
In Silico and In Vitro Study of Janus Kinases Inhibitors from Naphthoquinones
 , ,
, ,  , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Cytotoxicity on Leukemia Cell Lines
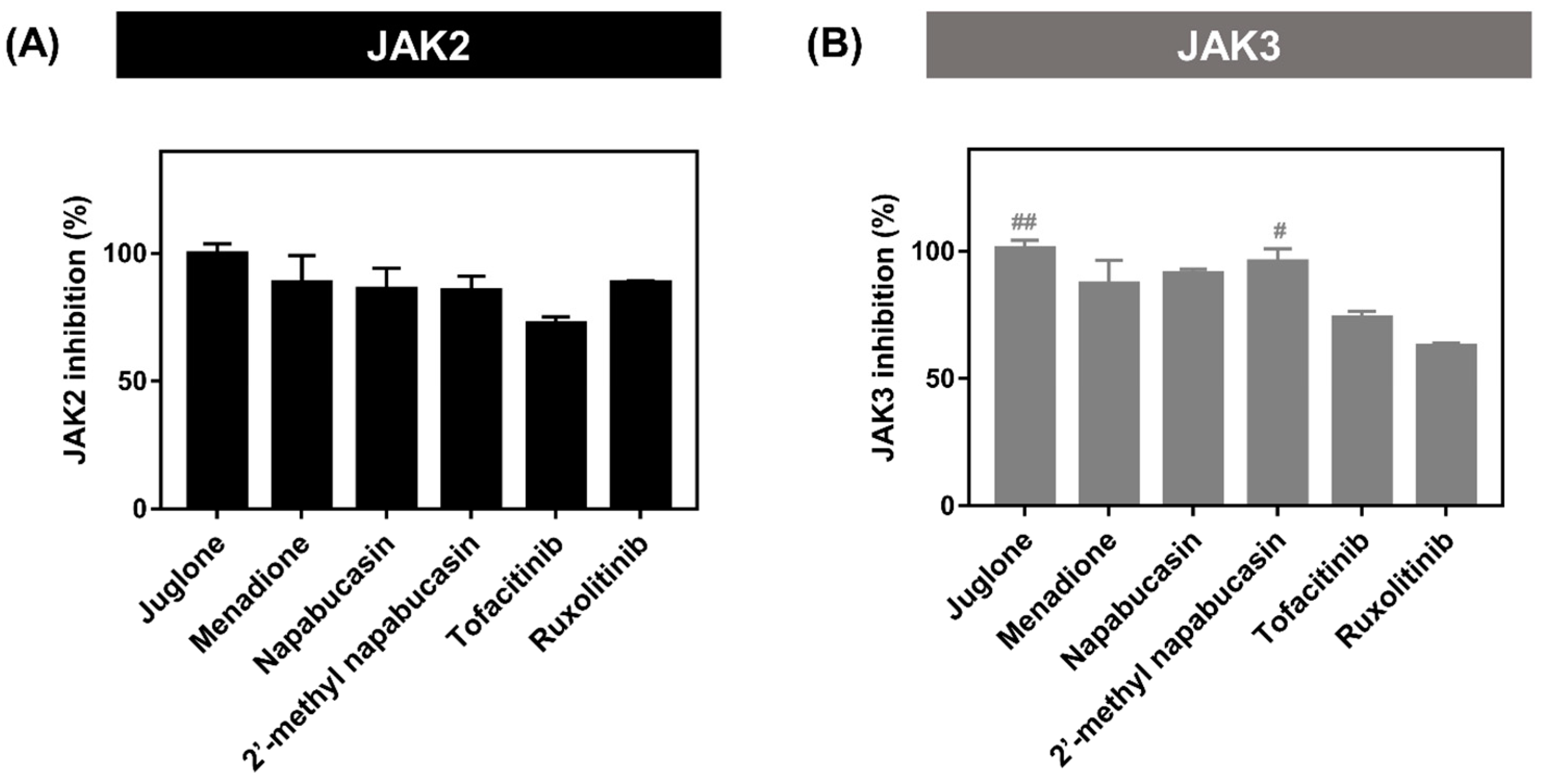
2.2. Kinase Inhibitory Activity of JAK2/3
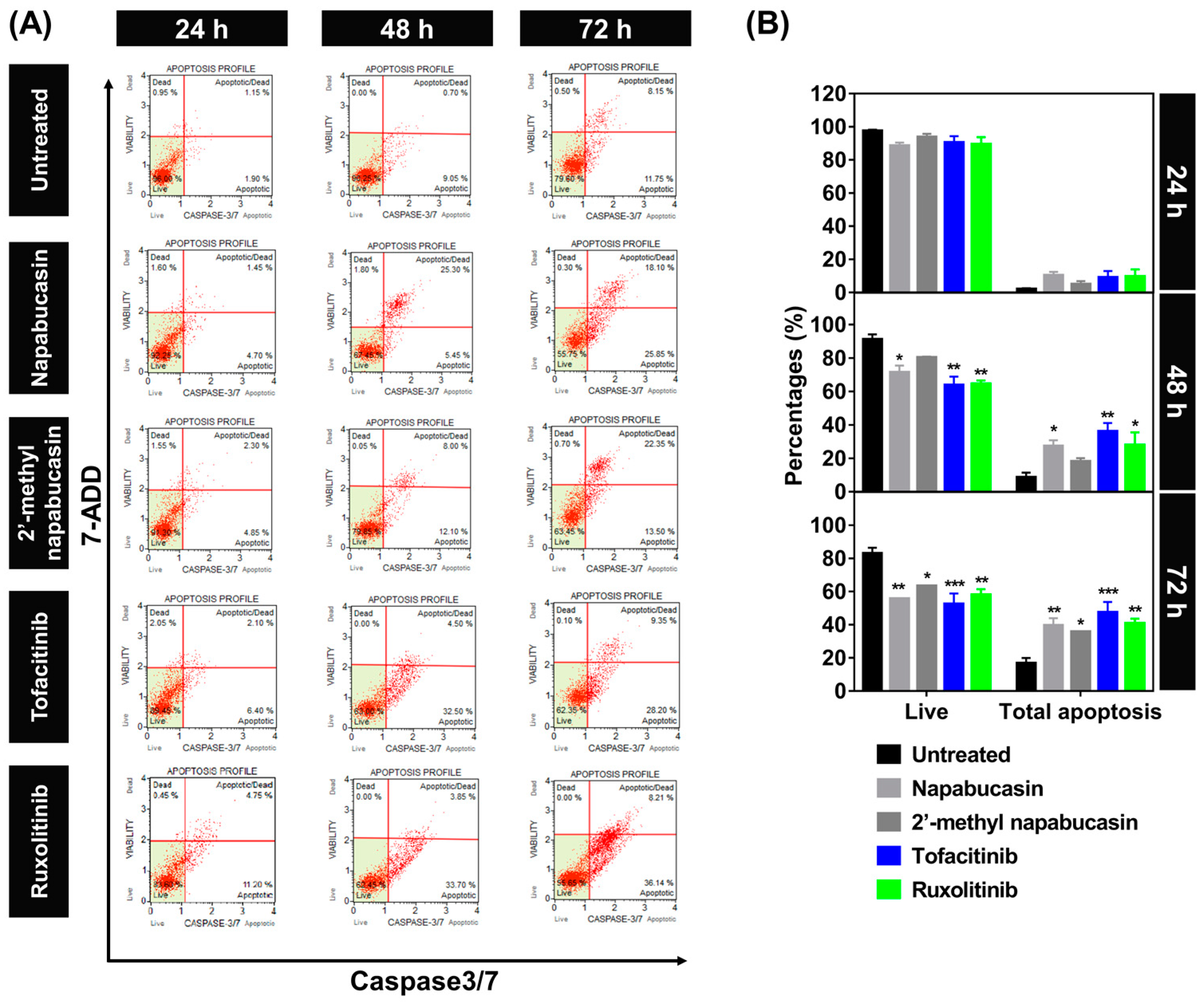
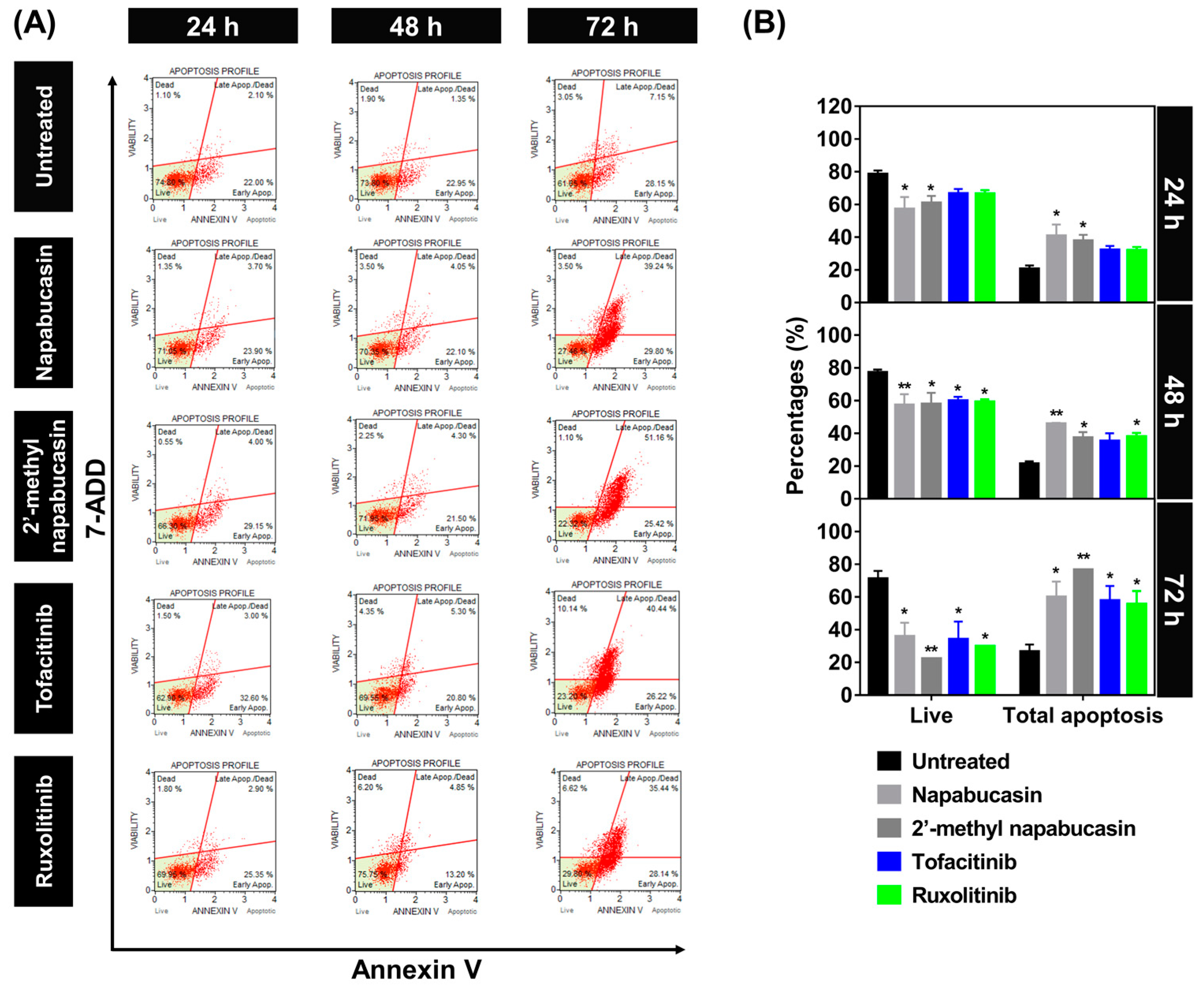
2.3. Apoptosis on TF1 Cell Treated with Napabucasin
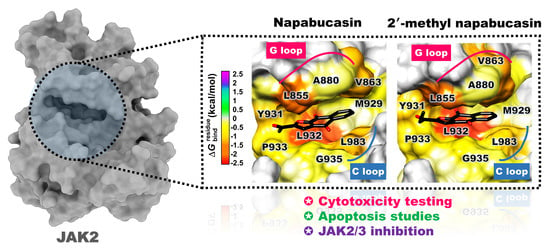
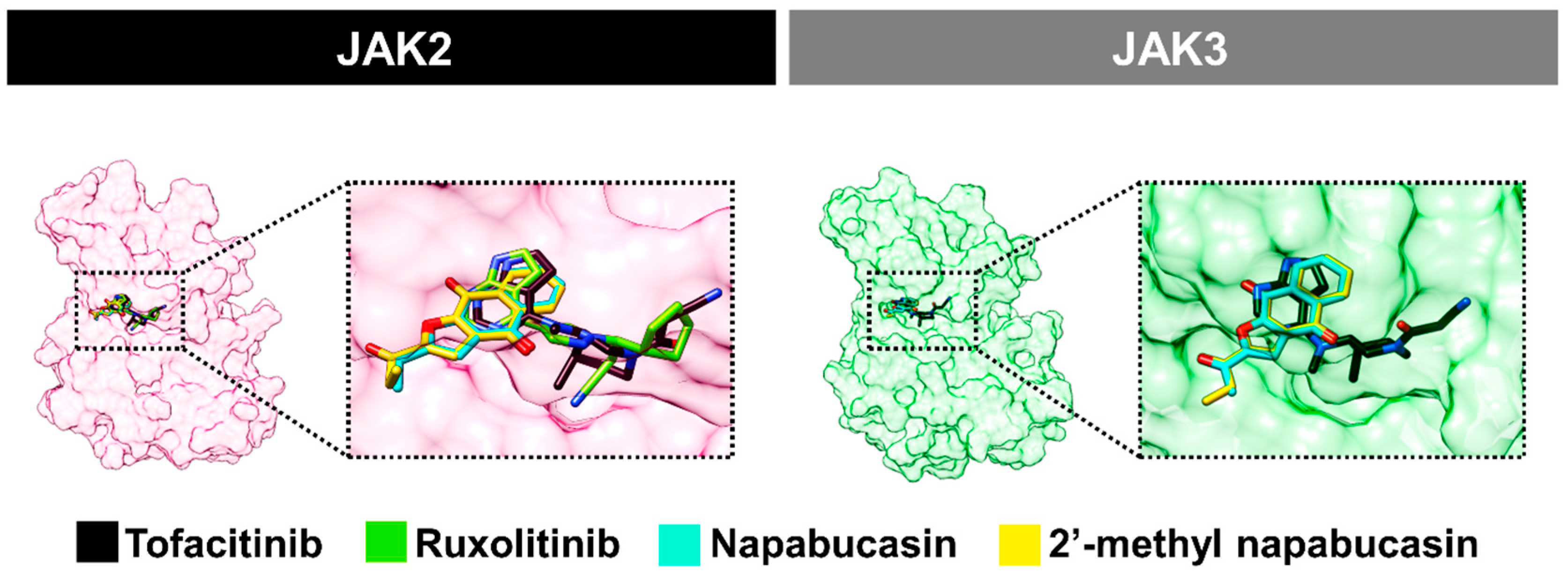
2.4. Binding Pattern of Napabucasin Series with JAK2/3 by Molecular Docking
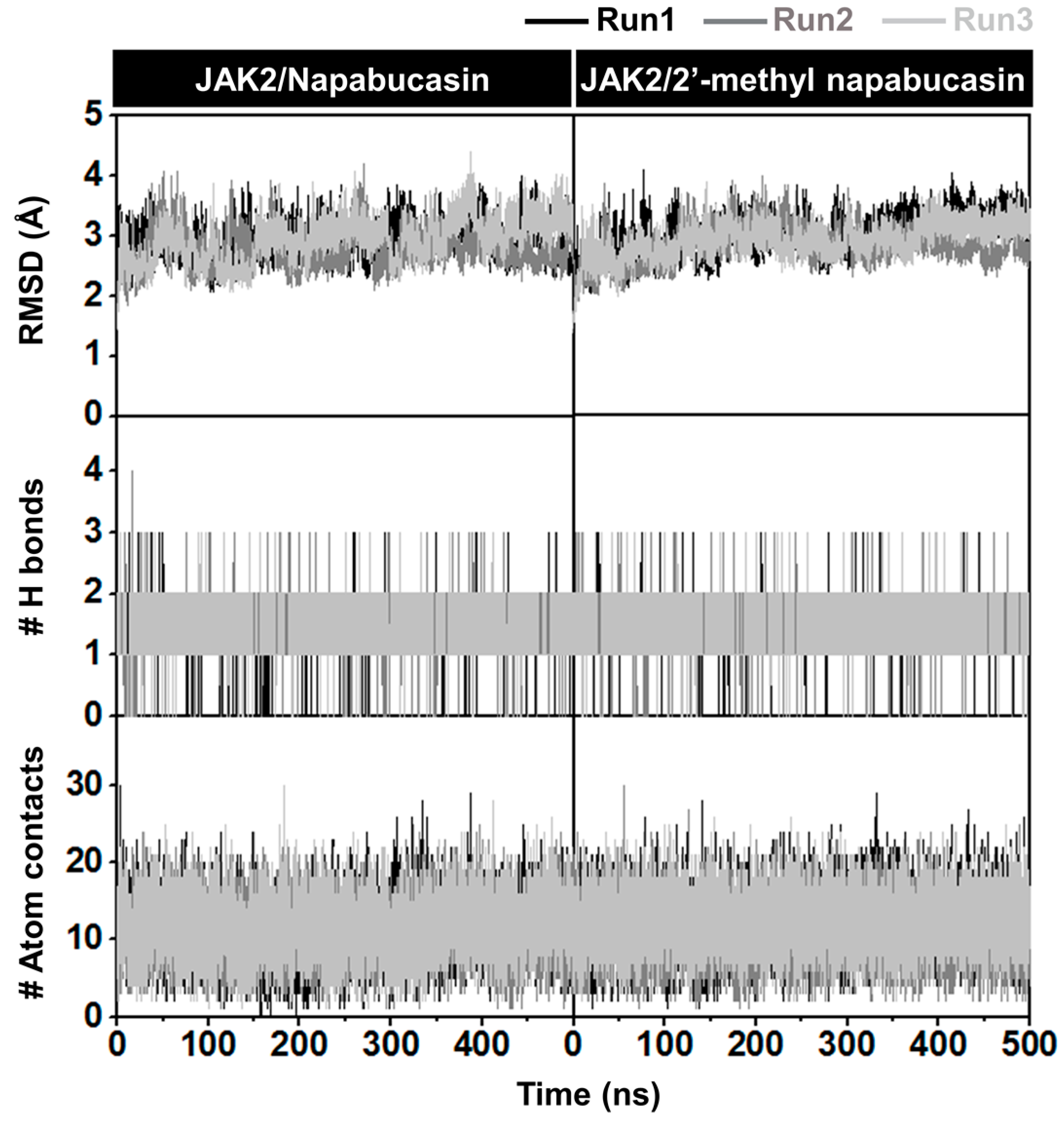
2.5. System Stability of JAK2 with Napabucasin Series
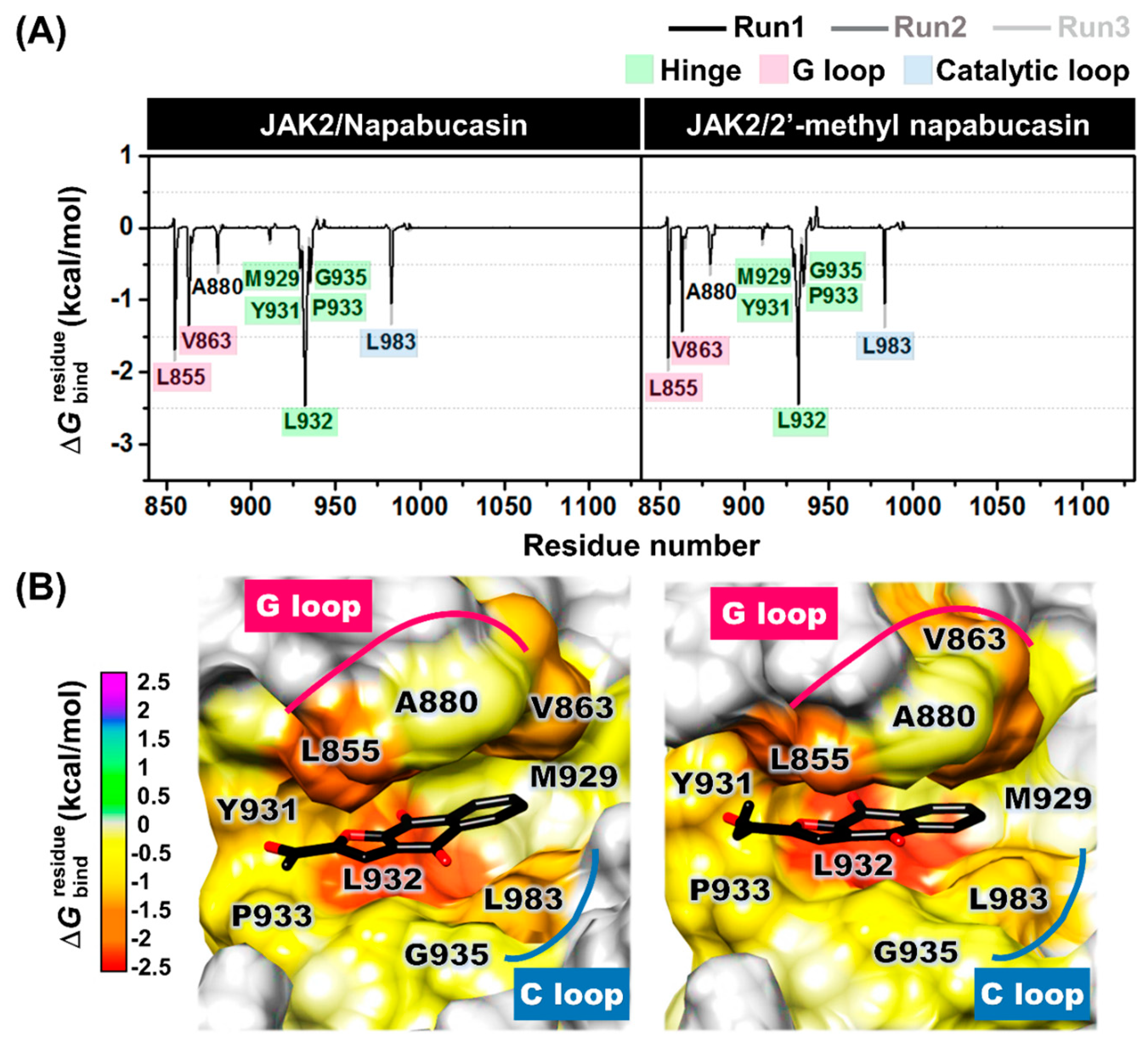
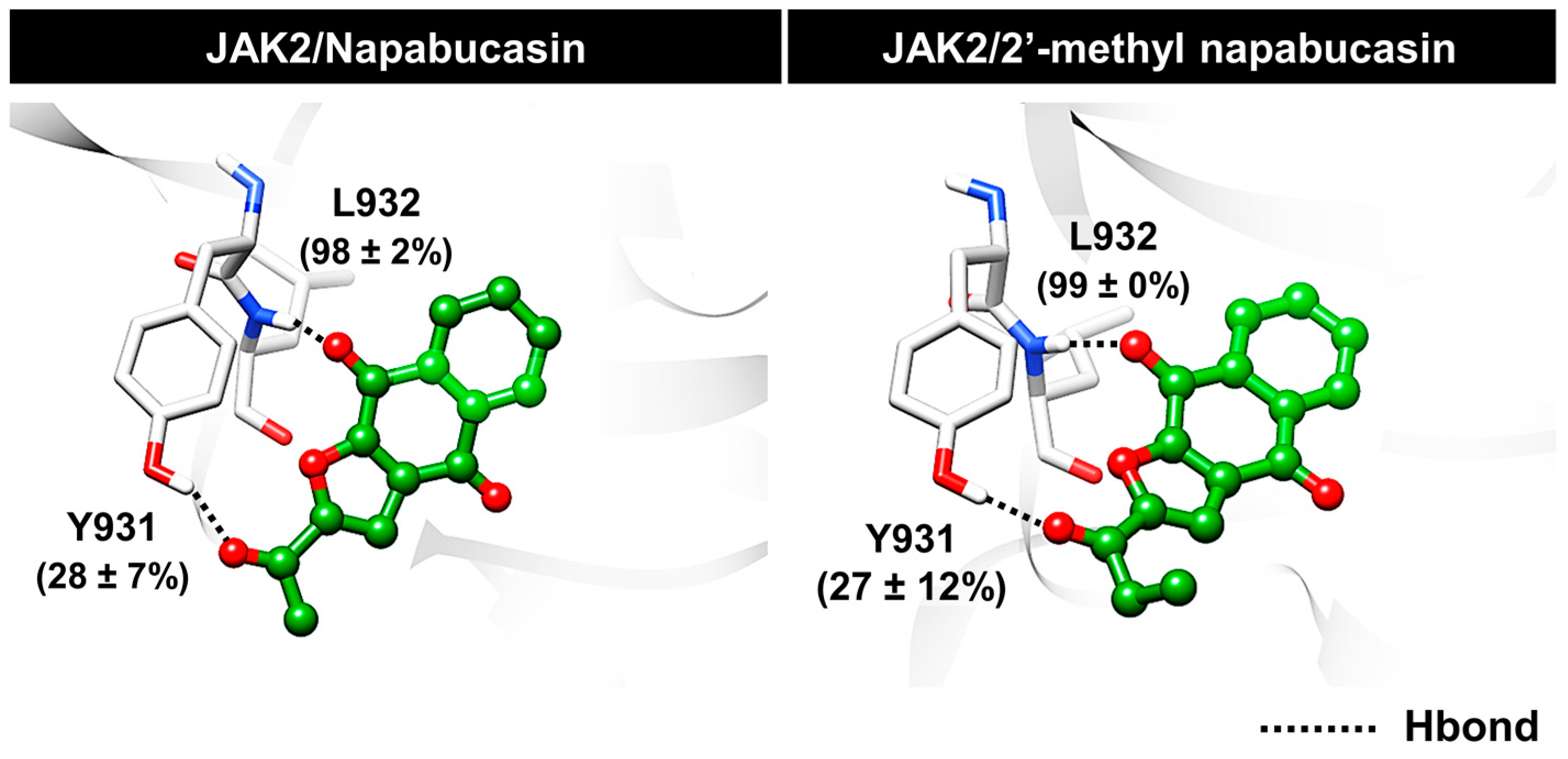
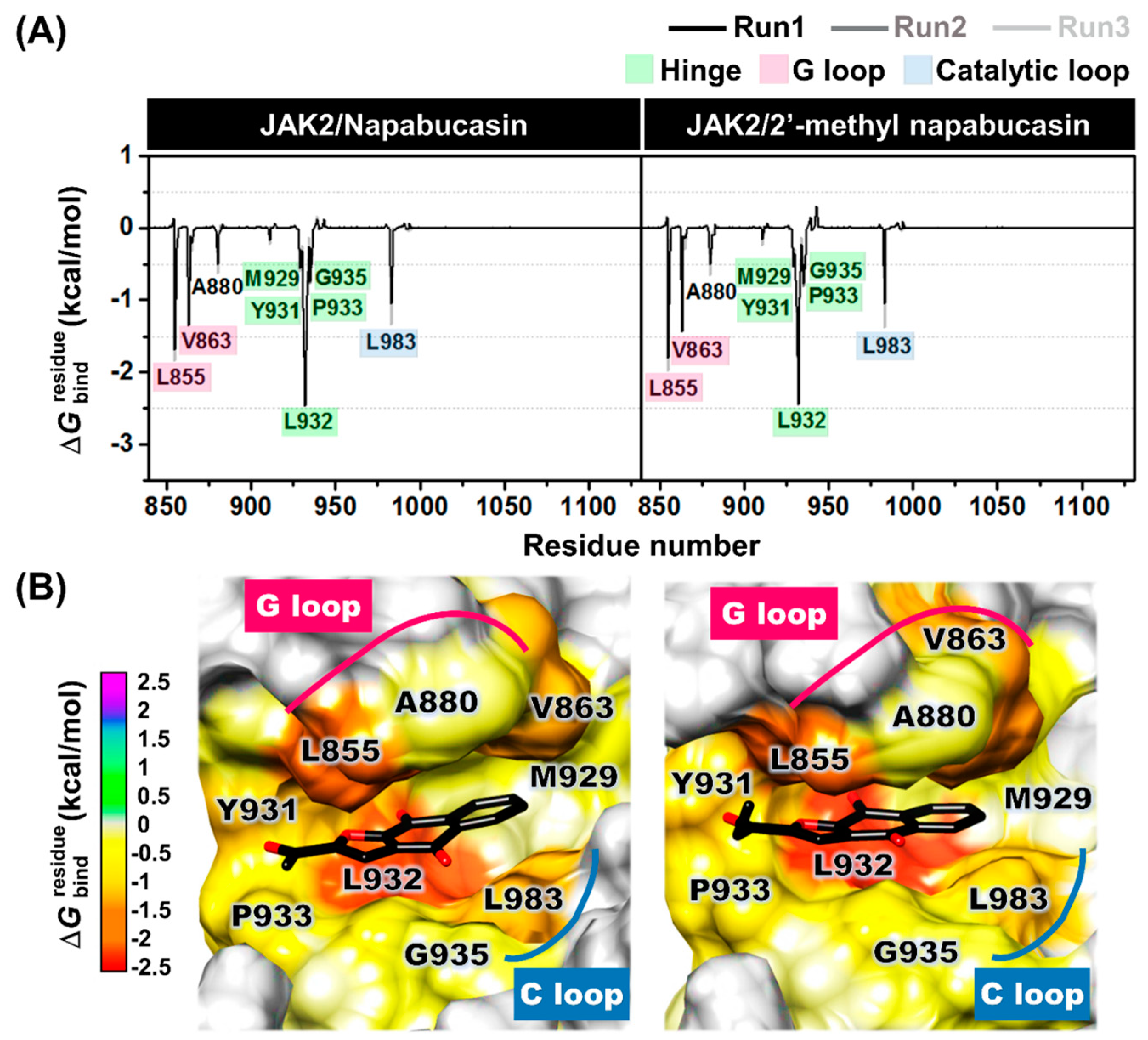
2.6. Binding Patterns of JAK2 with Napabucasin Series
3. Materials and Methods
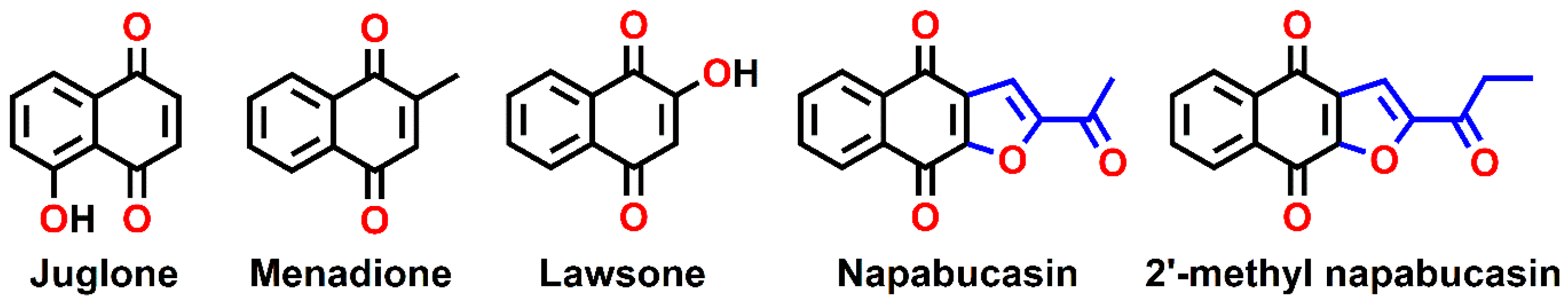
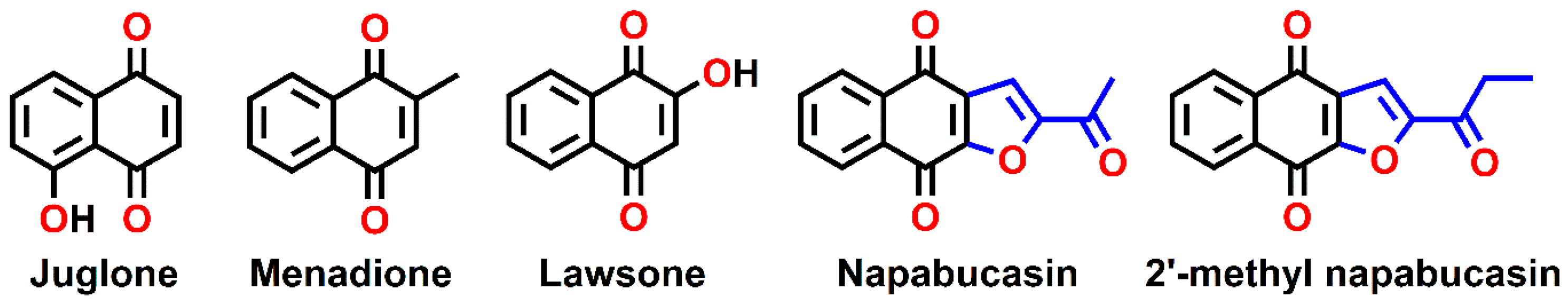
3.1. Preparation of Naphthoquinone Analogs
3.2. Cell Culture and Cytotoxicity of Leukemia Cell Lines
3.3. Kinase Inhibitory Activity of JAK2/3
3.4. Apoptosis Detection Assay
3.5. Caspase Assays
3.6. Statistical Analysis
3.7. Molecular Docking
3.8. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef]
- Murray, P.J. The JAK-STAT signaling pathway: Input and output integration. J. Immunol. 2007, 178, 2623–2629. [Google Scholar] [CrossRef] [Green Version]
- Bechman, K.; Yates, M.; Galloway, J.B. The new entries in the therapeutic armamentarium: The small molecule JAK inhibitors. Pharmacol. Res. 2019, 147, 104392. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Muller-Newen, G.; Schaper, F.; Graeve, L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998, 334, 297–314. [Google Scholar] [CrossRef] [Green Version]
- Jasuja, H.; Chadha, N.; Kaur, M.; Silakari, O. Dual inhibitors of Janus kinase 2 and 3 (JAK2/3): Designing by pharmacophore- and docking-based virtual screening approach. Mol. Divers. 2014, 18, 253–267. [Google Scholar] [CrossRef]
- Vainchenker, W.; Leroy, E.; Gilles, L.; Marty, C.; Plo, I.; Constantinescu, S.N. JAK inhibitors for the treatment of myeloproliferative neoplasms and other disorders. F1000Research 2018, 7, 82. [Google Scholar] [CrossRef] [Green Version]
- Verstovsek, S.; Kantarjian, H.; Mesa, R.A.; Pardanani, A.D.; Cortes-Franco, J.; Thomas, D.A.; Estrov, Z.; Fridman, J.S.; Bradley, E.C.; Erickson-Viitanen, S.; et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N. Engl. J. Med. 2010, 363, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R., Jr. Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol. Res. 2016, 111, 784–803. [Google Scholar] [CrossRef]
- Dowty, M.E.; Jesson, M.I.; Ghosh, S.; Lee, J.; Meyer, D.M.; Krishnaswami, S.; Kishore, N. Preclinical to clinical translation of tofacitinib, a Janus kinase inhibitor, in rheumatoid arthritis. J. Pharmacol. Exp. Ther. 2014, 348, 165–173. [Google Scholar] [CrossRef] [Green Version]
- Chrencik, J.E.; Patny, A.; Leung, I.K.; Korniski, B.; Emmons, T.L.; Hall, T.; Weinberg, R.A.; Gormley, J.A.; Williams, J.M.; Day, J.E.; et al. Structural and thermodynamic characterization of the TYK2 and JAK3 kinase domains in complex with CP-690550 and CMP-6. J. Mol. Biol. 2010, 400, 413–433. [Google Scholar] [CrossRef]
- Hernandez Boluda, J.C.; Gomez, M.; Perez, A. JAK2 inhibitors. Med. Clin. 2016, 147, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Changelian, P.S.; Flanagan, M.E.; Ball, D.J.; Kent, C.R.; Magnuson, K.S.; Martin, W.H.; Rizzuti, B.J.; Sawyer, P.S.; Perry, B.D.; Brissette, W.H.; et al. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science 2003, 302, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.M.; Jesson, M.I.; Li, X.; Elrick, M.M.; Funckes-Shippy, C.L.; Warner, J.D.; Gross, C.J.; Dowty, M.E.; Ramaiah, S.K.; Hirsch, J.L.; et al. Anti-inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP-690,550, in rat adjuvant-induced arthritis. J. Inflamm. 2010, 7, 41. [Google Scholar] [CrossRef] [Green Version]
- Guerra, B.; Martin-Rodriguez, P.; Diaz-Chico, J.C.; McNaughton-Smith, G.; Jimenez-Alonso, S.; Hueso-Falcon, I.; Montero, J.C.; Blanco, R.; Leon, J.; Rodriguez-Gonzalez, G.; et al. CM363, a novel naphthoquinone derivative which acts as multikinase modulator and overcomes imatinib resistance in chronic myelogenous leukemia. Oncotarget 2017, 8, 29679–29698. [Google Scholar] [CrossRef]
- Molinari, A.; Oliva, A.; Arismendi-Macuer, M.; Guzman, L.; Acevedo, W.; Aguayo, D.; Vinet, R.; San Feliciano, A. Antiproliferative Benzoindazolequinones as Potential Cyclooxygenase-2 Inhibitors. Molecules 2019, 24, 2261. [Google Scholar] [CrossRef] [Green Version]
- Blair, H.A. Daunorubicin/Cytarabine Liposome: A Review in Acute Myeloid Leukaemia. Drugs 2018, 78, 1903–1910. [Google Scholar] [CrossRef] [Green Version]
- Su, D.; Gao, Y.Q.; Deng, Y.J.; Zhang, H.H.; Wu, Y.R.; Hu, Y.; Mei, Q.X. Identification of Chinese Herbal Compounds with Potential as JAK3 Inhibitors. Evid. Based Complement. Alternat. Med. 2019, 2019, 4982062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, K.L.; Su, J.C.; Chien, C.M.; Tseng, C.H.; Chen, Y.L.; Chang, L.S.; Lin, S.R. Naphtho[1,2-b]furan-4,5-dione disrupts Janus kinase-2 and induces apoptosis in breast cancer MDA-MB-231 cells. Toxicol. Vitr. 2010, 24, 1158–1167. [Google Scholar] [CrossRef] [PubMed]
- Kuang, S.; Qi, C.; Liu, J.; Sun, X.; Zhang, Q.; Sima, Z.; Liu, J.; Li, W.; Yu, Q. 2-Methoxystypandrone inhibits signal transducer and activator of transcription 3 and nuclear factor-kappaB signaling by inhibiting Janus kinase 2 and IkappaB kinase. Cancer Sci. 2014, 105, 473–480. [Google Scholar] [CrossRef]
- Acuna, J.; Piermattey, J.; Caro, D.; Bannwitz, S.; Barrios, L.; Lopez, J.; Ocampo, Y.; Vivas-Reyes, R.; Aristizabal, F.; Gaitan, R.; et al. Synthesis, Anti-Proliferative Activity Evaluation and 3D-QSAR Study of Naphthoquinone Derivatives as Potential Anti-Colorectal Cancer Agents. Molecules 2018, 23, 186. [Google Scholar] [CrossRef]
- Mancini, I.; Vigna, J.; Sighel, D.; Defant, A. Hybrid Molecules Containing Naphthoquinone and Quinolinedione Scaffolds as Antineoplastic Agents. Molecules 2022, 27, 4948. [Google Scholar] [CrossRef]
- Binder, R.E.; Benson, M.E.; Flath, R.A. Eight 1,4-naphthoquinones from Juglans. Phytochemistry 1989, 28, 2799–2801. [Google Scholar] [CrossRef]
- Abd-el-Malek, Y.A.; el-Leithy, M.A.; Reda, F.A.; Khalil, M. Antimicrobial principles in leaves of Lawsonia inermis L. Zentralbl. Bakteriol. Parasitenkd. Infektionskr. Hyg. 1973, 128, 61–67. [Google Scholar] [CrossRef]
- Peraza-Sanchez, S.R.; Chavez, D.; Chai, H.B.; Shin, Y.G.; Garcia, R.; Mejia, M.; Fairchild, C.R.; Lane, K.E.; Menendez, A.T.; Farnsworth, N.R.; et al. Cytotoxic constituents of the roots of Ekmanianthe longiflora. J. Nat. Prod. 2000, 63, 492–495. [Google Scholar] [CrossRef]
- Tahara, T.; Watanabe, A.; Yutani, M.; Yamano, Y.; Sagara, M.; Nagai, S.; Saito, K.; Yamashita, M.; Ihara, M.; Iida, A. STAT3 inhibitory activity of naphthoquinones isolated from Tabebuia avellanedae. Bioorg. Med. Chem. 2020, 28, 115347. [Google Scholar] [CrossRef]
- Karnsomwan, W.; Netcharoensirisuk, P.; Rungrotmongkol, T.; De-Eknamkul, W.; Chamni, S. Synthesis, Biological Evaluation and Molecular Docking of Avicequinone C Analogues as Potential Steroid 5alpha-Reductase Inhibitors. Chem. Pharm. Bull. 2017, 65, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Reichstein, A.; Vortherms, S.; Bannwitz, S.; Tentrop, J.; Prinz, H.; Muller, K. Synthesis and structure-activity relationships of lapacho analogues. 1. Suppression of human keratinocyte hyperproliferation by 2-substituted naphtho[2,3-b]furan-4,9-diones, activation by enzymatic one- and two-electron reduction, and intracellular generation of superoxide. J. Med. Chem. 2012, 55, 7273–7284. [Google Scholar] [CrossRef]
- Petsri, K.; Thongsom, S.; Racha, S.; Chamni, S.; Jindapol, S.; Kaekratoke, N.; Zou, H.; Chanvorachote, P. Novel mechanism of napabucasin, a naturally derived furanonaphthoquinone: Apoptosis and autophagy induction in lung cancer cells through direct targeting on Akt/mTOR proteins. BMC Complement. Med. Ther. 2022, 22, 250. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, Y.; Wang, X.; Zhao, Z.; Cai, H.; Xie, M.; Jiang, X.; Zhang, L.; Cheng, J.; Yang, L.; et al. Acetylshikonin exerts anti-tumor effects on non-small cell lung cancer through dual inhibition of STAT3 and EGFR. Phytomedicine 2022, 101, 154109. [Google Scholar] [CrossRef]
- Li, Y.; Song, Z.; Han, Q.; Zhao, H.; Pan, Z.; Lei, Z.; Zhang, J. Targeted inhibition of STAT3 induces immunogenic cell death of hepatocellular carcinoma cells via glycolysis. Mol. Oncol. 2022, 16, 2861–2880. [Google Scholar] [CrossRef]
- Dorman, K.; Heinemann, V.; Kobold, S.; von Bergwelt-Baildon, M.; Boeck, S. Novel systemic treatment approaches for metastatic pancreatic cancer. Expert Opin. Investig. Drugs 2022, 31, 249–262. [Google Scholar] [CrossRef]
- Zhong, Y.; Deng, L.; Shi, S.; Huang, Q.Y.; Ou-Yang, S.M.; Mo, J.S.; Zhu, K.; Qu, X.M.; Liu, P.Q.; Wang, Y.X.; et al. The novel STAT3 inhibitor WZ-2-033 causes regression of human triple-negative breast cancer and gastric cancer xenografts. Acta Pharmacol. Sin. 2022, 43, 1013–1023. [Google Scholar] [CrossRef]
- Martin-Rodriguez, P.; Guerra, B.; Hueso-Falcon, I.; Aranda-Tavio, H.; Diaz-Chico, J.; Quintana, J.; Estevez, F.; Diaz-Chico, B.; Amesty, A.; Estevez-Braun, A.; et al. A Novel Naphthoquinone-Coumarin Hybrid That Inhibits BCR-ABL1-STAT5 Oncogenic Pathway and Reduces Survival in Imatinib-Resistant Chronic Myelogenous Leukemia Cells. Front. Pharmacol. 2018, 9, 1546. [Google Scholar] [CrossRef] [Green Version]
- Quentmeier, H.; MacLeod, R.A.; Zaborski, M.; Drexler, H.G. JAK2 V617F tyrosine kinase mutation in cell lines derived from myeloproliferative disorders. Leukemia 2006, 20, 471–476. [Google Scholar] [CrossRef] [Green Version]
- Senkevitch, E.; Durum, S. The promise of Janus kinase inhibitors in the treatment of hematological malignancies. Cytokine 2017, 98, 33–41. [Google Scholar] [CrossRef]
- Pissot-Soldermann, C.; Gerspacher, M.; Furet, P.; Gaul, C.; Holzer, P.; McCarthy, C.; Radimerski, T.; Regnier, C.H.; Baffert, F.; Drueckes, P.; et al. Discovery and SAR of potent, orally available 2,8-diaryl-quinoxalines as a new class of JAK2 inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 2609–2613. [Google Scholar] [CrossRef]
- Muto, A.; Hori, M.; Sasaki, Y.; Saitoh, A.; Yasuda, I.; Maekawa, T.; Uchida, T.; Asakura, K.; Nakazato, T.; Kaneda, T.; et al. Emodin has a cytotoxic activity against human multiple myeloma as a Janus-activated kinase 2 inhibitor. Mol. Cancer Ther. 2007, 6, 987–994. [Google Scholar] [CrossRef] [Green Version]
- Mahalapbutr, P.; Wonganan, P.; Chavasiri, W.; Rungrotmongkol, T. Butoxy Mansonone G Inhibits STAT3 and Akt Signaling Pathways in Non-Small Cell Lung Cancers: Combined Experimental and Theoretical Investigations. Cancers 2019, 11, 437. [Google Scholar] [CrossRef] [Green Version]
- Sanachai, K.; Mahalapbutr, P.; Tabtimmai, L.; Seetaha, S.; Kittikool, T.; Yotphan, S.; Choowongkomon, K.; Rungrotmongkol, T. Discovery of JAK2/3 Inhibitors from Quinoxalinone-Containing Compounds. ACS Omega 2022, 7, 33587–33598. [Google Scholar] [CrossRef]
- Sanachai, K.; Mahalapbutr, P.; Choowongkomon, K.; Poo-Arporn, R.P.; Wolschann, P.; Rungrotmongkol, T. Insights into the Binding Recognition and Susceptibility of Tofacitinib toward Janus Kinases. ACS Omega 2020, 5, 369–377. [Google Scholar] [CrossRef]
- Zhou, T.; Georgeon, S.; Moser, R.; Moore, D.J.; Caflisch, A.; Hantschel, O. Specificity and mechanism-of-action of the JAK2 tyrosine kinase inhibitors ruxolitinib and SAR302503 (TG101348). Leukemia 2014, 28, 471–472. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.H.; Khadka, D.B.; Cho, S.H.; Ju, H.K.; Lee, K.Y.; Han, H.J.; Lee, K.T.; Cho, W.J. Virtual screening and synthesis of quinazolines as novel JAK2 inhibitors. Bioorg. Med. Chem. 2011, 19, 968–977. [Google Scholar] [CrossRef]
- Li, J.J.; Cheng, P.; Tu, J.; Zhai, H.L.; Zhang, X.Y. Enhancing specificity in the Janus kinases: A study on the thienopyridine JAK2 selective mechanism combined molecular dynamics simulation. Mol. Biosyst. 2016, 12, 575–587. [Google Scholar] [CrossRef]
- Williams, N.K.; Bamert, R.S.; Patel, O.; Wang, C.; Walden, P.M.; Wilks, A.F.; Fantino, E.; Rossjohn, J.; Lucet, I.S. Dissecting Specificity in the Janus Kinases: The Structures of JAK-Specific Inhibitors Complexed to the JAK1 and JAK2 Protein Tyrosine Kinase Domains. J. Mol. Biol. 2009, 387, 219–232. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Rostkowski, M.; Olsson, M.H.M.; Sondergaard, C.R.; Jensen, J.H. Graphical analysis of pH-dependent properties of proteins predicted using PROPKA. BMC Struct. Biol. 2011, 11, 6. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.; Bernhard Schlegel, H.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Marvin was Used for Drawing, Displaying and Characterizing Chemical Structures, Substructures and Reactions, Marvin 17.21.0, ChemAxon. Available online: https://www.chemaxon.com (accessed on 1 February 2022).
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Sanachai, K.; Aiebchun, T.; Mahalapbutr, P.; Seetaha, S.; Tabtimmai, L.; Maitarad, P.; Xenikakis, I.; Geronikaki, A.; Choowongkomon, K.; Rungrotmongkol, T. Discovery of novel JAK2 and EGFR inhibitors from a series of thiazole-based chalcone derivatives. RSC Med. Chem. 2021, 12, 430–438. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- BIOVIA. Dassault Systèmes, Discovery Studio Visualizer Software, Version 2019 Client; Dassault Systèmes: San Diego, CA, USA, 2019; Available online: https://www.accelrys.com (accessed on 1 February 2022).
- Gotz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Aiebchun, T.; Mahalapbutr, P.; Auepattanapong, A.; Khaikate, O.; Seetaha, S.; Tabtimmai, L.; Kuhakarn, C.; Choowongkomon, K.; Rungrotmongkol, T. Identification of Vinyl Sulfone Derivatives as EGFR Tyrosine Kinase Inhibitor: In Vitro and In Silico Studies. Molecules 2021, 26, 2211. [Google Scholar] [CrossRef]
- Klaewkla, M.; Charoenwongpaiboon, T.; Mahalapbutr, P. Molecular basis of the new COVID-19 target neuropilin-1 in complex with SARS-CoV-2 S1 C-end rule peptide and small-molecule antagonists. J. Mol. Liq. 2021, 335, 116537. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.M.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- York, D.M.; Darden, T.A.; Pedersen, L.G. The Effect of Long-Range Electrostatic Interactions in Simulations of Macromolecular Crystals—A Comparison of the Ewald and Truncated List Methods. J. Chem. Phys. 1993, 99, 8345–8348. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 ± SEM (nM) | |
|---|---|---|
| JAK2 | JAK3 | |
| Napabucasin | 12.62 ± 2.12 *** | 14.84 ± 2.37 |
| 2′-methyl napabucasin | 11.11 ± 0.13 *** | 10.24 ± 0.66 ** |
| Tofacitinib | 32.10 ± 1.99 | 21.03 ± 0.97 |
| Ruxolitinib | 14.63 ± 0.81 | nd |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanachai, K.; Mahalapbutr, P.; Tabtimmai, L.; Seetaha, S.; Kaekratoke, N.; Chamni, S.; Azam, S.S.; Choowongkomon, K.; Rungrotmongkol, T. In Silico and In Vitro Study of Janus Kinases Inhibitors from Naphthoquinones. Molecules 2023, 28, 597. https://doi.org/10.3390/molecules28020597
Sanachai K, Mahalapbutr P, Tabtimmai L, Seetaha S, Kaekratoke N, Chamni S, Azam SS, Choowongkomon K, Rungrotmongkol T. In Silico and In Vitro Study of Janus Kinases Inhibitors from Naphthoquinones. Molecules. 2023; 28(2):597. https://doi.org/10.3390/molecules28020597
Chicago/Turabian StyleSanachai, Kamonpan, Panupong Mahalapbutr, Lueacha Tabtimmai, Supaphorn Seetaha, Nantawat Kaekratoke, Supakarn Chamni, Syed Sikander Azam, Kiattawee Choowongkomon, and Thanyada Rungrotmongkol. 2023. "In Silico and In Vitro Study of Janus Kinases Inhibitors from Naphthoquinones" Molecules 28, no. 2: 597. https://doi.org/10.3390/molecules28020597