscopy. Mar. Chem. 1996, 51, 325–346. [Google Scholar] [CrossRef]
- Goncalves-Araujo, R.; Granskog, M.A.; Bracher, A.; Azetsu-Scott, K.; Dodd, P.A.; Stedmon, C.A. Using fluorescent dissolved organic matter to trace and distinguish the origin of Arctic waters. Sci. Rep. 2016, 6, 33978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carstea, E.M.; Bridgeman, J.; Baker, A.; Reynolds, D.M. Fluorescence spectroscopy for wastewater monitoring: A review. Water Res. 2016, 95, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Yu, H.; Yan, Z.; Gao, H.; Zhang, Y. Tracking variations of fluorescent dissolved organic matter during wastewater treatment by accumulative fluorescence emission spectroscopy combined with principal component, second derivative and canonical correlation analysis. Chemosphere 2018, 194, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Tedetti, M.; Cuet, P.; Guigue, C.; Goutx, M. Characterization of dissolved organic matter in a coral reef ecosystem subjected to anthropogenic pressures (La Réunion Island, Indian Ocean) using multi-dimensional fluorescence spectroscopy. Sci. Total Environ. 2011, 409, 2198–2210. [Google Scholar] [CrossRef]
- Huang, C.-C.; Li, Y.-M.; Yang, H.; Sun, D.-Y.; Xu, L.-J.; Chen, X. Study of influencing factors to chromophoric dissolved organic matter absorption properties from fluorescence features in Taihu lake in autumn. J. Limnol. 2013, 72, 326–335. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Shi, H.; Jing, H.; Liu, R.; Cai, Q.; Takemura, M.; Haraguchi, S. Characterization and variations of dissolved organic matter in the Lake Taihu area of China. Water Sci. Technol. Water Supply 2012, 12, 439–450. [Google Scholar] [CrossRef]
- Goslan, E.H.; Voros, S.; Banks, J.; Wilson, D.; Hillis, P.; Campbell, A.T.; Parsons, S.A. A Model for Predicting Dissolved Organic Carbon Distribution in a Reservoir Water using Fluorescence Spectroscopy. Water Res. 2004, 38, 783–791. [Google Scholar] [CrossRef]
- Muller, C.L.; Baker, A.; Hutchinson, R.; Fairchild, I.; Kidd, C. Analysis of rainwater dissolved organic carbon compounds using fluorescence spectrophotometry. Atmos. Environ. 2008, 42, 8036–8045. [Google Scholar] [CrossRef]
- Costa, A.S.; Passos, E.D.A.; Garcia, C.A.B.; Alves, J.D.P.H. Characterization of Dissolved Organic Matter in the Piauí River Estuary, Northeast Brazil. J. Braz. Chem. Soc. 2011, 22, 2139–2147. [Google Scholar] [CrossRef] [Green Version]
- Carstea, E.M. Fluorescence Spectroscopy as a Potential Tool for In-Situ Monitoring of Dissolved Organic Matter in Surface Water Systems. In Water Pollution; Balkis, N., Ed.; IntechOpen: London, UK, 2012. [Google Scholar] [CrossRef]
- Kepkay, P.E.; Yeung, C.W.; Bugden, J.B.C.; Li, Z.; Lee, K. Ultraviolet Fluorescence Spectroscopy (UVFS): A new means of determining the effect of chemical dispersants on oil spills. In Proceedings of the International Oil spill Conference, Savannah, GA, USA, 4–8 May 2008; pp. 639–642. [Google Scholar]
- Nahorniak, M.L.; Booksh, K.S. Excitation-emission matrix fluorescence spectroscopy in conjuction with multiway analysis for PAH detection in complex matrices. Analyst 2006, 131, 1308–1315. [Google Scholar] [CrossRef]
- Uebel, U.; Kubitz, J.; Anders, A. Laser Induced Fluorescence Spectroscopy of Phytoplankton and Chemicals with Regard to an in situ Detection in Waters. J. Plant Physiol. 1996, 148, 586–592. [Google Scholar] [CrossRef]
- Baumann, T.; Haaszio, S.; Niessner, R. Applications of a Laser Induced Fluorescence Spectroscopy Sensor in aquatic systems. Water Res. 2000, 34, 1318–1326. [Google Scholar] [CrossRef]
- Sivaprakasam, V.; Killinger, D.K. Tunable ultraviolet laser-induced fluorescence detection of trace plastics and dissolved organic compounds in water. Appl. Opt. 2003, 42, 6739–6746. [Google Scholar] [CrossRef]
- Sivaprakasam, V.; Shannon, R.F.; Luo, C.; Coble, P.G.; Boehme, J.R.; Killinger, D.K. Development and initial Calibration of a portable Laser Induced Fluorescence system used for in situ measurements of trace plastics and organics in seawater and the Gulf of Mexico. Appl. Opt. 2003, 42, 6747. [Google Scholar] [CrossRef] [PubMed]
- Sivaprakasam, V.; Killinger, D.K. How Water Glows: Water Monitoring with Laser Fluorescence. Opt. Photonics News 2006, 17, 34–39. [Google Scholar] [CrossRef]
- Sharikova, A.V.; Killinger, D.K. Laser and UV-LED-induced fluorescence detection of dissolved organic compounds in water. In Proceedings of the SPIE 7666, Sensors, and Command, Control, Communications and Intelligence (C3I) Technologies for Homeland Security and Homeland Defence IX, 76661L, Orlando, FL, USA, 5–9 April 2010; SPIE Bellingham: Washington, DC, USA, 2010. [Google Scholar] [CrossRef]
- Sharikova, A.V.; Killinger, D.K. LIF detection of trace species in water Using different UV laser wavelengths. Int. J. High Speed Electron. Syst. 2007, 17, 689–695. [Google Scholar] [CrossRef]
- Gervase, L.; Carstea, E.M.; Pavelescu, G.; Savastru, D. Laser Induced Fluorescence Efficiency in water quality assessment. Rom. Rep. Phys. 2010, 62, 652–659. [Google Scholar]
- Carstea, E.M.; Pavelescu, G.; Baker, A.; Roman, C.; Ioja, C.; Savastru, D. Pollution analysis on the Arges River using fluorescence spectroscopy. J. Optoelectron. Adv. Mater. 2008, 10, 1489–1494. [Google Scholar]
- Du, R.; Yang, D.; Jiang, G.; Song, Y.; Yin, X. An Approach for In Situ Rapid Detection of Deep-Sea Aromatic Amino Acids Using Laser-Induced Fluorescence. Sensors 2020, 20, 1330. [Google Scholar] [CrossRef] [Green Version]
- Bukin, O.; Proschenko, D.; Alexey, C.; Korovetskiy, D.; Bukin, I.; Yurchik, V.; Sokolova, I.; Nadezhkin, A. New Solutions of Laser-Induced Fluorescence for Oil Pollution Monitoring at Sea. Photonics 2020, 7, 36. [Google Scholar] [CrossRef]
- Kang, J.; Li, R.; Wang, Y.; Chen, Y.; Yang, Y. Ultrasensitive detection of trace amounts of lead in water by LIBS-LIF using a wood-slice substrate as a water absorber. J. Anal. At. Spectrom. 2017, 32, 2292–2299. [Google Scholar] [CrossRef]
- Lui, S.; Godwal, Y.; Taschuk, M.; Tsui, Y.; Fedosejevs, R. Detection of Lead in Water Using Lasr-Induced Breakdown Spectroscopy and Laser-Induced Fluorescence. Anal. Chem. 2008, 80, 1995–2000. [Google Scholar] [CrossRef]
- Tian, H.; Jiao, L.; Dong, D. Rapid determination of trace cadmium in drinking water using laser-induced breakdown spectroscopy coupled with chelating resin enrichment. Sci. Rep. 2019, 9, 10443. [Google Scholar] [CrossRef] [Green Version]
- Yao, M.; Lin, J.; Liu, M.; Xu, Y. Detection of chromium in wastewater from refuse incineration power plant near Poyang Lake by laser induced breakdown spectroscopy. Appl. Opt. 2012, 51, 1552–1557. [Google Scholar] [CrossRef]
- Koch, S.; Garen, W.; Muller, M.; Neu, W. Detection of chromium in liquids by laser induced breakdown spectroscopy (LIBS). Appl. Phys. A Mater. Sci. Process 2004, 79, 1071–1073. [Google Scholar] [CrossRef]
- Xue, B.; Tian, Y.; Lu, Y.; Li, Y.; Zheng, R. Characteristics of the secondary breakdown of DP-LIBS in bulk water with different axial focusing arrangements and laser energies. Spectrochim. Acta B At. Spectrosc. 2019, 151, 20–25. [Google Scholar] [CrossRef]
- Li, Q.; Tian, Y.; Xue, B.; Li, N.; Ye, W.; Lu, Y.; Zheng, R. Improvement in the analytical performance of underwater LIBS signals by exploiting the plasma image information. J. Anal. At. Spectrom. 2020, 35, 366. [Google Scholar] [CrossRef]
- Guo, J.; Lu, Y.; Cheng, K.; Song, J.; Ye, W.; Li, N.; Zheng, R. Development of a compact underwater laser induced breakdown spectroscopy (LIBS) system and preliminary results in sea trials. Appl. Opt. 2017, 56, 8196. [Google Scholar] [CrossRef]
- Gabbarini, V.; Rossi, R.; Ciparisse, J.-F.; Malizia, A.; Divizia, A.; De Filippis, P.; Anselmi, M.; Carestia, M.; Palombi, L.; Divizia, M.; et al. Laser-induced fuorescence (LIF) as a smart method for fast environmental virological analyses: Validation on Picornaviruses. Sci. Rep. 2019, 9, 12598. [Google Scholar] [CrossRef] [Green Version]
- NAVFAC (Naval Facilities Engineering Command). Tech Data Sheet, Near-Real Time UV Fluorescence Technique for Characterization of PAHs in Marine Sediment, NFESC TDS-2075-ENV; NAVFAC: Washington, DC, USA, 10 June 2000; pp. 20374–25065. Available online: https://frtr.gov/pdf/uvfluorescence_2.pdf (accessed on 10 June 2022).
- Cristescu, L.; Pavelescu, G.; Cârstea, E.M.; Săvăstru, D. Evaluation of petroleum contaminants in soil by Fluorescence Spectroscopy. Environ. Eng. Manag. J. 2009, 8, 1269–1273. [Google Scholar] [CrossRef]
- Mielnik, L.; Kowalczuk, P. Optical characteristic of humic acids from lake sediments by excitation-emission matrix fluorescence with PARAFAC model. J. Soils Sediments 2018, 18, 2851–2862. [Google Scholar] [CrossRef] [Green Version]
- Santı’n, C.; Yamashita, Y.; Otero, X.L.; A’lvarez, M.A.; Jaffe, R. Characterizing humic substances from estuarine soils and sediments by excitation-emission matrix spectroscopy and parallel factor analysis. Biogeochemistry 2009, 96, 131–147. [Google Scholar] [CrossRef]
- Dang, D.H.; Lenoble, V.; Durrieu, G.; Mullot, J.-U.; Mounier, S.; Garnier, C. Sedimentary dynamics of coastal organic matter: An assessment of the porewater size/reactivity model by spectroscopic techniques. Estuar. Coast. Shelf Sci. 2014, 151, 100–111. [Google Scholar] [CrossRef]
- Derrien, M.; Lee, Y.K.; Ark, J.-E.; Li, P.; Chen, M.; Lee, S.H.; Lee, S.H.; Lee, J.-B.; Hur, J. Spectroscopic and molecular characterization of humic substances (HS) from soils and sediments in a watershed: Comparative study of HS chemical fractions and the origins. Environ. Sci. Pollut. Res. 2017, 24, 16933–16945. [Google Scholar] [CrossRef]
- Wei, D.; Li, Y.; Cai, S.-S.; Jin, L.; Li, Y.-M.; Wang, W.; Bai, Y.; Hu, Y.; Clarke, N. Fluorescence Characteristics of Humic Acid in Chinese Black Soil under Long-Term Fertilization. Adv. Polym. Technol. 2019, 2019, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Fasurova, N.; Pospisilova, L. Characterization of soil humic substances by Ultraviolet-Visible and Synchronous Fluorescence Spectroscopy. J. Cent. Eur. Agric. 2010, 11, 351–358. [Google Scholar]
- Perez, M.G.; Milori, D.M.B.P.; Martin-Neto, L.; Colnago, L.A.; de Camargo, O.A.; Berton, R.; Bettiol, W. Laser-Induced Fluorescence of organic matter from a Brazilian oxisol under sewage-sludge applications. Sci. Agric. 2006, 63, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Milori, D.M.B.P.; Galeti, H.V.A.; Martin-Neto, L.; Dieckow, J.; Gonzalez-Perez, M.; Bayer, C.; Salton, J. Organic matter study of whole soil samples using laser-induced fluorescence spectroscopy. Soil Sci. Soc. Am. J. 2006, 70, 57–63. [Google Scholar] [CrossRef]
- Segnini, A.; Posadas, A.; Quiroz, R.; Milori, D.M.B.P.; Saab, S.C.; Martin-Neto, L.; Vaz, C.M.P. Spectroscopic assessment of soil organic matter in wetlands from high Andes. Soil Sci. Soc. Am. J. 2010, 74, 2246–2253. [Google Scholar] [CrossRef]
- Ferreira, E.C.; Ferreira, E.J.; Villas-Boas, P.R.; Senesi, G.S.; Carvalho, C.M.; Romano, R.A.; Martin-Neto, L.; Milori, D.M.B.P. Novel estimation of the humification degree of soil organic matter by laser induced breakdown spectroscopy. Spectrochim. Acta B 2014, 99, 76–81. [Google Scholar] [CrossRef]
- Senesi, G.S.; Martin-Neto, L.; Villas-Boas, P.R.; Nicolodelli, G.; Milori, D.M.B.P. Laser-based spectroscopic methods to evaluate the humification degree of soil organic matter in whole soils: A review. J Soils Sediments 2018, 18, 1292–1302. [Google Scholar] [CrossRef]
- da Silva, J.M.; Utkin, A.B. Application of Laser-Induced Fluorescence in Functional Studies of Photosynthetic Biofilms. Processes 2018, 6, 227. [Google Scholar] [CrossRef] [Green Version]
- Nunes, R.A.; Tabares, R.H.; Bunkin, A.; Persh, S. Compact lidar for remote sensing of water pollution. Trans. Ecol. Environ. 1997, 14, 401–408. [Google Scholar]
- Babichenko, S. Laser Remote Sensing of the European Marine Environment: LIF Technology and Applications. In Remote Sensing of the European Seas; Barale, V., Gade, M., Eds.; Springer Science+Business Media B.V.: Berlin/Heidelberg, Germany, 2008; pp. 189–204. [Google Scholar]
- Pavelescu, G.; Vasilescu, J.-G.; Strechie, C.; Babichenko, S.; Lisin, A.; Onciu, T.; Belegante, L. Water pollution analysis from LIDAR investigations on the Romanian Black Sea coast. In Proceedings of the SPIE—Volume 6743 Remote Sensing of the Ocean, Sea Ice, and Large Water Regions 2007, Florence, Italy, 17–18 September 2007. [Google Scholar] [CrossRef]
- Vasilescu, J.; Belegante, L.; Sliwinski, C.; Cristescu, C.P. Using Active Remote Sesnsing to assess the seawater quality. UPB Sci. Bull. 2008, 70, 41–48. [Google Scholar]
- Vasilescu, J.; Onciu, T.; Jugaru, L.; Belegante, L. Remote estimation of fluorescence marine components distribution. Rom. Rep. Phys. 2009, 61, 721–729. [Google Scholar]
- Utkin, A.; Lavrov, A.; Vilar, R.; Babichenko, S.; Shchemelyov, S.; Sobolev, I.; Bastos, L.; Deurloo, R.; Palenzuela, J.T.; Yarovenko, N.; et al. Optical methods for water pollution monitoring, Spatial and Organizational Dynamics Discussion Papers. In CIEO-Research Centre for Spatial and Organizational Dynamics; University of Algarve: Faro, Portugal, 2011. [Google Scholar]
- Foldager, F.F.; Pedersen, J.M.; Skov, E.H.; Evgrafova, A.; Green, O. LiDAR-Based 3D Scans of Soil Surfaces and Furrows in Two Soil Types. Sensors 2019, 19, 661. [Google Scholar] [CrossRef] [Green Version]
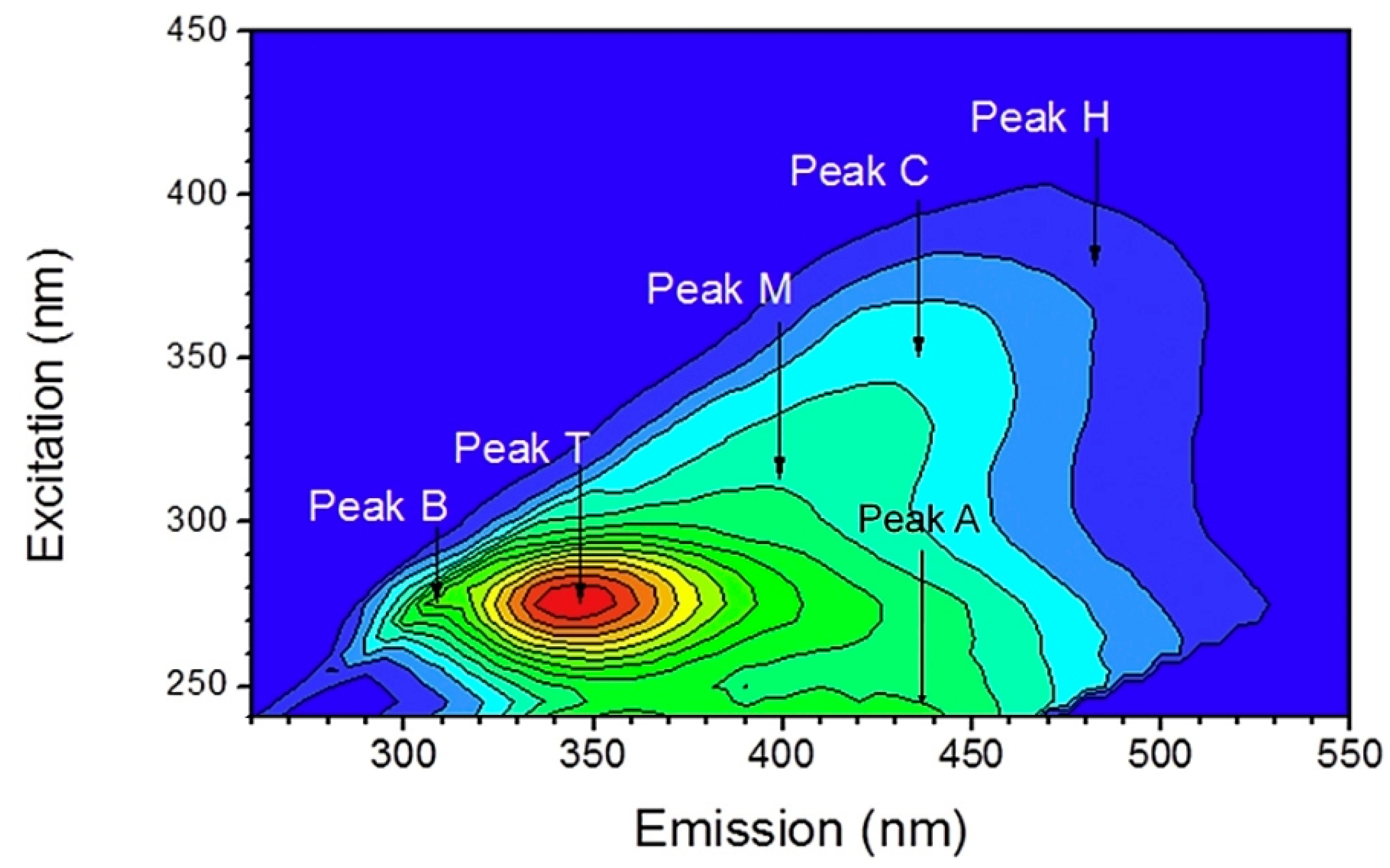


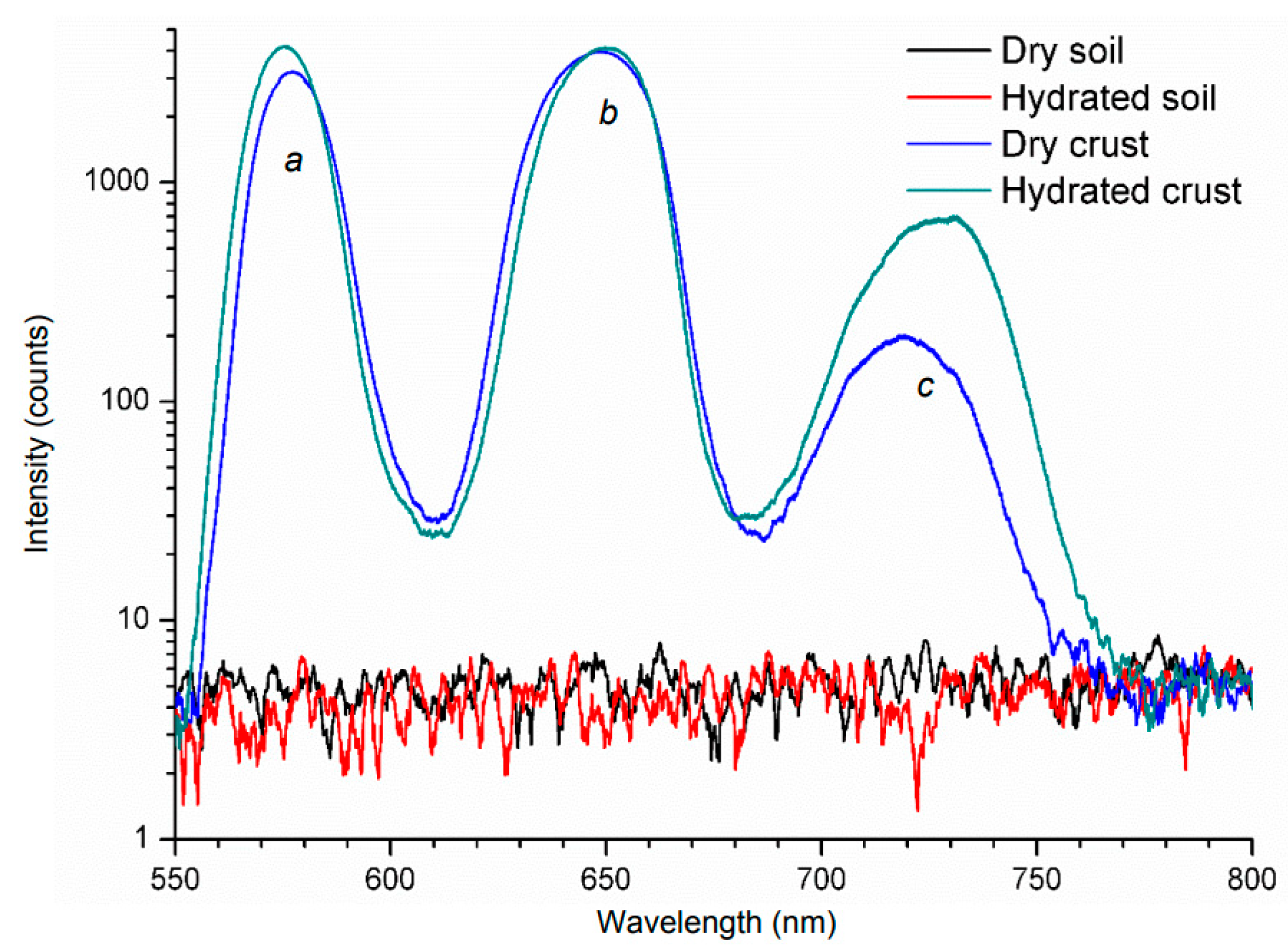
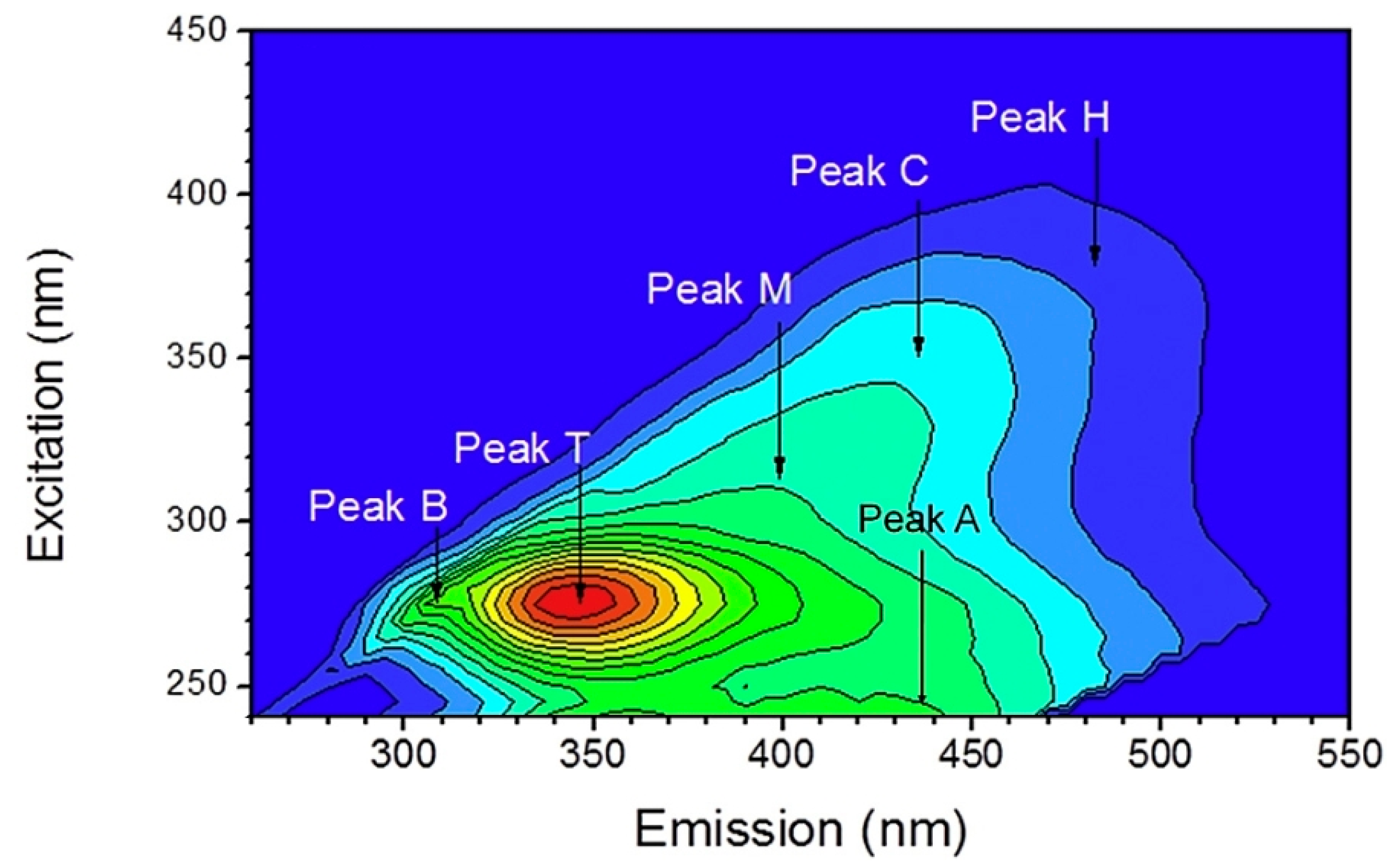
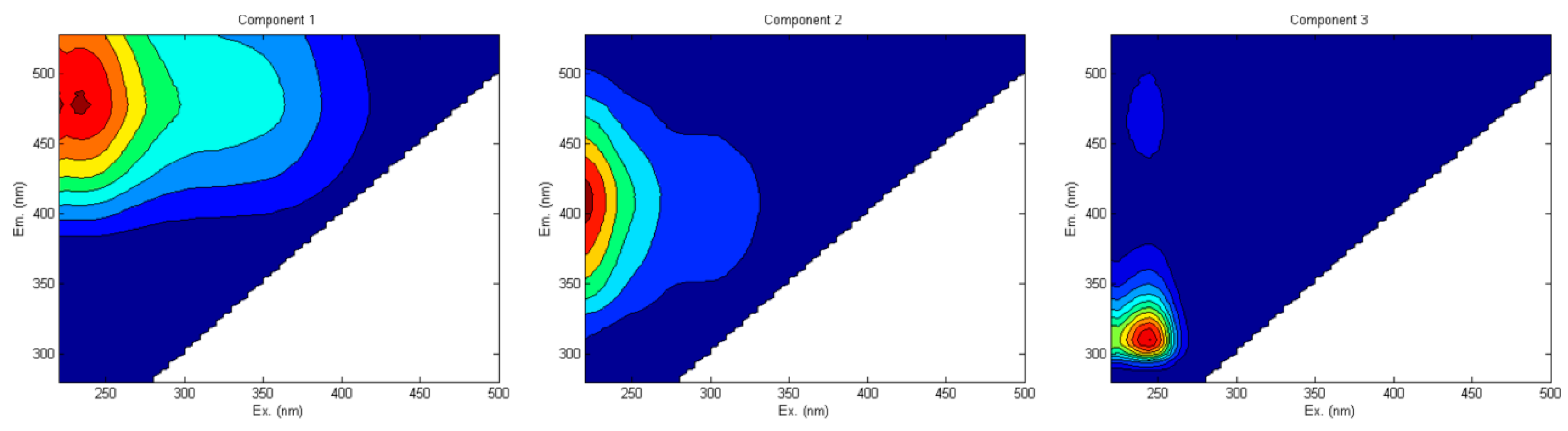
| Name | Letter | Ex (nm)/Em (nm) |
|---|---|---|
| Humic-like | C | 340–360/420–480 |
| Fulvic-like | A | 240–260/380–460 |
| Tyrosine-like | B (B1, B2) | 265–285/290–310 |
| Tryptophan-like | T (T1, T2) | 265–285/290–340 |
| Microbial-like (Marine-like) | M | 310–330/390–410 |
| Humic-like | H | 370–390/480–500 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zacharioudaki, D.-E.; Fitilis, I.; Kotti, M. Review of Fluorescence Spectroscopy in Environmental Quality Applications. Molecules 2022, 27, 4801. https://doi.org/10.3390/molecules27154801
Zacharioudaki D-E, Fitilis I, Kotti M. Review of Fluorescence Spectroscopy in Environmental Quality Applications. Molecules. 2022; 27(15):4801. https://doi.org/10.3390/molecules27154801
Chicago/Turabian StyleZacharioudaki, Despoina-Eleni, Ioannis Fitilis, and Melina Kotti. 2022. "Review of Fluorescence Spectroscopy in Environmental Quality Applications" Molecules 27, no. 15: 4801. https://doi.org/10.3390/molecules27154801
APA StyleZacharioudaki, D.-E., Fitilis, I., & Kotti, M. (2022). Review of Fluorescence Spectroscopy in Environmental Quality Applications. Molecules, 27(15), 4801. https://doi.org/10.3390/molecules27154801