

Synthesis and Study of Dibenzo[b, f]oxepine Combined with Fluoroazobenzenes—New Photoswitches for Application in Biological Systems
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis
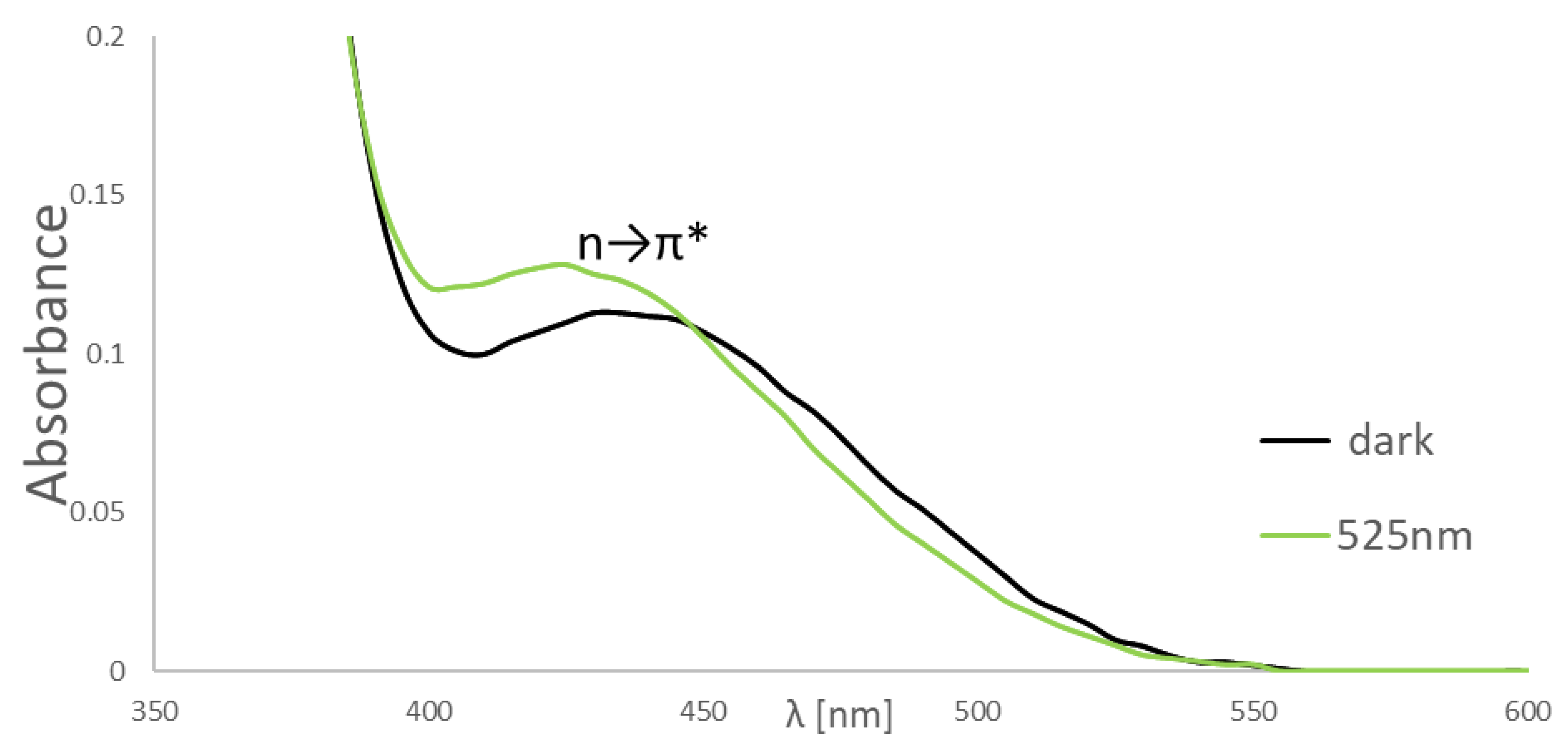
2.2. NMR and UV-VIS Spectra
3. Experimental Section
3.1. Nuclear Magnetic Resonance (NMR) Spectroscopy
3.2. Mass Spectrometry (MS)
3.3. Photoisomerization Studies by UV-VIS Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Ugwu, D.I.; Eze, F.U.; Ezugwu, J.A.; Attah, S.I.; Ugwu, M.C.; Ekoh, O.C.; Nyoyoko, I.F. Chemotherapeutic Importance of Oxepines. Int. J. Chem. Sci. 2021, 19, 401. [Google Scholar] [CrossRef]
- Mekala, A.B.; Satyal, P.; Setzer, W.N. Phytochemicals from the Bark of Rhamnus caroliniana. Nat. Prod. Commun. 2017, 12, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Numata, A.; Iwamoto, C.; Usami, Y.; Yamada, T.; Ohishi, H.; Cragg, G.M. Antineoplastic agents. 551. Isolation and structures of bauhiniastatins 1−4 from Bauhinia purpurea. J. Nat. Prod. 2006, 69, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Acton, D.; Hill, G.; Tait, B.S. Tricyclic triarylethylene antiestrogens: Dibenz [b,f] oxepins, dibenzo [b,f] thiepins, dibenzo [a,e] cyclooctenes, and dibenzo [b,f] thiocins. J. Med. Chem. 1983, 26, 1131–1137. [Google Scholar] [CrossRef]
- Agnew, M.N.; Rizwaniuk, A.; Ong, H.H.; Wichmann, J.K. Nuclear hydroxylated metabolite of fluradoline (2-fluoro-11-[(β-methylamino) ethylthio] dibenz [b,f] oxepin hydrochloride). Identification and synthesis. J. Heterocycl. Chem. 1986, 23, 265–269. [Google Scholar] [CrossRef]
- Lu, Y.-H.; Lin, C.-N.; Ko, H.-H.; Yang, S.-Z.; Tsao, L.-T.; Wang, J.-P. Novel Anti-Inflammatory Constituents of Artocarpus rigida. Helv. Chim. Acta 2003, 86, 2566–2572. [Google Scholar] [CrossRef]
- Rupčić, R.; Modrić, M.; Hutinec, A.; Čikoš, A.; Stanić, B.; Mesić, M.; Pešić, D.; Merćep, M. Novel tetracyclic imidazole derivatives: Synthesis, dynamic NMR study, and anti-inflammatory evaluation. J. Heterocycl. Chem. 2010, 47, 640–656. [Google Scholar] [CrossRef]
- Kiyama, R.; Honma, T.; Hayashi, K.; Ogawa, M.; Hara, M.; Fujimoto, M.; Fujishita, T. Novel Angiotensin II Receptor Antagonists. Design, Synthesis, and in Vitro Evaluation of Dibenzo [a,d] cycloheptene and Dibenzo [b,f] oxepin Derivatives. Searching for Bioisosteres of Biphenyltetrazole Using a Three-Dimensional Search Technique. J. Med. Chem. 1995, 38, 2728–2741. [Google Scholar] [CrossRef]
- Jinno, S.; Okita, T. Synthesis of an Antioxidant Having a Dibenz [b,f] oxepine Skeleton. Heterocycles 1999, 51, 303–314. [Google Scholar]
- Kittakoop, P.; Nopichai, S.; Thongon, N.; Charoenchai, P.; Thebtaranonth, Y. Bauhinoxepins A and B: New Antimycobacterial Dibenzo [b,f] oxepins from Bauhinia saccocalyx. Helv. Chim. Acta 2004, 87, 175–179. [Google Scholar] [CrossRef]
- Cloos, P.A.C.; Reissig, F.; Boissy, P.; Stahlhut, M. Coumpounds Modulating the Activity of Gapdh and/or Iamt. WO2004039773, 13 May 2004.
- Garbicz, D.; Mielecki, D.; Wrzesinski, M.; Pilzys, T.; Marcinkowski, M.; Piwowarski, J.; Debski, J.; Palak, E.; Szczecinski, P.; Krawczyk, H.; et al. Evaluation of Anti-cancer Activity of Stilbene and Methoxydibenzo[b,f]oxepin Derivatives. Curr. Cancer Drug Targets 2018, 18, 706–717. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, K.; Roggo, S.; Kragten, E.; Fürst, P.; Waldmeier, P. Synthesis of tools for target identification of the anti-apoptotic compound CGP 3466, Part I. Bioorg. Med. Chem. Lett. 1998, 8, 1195–1200. [Google Scholar] [CrossRef]
- Zimmermann, K.; Waldmeier, P.C.; Tatton, W.G. Dibenzoxepines as treatments for neurodegenerative diseases. Pure Appl. Chem. 1999, 71, 2039–2046. [Google Scholar] [CrossRef]
- Nantermet, P.G.; Rajapakse, H.A. Tricyclic Beta-Secretase Inhibitors for the Treatment of Alzheimer’s Disease. WO2007019080, 15 February 2007.
- Garbicz, D.; Tobiasz, P.; Borys, F.; Poterała, M.; Grzesiuk, E.; Krawczyk, H. The stilbene and dibenzo [b,f] oxepine derivatives as anticancer compounds. Biomed. Pharmacother. 2020, 123, 109781. [Google Scholar] [CrossRef] [PubMed]
- Lerch, M.; Hansen, M.; van Dam, G.; Szymański, W.; Feringa, B.L. Emerging Targets in Photopharmacology. Angew. Chem. 2016, 55, 10978–10999. [Google Scholar] [CrossRef] [PubMed]
- Bleger, D.; Hecht, S. Visible-Light-Activated Molecular Switches. Angew. Chem. Int. Ed. 2015, 54, 11338–11349. [Google Scholar] [CrossRef] [PubMed]
- Bleger, D.; Schwarz, J.; Brouwer, A.M.; Hecht, S. o-Fluoroazobenzenes as Readily Synthesized Photoswitches Offering Nearly Quantitative Two-Way Isomerization with Visible Light. J. Am. Chem. Soc. 2012, 134, 20597–20600. [Google Scholar] [CrossRef] [PubMed]
- Lameijer, L.N.; Budzak, S.; Simeth, N.A.; Hansen, M.J.; Feringa, B.L.; Jacquemin, D.; Szymański, W. General Principles for the Design of Visible-Light-Responsive Photoswitches: Tetra-ortho-Chloro-Azobenzenes. Angew. Chem. 2020, 132, 21847–21854. [Google Scholar] [CrossRef]
- Wegener, M.; Hansen, M.J.; Driessen, A.J.M.; Szymanski, W.; Feringa, B.L. Photocontrol of Antibacterial Activity: Shifting from UV to Red Light Activation. J. Am. Chem. Soc. 2017, 139, 17979–17986. [Google Scholar] [CrossRef]
- Welleman, I.M.; Hoorens, M.W.H.; Feringa, B.L.; Boersma, H.H.; Szymański, W. Photoresponsive molecular tools for emerging applications of light in medicine. Chem. Sci. 2020, 11, 11672–11691. [Google Scholar] [CrossRef]
- Agnetta, L.; Bermudez, M.; Riefolo, F.; Matera, C.; Claro, E.; Messerer, R.; Littmann, T.; Wolber, G.; Holzgrabe, U.; Decker, M. Fluorination of Photoswitchable Muscarinic Agonists Tunes Receptor Pharmacology and Photochromic Properties. J. Med. Chem. 2019, 62, 3009–3020. [Google Scholar] [CrossRef] [PubMed]
- Goulet-Hanssens, A.; Eisenreich, F.; Hecht, S. Enlightening Materials with Photoswitches. Adv. Mater. 2020, 32, 1905966. [Google Scholar] [CrossRef] [PubMed]
- Kanj, A.B.; Bürck, J.; Vankova, N.; Li, C.; Mutruc, D.; Chandresh, A.; Hecht, S.; Heine, T.; Heinke, L. Chirality Remote Control in Nanoporous Materials by Circularly Polarized Light. J. Am. Chem. Soc. 2021, 143, 7059–7068. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Xue, C.; Weis, P.; Suzuki, Y.; Huang, S.; Koynov, K.; Auernhammer, G.K.; Berger, R.; Butt, H.-J.; Wu, S. Photoswitching of glass transition temperatures of azobenzene-containing polymers induces reversible solid-to-liquid transitions. Nat. Chem. 2017, 9, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Zha, R.H.; Vantomme, G.; Berrocal, J.A.; Gosens, R.; de Waal, B.; Meskers, S.; Meijer, E.W. Photoswitchable nanomaterials based on hierarchically organized siloxane oligomers. Adv. Funct. Mater. 2018, 28, 1703952. [Google Scholar] [CrossRef]
- Wei, J.; Jin, T.-T.; Yang, J.-X.; . Jiang, X.-M.; Liu, L.-J.; Zhan, T.-G.; Zhang, K.-D. A tetrachloroazobenzene based macrocycle featuring with red-light regulated encapsulation for aryl dianionic guests. Tetrahedron Lett. 2020, 61, 151389. [Google Scholar] [CrossRef]
- Trads, J.B.; Burgstaller, J.; Laprell, L.; Konrad, D.B.; De La Osa de La Rosa, L.; Weaver, C.D.; Baier, H.; Trauner, D.; Barber, D.M. Optical control of GIRK channels using visible light. Org. Biomol. Chem. 2017, 15, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Beharry, A.A.; Sadovski, O.; McCormick, T.M.; Babalhavaeji, A.; Tropepe, V.; Woolley, G.A. Photoswitching Azo Compounds in Vivo with Red Light. J. Am. Chem. Soc. 2013, 135, 9777–9784. [Google Scholar] [CrossRef] [PubMed]
- Hammill, M.L.; Islam, G.; Desaulniers, J.-P. Synthesis, Derivatization an Photochemical Control of ortho-Functionalized Tetrachlorinated Azobenzene-Modified siRNAs. Chembiochem 2020, 21, 2367–2372. [Google Scholar] [CrossRef]
- Kerckhoffs, A.; Langton, M.J. Reversible photo-control over transmembrane anion transport using visible-light responsive supramolecular carriers. Chem. Sci. 2020, 11, 6325–6331. [Google Scholar] [CrossRef]
- Krawczyk, H. Mini revive: The derivative of stilbene, nucleosides, and nucleosides modified by stilbene derivatives. Bioorg. Chem. 2019, 90, 103073. [Google Scholar] [CrossRef] [PubMed]
- Čermák, V.; Dostál, V.; Jelínek, M.; Libusová, L.; Kovář, J.; Rösel, D.; Brábek, J. Microtubule-targeting agents and their impact on cancer treatment. Eur. J. Cell Biol. 2020, 99, 151075. [Google Scholar] [CrossRef]
- Borys, F.; Joachimiak, E.; Krawczyk, H.; Fabczak, H. Intrinsic and Extrinsic Factors Affecting Microtubule Dynamics in Normal and Cancer Cells. Molecules 2021, 25, 3705. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, S.N.A.; Kumar, G.B.; Revankar, H.M.; Qin, H.-L. Development of combretastatins as potent tubulin polymerization inhibitors. Bioorg. Chem. 2017, 72, 130–147. [Google Scholar] [CrossRef]
- Song, W.; Tang, Z.; Zhang, D.; Yu, H.; Chen, X. Coadministration of Vascular Disrupting Agents and Nanomedicines to Eradicate Tumors from Peripheral and Central Regions. Small 2015, 11, 3755–3761. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, D.; Song, W.; Tang, Z.; Zhu, J.; Ma, Z.; Wang, X.; Chen, X.; Tong, T. A poly(l-glutamic acid)-combretastatin A4 conjugate for solid tumor therapy: Markedly improved therapeutic efficiency through its low tissue penetration in solid tumor. Acta Biomater. 2017, 53, 179–189. [Google Scholar] [CrossRef]
- Li, L.; Jiang, S.; Li, X.; Liu, Y.; Su, J.; Chen, J. Recent advances in trimethoxyphenyl (TMP) based tubulin inhibitors targeting the colchicine binding site. Eur. J. Med. Chem. 2018, 151, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Borowiak, M.; Nahaboo, W.; Reynders, M.; Nekolla, K.; Jalinot, P.; Hasserodt, J.; Rehberg, M.; Delattre, M.; Zahler, S.; Vollmar, A.; et al. Photoswitchable Inhibitors of Microtubule Dynamics Optically Control Mitosis and Cell Death. Cell 2015, 162, 403–411. [Google Scholar] [CrossRef]
- Sailer, A.; Ermer, F.; Kraus, Y.; Lutter, F.H.; Donau, C.; Bremerich, M.; Ahlfeld, J.; Thorn-Seshold, O. Hemithioindigos for Cellular Photopharmacology: Desymmetrised Molecular Switch Scaffolds Enabling Design Control over the Isomer-Dependency of Potent Antimitotic Bioactivity. ChemBioChem 2019, 20, 1305–1314. [Google Scholar] [CrossRef]
- Sailer, A.; Ermer, F.; Kraus, Y.; Bingham, R.; Lutter, F.H.; Ahlfeld, J.; Thorn-Seshold, O. Potent hemithioindigo-based antimitotics photocontrol the microtubule cytoskeleton in cellulo. Beilstein J. Org. Chem. 2020, 16, 125–134. [Google Scholar] [CrossRef]
- Gao, L.; Meiring, J.C.M.; Varady, A.; Ruider, I.R.; Heise, C.; Wranik, M.; Velasco, C.D.; Taylor, J.A.; Terni, B.; Weinert, T.; et al. In Vivo Photocontrol of Microtubule Dynamics and Integrity, Migration and Mitosis, by the Potent GFP-Imaging-Compatible Photoswitchable Reagents SBTubA4P and SBTub2M. J. Am. Chem. Soc. 2022, 144, 5614–5628. [Google Scholar] [CrossRef] [PubMed]
- Müller-Deku, A.; Meiring, J.C.M.; Loy, K.; Kraus, Y.; Heise, C.; Bingham, R.; Jansen, K.I.; Qu, X.; Bartolini, F.; Kapitein, L.C.; et al. Photoswitchable paclitaxel-based microtubule stabilisers allow optical control over the microtubule cytoskeleton. Nat. Commun. 2020, 11, 4640. [Google Scholar] [CrossRef]
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef] [PubMed]
- Borowiak, M.; Küllmer, F.; Gegenfurtner, F.; Peil, S.; Nasufovic, V.; Zahler, S.; Thorn-Seshold, O.; Trauner, D.; Arndt, H.-D. Optical Manipulation of F-Actin with Photoswitchable Small Molecules. J. Am. Chem. Soc. 2020, 142, 9240–9249. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, S.; Pianowski, Z. Review: Photopharmacology of Antimitotic Agents. Int. J. Mol. Sci. 2022, 23, 5657. [Google Scholar] [CrossRef]
- Tobiasz, P.; Borys, F.; Borecka, M.; Krawczyk, H. Synthesis of building blocks with dibenzo [b,f] oxepin for use in photopharmacology. Int. J. Mol. Sci. 2021, 22, 11033. [Google Scholar] [CrossRef]
- RCSB Protein Data Bank—RCSB PDB. Available online: http://www.rcsb.org/pdb/home/home.do (accessed on 23 February 2004).
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera? A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Ravelli, R.B.G.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. [Google Scholar] [CrossRef]
- Tron, G.C.; Pirali, T.; Sorba, G.; Pagliai, F.; Busacca, S.; Genazzani, A.A. Medicinal chemistry of combretastatin A4: Present and future directions. J. Med. Chem. 2006, 49, 3033–3044. [Google Scholar] [CrossRef]
- Zhang, Q.; Peng, Y.Y.; Wang, X.I.; Keenan, S.M.; Arora, S.; Welsh, W.J. Highly potent triazole-based tubulin polymerization inhibitors. J. Med. Chem. 2007, 50, 749–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massarotti, A.; Coluccia, A.; Silvestri, R.; Sorba, G.; Brancale, A. The Tubulin Colchicine Domain: A Molecular Modeling Perspective. Chem. Med. Chem. 2012, 7, 33–42. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Total Energy [a.u.] of E Isomer | Total Energy [a.u.] of Z Isomer | Δ Total Energy of E/Z [a.u.] | Δ Total Energy of E/Z [kJ/mol] |
|---|---|---|---|---|
| 4a | −1469.512551 | −1469.494105 | 0.018446 | 48.7 |
| 4b | −1568.749191 | −1568.727354 | 0.021837 | 57.7 |
| 4c | −1667.971636 | −1667.953644 | 0.017992 | 47.5 |
| 4d | −1667.970488 | −1667.949743 | 0.020745 | 54.8 |
| 4e | −1469.520836 | −1469.499351 | 0.021485 | 56.7 |
| 4f | −1568.754537 | −1568.732414 | 0.022123 | 58.4 |
| 4g | −1667.979888 | −1667.961826 | 0.018062 | 47.7 |
| 4h | −1667.975848 | −1667.959131 | 0.016717 | 44.1 |
| 5a | −1469.514979 | −1469.493685 | 0.021294 | 56.2 |
| 5b | −1568.748705 | −1568.726548 | 0.022157 | 58.5 |
| 5c | −1667.977288 | −1667.953766 | 0.023522 | 62.0 |
| 5d | −1667.970230 | −1667.953343 | 0.016887 | 44.6 |
| 5e | −1469.520231 | −1469.498987 | 0.021244 | 56.1 |
| 5f | −1568.753982 | −1568.731863 | 0.022119 | 58.4 |
| 5g | −1667.982627 | −1667.958113 | 0.024514 | 64.7 |
| 5h (anti form) | −1667.975565 | −1667.957738 | 0.017827 | 47.1 |
| 5h (syn form) | −1667.968507 | - | - | - |
| Compound | The Energy of HOMO-LUMO Orbitals/eV | ΔE[eV] | |
|---|---|---|---|
| HOMO | LUMO | ||
| 4a E | −6.430 | −2.616 | 3.814 |
| 4a Z | −6.100 | −2.399 | 3.701 |
| 4b E | −6.478 | −2.627 | 3.851 |
| 4b Z | −6.107 | −2.415 | 3.692 |
| 4c E | −6.410 | −2.641 | 3.769 |
| 4c Z | −6.228 | −2.517 | 3.711 |
| 4d E | −6.413 | −2.712 | 3.701 |
| 4d Z | −6.334 | −2.587 | 3.747 |
| 4e E | −6.434 | −2.618 | 3.816 |
| 4e Z | −6.081 | −2.415 | 3.666 |
| 4f E | −6.480 | −2.629 | 3.851 |
| 4f Z | −6.096 | −2.433 | 3.663 |
| 4g E | −6.409 | −2.639 | 3.77 |
| 4g Z | −6.220 | −2.525 | 3.695 |
| 4h E | −6.422 | −2.717 | 3.705 |
| 4h Z | −6.311 | −2.577 | 3.734 |
| 5a E | −6.403 | −2.465 | 3.938 |
| 5a Z | −6.075 | −2.280 | 3.795 |
| 5b E | −6.452 | −2.479 | 3.973 |
| 5b Z | −6.088 | −2.315 | 3.773 |
| 5c E | −6.529 | −2.592 | 3.937 |
| 5c Z | −6.314 | −2.230 | 4.084 |
| 5d E | −6.418 | −2.601 | 3.817 |
| 5d Z | −6.328 | −2.448 | 3.88 |
| 5e E | −6.408 | −2.466 | 3.942 |
| 5e Z | −6.067 | −2.287 | 3.78 |
| 5f E | −6.453 | −2.48 | 3.973 |
| 5f Z | −6.091 | −2.309 | 3.782 |
| 5g E | −6.532 | −2.593 | 3.939 |
| 5g Z | −6.434 | −2.347 | 4.087 |
| 5h E | −6.401 | −2.595 | 3.806 |
| 5h Z | −6.442 | −2.416 | 4.026 |
| Compound | Binding Pose and Interactions | Type of Interaction | Active Residues |
|---|---|---|---|
| 4cZ |  | Hydrogen Bonds | Gly144 Lys253 |
| Halogen Bond | Glyn11 | ||
| 4dE |  | Hydrogen Bonds | Asn249 Lys254 |
| Halogen Bond | Cys241 | ||
| 4hE |  | Hydrogen Bonds | Asn101 Lys254 |
| π – interaction | Lys254 | ||
| Halogen Bond | Asn101 | ||
| 5bE |  | Hydrogen Bonds | Asn101 Lys254 Lys352 |
| Halogen Bond | Thr202 | ||
| 5fE |  | Hydrogen Bonds | Ala101 Lys254 Lys352 |
| Halogen Bond | Thr202 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borys, F.; Tobiasz, P.; Sobel, J.; Krawczyk, H. Synthesis and Study of Dibenzo[b, f]oxepine Combined with Fluoroazobenzenes—New Photoswitches for Application in Biological Systems. Molecules 2022, 27, 5836. https://doi.org/10.3390/molecules27185836
Borys F, Tobiasz P, Sobel J, Krawczyk H. Synthesis and Study of Dibenzo[b, f]oxepine Combined with Fluoroazobenzenes—New Photoswitches for Application in Biological Systems. Molecules. 2022; 27(18):5836. https://doi.org/10.3390/molecules27185836
Chicago/Turabian StyleBorys, Filip, Piotr Tobiasz, Jakub Sobel, and Hanna Krawczyk. 2022. "Synthesis and Study of Dibenzo[b, f]oxepine Combined with Fluoroazobenzenes—New Photoswitches for Application in Biological Systems" Molecules 27, no. 18: 5836. https://doi.org/10.3390/molecules27185836