
Synthesis and Investigation of the G-Quadruplex Binding Properties of Kynurenic Acid Derivatives with a Dihydroimidazoquinoline-3,5-dione Core

 , , , , , and
, , , , , and
Abstract
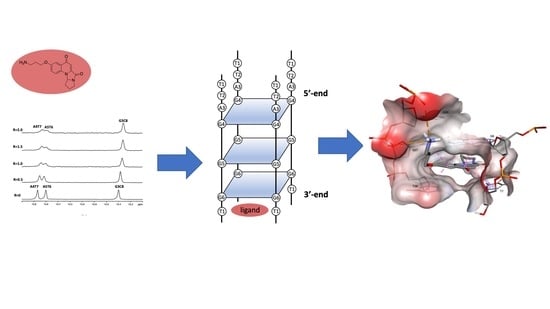
:
1. Introduction
2. Results and Discussion
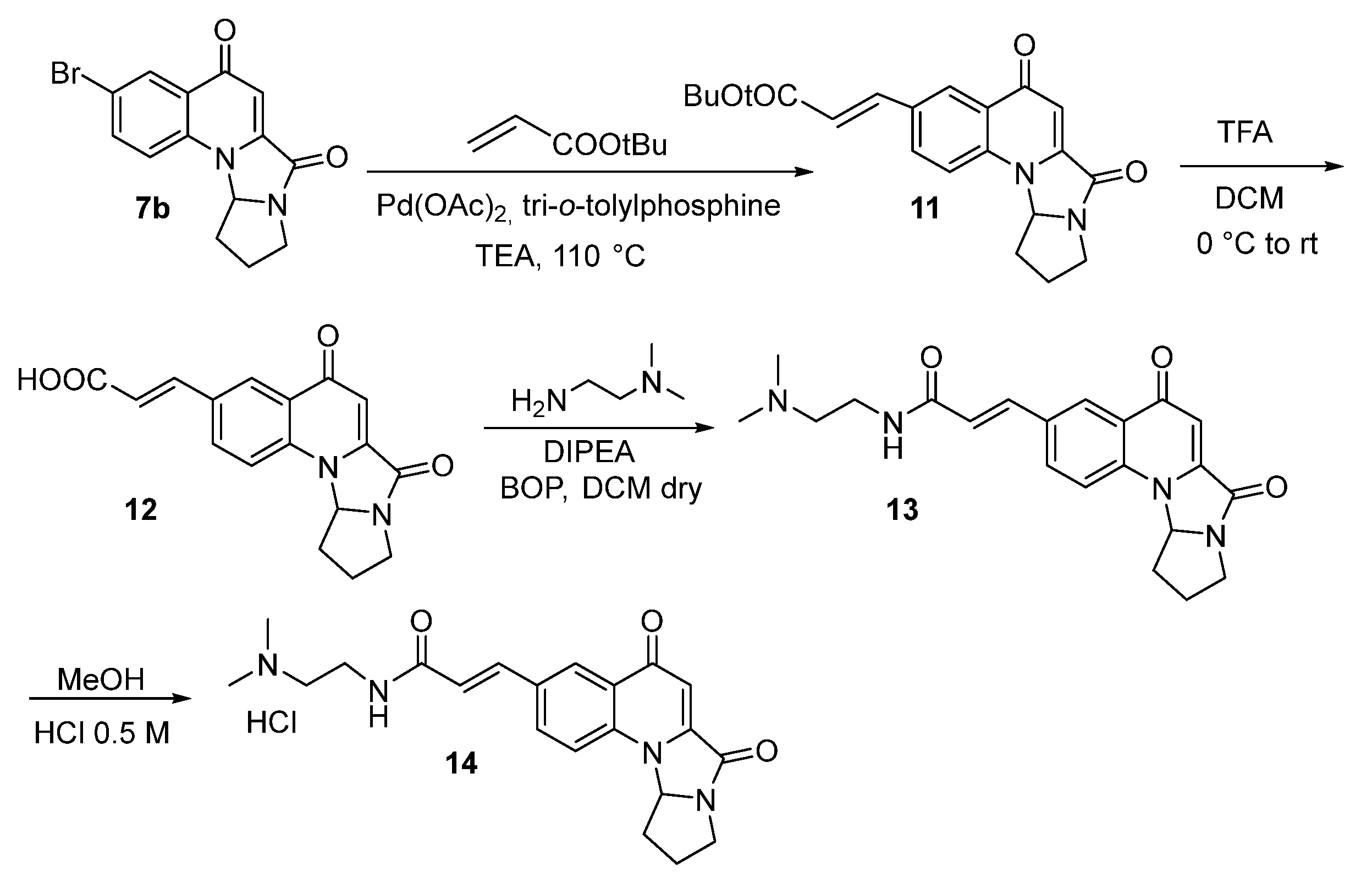
2.1. Synthesis
2.2. Biological Activity
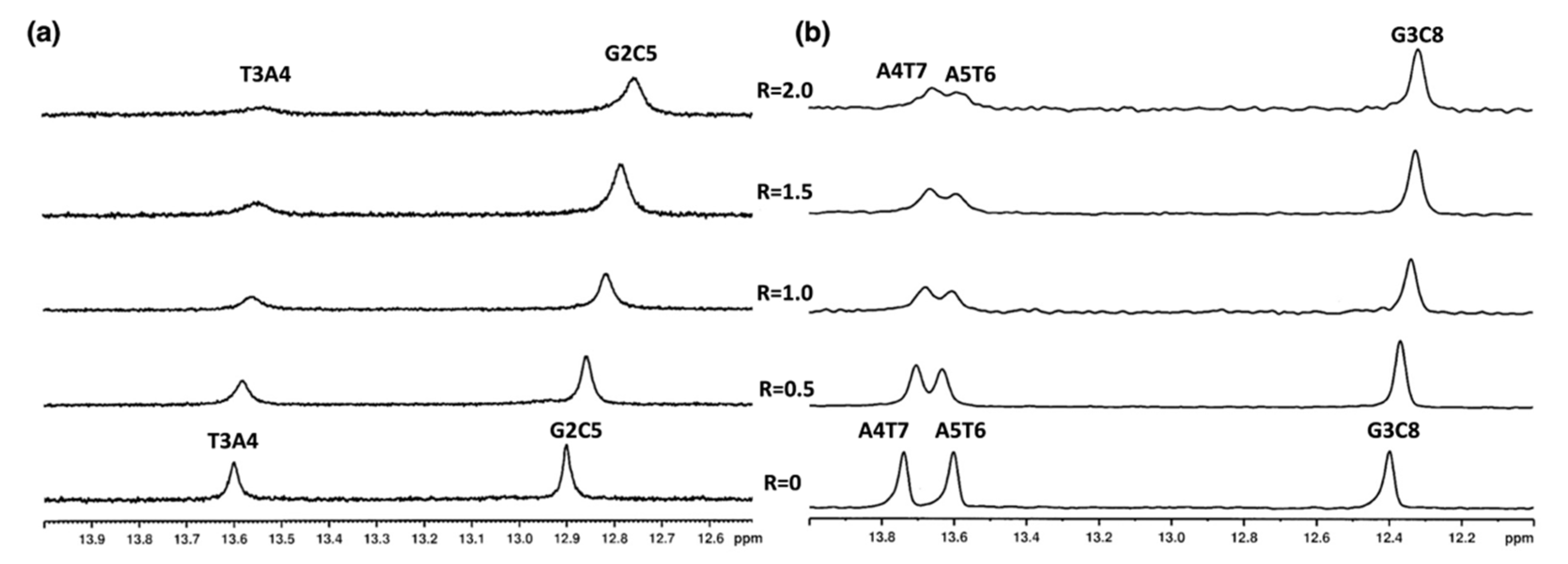
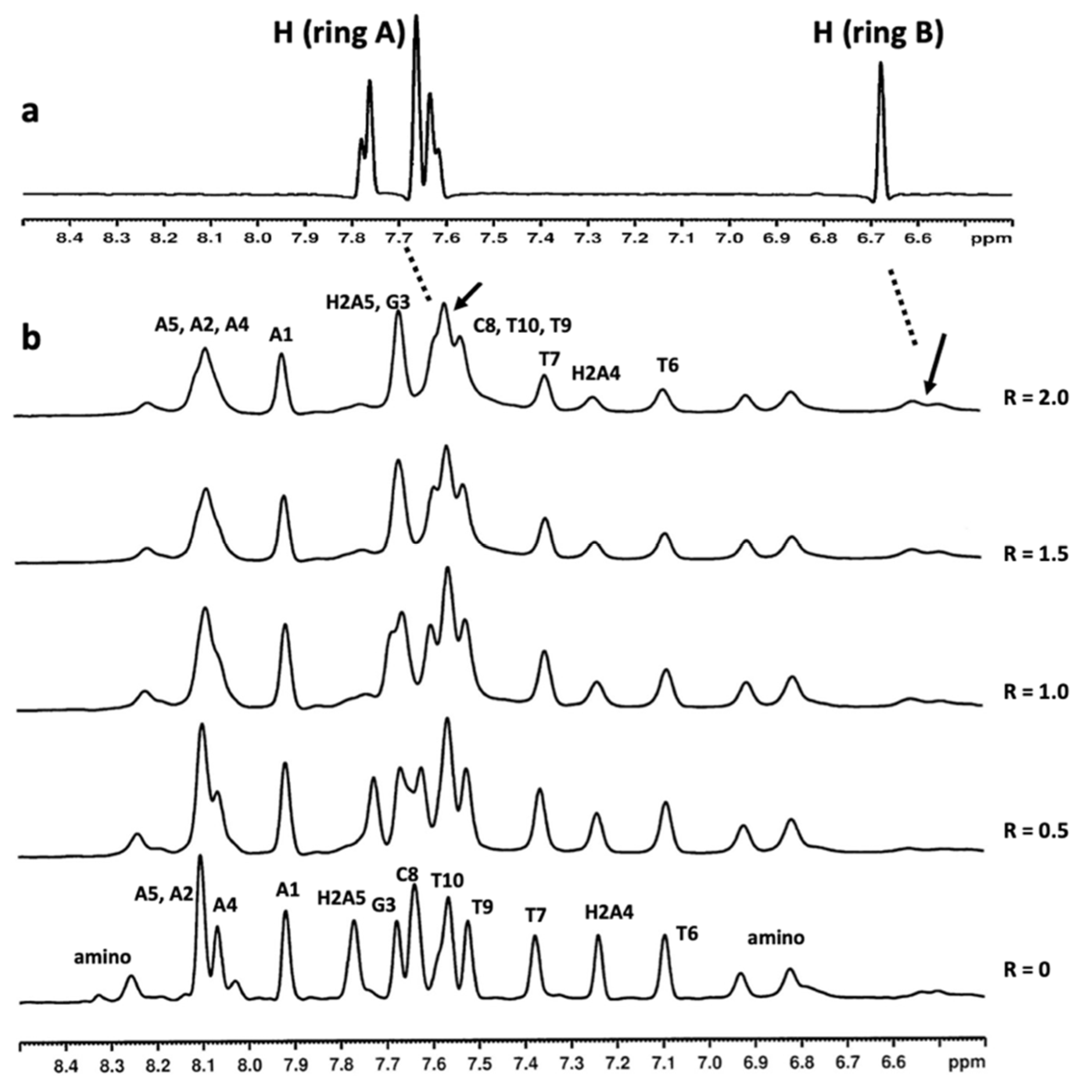
2.3. Mode of Binding of Compound 9 with Duplex and G-Quadruplex Structures by NMR Spectroscopy
3. Material and Methods
3.1. Chemistry
3.2. NMR Studies
3.3. Cytotoxic Assay
3.4. Molecular Modeling Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Walczak, K.; Wnorowski, A.; Turski, W.A.; Plech, T. Kynurenic Acid and Cancer: Facts and Controversies. Cell. Mol. Life Sci. 2020, 77, 1531–1550. [Google Scholar] [CrossRef] [Green Version]
- Moroni, F.; Cozzi, A.; Sili, M.; Mannaioni, G. Kynurenic Acid: A Metabolite with Multiple Actions and Multiple Targets in Brain and Periphery. J. Neural Transm. 2012, 119, 133–139. [Google Scholar] [CrossRef]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.-Q. Kynurenines in the Mammalian Brain: When Physiology Meets Pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef]
- Wirthgen, E.; Hoeflich, A.; Rebl, A.; Günther, J. Kynurenic Acid: The Janus-Faced Role of an Immunomodulatory Tryptophan Metabolite and Its Link to Pathological Conditions. Front. Immunol. 2018, 8, 1957. [Google Scholar] [CrossRef] [Green Version]
- Małaczewska, J.; Siwicki, A.K.; Wójcik, R.M.; Turski, W.A.; Kaczorek, E. The Effect of Kynurenic Acid on the Synthesis of Selected Cytokines by Murine Splenocytes—In Vitro and Ex Vivo Studies. Cent. Eur. J. Immunol. 2016, 41, 39–46. [Google Scholar] [CrossRef]
- Resta, F.; Masi, A.; Sili, M.; Laurino, A.; Moroni, F.; Mannaioni, G. Kynurenic Acid and Zaprinast Induce Analgesia by Modulating HCN Channels through GPR35 Activation. Neuropharmacology 2016, 108, 136–143. [Google Scholar] [CrossRef]
- Cosi, C.; Mannaioni, G.; Cozzi, A.; Carlà, V.; Sili, M.; Cavone, L.; Maratea, D.; Moroni, F. G-Protein Coupled Receptor 35 (GPR35) Activation and Inflammatory Pain: Studies on the Antinociceptive Effects of Kynurenic Acid and Zaprinast. Neuropharmacology 2011, 60, 1227–1231. [Google Scholar] [CrossRef]
- Glavin, G.B.; Pinsky, C. Kynurenic Acid Attenuates Experimental Ulcer Formation and Basal Gastric Acid Secretion in Rats. Res. Commun. Chem. Pathol. Pharmacol. 1989, 64, 111–119. [Google Scholar]
- Glavin, G.B.; Bose, R.; Pinsky, C. Kynurenic Acid Protects against Gastroduodenal Ulceration in Mice Injected with Extracts from Poisonous Atlantic Shellfish. Prog. Neuro Psychopharmacol. Biol. Psychiatry 1989, 13, 569–572. [Google Scholar] [CrossRef]
- Turski, M.P.; Turska, M.; Paluszkiewicz, P.; Parada-Turska, J.; Oxenkrug, G.F. Kynurenic Acid in the Digestive System–-New Facts, New Challenges. Int. J. Tryptophan Res. 2013, 6, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Pawlak, K.; Myśliwiec, M.; Pawlak, D. Kynurenine Pathway—A New Link between Endothelial Dysfunction and Carotid Atherosclerosis in Chronic Kidney Disease Patients. Adv. Med. Sci. 2010, 55, 196–203. [Google Scholar] [CrossRef]
- Lugo-Huitrón, R.; Blanco-Ayala, T.; Ugalde-Muñiz, P.; Carrillo-Mora, P.; Pedraza-Chaverrí, J.; Silva-Adaya, D.; Maldonado, P.D.; Torres, I.; Pinzón, E.; Ortiz-Islas, E.; et al. On the Antioxidant Properties of Kynurenic Acid: Free Radical Scavenging Activity and Inhibition of Oxidative Stress. Neurotoxicol. Teratol. 2011, 33, 538–547. [Google Scholar] [CrossRef]
- Marciniak, S.; Wnorowski, A.; Smolińska, K.; Walczyna, B.; Turski, W.; Kocki, T.; Paluszkiewicz, P.; Parada-Turska, J. Kynurenic Acid Protects against Thioacetamide-Induced Liver Injury in Rats. Anal. Cell. Pathol. 2018, 2018, 1270483. [Google Scholar] [CrossRef] [Green Version]
- Walczak, K.; Deneka-Hannemann, S.; Jarosz, B.; Zgrajka, W.; Stoma, F.; Trojanowski, T.; Turski, W.A.; Rzeski, W. Kynurenic Acid Inhibits Proliferation and Migration of Human Glioblastoma T98G Cells. Pharmacol. Rep. 2014, 66, 130–136. [Google Scholar] [CrossRef]
- Walczak, K.; Dąbrowski, W.; Langner, E.; Zgrajka, W.; Piłat, J.; Kocki, T.; Rzeski, W.; Turski, W.A. Kynurenic Acid Synthesis and Kynurenine Aminotransferases Expression in Colon Derived Normal and Cancer Cells. Scand. J. Gastroenterol. 2011, 46, 903–912. [Google Scholar] [CrossRef]
- Walczak, K.; Żurawska, M.; Kiś, J.; Starownik, R.; Zgrajka, W.; Bar, K.; Turski, W.A.; Rzeski, W. Kynurenic Acid in Human Renal Cell Carcinoma: Its Antiproliferative and Antimigrative Action on Caki-2 Cells. Amino Acids 2012, 43, 1663–1670. [Google Scholar] [CrossRef]
- Walczak, K.; Turski, W.A.; Rajtar, G. Kynurenic Acid Inhibits Colon Cancer Proliferation in Vitro: Effects on Signaling Pathways. Amino Acids 2014, 46, 2393–2401. [Google Scholar] [CrossRef] [Green Version]
- Walczak, K.; Turski, W.A.; Rzeski, W. Kynurenic Acid Enhances Expression of P21 Waf1/Cip1 in Colon Cancer HT-29 Cells. Pharmacol. Rep. 2012, 64, 745–750. [Google Scholar] [CrossRef] [Green Version]
- Dankers, A.C.A.; Mutsaers, H.A.M.; Dijkman, H.B.P.M.; van den Heuvel, L.P.; Hoenderop, J.G.; Sweep, F.C.G.J.; Russel, F.G.M.; Masereeuw, R. Hyperuricemia Influences Tryptophan Metabolism via Inhibition of Multidrug Resistance Protein 4 (MRP4) and Breast Cancer Resistance Protein (BCRP). Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1715–1722. [Google Scholar] [CrossRef] [Green Version]
- Mutsaers, H.A.M.; van den Heuvel, L.P.; Ringens, L.H.J.; Dankers, A.C.A.; Russel, F.G.M.; Wetzels, J.F.M.; Hoenderop, J.G.; Masereeuw, R. Uremic Toxins Inhibit Transport by Breast Cancer Resistance Protein and Multidrug Resistance Protein 4 at Clinically Relevant Concentrations. PLoS ONE 2011, 6, e18438. [Google Scholar] [CrossRef] [Green Version]
- Beretta, G.; Artali, R.; Caneva, E.; Orlandini, S.; Centini, M.; Facino, R.M. Quinoline Alkaloids in Honey: Further Analytical (HPLC-DAD-ESI-MS, Multidimensional Diffusion-Ordered NMR Spectroscopy), Theoretical and Chemometric Studies. J. Pharm. Biomed. Anal. 2009, 50, 432–439. [Google Scholar] [CrossRef]
- Beretta, G.; Moretti, R.M.; Nasti, R.; Cincinelli, R.; Dallavalle, S.; Montagnani Marelli, M. Apoptosis-Mediated Anticancer Activity in Prostate Cancer Cells of a Chestnut Honey (Castanea sativa L.) Quinoline–Pyrrolidine Gamma-Lactam Alkaloid. Amino Acids 2021, 53, 869–880. [Google Scholar] [CrossRef]
- Cincinelli, R.; Beretta, G.; Dallavalle, S. Total Synthesis of Tetracyclic Kynurenic Acid Analogues Isolated from Chestnut Honey. Tetrahedron Lett. 2018, 59, 163–166. [Google Scholar] [CrossRef]
- Cincinelli, R.; Musso, L.; Beretta, G.; Dallavalle, S. 4-Quinolone Fused Heterocyclic Ring Systems by Intramolecular Reactions of 4-Quinolone-2-Carboxamides. Tetrahedron 2014, 70, 9797–9804. [Google Scholar] [CrossRef]
- Mazzini, S.; Gargallo, R.; Musso, L.; de Santis, F.; Aviñó, A.; Scaglioni, L.; Eritja, R.; di Nicola, M.; Zunino, F.; Amatulli, A.; et al. Stabilization of C-KIT G-Quadruplex DNA Structures by the RNA Polymerase I Inhibitors BMH-21 and BA-41. Int. J. Mol. Sci. 2019, 20, 4927. [Google Scholar] [CrossRef] [Green Version]
- Musso, L.; Mazzini, S.; Rossini, A.; Castagnoli, L.; Scaglioni, L.; Artali, R.; di Nicola, M.; Zunino, F.; Dallavalle, S. C-MYC G-Quadruplex Binding by the RNA Polymerase I Inhibitor BMH-21 and Analogues Revealed by a Combined NMR and Biochemical Approach. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 615–629. [Google Scholar] [CrossRef]
- Dallavalle, S.; Musso, L.; Artali, R.; Aviñó, A.; Scaglioni, L.; Eritja, R.; Gargallo, R.; Mazzini, S. G-Quadruplex Binding Properties of a Potent PARP-1 Inhibitor Derived from 7-Azaindole-1-Carboxamide. Sci. Rep. 2021, 11, 3869. [Google Scholar] [CrossRef]
- Davis, J.T. G-Quartets 40 Years Later: From 5′-GMP to Molecular Biology and Supramolecular Chemistry. Angew. Chem. Int. Ed. 2004, 43, 668–698. [Google Scholar] [CrossRef]
- Valton, A.-L.; Prioleau, M.-N. G-Quadruplexes in DNA Replication: A Problem or a Necessity? Trends Genet. 2016, 32, 697–706. [Google Scholar] [CrossRef]
- Pavlova, A.V.; Kubareva, E.A.; Monakhova, M.V.; Zvereva, M.I.; Dolinnaya, N.G. Impact of G-Quadruplexes on the Regulation of Genome Integrity, DNA Damage and Repair. Biomolecules 2021, 11, 1284. [Google Scholar] [CrossRef]
- van Kregten, M.; Tijsterman, M. The Repair of G-Quadruplex-Induced DNA Damage. Exp. Cell Res. 2014, 329, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Rankin, S.; Reszka, A.P.; Huppert, J.; Zloh, M.; Parkinson, G.N.; Todd, A.K.; Ladame, S.; Balasubramanian, S.; Neidle, S. Putative DNA Quadruplex Formation within the Human C-Kit Oncogene. J. Am. Chem. Soc. 2005, 127, 10584–10589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D. Facilitation of a Structural Transition in the Polypurine/Polypyrimidine Tract within the Proximal Promoter Region of the Human VEGF Gene by the Presence of Potassium and G-Quadruplex-Interactive Agents. Nucleic Acids Res. 2005, 33, 6070–6080. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Chen, D.; Jones, R.A.; Hurley, L.H.; Yang, D. NMR Solution Structure of the Major G-Quadruplex Structure Formed in the Human BCL2 Promoter Region. Nucleic Acids Res. 2006, 34, 5133–5144. [Google Scholar] [CrossRef]
- Qin, Y.; Rezler, E.M.; Gokhale, V.; Sun, D.; Hurley, L.H. Characterization of the G-Quadruplexes in the Duplex Nuclease Hypersensitive Element of the PDGF-A Promoter and Modulation of PDGF-A Promoter Activity by TMPyP4. Nucleic Acids Res. 2007, 35, 7698–7713. [Google Scholar] [CrossRef] [Green Version]
- Pirota, V.; Stasi, M.; Benassi, A.; Doria, F. An Overview of Quadruplex Ligands: Their Common Features and Chemotype Diversity. Annu. Rep. Med. Chem. 2020, 54, 163–196. [Google Scholar]
- Sun, Z.-Y.; Wang, X.-N.; Cheng, S.-Q.; Su, X.-X.; Ou, T.-M. Developing Novel G-Quadruplex Ligands: From Interaction with Nucleic Acids to Interfering with Nucleic Acid–Protein Interaction. Molecules 2019, 24, 396. [Google Scholar] [CrossRef] [Green Version]
- Santos, T.; Salgado, G.F.; Cabrita, E.J.; Cruz, C. G-Quadruplexes and Their Ligands: Biophysical Methods to Unravel G-Quadruplex/Ligand Interactions. Pharmaceuticals 2021, 14, 769. [Google Scholar] [CrossRef]
- Feigon, J.; Denny, W.A.; Leupin, W.; Kearns, D.R. Interactions of Antitumor Drugs with Natural DNA: Proton NMR Study of Binding Mode and Kinetics. J. Med. Chem. 1984, 27, 450–465. [Google Scholar] [CrossRef]
- Mazzini, S.; Mondelli, R.; Ragg, E. Structure and Dynamics of Intercalation Complexes of Anthracyclines with d(CGATCG)2 and d(CGTACG)2. 2D-1H and 31P NMR Investigations. J. Chem. Soc. Perkin Trans. 2 1998, 9, 1983–1992. [Google Scholar] [CrossRef]
- Mazzini, S.; Bellucci, M.C.; Mondelli, R. Mode of Binding of the Cytotoxic Alkaloid Berberine with the Double Helix Oligonucleotide d(AAGAATTCTT)2. Bioorg. Med. Chem. 2003, 11, 505–514. [Google Scholar] [CrossRef]
- Sundquist, W.I.; Klug, A. Telomeric DNA Dimerizes by Formation of Guanine Tetrads between Hairpin Loops. Nature 1989, 342, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Raymond, E.; Soria, J.-C.; lzbicka, E.; Boussin, F.; Hurley, L.; von Hoff, D.D. DNA G-Quadruplexes, Telomere-Specific Proteins and Telomere-Associated Enzymes as Potential Targets for New Anticancer Drugs. Investig. New Drugs 2000, 18, 123–137. [Google Scholar] [CrossRef]
- Gavathiotis, E.; Heald, R.A.; Stevens, M.F.G.; Searle, M.S. Recognition and Stabilization of Quadruplex DNA by a Potent New Telomerase Inhibitor: NMR Studies of the 2:1 Complex of a Pentacyclic Methylacridinium Cation with d(TTAGGGT)4. Angew. Chem. Int. Ed. 2001, 40, 4749–4751. [Google Scholar] [CrossRef]
- Riva, B.; Ferreira, R.; Musso, L.; Artali, R.; Scaglioni, L.; Mazzini, S. Molecular Recognition in Naphthoquinone Derivatives—G-Quadruplex Complexes by NMR. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 673–680. [Google Scholar] [CrossRef]
- Scaglioni, L.; Mondelli, R.; Artali, R.; Sirtori, F.R.; Mazzini, S. Nemorubicin and Doxorubicin Bind the G-Quadruplex Sequences of the Human Telomeres and of the c-MYC Promoter Element Pu22. Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 1129–1138. [Google Scholar] [CrossRef]
- Hou, D.-R.; Cheng, H.-Y.; Wang, E.-C. Efficient Syntheses of Oncinotine and Neooncinotine. J. Org. Chem. 2004, 69, 6094–6099. [Google Scholar] [CrossRef]
- Gavathiotis, E.; Searle, M.S. Structure of the Parallel-Stranded DNA Quadruplex d(TTAGGGT)4 Containing the Human Telomeric Repeat: Evidence for A-Tetrad Formation from NMR and Molecular Dynamics Simulations. Org. Biomol. Chem. 2003, 1, 1650–1656. [Google Scholar] [CrossRef]
- Stewart, J.J.P. MOPAC: A Semiempirical Molecular Orbital Program. J. Comput. Aided Mol. Des. 1990, 4, 1–103. [Google Scholar] [CrossRef]
- Hounsou, C.; Guittat, L.; Monchaud, D.; Jourdan, M.; Saettel, N.; Mergny, J.-L.; Teulade-Fichou, M.-P. G-Quadruplex Recognition by Quinacridines: A SAR, NMR, and Biological Study. ChemMedChem Chem. Enabling Drug Discov. 2007, 2, 655–666. [Google Scholar] [CrossRef]
- Ferreira, R.; Artali, R.; Benoit, A.; Gargallo, R.; Eritja, R.; Ferguson, D.M.; Sham, Y.Y.; Mazzini, S. Structure and Stability of Human Telomeric G-Quadruplex with Preclinical 9-Amino Acridines. PLoS ONE 2013, 8, e57701. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasteiger, J.; Marsili, M. Iterative Partial Equalization of Orbital Electronegativity—A Rapid Access to Atomic Charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A Programming Language for Software Integration and Development. J. Mol. Graph. Model 1999, 17, 57–61. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cell Line (IC50 μM) | |
|---|---|---|
| IGROV-1 | RWPE1 | |
| Cisplatin | 0.38 ± 0.049 | - |
| 9 | 14.5 ± 5 | >100 |
| 10 | >30 | - |
| 14 | >30 | >100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazzini, S.; Princiotto, S.; Musso, L.; Passarella, D.; Beretta, G.L.; Perego, P.; Dallavalle, S. Synthesis and Investigation of the G-Quadruplex Binding Properties of Kynurenic Acid Derivatives with a Dihydroimidazoquinoline-3,5-dione Core. Molecules 2022, 27, 2791. https://doi.org/10.3390/molecules27092791
Mazzini S, Princiotto S, Musso L, Passarella D, Beretta GL, Perego P, Dallavalle S. Synthesis and Investigation of the G-Quadruplex Binding Properties of Kynurenic Acid Derivatives with a Dihydroimidazoquinoline-3,5-dione Core. Molecules. 2022; 27(9):2791. https://doi.org/10.3390/molecules27092791
Chicago/Turabian StyleMazzini, Stefania, Salvatore Princiotto, Loana Musso, Daniele Passarella, Giovanni Luca Beretta, Paola Perego, and Sabrina Dallavalle. 2022. "Synthesis and Investigation of the G-Quadruplex Binding Properties of Kynurenic Acid Derivatives with a Dihydroimidazoquinoline-3,5-dione Core" Molecules 27, no. 9: 2791. https://doi.org/10.3390/molecules27092791