
Electrocatalytic CO2 Reduction and H2 Evolution by a Copper (II) Complex with Redox-Active Ligand

Abstract
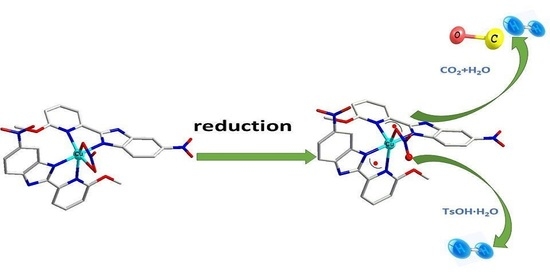
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
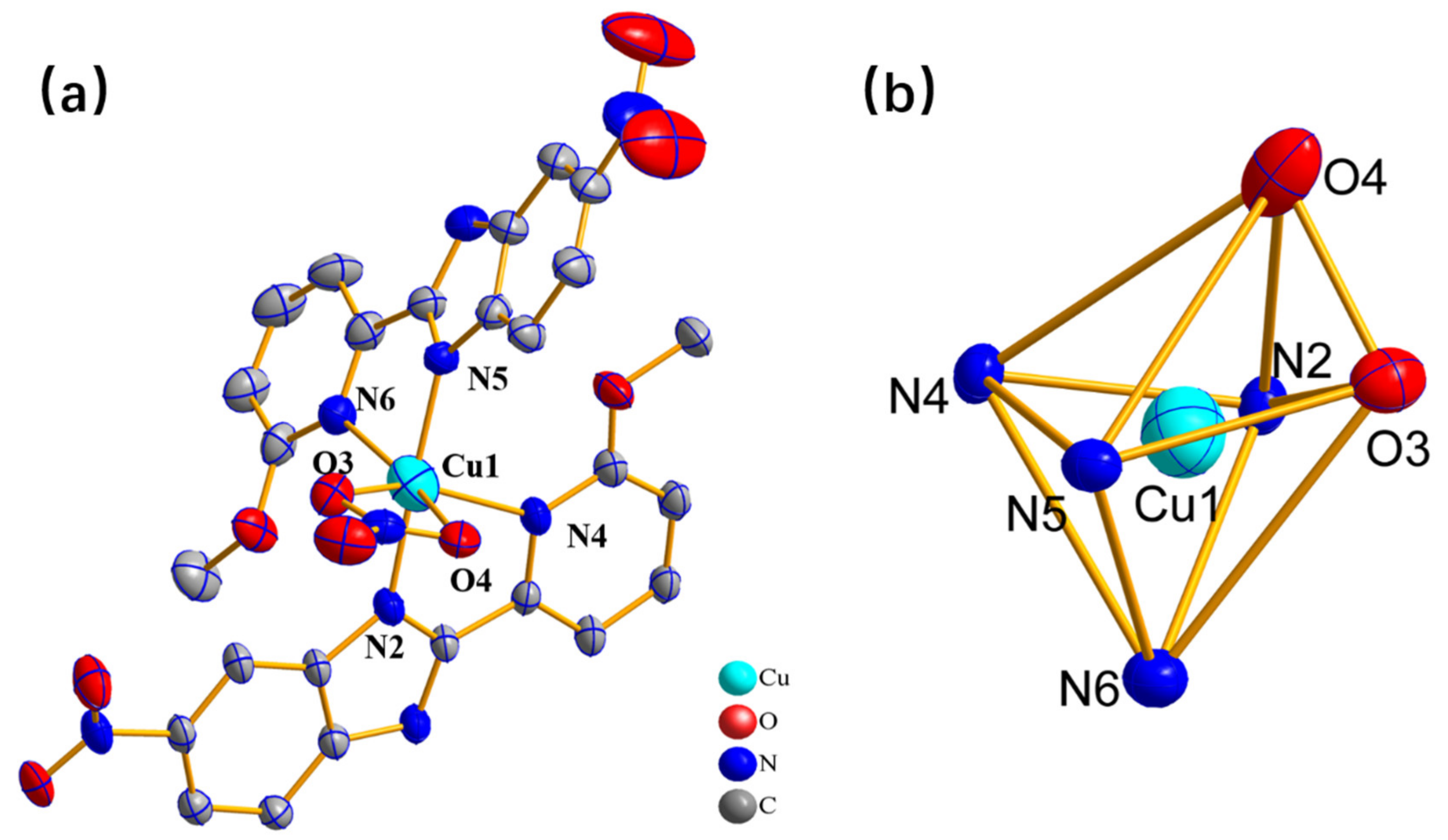
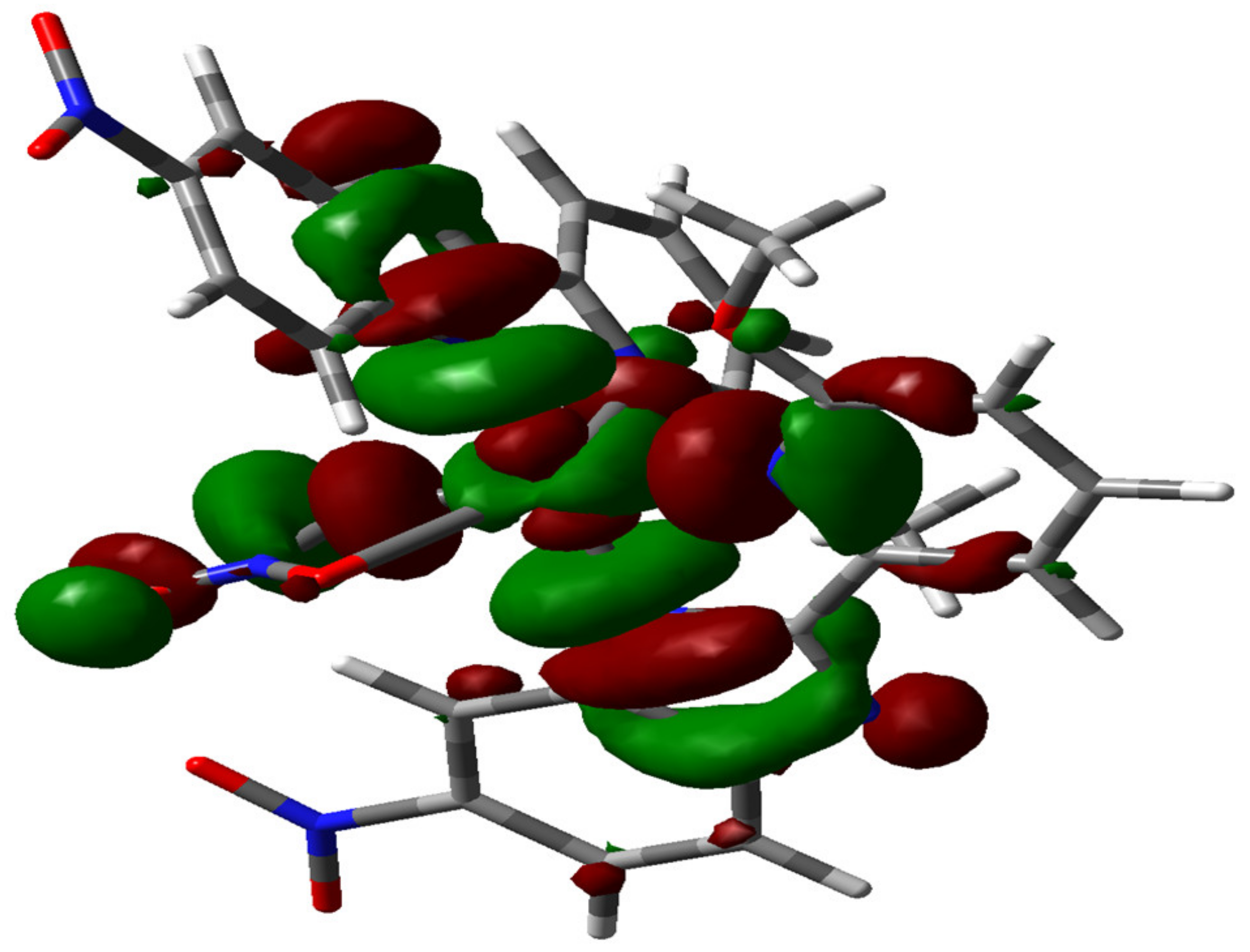
2.1. The Nature and Character of the Complex
2.2. Electrochemistry under Atmosphere of 1 atm Ar
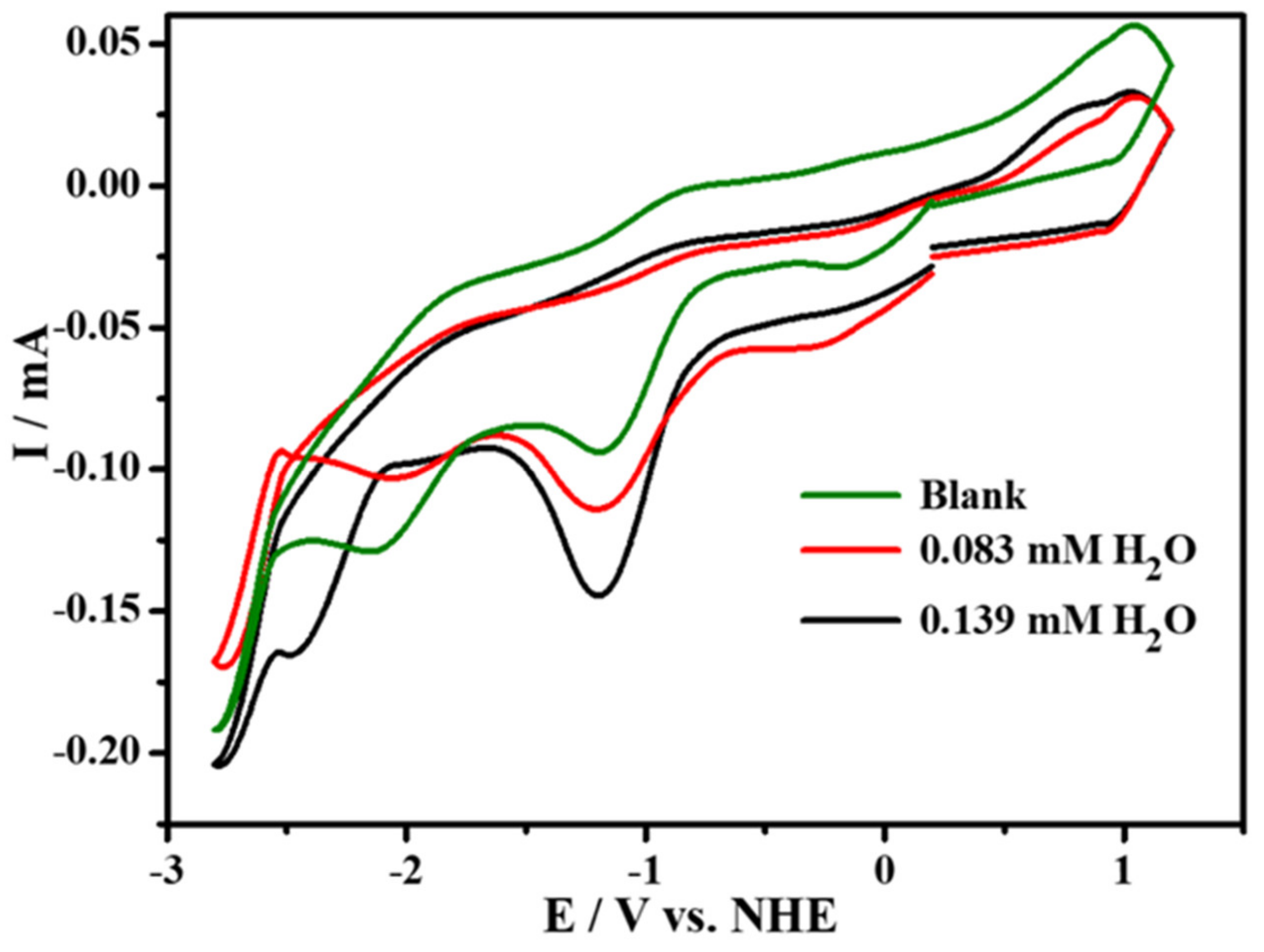
2.3. Electrochemistry in the Presence of CO2
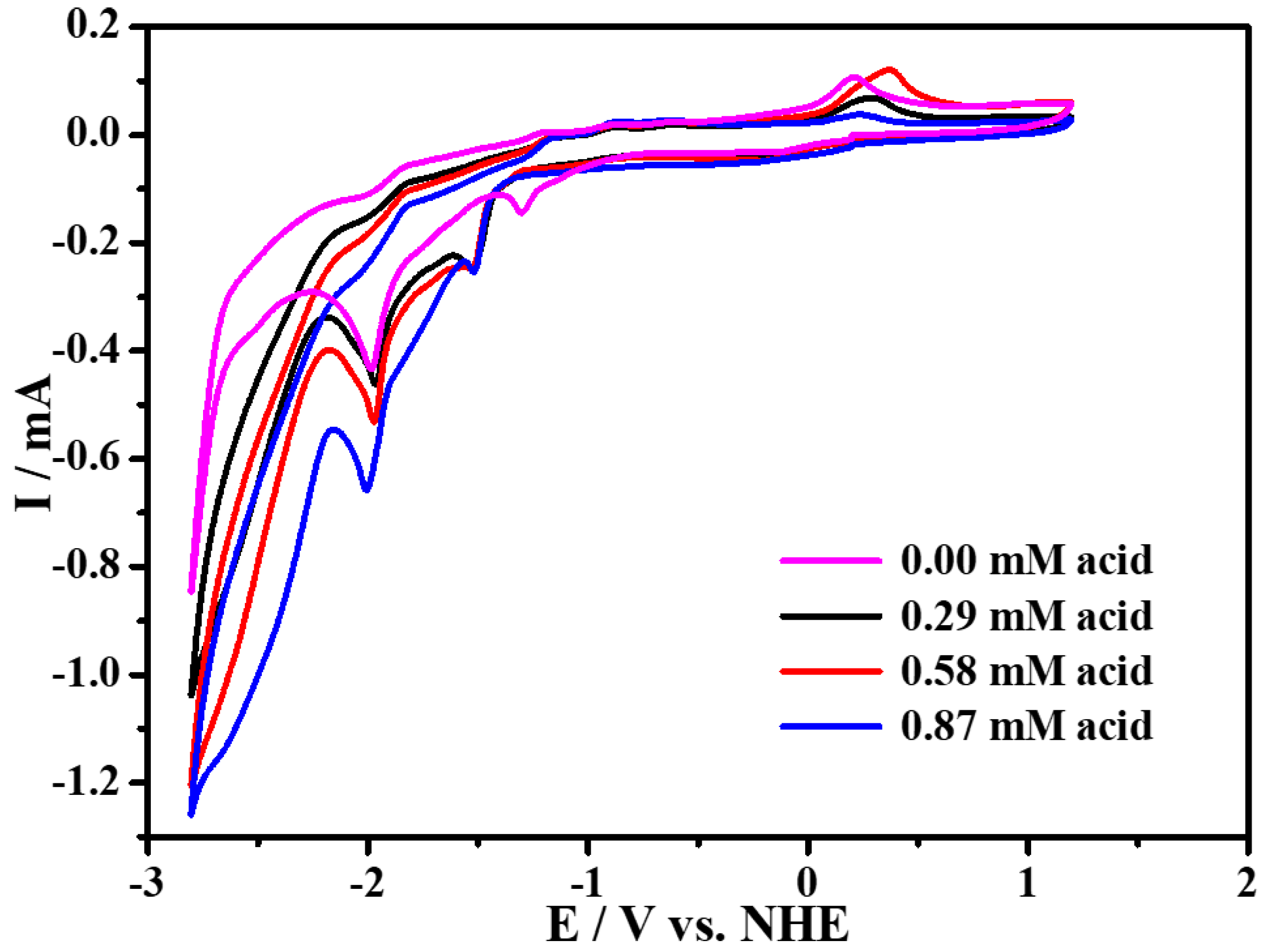
2.4. Electrocatalytic Property for Hydrogen Evolution
3. Materials and Methods
3.1. Synthesis
3.2. General Materials and Characterization
3.3. Crystal Structure Determination
3.4. Electrochemical Measurement and Electrolytic Product Analysis
3.5. Density Functional Theory Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Gurney, K.R.; Mendoza, D.L.; Zhou, Y.; Fischer, M.L.; Miller, C.C.; Geethakumar, S.; de la Rue du Can, S. High resolution fossil Fuel Combustion CO2 emission fluxes for combustionthe united states. Environ. Sci. Technol. 2009, 43, 5535–5541. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Wu, T.; Sun, L.; Nsanzimana, J.M.V.; Fisher, A.C.; Wang, X. Selective electrochemical reduction of CO2 to ethylene on Nanopores-Modified copper electrodes in aqueous solution. ACS Appl. Mater. Inter. 2017, 9, 32782–32789. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Xie, H.; Chen, M.; D’Aloia, A.; Cho, J.; Wu, G.; Li, Q. Precious metal-free approach to hydrogen electrocatalysis for energy conversion: From mechanism understanding to catalyst design. Nano Energy 2017, 42, 69–89. [Google Scholar] [CrossRef]
- Anantharaj, S.; Kundu, S. Do the evaluation parameters reflect intrinsic activity of electrocatalysts in electrochemical water splitting? ACS Energy Lett. 2019, 4, 1260–1264. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Wang, Y.; Qian, L.; Al-Enizi, A.M.; Zhang, L.; Zheng, G. Heterogeneous electrocatalysts for CO2 reduction. ACS Appl. Energy Mater. 2021, 4, 1034–1044. [Google Scholar] [CrossRef]
- Xiong, B.; Yang, Y.; Liu, J.; Ding, J.; Yang, Y. Electrochemical conversion of CO2 to syngas over Cu-M (M = Cd, Zn, Ni, Ag, and Pd) bimetal catalysts. Fuel 2021, 304, 121341. [Google Scholar] [CrossRef]
- Zhang, L.; Li, X.; Lang, Z.; Liu, Y.; Liu, J.; Yuan, L.; Lu, W.; Xia, Y.; Dong, L.; Yuan, D.; et al. Enhanced cuprophilic interactions in crystalline catalysts facilitate the highly selective electroreduction of CO2 to CH4. J. Am. Chem. Soc. 2021, 143, 3808–3816. [Google Scholar] [CrossRef]
- Drosou, M.; Kamatsos, F.; Ioannidis, G.; Zarkadoulas, A.; Mitsopoulou, C.A.; Papatriantafyllopoulou, C.; Tzeli, D. Reactivity and mechanism of photo- and electrocatalytic hydrogen evolution by a diimine Copper(I) complex. Catalysts 2020, 10, 1302. [Google Scholar] [CrossRef]
- Liu, D.; Ke, M.; Ru, T.; Ning, Y.; Chen, F. Room-temperature Pd-catalyzed methoxycarbonylation of terminal alkynes with high branched selectivity enabled by bisphosphine-picolinamide ligand. Chem. Commun. 2017, 27, 9359–9363. [Google Scholar] [CrossRef]
- Queyriaux, N. Redox-Active ligands in electroassisted catalytic H+ and CO2 reductions: Benefits and risks. ACS Catal. 2021, 11, 4024–4035. [Google Scholar] [CrossRef]
- Lyaskovskyy, V.; de Bruin, B. Redox Non-Innocent ligands: Versatile new tools to control catalytic reactions. ACS Catal. 2012, 2, 270–279. [Google Scholar] [CrossRef]
- Wang, M.; England, J.; Weyhermüller, T.; Kokatam, S.; Pollock, C.J.; Debeer, S.; Shen, J.; Yap, G.P.A.; Theopold, K.H.; Wieghardt, K. New complexes of Chromium (III) containing organic π-Radical ligands: An experimental and density functional theory study. Inorg. Chem. 2013, 52, 4472–4487. [Google Scholar] [CrossRef]
- Natesakhawat, S.; Lekse, J.W.; Baltrus, J.P.; Ohodnicki, P.R.; Howard, B.H.; Deng, X.; Matranga, C. Active sites and structure–activity relationships of Copper-Based catalysts for carbon dioxide hydrogenation to methanol. ACS Catal. 2012, 2, 1667–1676. [Google Scholar] [CrossRef]
- Su, X.; Mccardle, K.M.; Chen, L.; Panetier, J.A.; Jurss, J.W. Robust and selective cobalt catalysts bearing Redox-Active Bipyridyl-N-heterocyclic carbene frameworks for electrochemical CO2 reduction in aqueous solutions. ACS Catal. 2019, 9, 7398–7408. [Google Scholar] [CrossRef]
- Shi, N.N.; Xie, W.J.; Gao, W.S.; Wang, J.M.; Zhang, S.F.; Fan, Y.H.; Wang, M. Effect of PDI ligand binding pattern on the electrocatalytic activity of two Ru (II) complexes for CO2 reduction. Appl. Organomet. Chem. 2020, 34, e5551. [Google Scholar] [CrossRef]
- Tok, G.C.; Reiter, S.; Freiberg, A.T.S.; Reinschlüssel, L.; Gasteiger, H.A.; de Vivie-Riedle, R.; Hess, C.R. H2 Evolution from Electrocatalysts with Redox-Active Ligands: Mechanistic Insights from Theory and Experiment vis-à-vis Co-Mabiq. Inorg. Chem. 2021, 60, 13888–13902. [Google Scholar] [CrossRef]
- Orchanian, N.M.; Hong, L.E.; Velazquez, D.A.; Marinescu, S.C. Electrocatalytic syngas generation with a redox non-innocent cobalt 2-phosphinobenzenethiolate complex. Dalton Trans. Int. J. Inorg. Chem. 2021, 50, 10779–10788. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, C.; Zhu, J.; Zhou, Q.; Yu, R.; Wang, Y.; An, P.; Zhang, J.; Qiu, M.; Zhou, L.; et al. Dynamic restructuring of coordinatively unsaturated copper paddle wheel clusters to boost electrochemical CO2 reduction to hydrocarbons**. Angew. Chem. Int. Ed. 2021, 61, e5551. [Google Scholar]
- Jiang, Y.; Deng, Y.; Liang, R.; Fu, J.; Gao, R.; Luo, D.; Bai, Z.; Hu, Y.; Yu, A.; Chen, Z. D-Orbital steered active sites through ligand editing on heterometal imidazole frameworks for rechargeable zinc-air battery. Nat. Commun. 2020, 11, 5858. [Google Scholar] [CrossRef]
- Ghosh, P.; Vos, S.; Lutz, M.; Gloaguen, F.; Schollhammer, P.; Moret, M.E.; Klein Gebbink, R.J.M. Electrocatalytic proton reduction by a cobalt complex containing a proton-responsive bis(alkylimdazole)methane ligand: Involvement of a C−H bond in H2 formation. Chem. Eur. J. 2020, 26, 12560–12569. [Google Scholar] [CrossRef]
- Thorp, W.L.A.H. Bond valence sum analysis of Metal-Ligand bond lengths in metalloenzymes and model. Inorg. Chem. 1993, 19, 4102–4105. [Google Scholar] [CrossRef]
- Bersuker, I.B. Pseudo-Jahn–Teller effect—A Two-State paradigm in formation, deformation, and transformation of molecular systems and solids. Chem. Rev. 2013, 113, 1351–1390. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.; Avnir, D.; Llunell, M.; Pinsky, M. Continuous symmetry maps and shape classification. The case of six-coordinated metal compoundsElectronic supplementary information (ESI) available: Tables of CSD refcodes, structural parameters and symmetry measures for the studied compounds. New J. Chem. 2002, 26, 996–1009. [Google Scholar] [CrossRef]
- Pinsky, M.; Avnir, D. Continuous Symmetry Measures. 5. The Classical Polyhedra. Inorg. Chem. 1998, 37, 5575–5582. [Google Scholar] [CrossRef]
- Pandey, S.; Kumar, G.; Gupta, R. Postfunctionalized Metalloligand-Based catenated coordination polymers: Syntheses, structures, and effect of labile sites on catalysis. Cryst. Growth Des. 2019, 19, 2723–2735. [Google Scholar] [CrossRef]
- Schlagintweit, J.F.; Dyckhoff, F.; Nguyen, L.; Jakob, C.H.G.; Reich, R.M.; Kühn, F.E. Mixed tetradentate NHC/1,2,3-triazole iron complexes bearing cis labile coordination sites as highly active catalysts in Lewis and Brønsted acid mediated olefin epoxidation. J. Catal. 2020, 383, 144–152. [Google Scholar] [CrossRef]
- Biswas, S.; Chowdhury, S.N.; Lepcha, P.; Biswas, A.N. Pentadentate Copper (II)-amidate complex as a precatalyst for electrocatalytic proton reduction. Int. J. Hydrog. Energ. 2021, 46, 21542–21548. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhang, Y.; Zhai, R.; Gu, Z.; Zhang, J. Helical copper-porphyrinic framework nanoarrays for highly efficient CO2 electroreduction. Sci. China Mater. 2021, 1–7. [Google Scholar] [CrossRef]
- Lu, Y.F.; Dong, L.Z.; Liu, J.; Yang, R.X.; Liu, J.J.; Zhang, Y.; Zhang, L.; Wang, Y.R.; Li, S.L.; Lan, Y.Q. Predesign of catalytically active sites via stable coordination cluster model system for electroreduction of CO2 to ethylene. Angew. Chem. Int. Ed. 2021, 60, 26210–26217. [Google Scholar] [CrossRef]
- Li, G.; Zhao, Y.; Li, J.P.H.; Chen, W.; Li, S.; Dong, X.; Song, Y.; Yang, Y.; Wei, W.; Sun, Y. Insight into Composition and Intermediate Evolutions of Copper-Based Catalysts during Gas-Phase CO2 Electroreduction to Multicarbon Oxygenates. Catalysts 2021, 11, 1502. [Google Scholar] [CrossRef]
- Wei, L.; Ye, B. Efficient Conversion of CO2 via Grafting Urea Group into a [Cu2(COO)4]-Based Metal–Organic Framework with Hierarchical Porosity. Inorg. Chem. 2019, 58, 4385–4393. [Google Scholar] [CrossRef]
- Zhang, Y.; Cao, Q.; Wu, X.; Xiao, Y.; Meng, A.; Zhang, Q.; Yu, Y.; Zhang, W. Efficient photocatalytic H2 evolution and α-methylation of ketones from copper complex modified polymeric carbon nitride. Chem. Eng. J. 2022, 427, 132042. [Google Scholar] [CrossRef]
- Datta, A.; Das, K.; Beyene, B.B.; Garribba, E.; Gajewska, M.J.; Hung, C. EPR interpretation and electrocatalytic H2 evolution study of bis(3,5-di-methylpyrazol-1-yl) acetate anchored Cu (II) and Mn (II) complexes. Mol. Catal. 2017, 439, 81–90. [Google Scholar] [CrossRef]
- Lei, H.; Fang, H.; Han, Y.; Lai, W.; Fu, X.; Cao, R. Reactivity and mechanism studies of hydrogen evolution catalyzed by copper corroles. ACS Catal. 2015, 5, 5145–5153. [Google Scholar] [CrossRef]
- Chen, S.; Fan, R.; Wang, X.; Yang, Y. Novel bright blue emissions IIB group complexes constructed with various polyhedron-induced 2-[2′-(6-methoxy-pyridyl)]-benzimidazole derivatives. Crystengcomm 2014, 16, 6114. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colic-Salvetti correlation-energy into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Cometto, C.; Chen, L.; Lo, P.; Guo, Z.; Lau, K.; Anxolabéhère-Mallart, E.; Fave, C.; Lau, T.; Robert, M. Highly selective molecular catalysts for the CO2-to-CO electrochemical conversion at very low overpotential. Contrasting Fe vs Co quaterpyridine complexes upon mechanistic studies. ACS Catal. 2018, 8, 3411–3417. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Zhang, S.; Wang, J.; Yin, X.; Han, Z.; Chen, G.; Zhang, D.; Wang, M. Electrocatalytic CO2 Reduction and H2 Evolution by a Copper (II) Complex with Redox-Active Ligand. Molecules 2022, 27, 1399. https://doi.org/10.3390/molecules27041399
Li J, Zhang S, Wang J, Yin X, Han Z, Chen G, Zhang D, Wang M. Electrocatalytic CO2 Reduction and H2 Evolution by a Copper (II) Complex with Redox-Active Ligand. Molecules. 2022; 27(4):1399. https://doi.org/10.3390/molecules27041399
Chicago/Turabian StyleLi, Jingjing, Shifu Zhang, Jinmiao Wang, Xiaomeng Yin, Zhenxing Han, Guobo Chen, Dongmei Zhang, and Mei Wang. 2022. "Electrocatalytic CO2 Reduction and H2 Evolution by a Copper (II) Complex with Redox-Active Ligand" Molecules 27, no. 4: 1399. https://doi.org/10.3390/molecules27041399