
Site Density Functional Theory and Structural Bioinformatics Analysis of the SARS-CoV Spike Protein and hACE2 Complex

 ,
,  , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
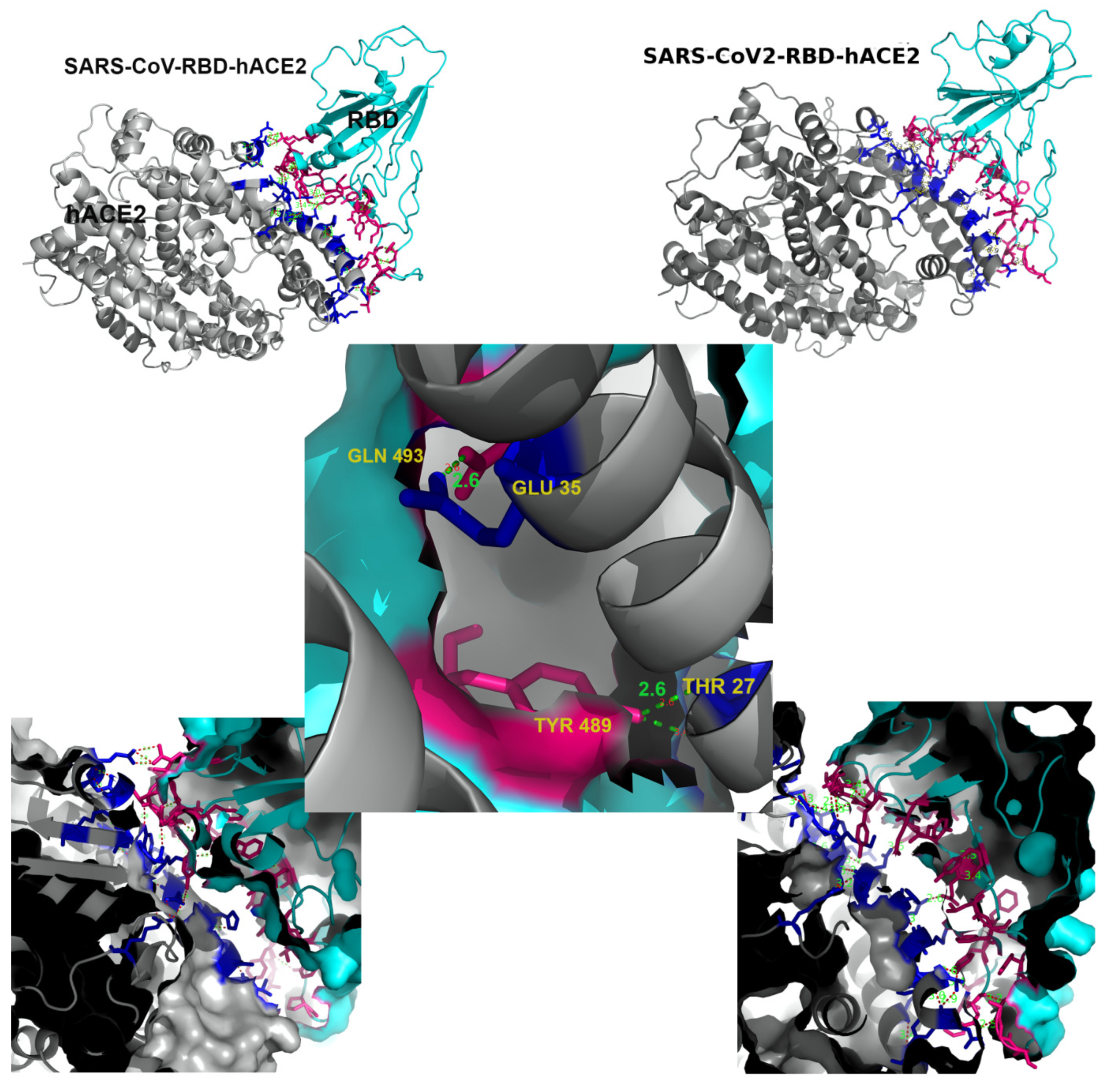
2.1. Preliminary Bioinformatics Treatment
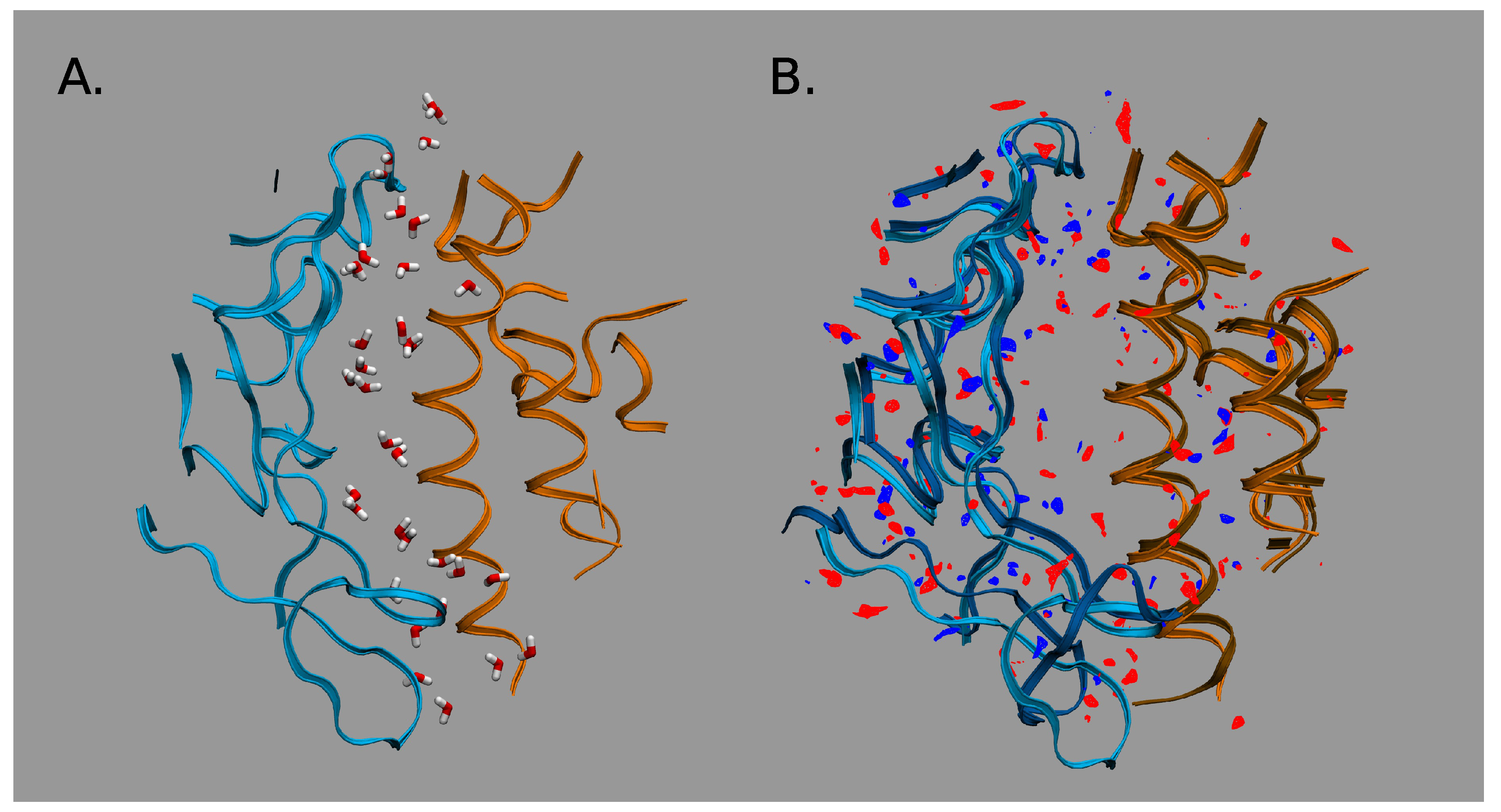
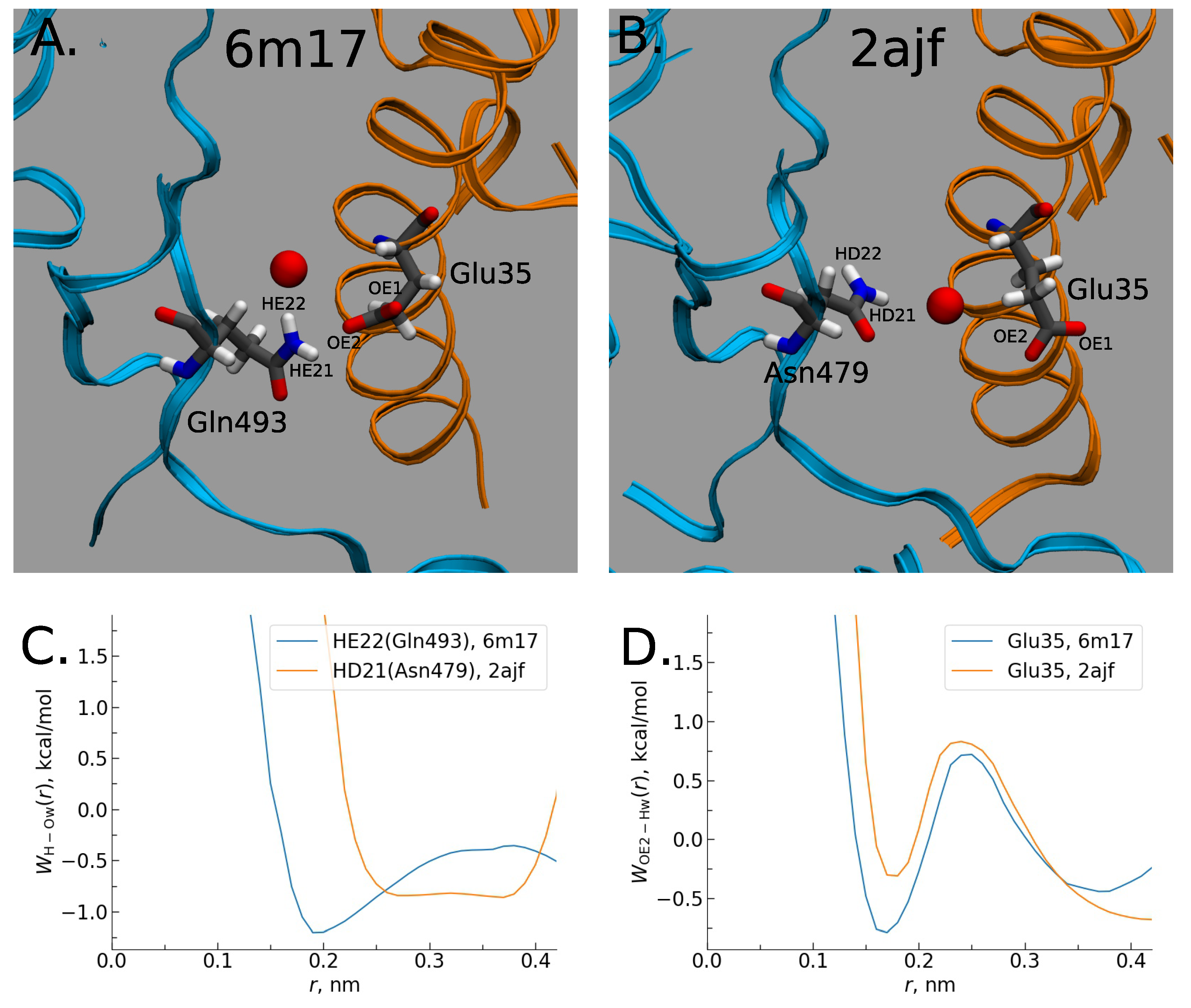
2.2. 3DRISM Studies of Water-Mediated Interactions in SARS-CoV-1-hACE2 and SARS-CoV-2-hACE2 Complexes
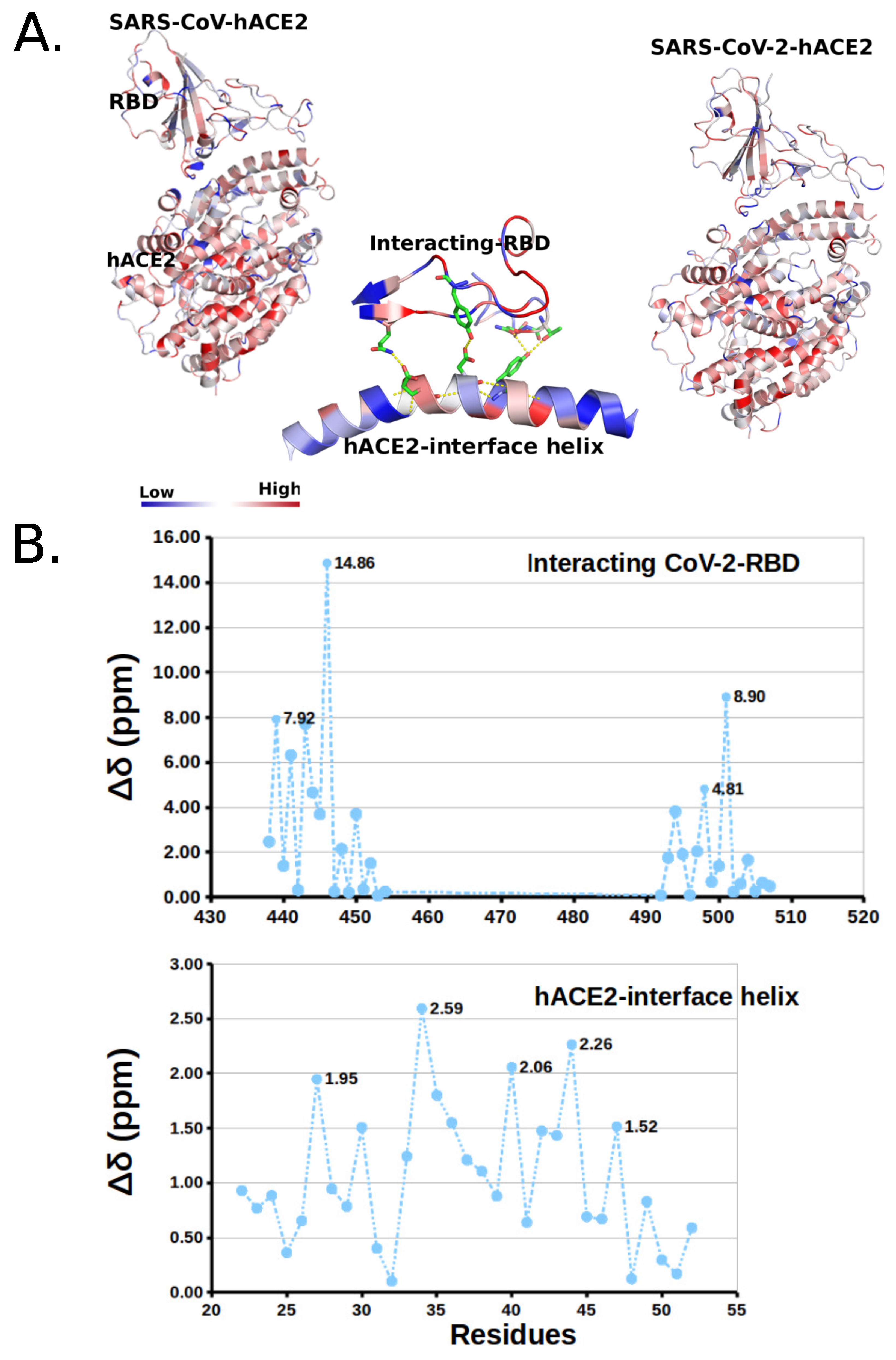
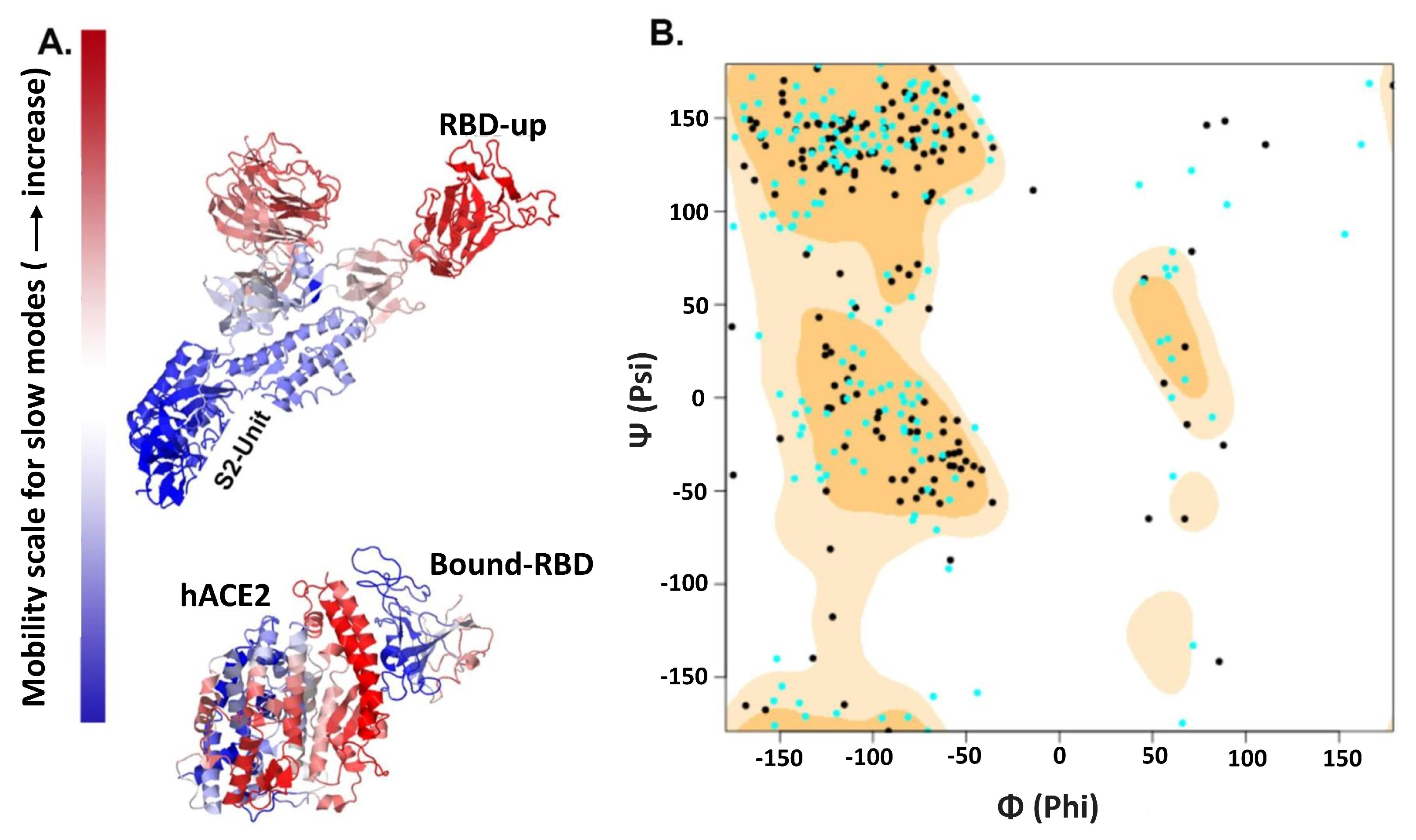
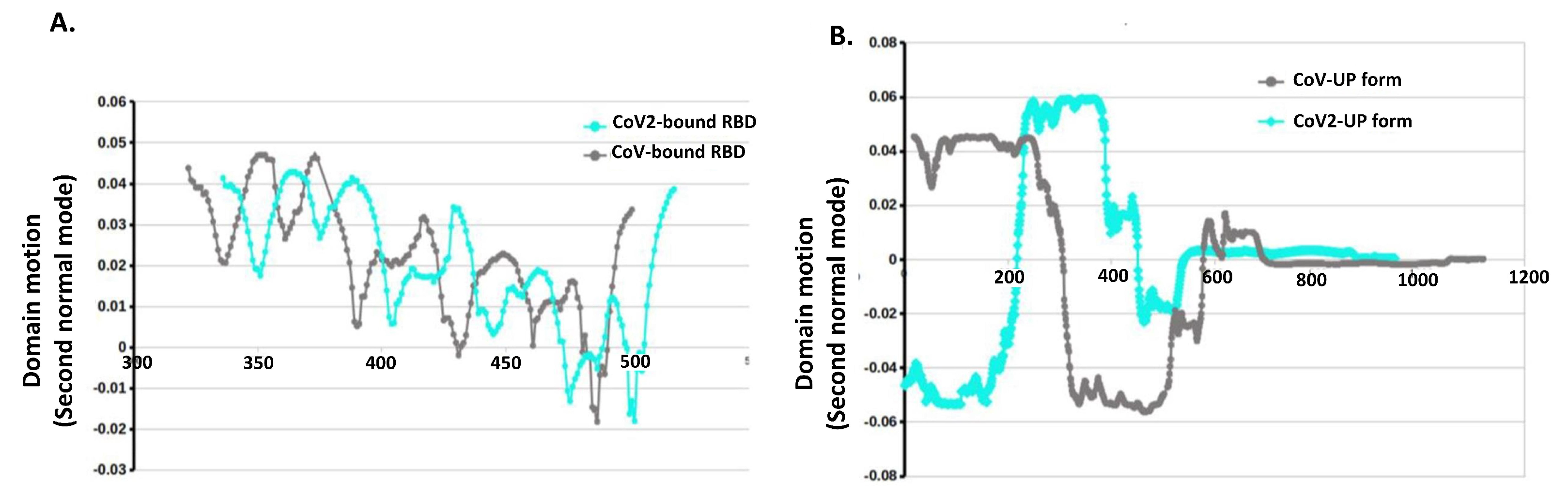
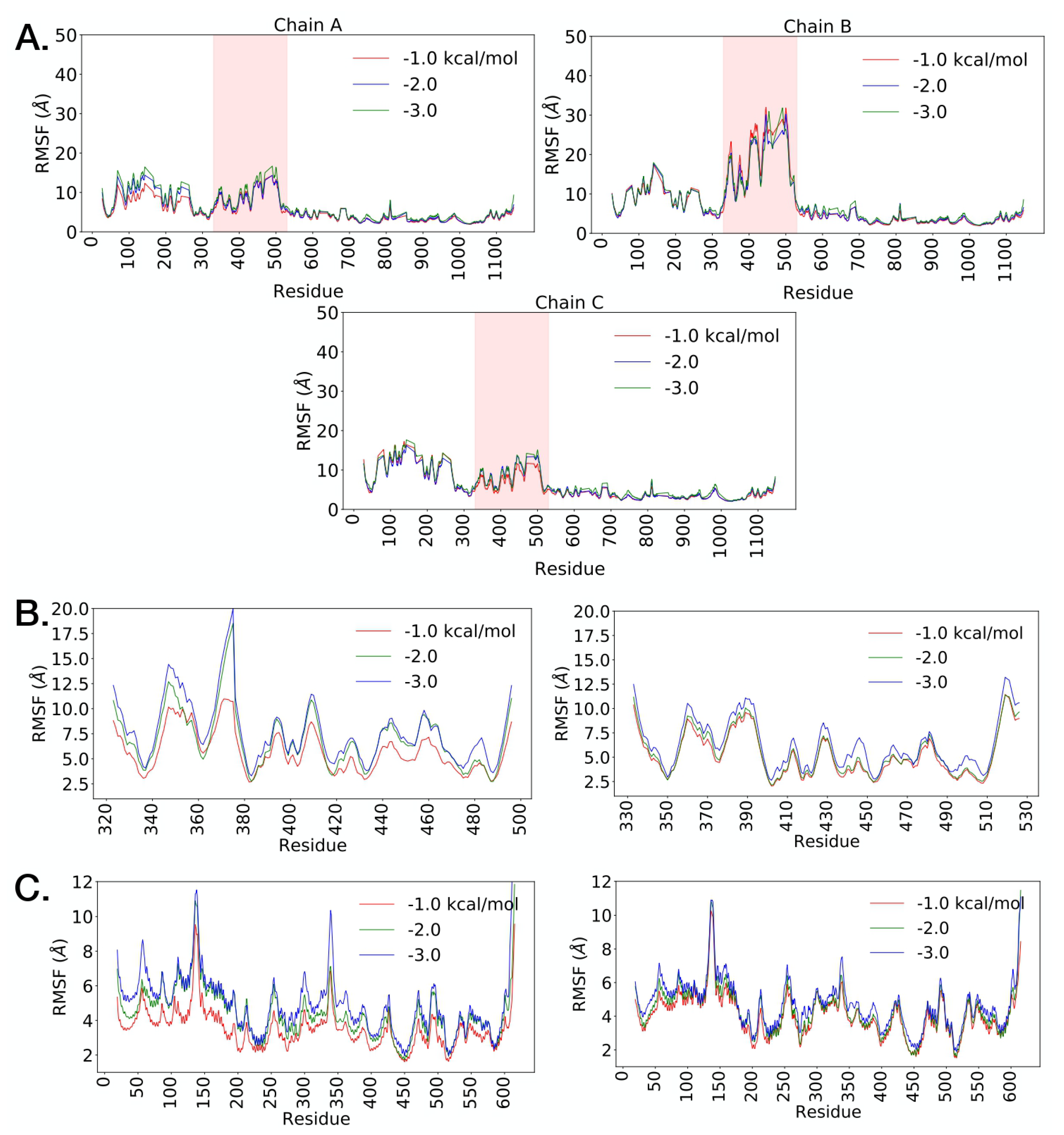
2.3. Posterior Bioinformatics Treatment by NMA and CGS
3. Discussion and Conclusions
4. Methods
4.1. Classical Site Density Functional Theory
4.2. Bioinformatics Tools
4.2.1. Root-Mean-Squared Deviation (RMSD) of C-α Atoms
4.2.2. Normal Mode Analysis
4.2.3. Rigidity Analysis
4.2.4. Constrained Geometric Simulations for Conformational Sampling
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yao, H.; Song, Y.; Chen, Y.; Wu, N.; Xu, J.; Sun, C.; Zhang, J.; Weng, T.; Zhang, Z.; Wu, Z.; et al. Molecular Architecture of the SARS-CoV-2 Virus. Cell 2020, 183, 730–738.e13. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Qing, E.; Gallagher, T. SARS Coronavirus Redux. Trends Immunol. 2020, 41, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of the SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F. Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Challenges for Density Functional Theory. Chem. Rev. 2012, 112, 289–320. [Google Scholar] [CrossRef]
- Jones, R.O. Density functional theory: Its origins, rise to prominence, and future. Rev. Mod. Phys. 2015, 87, 897–923. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.P.; McDonald, I.R. Theory of Simple Liquids: With Applications to Soft Matter; Elsevier Academic Press: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Ding, L.; Levesque, M.; Borgis, D.; Belloni, L. Efficient molecular density functional theory using generalized spherical harmonics expansions. J. Chem. Phys. 2017, 147, 094107. [Google Scholar] [CrossRef] [Green Version]
- Ishizuka, R.; Yoshida, N. Application of efficient algorithm for solving six-dimensional molecular Ornstein-Zernike equation. J. Chem. Phys. 2012, 136, 114106. [Google Scholar] [CrossRef]
- Chandler, D.; Andersen, H.C. Optimized Cluster Expansions for Classical Fluids. II. Theory of Molecular Liquids. J. Chem. Phys. 1972, 57, 1930–1937. [Google Scholar] [CrossRef]
- Chandler, D.; Mccoy, J.D.; Singer, S.J. Density Functional Theory of Nonuniform Polyatomic Systems. 1. General Formulation. J. Chem. Phys. 1986, 85, 5971–5976. [Google Scholar] [CrossRef]
- Chandler, D.; Mccoy, J.D.; Singer, S.J. Density Functional Theory of Nonuniform Polyatomic Systems. 2. Rational Closures for Integral-Equations. J. Chem. Phys. 1986, 85, 5977–5982. [Google Scholar] [CrossRef]
- Seok, C.; Oxtoby, D.W. Nucleation in n-alkanes: A density-functional approach. J. Chem. Phys. 1998, 109, 7982–7990. [Google Scholar] [CrossRef] [Green Version]
- Sumi, T.; Sekino, H. An interaction site model integral equation study of molecular fluids explicitly considering the molecular orientation. J. Chem. Phys. 2006, 125, 034509. [Google Scholar] [CrossRef] [PubMed]
- Lischner, J.; Arias, T.A. Kohn-Sham-Like Approach toward a Classical Density-Functional Theory of Inhomogeneous Polar Molecular Liquids: An Application to Liquid Hydrogen Chloride. Phys. Rev. Lett. 2008, 101, 216401. [Google Scholar] [CrossRef] [Green Version]
- Lischner, J.; Arias, T.A. Classical Density-Functional Theory of Inhomogeneous Water Including Explicit Molecular Structure and Nonlinear Dielectric Response. J. Phys. Chem. B 2010, 114, 1946–1953. [Google Scholar] [CrossRef] [Green Version]
- Chong, S.H.; Ham, S. Aqueous interaction site integral-equation theory that exactly takes into account intramolecular correlations. J. Chem. Phys. 2012, 137, 154101. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, S.; Wu, J. A Site Density Functional Theory for Water: Application to Solvation of Amino Acid Side Chains. J. Chem. Theory Comput. 2013, 9, 1896–1908. [Google Scholar] [CrossRef]
- Sundararaman, R.; Arias, T. Efficient classical density-functional theories of rigid-molecular fluids and a simplified free energy functional for liquid water. Comput. Phys. Commun. 2014, 185, 818–825. [Google Scholar] [CrossRef] [Green Version]
- Sheng, S.; Miller, M.; Wu, J. Molecular Theory of Hydration at Different Temperatures. J. Phys. Chem. B 2017, 121, 6898–6908. [Google Scholar] [CrossRef]
- Chuev, G.N.; Fedotova, M.V.; Valiev, M. Chemical bond effects in classical site density functional theory of inhomogeneous molecular liquids. J. Chem. Phys. 2020, 152, 041101. [Google Scholar] [CrossRef] [PubMed]
- Chuev, G.N.; Fedotova, M.V.; Valiev, M. Renormalized site density functional theory. J. Stat. Mech. 2021, 2021, 033205. [Google Scholar] [CrossRef]
- Chuev, G.N.; Fedotova, M.V.; Valiev, M. Renormalized site density functional theory for models of ion hydration. J. Chem. Phys. 2021, 155, 064501. [Google Scholar] [CrossRef] [PubMed]
- Beglov, D.; Roux, B. An integral equation to describe the solvation of polar moleculesin liquid water. J. Phys. Chem. B 1997, 101, 7821–7826. [Google Scholar] [CrossRef]
- Du, Q.H.; Beglov, D.; Roux, B. Solvation free energy of polar and nonpolar molecules in water: Anextended interaction site integral equation theory in three dimensions. J. Phys. Chem. B 2000, 104, 796–805. [Google Scholar] [CrossRef]
- Howard, J.J.; Perkyns, J.S.; Choudhury, N.; Pettitt, B.M. Integral Equation Study of the Hydrophobic Interaction between Graphene Plates. J. Chem. Theory Comput. 2008, 4, 1928–1939. [Google Scholar] [CrossRef] [Green Version]
- Kast, S.M. Combinations of simulation and integral equation theory. Phys. Chem. Chem. Phys. 2001, 3, 5087–5092. [Google Scholar] [CrossRef]
- Chuev, G.N.; Fedorov, M.V. Reference interaction site model study of self-aggregating cyanine dyes. J. Chem. Phys. 2009, 131, 074503. [Google Scholar] [CrossRef]
- Genheden, S.; Luchko, T.; Gusarov, S.; Kovalenko, A.; Ryde, U. An MM/3D-RISM Approach for Ligand Binding Affinities. J. Phys. Chem. B 2010, 114, 8505–8516. [Google Scholar] [CrossRef]
- Luchko, T.; Gusarov, S.; Roe, D.R.; Simmerling, C.; Case, D.A.; Tuszynski, J.; Kovalenko, A. Three-Dimensional Molecular Theory of Solvation Coupled with Molecular Dynamics in Amber. J. Chem. Theory Comput. 2010, 6, 607–624. [Google Scholar] [CrossRef] [Green Version]
- Perkyns, J.S.; Lynch, G.C.; Howard, J.J.; Pettitt, B.M. Protein solvation from theory and simulation: Exact treatment of Coulomb interactions in three-dimensional theories. J. Chem. Phys. 2010, 132, 064106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, D.S.; Chuev, G.N.; Ratkova, E.L.; Fedorov, M.V. In Silico Screening of Bioactive and Biomimetic Solutes Using Molecular Integral Equation Theory. Curr. Pharm. Des. 2011, 17, 1695–1708. [Google Scholar] [CrossRef] [PubMed]
- Sindhikara, D.J.; Yoshida, N.; Hirata, F. Placevent: An algorithm for prediction of explicit solvent atom distribution—Application to HIV-1 protease and F-ATP synthase. J. Comput. Chem. 2012, 33, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Gusarov, S.; Pujari, B.S.; Kovalenko, A. Efficient treatment of solvation shells in 3D molecular theory of solvation. J. Comput. Chem. 2012, 33, 1478–1494. [Google Scholar] [CrossRef] [PubMed]
- Fedotova, M.V.; Kruchinin, S.E. 1D-RISM study of glycine zwitterion hydration and ion-molecular complex formation in aqueous NaCl solutions. J. Mol. Liq. 2012, 169, 1–7. [Google Scholar] [CrossRef]
- Fedotova, M.V.; Kruchinin, S.E. Hydration of para-aminobenzoic acid (PABA) and its anion-The view from statistical mechanics. J. Mol. Liq. 2013, 186, 90–97. [Google Scholar] [CrossRef]
- Fedotova, M.V.; Kruchinin, S.E. Ion-binding of glycine zwitterion with inorganic ions in biologically relevant aqueous electrolyte solutions. Biophys. Chem. 2014, 190, 25–31. [Google Scholar] [CrossRef]
- Fedotova, M.V.; Dmitrieva, O. Ion-selective interactions of biologically relevant in organic ions with alanine zwitterion: A 3D-RISM study. Amino Acids 2015, 47, 1015–1023. [Google Scholar] [CrossRef]
- Fedotova, M.V.; Dmitrieva, O.A. Characterization of selective binding of biologically relevant inorganic ions with the proline zwitterion by 3D-RISM theory. New J. Chem. 2015, 39, 8594–8601. [Google Scholar] [CrossRef]
- Eiberweiser, A.; Nazet, A.; Kruchinin, S.E.; Fedotova, M.V.; Buchner, R. Hydration and ion binding of the osmolyte ectoine. J. Phys. Chem. B 2015, 119, 15203–15211. [Google Scholar] [CrossRef]
- Fedotova, M.V.; Dmitrieva, O. Proline hydration at low temperatures: Its role in the protection of cell from freeze-induced stress. Amino Acids 2016, 48, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Fedotova, M.V.; Kruchinin, S.E.; Chuev, G.N. Local ion hydration structure in aqueous imidazolium-based ionic liquids: The effects of concentration and anion nature. J. Mol. Liq. 2017, 247, 100–108. [Google Scholar] [CrossRef]
- Fedotova, M.V.; Kruchinin, S.E.; Chuev, G.N. Hydration structure of osmolyte TMAO: Concentration/pressure-induced response. New J. Chem. 2017, 41, 1219–1228. [Google Scholar] [CrossRef]
- Fedotova, M.V.; Kruchinin, S.E. Hydration and ion-binding of glycine betaine: How they may be involved into protection of proteins under abiotic stresses. J. Mol. Liq. 2017, 244, 489–498. [Google Scholar] [CrossRef]
- Güssregen, S.; Matter, H.; Hessler, G.; Lionta, E.; Heil, J.; Kast, S.M. Thermodynamic Characterization of Hydration Sites from Integral Equation-Derived Free Energy Densities: Application to Protein Binding Sites and Ligand Series. J. Chem. Inf. Model. 2017, 57, 1652–1666. [Google Scholar] [CrossRef]
- Dmitrieva, O.A.; Fedotova, M.V.; Buchner, R. Evidence for cooperative Na+ and Cl- binding by strongly hydrated L-proline. Phys. Chem. Chem. Phys. 2017, 19, 20474–20483. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, N. Role of Solvation in Drug Design as Revealed by the Statistical Mechanics Integral Equation Theory of Liquids. J. Chem. Inf. Model. 2017, 57, 2646–2656. [Google Scholar] [CrossRef]
- Fedotova, M.V. Compatible osmolytes—Bioprotectants: Is there a common link between their hydration and their protective action under abiotic stresses? J. Mol. Liq. 2019, 292, 111339. [Google Scholar] [CrossRef]
- Fedotova, M.V.; Kruchinin, S.E.; Chuev, G.N. Features of local ordering of biocompatible ionic liquids: The case of choline-based amino acid ionic liquids. J. Mol. Liq. 2019, 296, 112081. [Google Scholar] [CrossRef]
- Fedotova, M.V.; Kruchinin, S.E.; Chuev, G.N. Molecular insight on ion hydration and association in aqueous choline chloride solutions. J. Mol. Liq. 2020, 313, 113563. [Google Scholar] [CrossRef]
- Fedotova, M.V.; Kruchinin, S.E.; Chuev, G.N. Hydration features of the neurotransmitter acetylcholine. J. Mol. Liq. 2020, 304, 112757. [Google Scholar] [CrossRef]
- Friesen, S.; Fedotova, M.V.; Kruchinin, S.E.; Buchner, R. Hydration and dynamics of l-glutamate ion in aqueous solution. Phys. Chem. Chem. Phys. 2021, 23, 1590–1600. [Google Scholar] [CrossRef] [PubMed]
- Kruchinin, S.E.; Fedotova, M.V. Ion Pairing of the Neurotransmitters Acetylcholine and Glutamate in Aqueous Solutions. J. Phys. Chem. B 2021, 125, 11219–11231. [Google Scholar] [CrossRef]
- Olson, B.; Cruz, A.; Chen, L.; Ghattas, M.; Ji, Y.; Huang, K.; Ayoub, S.; Luchko, T.; McKay, D.J.; Kurtzman, T. An online repository of solvation thermodynamic and structural maps of SARS-CoV-2 targets. J. Comput. Aid. Mol. Des. 2020, 34, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Kobryn, A.E.; Maruyama, Y.; Velázquez-Martínez, C.A.; Yoshida, N.; Gusarov, S. Modeling the interaction of SARS-CoV-2 binding to the ACE2 receptor via molecular theory of solvation. New J. Chem. 2021, 45, 15448–15457. [Google Scholar] [CrossRef]
- Kinoshita, M.; Okamoto, Y.; Hirata, F. First-principle determination of peptide conformations in solvents:Combination of Monte Carlo simulated annealing and RISM theory. J. Am. Chem. Soc. 1998, 120, 1855–1863. [Google Scholar] [CrossRef]
- Lavecchia, A.; Di Giovanni, C. Virtual Screening Strategies in Drug Discovery: A Critical Review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef]
- Wang, E.; Weng, G.; Sun, H.; Du, H.; Zhu, F.; Chen, F.; Wang, Z.; Hou, T. Assessing the performance of the MM/PBSA and MM/GBSA methods. 10. Impacts of enhanced sampling and variable dielectric model on protein–protein Interactions. Phys. Chem. Chem. Phys. 2019, 21, 18958–18969. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV-1 spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Kirchdoerfer, R.; Wang, N.; Pallesen, J.; Wrapp, D.; Turner, H.; Cottrell, C.; Corbett, K.; Graham, B.; McLellan, J.; Ward, A. Stabilized coronavirus spikes are resistant to conformational changes induced by receptor recognition or proteolysis. Sci. Rep. 2018, 8, 15701. [Google Scholar] [CrossRef] [Green Version]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, W.; Farzan, M.; Harrison, S. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, M.H.; Bastidas, O.; Kokron, D.S.; Henze, C. Static all-atom energetic mappings of the SARS-Cov-2 spike protein and dynamic stability analysis of “Up” versus “Down” protomer states. PLoS ONE 2020, 15, e0241168. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Rodrigues, J.; Kastritis, P.; Amjj, B.; Vangone, A. PRODIGY: A web-server for predicting the binding affinity in protein-protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.; Zhu, Z.; Shi, Y.; Wang, X.; Mu, K.; Yang, Y.; Zhang, X.; Xu, Z.; Zhu, W. Computational Insights into the Conformational Accessibility and Binding Strength of SARS-CoV-2 Spike Protein to Human Angiotensin-Converting Enzyme 2. J. Phys. Chem. Lett. 2020, 11, 10482–10488. [Google Scholar] [CrossRef]
- Bai, C.; Warshel, A. Critical Differences between the Binding Features of the Spike Proteins of SARS-CoV-2 and SARS-CoV. J. Phys. Chem. B 2020, 124, 5907–5912. [Google Scholar] [CrossRef]
- Han, B.; Liu, Y.; Ginzinger, S.W.; Wishart, D. SHIFTX2: Significantly improved protein chemical shift prediction. J. Biomol. NMR 2011, 50, 43–57. [Google Scholar] [CrossRef] [Green Version]
- Malik, A.; Prahlad, D.; Kulkarni, N.; Kayal, A. Interfacial Water Molecules Make RBD of SPIKE Protein and Human ACE2 to Stick Together. bioRxiv 2020. [Google Scholar] [CrossRef]
- Atilgan, A.R.; Durell, S.R.; Jernigan, R.L.; Demirel, M.C.; Keskin, O.; Bahar, I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 2001, 80, 505–515. [Google Scholar] [CrossRef] [Green Version]
- López-Blanco, J.R.; Aliaga, J.I.; Quintana-Ortí, E.S.; Chacón, P. iMODS: Internal coordinates normal mode analysis server. Nucleic Acids Res. 2014, 42, W271–W276. [Google Scholar] [CrossRef] [PubMed]
- Farrell, D.W.; Speranskiy, K.; Thorpe, M.F. Generating stereochemically acceptable protein pathways. Proteins 2010, 78, 2908–2921. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Shala, A.; Bezginov, A.; Sljoka, A.; Audette, G.; Wilson, D. Hyperphosphorylation of intrinsically disordered tau protein induces an amyloidogenic shift in its conformational ensemble. PLoS ONE 2015, 10, e0120416. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, M.I.; Porter, J.R.; Ward, M.D.; Singh, S.; Vithani, N.; Meller, A.; Mallimadugula, U.L.; Kuhn, C.E.; Borowsky, J.H.; Bowman, G.R. SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nat. Chem. 2021, 13, 651–659. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020, 183, 739–751.e8. [Google Scholar] [CrossRef]
- Chan, K.H.; Peiris, J.S.; Lam, S.Y.; Poon, L.L.; Yuen, K.Y.; Seto, W.H. The effects of temperature and relative humidity on the viability of the sars coronavirus. Adv. Virol. 2011, 2011, 734690. [Google Scholar] [CrossRef]
- Kumar, S.; Paul, A.; Chatterjee, S.; Putz, S.; Nehra, N.; Wang, D.S.; Nisar, A.; Jennings, C.M.; Parekh, S.H. Effect of ambient temperature on respiratory tract cells exposed to sars-cov-2 viral mimicking nanospheres-an experimental study. Biointerphases 2021, 16, 011006. [Google Scholar] [CrossRef]
- Jacobs, D.J.; Rader, A.J.; Kuhn, L.A.; Thorpe, M.F. Protein flexibility predictions using graph theory. Proteins 2001, 44, 150–165. [Google Scholar] [CrossRef]
- Sljoka, A. Structural and Functional Analysis of Proteins Using Rigidity Theory. In Sublinear Computation Paradigm; Katoh, N., Higashikawa, Y., Ito, H., Nagao, A., Shibuya, T., Sljoka, A., Tanaka, K., Uno, Y., Eds.; Springer: Singapore, 2022; pp. 337–367. [Google Scholar]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [Green Version]
- Shang, W.; Yang, Y.; Rao, Y.; Rao, X. The outbreak of SARS-CoV-2 pneumonia calls for viral vaccines. Npj Vaccines 2020, 5, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Fuentes, L.; Drummond, C.; Faraudo, J.; Bastos-González, D. Interaction of organic ions with proteins. Soft Matter 2017, 13, 1120–1131. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.I.; Consta, S. What factors determine the stability of a weak protein–protein interaction in a charged aqueous droplet? Phys. Chem. Chem. Phys. 2017, 19, 31965–31981. [Google Scholar] [CrossRef] [PubMed]
- Valiev, M.; Chuev, G.N. Site density models of inhomogeneous classical molecular liquids. J. Stat. Mech. 2018, 2018, 093201. [Google Scholar] [CrossRef]
- Kezic, B.; Perera, A. Towards a more accurate reference interaction site model integral equation theory for molecular liquids. J. Chem. Phys. 2011, 135, 234104. [Google Scholar] [CrossRef]
- Chuev, G.N.; Vyalov, I.; Georgi, N. Extraction of atom-atom bridge and direct correlation functions from molecular simulations: A test for ambient water. Chem. Phys. Lett. 2013, 561–562, 175–178. [Google Scholar] [CrossRef]
- Chuev, G.N.; Vyalov, I.; Georgi, N. Extraction of site–site bridge functions and effective pair potentials from simulations of polar molecular liquids. J. Comput. Chem. 2014, 35, 1010–1023. [Google Scholar] [CrossRef]
- Lue, L.; Blankschtein, D. Liquid-state theory of hydrocarbon-water systems: Application to methane, ethane, and propane. J. Phys. Chem. 1992, 96, 8582–8594. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | CoV-1-RBD-hACE2 | CoV-2-RBD-hACE2 |
|---|---|---|
| structure | ||
| Bound-RBD surface area (Å2) | 18,435.6 | 19,866.4 |
| Interface area (Å2) | 796.7 | 827.7 |
| thermodynamics | ||
| Binding Energy (kcal/mol) | ||
| PRODIGY | −11.1 | −10.8 |
| EXP [67] | −10.7 | −11.8 |
| MM/GBSA [68] | −10.0 | −24.9 |
| CG [69] | −66.4 | −70.7 |
| 3DRISM | −50.1 | −57.2 |
| ULJ (kcal/mol) a | −281 | −261 |
| UC (kcal/mol) a | −4887 | −5160 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumawat, N.; Tucs, A.; Bera, S.; Chuev, G.N.; Valiev, M.; Fedotova, M.V.; Kruchinin, S.E.; Tsuda, K.; Sljoka, A.; Chakraborty, A. Site Density Functional Theory and Structural Bioinformatics Analysis of the SARS-CoV Spike Protein and hACE2 Complex. Molecules 2022, 27, 799. https://doi.org/10.3390/molecules27030799
Kumawat N, Tucs A, Bera S, Chuev GN, Valiev M, Fedotova MV, Kruchinin SE, Tsuda K, Sljoka A, Chakraborty A. Site Density Functional Theory and Structural Bioinformatics Analysis of the SARS-CoV Spike Protein and hACE2 Complex. Molecules. 2022; 27(3):799. https://doi.org/10.3390/molecules27030799
Chicago/Turabian StyleKumawat, Nitesh, Andrejs Tucs, Soumen Bera, Gennady N. Chuev, Marat Valiev, Marina V. Fedotova, Sergey E. Kruchinin, Koji Tsuda, Adnan Sljoka, and Amit Chakraborty. 2022. "Site Density Functional Theory and Structural Bioinformatics Analysis of the SARS-CoV Spike Protein and hACE2 Complex" Molecules 27, no. 3: 799. https://doi.org/10.3390/molecules27030799