
Peptides as Therapeutic Agents: Challenges and Opportunities in the Green Transition Era
 ,
,  , ,
, ,  , and
, and
Abstract
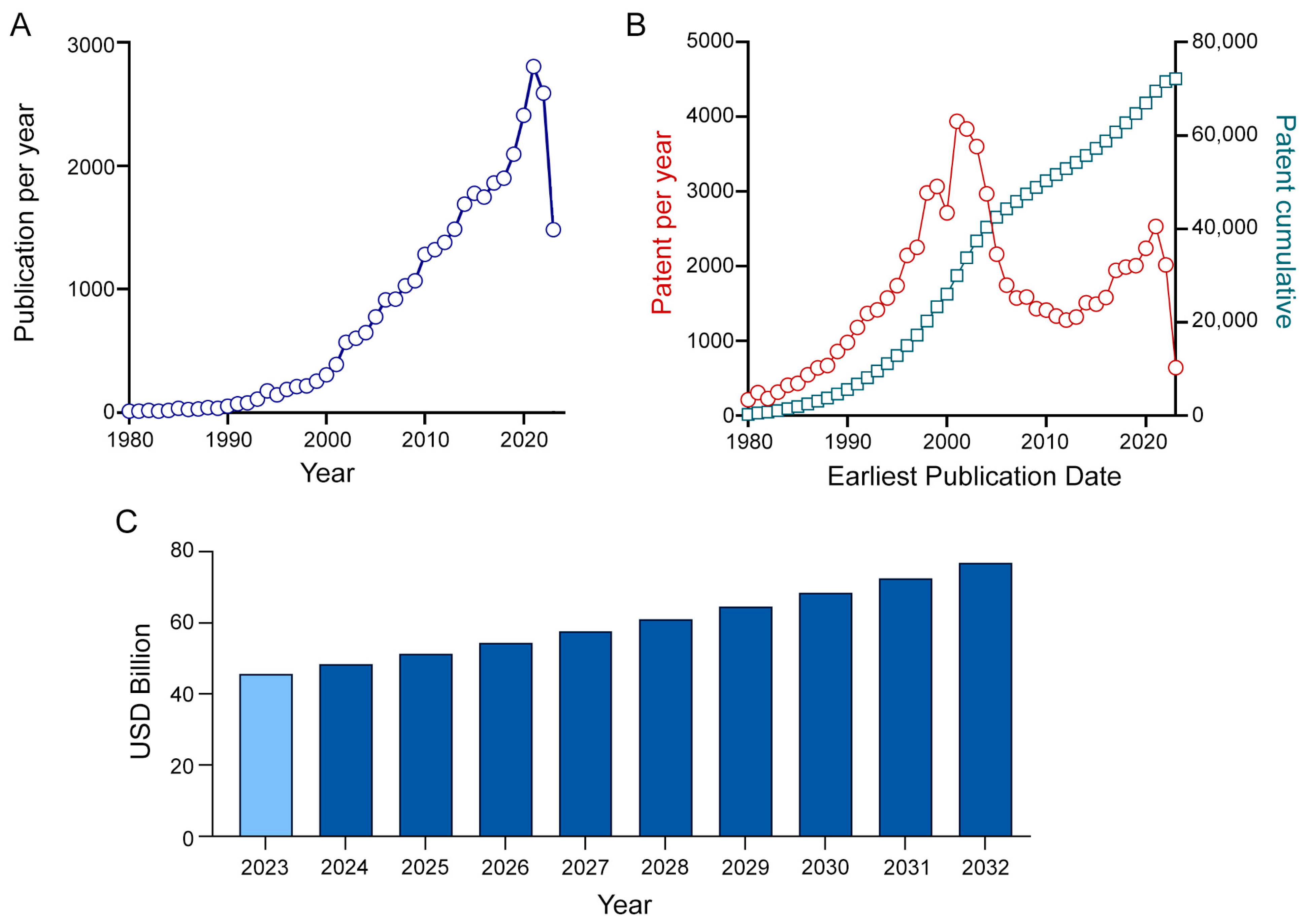
:1. Introduction
2. Peptides as Therapeutic Agents
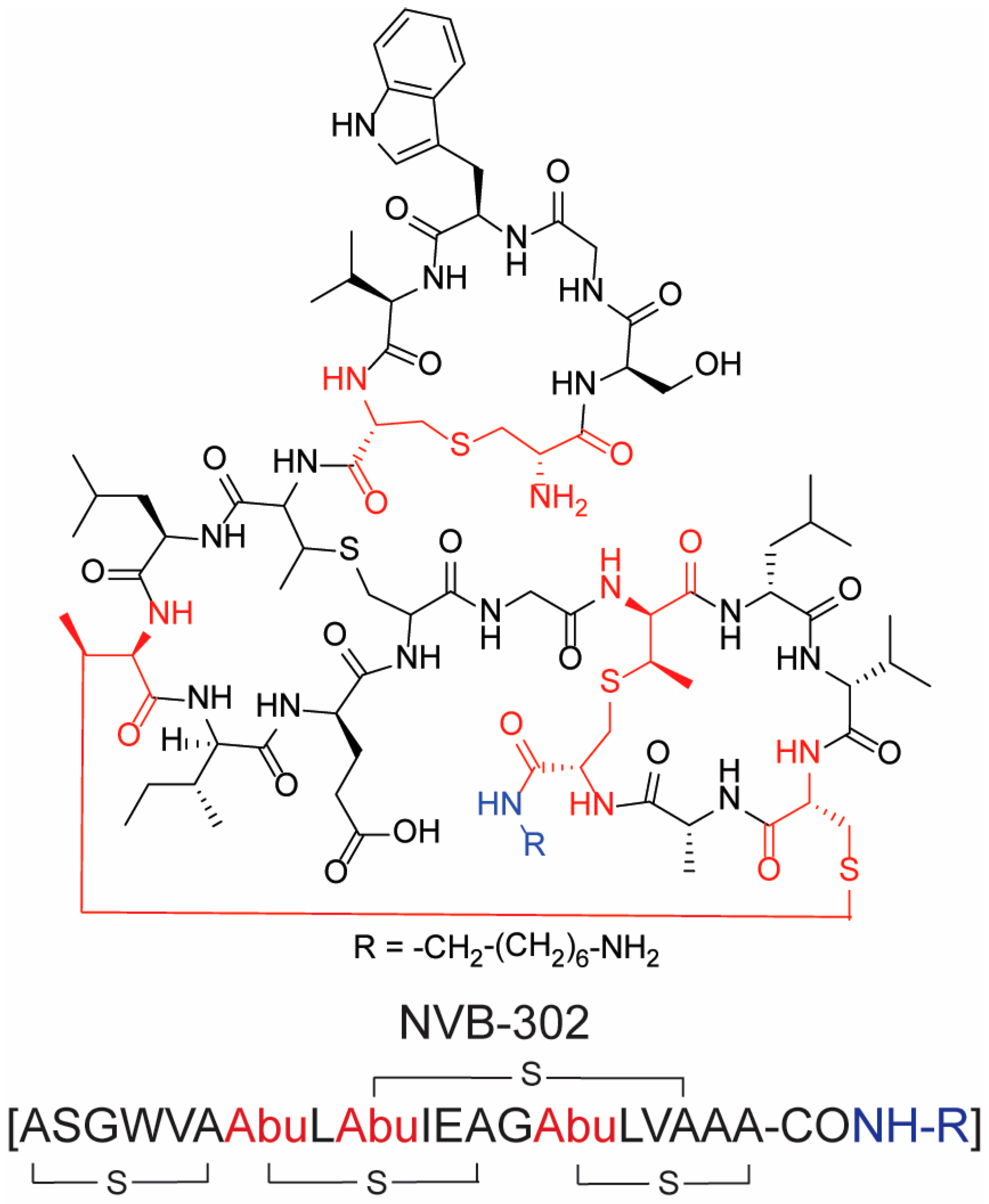
2.1. Antimicrobial Peptides
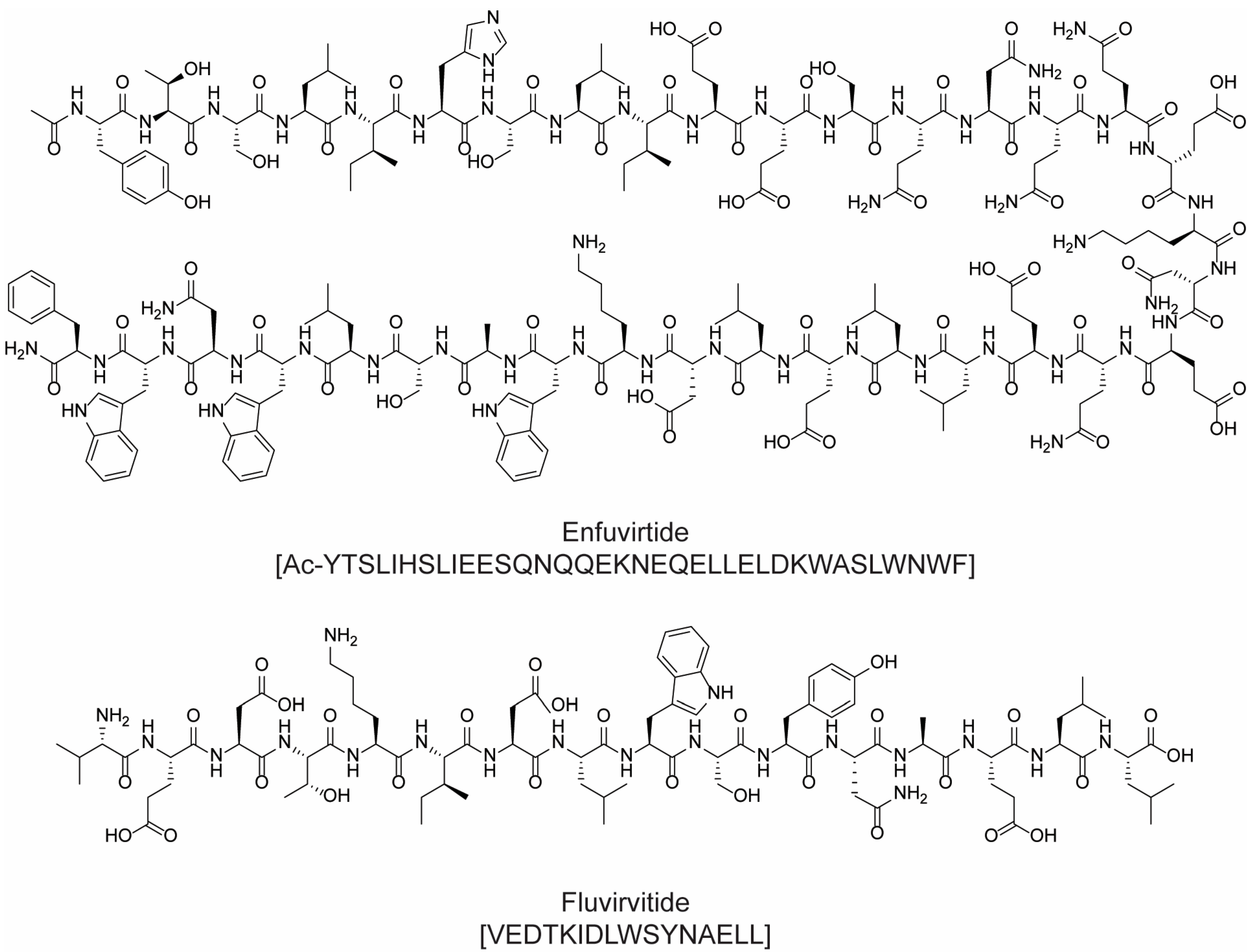
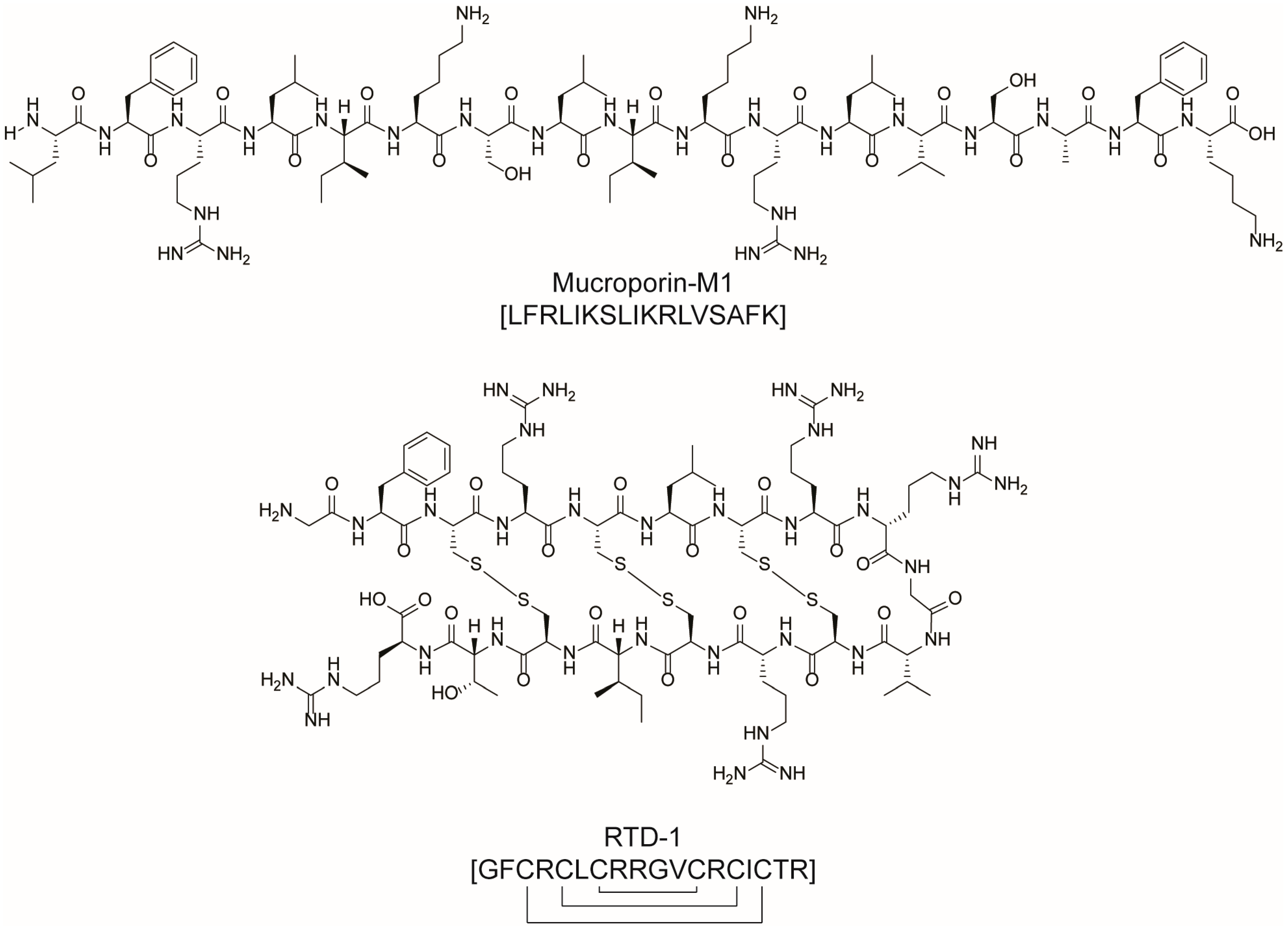
2.2. Antiviral Drugs
2.3. Anti-Neoplastic Agents
2.4. Others
3. Peptide Synthesis: Challenges and Opportunities
3.1. Solid-Phase Peptide Synthesis (SPPS)
3.1.1. Green Solvents
3.1.2. Green Coupling Agents
3.2. Microwave-Assisted Peptide Synthesis
3.3. Liquid-Phase Peptide Synthesis (LPPS)
3.4. Chemoenzymatic Peptide Synthesis (CEPS)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sims, E.K.; Carr, A.L.J.; Oram, R.A.; DiMeglio, L.A.; Evans-Molina, C. 100 Years of Insulin: Celebrating the Past, Present and Future of Diabetes Therapy. Nat. Med. 2021, 27, 1154–1164. [Google Scholar] [CrossRef]
- Erak, M.; Bellmann-Sickert, K.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide Chemistry Toolbox—Transforming Natural Peptides into Peptide Therapeutics. Bioorg. Med. Chem. 2018, 26, 2759–2765. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic Peptides: Historical Perspectives, Current Development Trends, and Future Directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Al Musaimi, O.; Al Shaer, D.; Albericio, F.; de la Torre, B.G. 2022 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2023, 16, 336. [Google Scholar] [CrossRef]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in Peptide Drug Discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic Peptides: Current Applications and Future Directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Bojarska, J.; Chai, T.-T.; Elnagdy, S.; Kaczmarek, K.; Matsoukas, J.; New, R.; Parang, K.; Lopez, O.P.; Parhiz, H.; et al. A Global Review on Short Peptides: Frontiers and Perspectives. Molecules 2021, 26, 430. [Google Scholar] [CrossRef]
- Peptide Therapeutics Market Size and Share [2023 Report]. Available online: https://www.grandviewresearch.com/industry-analysis/peptide-therapeutics-market (accessed on 3 August 2023).
- Enayathullah, M.G.; Parekh, Y.; Banu, S.; Ram, S.; Nagaraj, R.; Kumar, B.K.; Idris, M.M. Gramicidin S and Melittin: Potential Anti-Viral Therapeutic Peptides to Treat SARS-CoV-2 infection. Sci. Rep. 2022, 12, 3446. [Google Scholar] [CrossRef]
- Bobde, S.S.; Alsaab, F.M.; Wang, G.; Van Hoek, M.L. Ab Initio Designed Antimicrobial Peptides Against Gram-Negative Bacteria. Front. Microbiol. 2021, 12, 3460. [Google Scholar] [CrossRef]
- Jitendra; Sharma, P.K.; Bansal, S.; Banik, A. Noninvasive Routes of Proteins and Peptides Drug Delivery. Indian J. Pharm. Sci. 2011, 73, 367–375. [Google Scholar] [CrossRef]
- Gentilucci, L.; Tolomelli, A.; Squassabia, F. Peptides and Peptidomimetics in Medicine, Surgery and Biotechnology. Curr. Med. Chem. 2006, 13, 2449–2466. [Google Scholar] [CrossRef]
- Grauer, A.; König, B. Peptidomimetics—A Versatile Route to Biologically Active Compounds. Eur. J. Org. Chem. 2009, 2009, 5099–5111. [Google Scholar] [CrossRef]
- Rodbard, H.W.; Rodbard, D. Biosynthetic Human Insulin and Insulin Analogs. Am. J. Ther. 2020, 27, e42–e51. [Google Scholar] [CrossRef]
- Amorim-Carmo, B.; Parente, A.M.S.; Souza, E.S.; Silva-Junior, A.A.; Araújo, R.M.; Fernandes-Pedrosa, M.F. Antimicrobial Peptide Analogs from Scorpions: Modifications and Structure-Activity. Front. Mol. Biosci. 2022, 9, 887763. [Google Scholar] [CrossRef]
- Salas-Ambrosio, P.; Tronnet, A.; Verhaeghe, P.; Bonduelle, C. Synthetic Polypeptide Polymers as Simplified Analogues of Antimicrobial Peptides. Biomacromolecules 2021, 22, 57–75. [Google Scholar] [CrossRef]
- Rai, J. Peptide and Protein Mimetics by Retro and Retroinverso Analogs. Chem. Biol. Drug Des. 2019, 93, 724–736. [Google Scholar] [CrossRef]
- Li Petri, G.; Di Martino, S.; De Rosa, M. Peptidomimetics: An Overview of Recent Medicinal Chemistry Efforts toward the Discovery of Novel Small Molecule Inhibitors. J. Med. Chem. 2022, 65, 7438–7475. [Google Scholar] [CrossRef]
- Gomari, M.M.; Abkhiz, S.; Pour, T.G.; Lotfi, E.; Rostami, N.; Monfared, F.N.; Ghobari, B.; Mosavi, M.; Alipour, B.; Dokholyan, N.V. Peptidomimetics in Cancer Targeting. Mol. Med. Camb. Mass 2022, 28, 146. [Google Scholar] [CrossRef]
- Ding, D.; Xu, S.; da Silva-Júnior, E.F.; Liu, X.; Zhan, P. Medicinal Chemistry Insights into Antiviral Peptidomimetics. Drug Discov. Today 2023, 28, 103468. [Google Scholar] [CrossRef]
- Isidro-Llobet, A.; Kenworthy, M.N.; Mukherjee, S.; Kopach, M.E.; Wegner, K.; Gallou, F.; Smith, A.G.; Roschangar, F. Sustainability Challenges in Peptide Synthesis and Purification: From R&D to Production. J. Org. Chem. 2019, 84, 4615–4628. [Google Scholar] [CrossRef]
- Sheldon, R.A. The E Factor 25 Years on: The Rise of Green Chemistry and Sustainability. Green Chem. 2017, 19, 18–43. [Google Scholar] [CrossRef]
- Samreen; Ahmad, I.; Malak, H.A.; Abulreesh, H.H. Environmental Antimicrobial Resistance and its Drivers: A Potential Threat to Public Health. J. Glob. Antimicrob. Resist. 2021, 27, 101–111. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; Government of the United Kingdom: London, UK, 2016.
- Chen, C.H.; Lu, T.K. Development and Challenges of Antimicrobial Peptides for Therapeutic Applications. Antibiotics 2020, 9, 24. [Google Scholar] [CrossRef]
- Vila, J.; Moreno-Morales, J.; Ballesté-Delpierre, C. Current Landscape in the Discovery of Novel Antibacterial Agents. Clin. Microbiol. Infect. 2020, 26, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Molchanova, N.; Hansen, P.R.; Franzyk, H. Advances in Development of Antimicrobial Peptidomimetics as Potential Drugs. Molecules 2017, 22, 1430. [Google Scholar] [CrossRef] [PubMed]
- Ongey, E.L.; Neubauer, P. Lanthipeptides: Chemical Synthesis versus In Vivo Biosynthesis as Tools for Pharmaceutical Production. Microb. Cell Factories 2016, 15, 97. [Google Scholar] [CrossRef]
- Petrosillo, N.; Granata, G.; Cataldo, M.A. Novel Antimicrobials for the Treatment of Clostridium Difficile Infection. Front. Med. 2018, 5, 96. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Huang, Y.; Chen, M.; Li, G.; Chen, Y. Functional Synergy of α-Helical Antimicrobial Peptides and Traditional Antibiotics against Gram-Negative and Gram-Positive Bacteria In Vitro and In Vivo. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Protelight Pharmaceuticals Australia PTY LTD. A Phase 1 Study to Evaluate the Safety, Tolerability and Pharmacokinetics of Single and Multiple Ascending Doses of Antimicrobial Peptide PL-18 Vaginal Suppositories in Healthy Adult Subjects. 2022. Available online: ClinicalTrials.gov (accessed on 7 September 2023).
- Mandel, S.; Michaeli, J.; Nur, N.; Erbetti, I.; Zazoun, J.; Ferrari, L.; Felici, A.; Cohen-Kutner, M.; Bachnoff, N. OMN6 a Novel Bioengineered Peptide for the Treatment of Multidrug Resistant Gram Negative Bacteria. Sci. Rep. 2021, 11, 6603. [Google Scholar] [CrossRef] [PubMed]
- Harper, A.; Vijayakumar, V.; Ouwehand, A.C.; ter Haar, J.; Obis, D.; Espadaler, J.; Binda, S.; Desiraju, S.; Day, R. Viral Infections, the Microbiome, and Probiotics. Front. Cell. Infect. Microbiol. 2021, 10, 596166. [Google Scholar] [CrossRef]
- Piret, J.; Boivin, G. Pandemics Throughout History. Front. Microbiol. 2021, 11, 631736. [Google Scholar] [CrossRef] [PubMed]
- Nathanson, N. The Human Toll of Viral Diseases. In Viral Pathogenesis; Academic Press: Cambridge, MA, USA, 2016; pp. 3–16. [Google Scholar] [CrossRef]
- Vilas Boas, L.C.P.; Campos, M.L.; Berlanda, R.L.A.; de Carvalho Neves, N.; Franco, O.L. Antiviral Peptides as Promising Therapeutic Drugs. Cell. Mol. Life Sci. 2019, 76, 3525–3542. [Google Scholar] [CrossRef] [PubMed]
- Al-Azzam, S.; Ding, Y.; Liu, J.; Pandya, P.; Ting, J.P.; Afshar, S. Peptides to Combat Viral Infectious Diseases. Peptides 2020, 134, 170402. [Google Scholar] [CrossRef] [PubMed]
- Mammari, N.; Krier, Y.; Albert, Q.; Devocelle, M.; Varbanov, M. Plant-Derived Antimicrobial Peptides as Potential Antiviral Agents in Systemic Viral Infections. Pharmaceuticals 2021, 14, 774. [Google Scholar] [CrossRef]
- Ahmadi, K.; Farasat, A.; Rostamian, M.; Johari, B.; Madanchi, H. Enfuvirtide, an HIV-1 Fusion Inhibitor Peptide, Can Act as a Potent SARS-CoV-2 Fusion Inhibitor: An In Silico Drug Repurposing Study. J. Biomol. Struct. Dyn. 2022, 40, 5566–5576. [Google Scholar] [CrossRef]
- Naesens, L.; Stevaert, A.; Vanderlinden, E. Antiviral Therapies on the Horizon for Influenza. Curr. Opin. Pharmacol. 2016, 30, 106–115. [Google Scholar] [CrossRef]
- Bai, Y.; Jones, J.C.; Wong, S.-S.; Zanin, M. Antivirals Targeting the Surface Glycoproteins of Influenza Virus: Mechanisms of Action and Resistance. Viruses 2021, 13, 624. [Google Scholar] [CrossRef]
- Freitas, E.D.; Bataglioli, R.A.; Oshodi, J.; Beppu, M.M. Antimicrobial Peptides and Their Potential Application in Antiviral Coating Agents. Colloids Surf. B Biointerfaces 2022, 217, 112693. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, X.; Li, B.; Lu, C.; Yang, S.; Long, J.; Chen, H.; Huang, J.; He, B. A Database of Anti-Coronavirus Peptides. Sci. Data 2022, 9, 294. [Google Scholar] [CrossRef]
- Madhavan, M.; AlOmair, L.A.; Ks, D.; Mustafa, S. Exploring Peptide Studies Related to SARS-CoV to Accelerate the Development of Novel Therapeutic and Prophylactic Solutions Against COVID-19. J. Infect. Public Health 2021, 14, 1106–1119. [Google Scholar] [CrossRef]
- Souza, F.R.; Moura, P.G.; Costa, R.K.M.; Silva, R.S.; Pimentel, A.S. Absolute Binding Free Energies of Mucroporin and Its Analog Mucroporin-M1 with the Heptad Repeat 1 Domain and RNA-Dependent RNA Polymerase of SARS-CoV-2. J. Biomol. Struct. Dyn. 2023, 41, 6957–6968. [Google Scholar] [CrossRef]
- Mahendran, A.S.K.; Lim, Y.S.; Fang, C.-M.; Loh, H.-S.; Le, C.F. The Potential of Antiviral Peptides as COVID-19 Therapeutics. Front. Pharmacol. 2020, 11, 575444. [Google Scholar] [CrossRef]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (Previously 2019-nCoV) Infection by a Highly Potent Pan-Coronavirus Fusion Inhibitor Targeting Its Spike Protein That Harbors a High Capacity to Mediate Membrane Fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Li, C.M.; Haratipour, P.; Lingeman, R.G.; Perry, J.J.P.; Gu, L.; Hickey, R.J.; Malkas, L.H. Novel Peptide Therapeutic Approaches for Cancer Treatment. Cells 2021, 10, 2908. [Google Scholar] [CrossRef]
- Chiangjong, W.; Chutipongtanate, S.; Hongeng, S. Anticancer Peptide: Physicochemical Property, Functional Aspect and Trend in Clinical Application (Review). Int. J. Oncol. 2020, 57, 678–696. [Google Scholar] [CrossRef] [PubMed]
- Trinidad-Calderón, P.A.; Varela-Chinchilla, C.D.; García-Lara, S. Natural Peptides Inducing Cancer Cell Death: Mechanisms and Properties of Specific Candidates for Cancer Therapeutics. Molecules 2021, 26, 7453. [Google Scholar] [CrossRef]
- Negi, B.; Kumar, D.; Rawat, D.S. Marine Peptides as Anticancer Agents: A Remedy to Mankind by Nature. Curr. Protein Pept. Sci. 2017, 18, 885–904. [Google Scholar] [CrossRef]
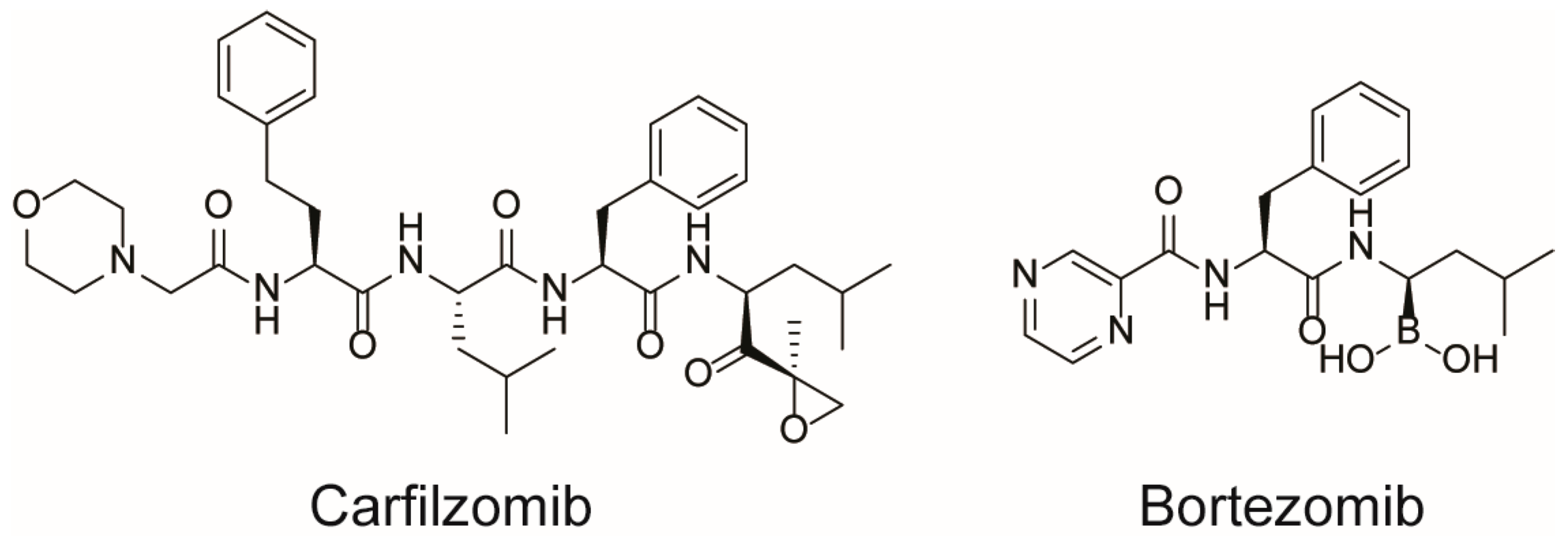
- Jayaweera, S.P.E.; Wanigasinghe Kanakanamge, S.P.; Rajalingam, D.; Silva, G.N. Carfilzomib: A Promising Proteasome Inhibitor for the Treatment of Relapsed and Refractory Multiple Myeloma. Front. Oncol. 2021, 11, 740796. [Google Scholar] [CrossRef]
- Hilchie, A.L.; Hoskin, D.W.; Power Coombs, M.R. Anticancer Activities of Natural and Synthetic Peptides. Adv. Exp. Med. Biol. 2019, 1117, 131–147. [Google Scholar] [CrossRef] [PubMed]
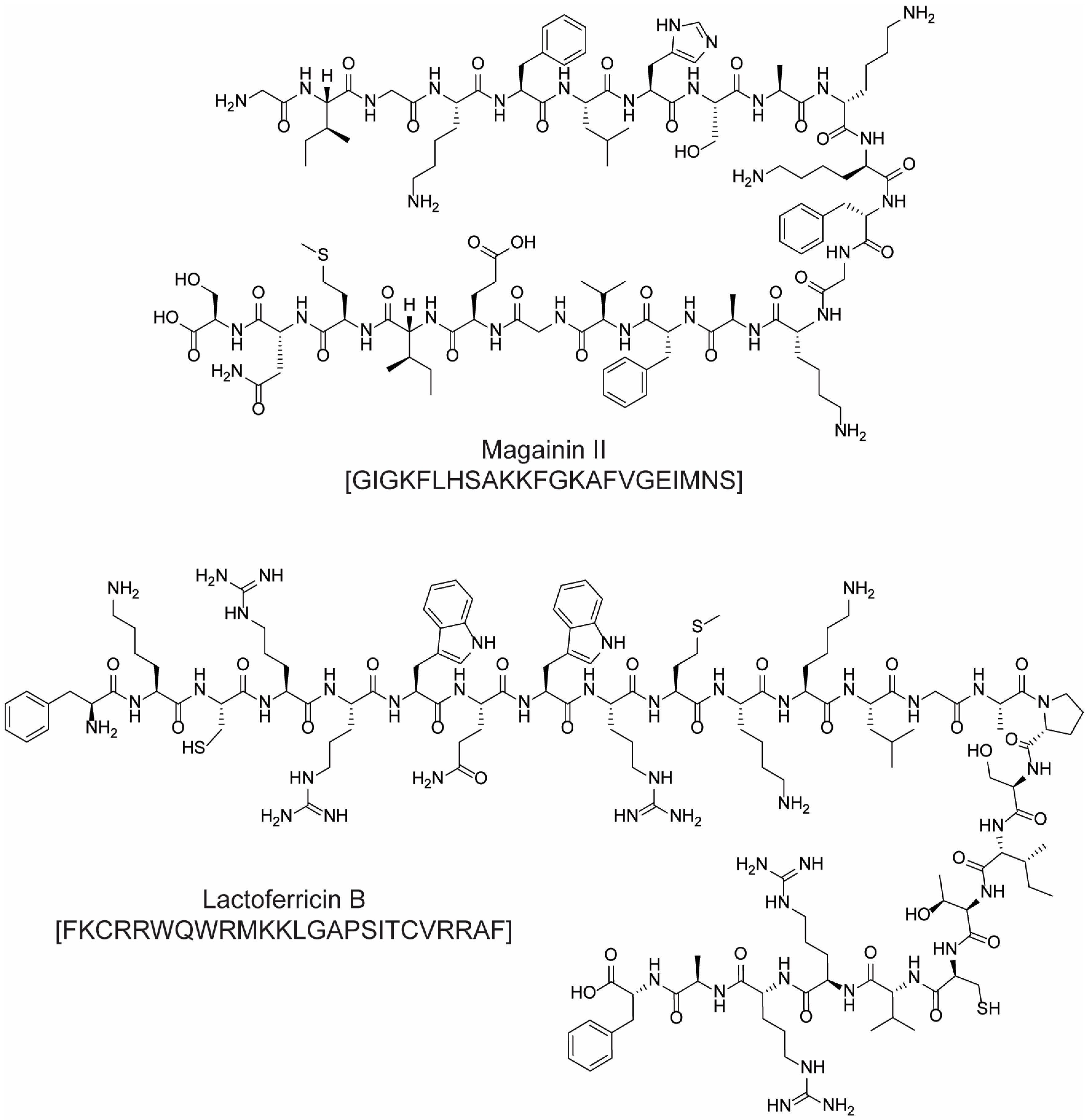
- Lehmann, J.; Retz, M.; Sidhu, S.S.; Suttmann, H.; Sell, M.; Paulsen, F.; Harder, J.; Unteregger, G.; Stöckle, M. Antitumor Activity of the Antimicrobial Peptide Magainin II against Bladder Cancer Cell Lines. Eur. Urol. 2006, 50, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Eliassen, L.T.; Berge, G.; Leknessund, A.; Wikman, M.; Lindin, I.; Løkke, C.; Ponthan, F.; Johnsen, J.I.; Sveinbjørnsson, B.; Kogner, P.; et al. The Antimicrobial Peptide, Lactoferricin B, Is Cytotoxic to Neuroblastoma Cells In Vitro and Inhibits Xenograft Growth In Vivo. Int. J. Cancer 2006, 119, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Hilchie, A.L.; Doucette, C.D.; Pinto, D.M.; Patrzykat, A.; Douglas, S.; Hoskin, D.W. Pleurocidin-Family Cationic Antimicrobial Peptides Are Cytolytic for Breast Carcinoma Cells and Prevent Growth of Tumor Xenografts. Breast Cancer Res. 2011, 13, R102. [Google Scholar] [CrossRef]
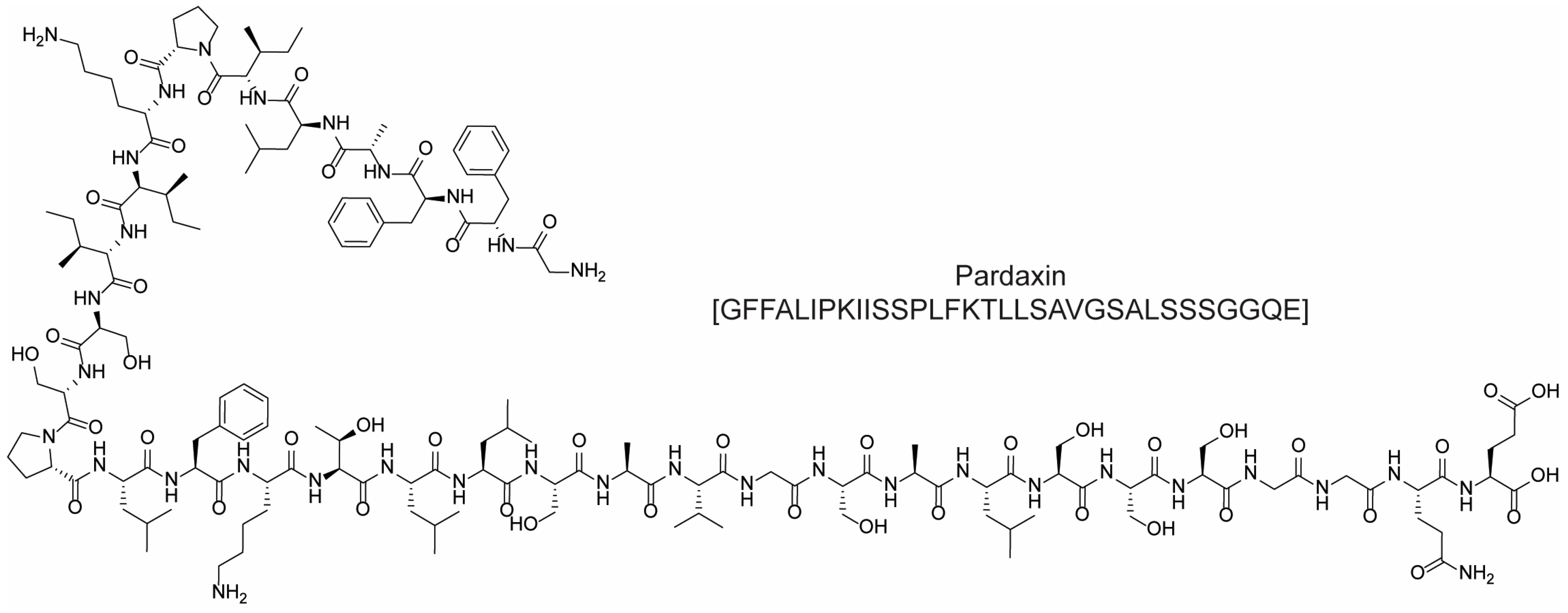
- Han, Y.; Cui, Z.; Li, Y.-H.; Hsu, W.-H.; Lee, B.-H. In Vitro and In Vivo Anticancer Activity of Pardaxin against Proliferation and Growth of Oral Squamous Cell Carcinoma. Mar. Drugs 2016, 14, 2. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Park, C.B.; Kim, J.M.; Jang, S.A.; Park, I.Y.; Kim, M.S.; Cho, J.H.; Kim, S.C. Mechanism of Anticancer Activity of Buforin IIb, a Histone H2A-Derived Peptide. Cancer Lett. 2008, 271, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Sheu, M.J.; Baldwin, W.W.; Brunson, K.W. Cytotoxicity of Rabbit Macrophage Peptides MCP-1 and MCP-2 for Mouse Tumor Cells. Antimicrob. Agents Chemother. 1985, 28, 626–629. [Google Scholar] [CrossRef]
- Pan, F.; Li, Y.; Ding, Y.; Lv, S.; You, R.; Hadianamrei, R.; Tomeh, M.A.; Zhao, X. Anticancer Effect of Rationally Designed α-Helical Amphiphilic Peptides. Colloids Surf. B Biointerfaces 2022, 220, 112841. [Google Scholar] [CrossRef]
- Kordi, M.; Borzouyi, Z.; Chitsaz, S.; hadi Asmaei, M.; Salami, R.; Tabarzad, M. Antimicrobial Peptides with Anticancer Activity: Today Status, Trends and Their Computational Design. Arch. Biochem. Biophys. 2023, 733, 109484. [Google Scholar] [CrossRef]
- Ye, N. Survey of In-Silico Prediction of Anticancer Peptides. Curr. Top. Med. Chem. 2021, 21, 1310–1318. [Google Scholar] [CrossRef]
- Shoombuatong, W.; Schaduangrat, N.; Nantasenamat, C. Unraveling the Bioactivity of Anticancer Peptides as Deduced from Machine Learning. EXCLI J. 2018, 17, 734–752. [Google Scholar] [CrossRef]
- Hadianamrei, R.; Tomeh, M.A.; Brown, S.; Wang, J.; Zhao, X. Rationally Designed Short Cationic α-Helical Peptides with Selective Anticancer Activity. J. Colloid Interface Sci. 2022, 607, 488–501. [Google Scholar] [CrossRef]
- Wan, X.; Liu, H.; Sun, Y.; Zhang, J.; Chen, X.; Chen, N. Lunasin: A Promising Polypeptide for the Prevention and Treatment of Cancer (Review). Oncol. Lett. 2017, 13, 3997–4001. [Google Scholar] [CrossRef] [PubMed]
- Rusdi, N.; Purwaningsih, E.; Hestiantoro, A.; Elya, B.; Kusmardi, K.; Kusmardi, K. In Vivo Antimammary Tumor Effects of Soybean Extract with Targeted Lunasin (ET-Lun). Pharmacogn. J. 2021, 13, 1269–1276. [Google Scholar] [CrossRef]
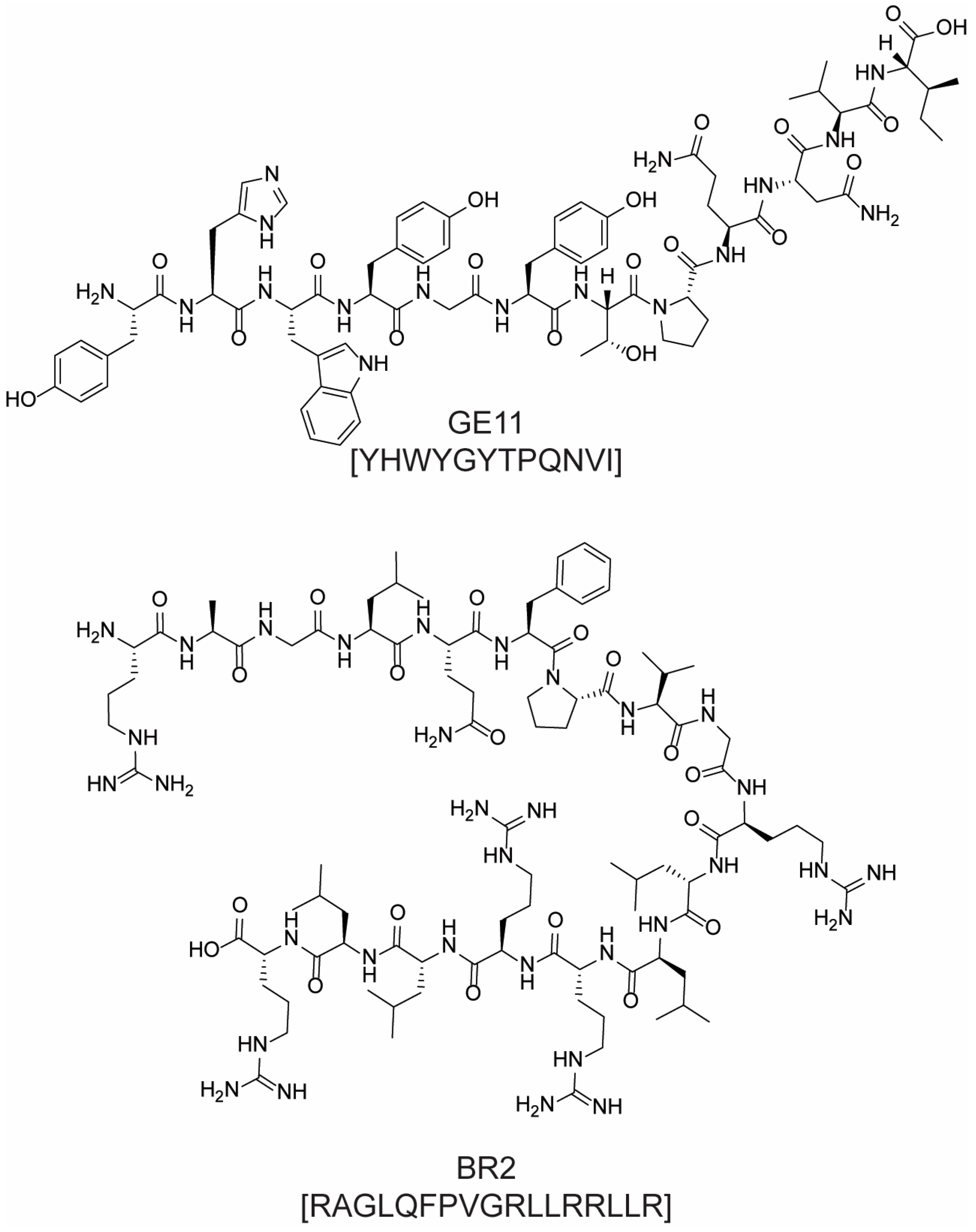
- Li, K.; Pang, L.; Pan, X.; Fan, S.; Wang, X.; Wang, Q.; Dai, P.; Gao, W.; Gao, J. GE11 Modified PLGA/TPGS Nanoparticles Targeting Delivery of Salinomycin to Breast Cancer Cells. Technol. Cancer Res. Treat. 2021, 20, 15330338211004954. [Google Scholar] [CrossRef] [PubMed]
- Genta, I.; Chiesa, E.; Colzani, B.; Modena, T.; Conti, B.; Dorati, R. GE11 Peptide as an Active Targeting Agent in Antitumor Therapy: A Minireview. Pharmaceutics 2018, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Gao, C.; Zhou, L.; Liu, M.; Xie, C.; Lu, W. EGFR-Targeted Poly(Ethylene Glycol)-Distearoylphosphatidylethanolamine Micelle Loaded with Paclitaxel for Laryngeal Cancer: Preparation, Characterization and In Vitro Evaluation. Drug Deliv. 2015, 22, 785–794. [Google Scholar] [CrossRef]
- Lim, K.J.; Sung, B.H.; Shin, J.R.; Lee, Y.W.; Kim, D.J.; Yang, K.S.; Kim, S.C. A Cancer Specific Cell-Penetrating Peptide, BR2, for the Efficient Delivery of an scFv into Cancer Cells. PLoS ONE 2013, 8, e66084. [Google Scholar] [CrossRef]
- Muggia, F.M.; Bonetti, A.; Hoeschele, J.D.; Rozencweig, M.; Howell, S.B. Platinum Antitumor Complexes: 50 Years Since Barnett Rosenberg’s Discovery. J. Clin. Oncol. 2015, 33, 4219–4226. [Google Scholar] [CrossRef]
- Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B. Mechanisms of Cisplatin Nephrotoxicity. Toxins 2010, 2, 2490–2518. [Google Scholar] [CrossRef]
- Boscutti, G.; Nardon, C.; Marchiò, L.; Crisma, M.; Biondi, B.; Dalzoppo, D.; Dalla Via, L.; Formaggio, F.; Casini, A.; Fregona, D. Anticancer Gold(III) Peptidomimetics: From Synthesis to In Vitro and Ex Vivo Biological Evaluations. ChemMedChem 2018, 13, 1131–1145. [Google Scholar] [CrossRef] [PubMed]
- Mauriello, A.; Cavalluzzo, B.; Manolio, C.; Ragone, C.; Luciano, A.; Barbieri, A.; Tornesello, M.L.; Buonaguro, F.M.; Tagliamonte, M.; Buonaguro, L. Long-Term Memory T Cells as Preventive Anticancer Immunity Elicited by TuA-Derived Heteroclitic Peptides. J. Transl. Med. 2021, 19, 526. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in Cancer: Biological Implications and Therapeutic Opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The First Anti-Angiogenic Small Molecule Drug Candidate Design, Synthesis and Clinical Evaluation. Anticancer Agents Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.-K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide Combined with Standard Treatment for Patients with Newly Diagnosed Glioblastoma with Methylated MGMT Promoter (CENTRIC EORTC 26071-22072 Study): A Multicentre, Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Alipour, M.; Baneshi, M.; Hosseinkhani, S.; Mahmoudi, R.; Jabari Arabzadeh, A.; Akrami, M.; Mehrzad, J.; Bardania, H. Recent Progress in Biomedical Applications of RGD-Based Ligand: From Precise Cancer Theranostics to Biomaterial Engineering: A Systematic Review. J. Biomed. Mater. Res. A 2020, 108, 839–850. [Google Scholar] [CrossRef]
- Arosio, D.; Casagrande, C. Advancement in Integrin Facilitated Drug Delivery. Adv. Drug Deliv. Rev. 2016, 97, 111–143. [Google Scholar] [CrossRef]
- Battistini, L.; Bugatti, K.; Sartori, A.; Curti, C.; Zanardi, F. RGD Peptide-Drug Conjugates as Effective Dual Targeting Platforms: Recent Advances. Eur. J. Org. Chem. 2021, 2021, 2506–2528. [Google Scholar] [CrossRef]
- Serra, M.; Tambini, S.M.; Di Giacomo, M.; Peviani, E.G.; Belvisi, L.; Colombo, L. Synthesis of Easy-to-Functionalize Azabicycloalkane Scaffolds as Dipeptide Turn Mimics En Route to cRGD-Based Bioconjugates. Eur. J. Org. Chem. 2015, 2015, 7557–7570. [Google Scholar] [CrossRef]
- Serra, M.; Bernardi, E.; Lorenzi, E.D.; Colombo, L. Synthesis of Functionalized 6,5- and 7,5-Azabicycloalkane Amino Acids by Metathesis Reactions. J. Org. Chem. 2019, 84, 15726–15734. [Google Scholar] [CrossRef] [PubMed]
- Pilkington-Miksa, M.; Arosio, D.; Battistini, L.; Belvisi, L.; De Matteo, M.; Vasile, F.; Burreddu, P.; Carta, P.; Rassu, G.; Perego, P.; et al. Design, Synthesis, and Biological Evaluation of Novel cRGD–Paclitaxel Conjugates for Integrin-Assisted Drug Delivery. Bioconjug. Chem. 2012, 23, 1610–1622. [Google Scholar] [CrossRef]
- Lanzardo, S.; Conti, L.; Brioschi, C.; Bartolomeo, M.P.; Arosio, D.; Belvisi, L.; Manzoni, L.; Maiocchi, A.; Maisano, F.; Forni, G. A New Optical Imaging Probe Targeting αVβ3 Integrin In Glioblastoma Xenografts. Contrast Media Mol. Imaging 2011, 6, 449–458. [Google Scholar] [CrossRef]
- Manzoni, L.; Belvisi, L.; Arosio, D.; Bartolomeo, M.P.; Bianchi, A.; Brioschi, C.; Buonsanti, F.; Cabella, C.; Casagrande, C.; Civera, M.; et al. Synthesis of Gd and 68Ga Complexes in Conjugation with a Conformationally Optimized RGD Sequence as Potential MRI and PET Tumor-Imaging Probes. ChemMedChem 2012, 7, 1084–1093. [Google Scholar] [CrossRef]
- Battistini, L.; Burreddu, P.; Sartori, A.; Arosio, D.; Manzoni, L.; Paduano, L.; D’Errico, G.; Sala, R.; Reia, L.; Bonomini, S.; et al. Enhancement of the Uptake and Cytotoxic Activity of Doxorubicin in Cancer Cells by Novel cRGD-Semipeptide-Anchoring Liposomes. Mol. Pharm. 2014, 11, 2280–2293. [Google Scholar] [CrossRef] [PubMed]
- Pirota, V.; Bisbano, G.; Serra, M.; Torre, M.L.; Doria, F.; Bari, E.; Paolillo, M. cRGD-Functionalized Silk Fibroin Nanoparticles: A Strategy for Cancer Treatment with a Potent Unselective Naphthalene Diimide Derivative. Cancers 2023, 15, 1725. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.S. Peptides and Peptidomimetics as Potential Antiobesity Agents: Overview of Current Status. Front. Nutr. 2019, 6, 11. [Google Scholar] [CrossRef]
- Marthandam Asokan, S.; Hung, T.-H.; Chiang, W.-D.; Lin, W.-T. Lipolysis-Stimulating Peptide from Soybean Protects Against High Fat Diet-Induced Apoptosis in Skeletal Muscles. J. Med. Food 2018, 21, 225–232. [Google Scholar] [CrossRef]
- Kolonin, M.G.; Saha, P.K.; Chan, L.; Pasqualini, R.; Arap, W. Reversal of Obesity by Targeted Ablation of Adipose Tissue. Nat. Med. 2004, 10, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Barnhart, K.F.; Christianson, D.R.; Hanley, P.W.; Driessen, W.H.P.; Bernacky, B.J.; Baze, W.B.; Wen, S.; Tian, M.; Ma, J.; Kolonin, M.G.; et al. A Peptidomimetic Targeting White Fat Causes Weight Loss and Improved Insulin Resistance in Obese Monkeys. Sci. Transl. Med. 2011, 3, 108ra112. [Google Scholar] [CrossRef] [PubMed]
- Staquicini, F.I.; Cardó-Vila, M.; Kolonin, M.G.; Trepel, M.; Edwards, J.K.; Nunes, D.N.; Sergeeva, A.; Efstathiou, E.; Sun, J.; Almeida, N.F.; et al. Vascular Ligand-Receptor Mapping by Direct Combinatorial Selection in Cancer Patients. Proc. Natl. Acad. Sci. USA 2011, 108, 18637–18642. [Google Scholar] [CrossRef]
- M.D. Anderson Cancer Center. A First-in-Man, Phase I Evaluation of A Single Cycle of Prohibitin Targeting Peptide 1 in Patients with Metastatic Prostate Cancer and Obesity. 2019. Available online: ClinicalTrials.gov (accessed on 7 September 2023).
- Cardiovascular Diseases. Available online: https://www.who.int/health-topics/cardiovascular-diseases (accessed on 8 August 2023).
- Roth, G.A.; Forouzanfar, M.H.; Moran, A.E.; Barber, R.; Nguyen, G.; Feigin, V.L.; Naghavi, M.; Mensah, G.A.; Murray, C.J.L. Demographic and Epidemiologic Drivers of Global Cardiovascular Mortality. N. Engl. J. Med. 2015, 372, 1333–1341. [Google Scholar] [CrossRef]
- Fisher, E.A.; Feig, J.E.; Hewing, B.; Hazen, S.L.; Smith, J.D. High-Density Lipoprotein Function, Dysfunction, and Reverse Cholesterol Transport. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2813–2820. [Google Scholar] [CrossRef]
- Davidson, W.S.; Hazlett, T.; Mantulin, W.W.; Jonas, A. The Role of Apolipoprotein AI Domains in Lipid Binding. Proc. Natl. Acad. Sci. USA 1996, 93, 13605–13610. [Google Scholar] [CrossRef] [PubMed]
- Stoekenbroek, R.M.; Stroes, E.S.; Hovingh, G.K. ApoA-I Mimetics. Handb. Exp. Pharmacol. 2015, 224, 631–648. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, E.J.; Santos, R.D.; Asztalos, B.F. Marked HDL Deficiency and Premature Coronary Heart Disease. Curr. Opin. Lipidol. 2010, 21, 289–297. [Google Scholar] [CrossRef]
- Orsó, E.; Broccardo, C.; Kaminski, W.E.; Böttcher, A.; Liebisch, G.; Drobnik, W.; Götz, A.; Chambenoit, O.; Diederich, W.; Langmann, T.; et al. Transport of Lipids from Golgi to Plasma Membrane Is Defective in Tangier Disease Patients and Abc1-Deficient mice. Nat. Genet. 2000, 24, 192–196. [Google Scholar] [CrossRef]
- Vaisman, B.L.; Lambert, G.; Amar, M.; Joyce, C.; Ito, T.; Shamburek, R.D.; Cain, W.J.; Fruchart-Najib, J.; Neufeld, E.D.; Remaley, A.T.; et al. ABCA1 Overexpression Leads to Hyperalphalipoproteinemia and Increased Biliary Cholesterol Excretion in Transgenic Mice. J. Clin. Investig. 2001, 108, 303–309. [Google Scholar] [CrossRef]
- Uehara, Y.; Chiesa, G.; Saku, K. High-Density Lipoprotein-Targeted Therapy and Apolipoprotein A-I Mimetic Peptides. Circ. J. 2015, 79, 2523–2528. [Google Scholar] [CrossRef] [PubMed]
- Takata, K.; Imaizumi, S.; Kawachi, E.; Yahiro, E.; Suematsu, Y.; Shimizu, T.; Abe, S.; Matsuo, Y.; Nakajima, K.; Yasuno, T.; et al. The ApoA-I Mimetic Peptide FAMP Promotes Recovery from Hindlimb Ischemia through a Nitric Oxide (NO)-Related Pathway. Int. J. Cardiol. 2016, 207, 317–325. [Google Scholar] [CrossRef]
- Suematsu, Y.; Kawachi, E.; Idemoto, Y.; Matsuo, Y.; Kuwano, T.; Kitajima, K.; Imaizumi, S.; Kawamura, A.; Saku, K.; Uehara, Y.; et al. Anti-Atherosclerotic Effects of an Improved Apolipoprotein A-I Mimetic Peptide. Int. J. Cardiol. 2019, 297, 111–117. [Google Scholar] [CrossRef]
- Ferrazzano, L.; Catani, M.; Cavazzini, A.; Martelli, G.; Corbisiero, D.; Cantelmi, P.; Fantoni, T.; Mattellone, A.; Luca, C.D.; Felletti, S.; et al. Sustainability in Peptide Chemistry: Current Synthesis and Purification Technologies and Future Challenges. Green Chem. 2022, 24, 975–1020. [Google Scholar] [CrossRef]
- Pawlas, J.; Rasmussen, J.H. ReGreen SPPS: Enabling Circular Chemistry in Environmentally Sensible Solid-Phase Peptide Synthesis. Green Chem. 2019, 21, 5990–5998. [Google Scholar] [CrossRef]
- Lawrenson, S.B.; Arav, R.; North, M. The Greening of Peptide Synthesis. Green Chem. 2017, 19, 1685–1691. [Google Scholar] [CrossRef]
- Bryan, M.C.; Dunn, P.J.; Entwistle, D.; Gallou, F.; Koenig, S.G.; Hayler, J.D.; Hickey, M.R.; Hughes, S.; Kopach, M.E.; Moine, G.; et al. Key Green Chemistry Research Areas from a Pharmaceutical Manufacturers’ Perspective Revisited. Green Chem. 2018, 20, 5082–5103. [Google Scholar] [CrossRef]
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B.H. Synthesis of Proteins by Native Chemical Ligation. Science 1994, 266, 776–779. [Google Scholar] [CrossRef]
- Tam, J.P.; Yu, Q. Methionine Ligation Strategy in the Biomimetic Synthesis of Parathyroid Hormones. Biopolymers 1998, 46, 319–327. [Google Scholar] [CrossRef]
- Yin, H.; Lu, D.; Wang, S.; Wang, P. Development of Powerful Auxiliary-Mediated Ligation To Facilitate Rapid Protein Assembly. Org. Lett. 2019, 21, 5138–5142. [Google Scholar] [CrossRef] [PubMed]
- Raibaut, L.; El Mahdi, O.; Melnyk, O. Solid Phase Protein Chemical Synthesis. In Protein Ligation and Total Synthesis II; Liu, L., Ed.; Topics in Current Chemistry; Springer International Publishing: Cham, Switzerland, 2015; pp. 103–154. ISBN 978-3-319-19189-8. [Google Scholar]
- Agouridas, V.; Diemer, V.; Melnyk, O. Strategies and Open Questions in Solid-Phase Protein Chemical Synthesis. Curr. Opin. Chem. Biol. 2020, 58, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shelton, P.T.; Jensen, K.J. Linkers, Resins, and General Procedures for Solid-Phase Peptide Synthesis. In Peptide Synthesis and Applications; Jensen, K.J., Tofteng Shelton, P., Pedersen, S.L., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; pp. 23–41. ISBN 978-1-62703-544-6. [Google Scholar]
- Regulation (EC) No 1907/2006 of the European Parliament and of the Council of 18 December 2006 Concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), Establishing a European Chemicals Agency, Amending Directive 1999/45/EC and Repealing Council Regulation (EEC) No 793/93 and Commission Regulation (EC) No 1488/94 as Well as Council Directive 76/769/EEC and Commission Directives 91/155/EEC, 93/67/EEC, 93/105/EC and 2000/21/EC (Text with EEA Relevance). 2014.
- Acosta, G.A.; del Fresno, M.; Paradis-Bas, M.; Rigau-DeLlobet, M.; Côté, S.; Royo, M.; Albericio, F. Solid-Phase Peptide Synthesis Using Acetonitrile as a Solvent in Combination with PEG-Based Resins. J. Pept. Sci. 2009, 15, 629–633. [Google Scholar] [CrossRef]
- Pace, V.; Hoyos, P.; Castoldi, L.; Domínguez de María, P.; Alcántara, A.R. 2-Methyltetrahydrofuran (2-MeTHF): A Biomass-Derived Solvent with Broad Application in Organic Chemistry. ChemSusChem 2012, 5, 1369–1379. [Google Scholar] [CrossRef]
- Jad, Y.E.; Acosta, G.A.; Khattab, S.N.; de la Torre, B.G.; Govender, T.; Kruger, H.G.; El-Faham, A.; Albericio, F. 2-Methyltetrahydrofuran and Cyclopentyl Methyl Ether for Green Solid-Phase Peptide Synthesis. Amino Acids 2016, 48, 419–426. [Google Scholar] [CrossRef]
- Jad, Y.E.; Acosta, G.A.; Khattab, S.N.; de la Torre, B.G.; Govender, T.; Kruger, H.G.; El-Faham, A.; Albericio, F. Peptide Synthesis beyond DMF: THF and ACN as Excellent and Friendlier Alternatives. Org. Biomol. Chem. 2015, 13, 2393–2398. [Google Scholar] [CrossRef]
- Jad, Y.E.; Govender, T.; Kruger, H.G.; El-Faham, A.; de la Torre, B.G.; Albericio, F. Green Solid-Phase Peptide Synthesis (GSPPS) 3. Green Solvents for Fmoc Removal in Peptide Chemistry. Org. Process Res. Dev. 2017, 21, 365–369. [Google Scholar] [CrossRef]
- Lopez, J.; Pletscher, S.; Aemissegger, A.; Bucher, C.; Gallou, F. N-Butylpyrrolidinone as Alternative Solvent for Solid-Phase Peptide Synthesis. Org. Process Res. Dev. 2018, 22, 494–503. [Google Scholar] [CrossRef]
- Martelli, G.; Cantelmi, P.; Tolomelli, A.; Corbisiero, D.; Mattellone, A.; Ricci, A.; Fantoni, T.; Cabri, W.; Vacondio, F.; Ferlenghi, F.; et al. Steps towards Sustainable Solid Phase Peptide Synthesis: Use and Recovery of N-Octyl Pyrrolidone. Green Chem. 2021, 23, 4095–4106. [Google Scholar] [CrossRef]
- Hojo, K.; Hara, A.; Kitai, H.; Onishi, M.; Ichikawa, H.; Fukumori, Y.; Kawasaki, K. Development of a Method for Environmentally Friendly Chemical Peptide Synthesis in Water Using Water-Dispersible Amino Acid Nanoparticles. Chem. Cent. J. 2011, 5, 49. [Google Scholar] [CrossRef] [PubMed]
- Hojo, K.; Shinozaki, N.; Hidaka, K.; Tsuda, Y.; Fukumori, Y.; Ichikawa, H.; Wade, J.D. Aqueous Microwave-Assisted Solid-Phase Peptide Synthesis Using Fmoc Strategy. III: Racemization Studies and Water-Based Synthesis of Histidine-Containing Peptides. Amino Acids 2014, 46, 2347–2354. [Google Scholar] [CrossRef]
- Hojo, K.; Fujiwara, S.; Inai, H.; Manabe, Y.; Tsuda, Y. Environmentally Conscious In-Water Peptide Synthesis Using Boc Strategy. Int. J. Pept. Res. Ther. 2022, 28, 51. [Google Scholar] [CrossRef]
- Gabriel, C.M.; Keener, M.; Gallou, F.; Lipshutz, B.H. Amide and Peptide Bond Formation in Water at Room Temperature. Org. Lett. 2015, 17, 3968–3971. [Google Scholar] [CrossRef] [PubMed]
- Lipshutz, B.H.; Ghorai, S.; Cortes-Clerget, M. The Hydrophobic Effect Applied to Organic Synthesis: Recent Synthetic Chemistry “in Water”. Chem.—Eur. J. 2018, 24, 6672–6695. [Google Scholar] [CrossRef]
- Sharma, S.; Buchbinder, N.W.; Braje, W.M.; Handa, S. Fast Amide Couplings in Water: Extraction, Column Chromatography, and Crystallization Not Required. Org. Lett. 2020, 22, 5737–5740. [Google Scholar] [CrossRef]
- Ferrazzano, L.; Corbisiero, D.; Martelli, G.; Tolomelli, A.; Viola, A.; Ricci, A.; Cabri, W. Green Solvent Mixtures for Solid-Phase Peptide Synthesis: A Dimethylformamide-Free Highly Efficient Synthesis of Pharmaceutical-Grade Peptides. ACS Sustain. Chem. Eng. 2019, 7, 12867–12877. [Google Scholar] [CrossRef]
- Jadhav, S.; Martin, V.; Egelund, P.H.G.; Castro, H.J.; Krüger, T.; Richner, F.; Quement, S.T.L.; Albericio, F.; Dettner, F.; Lechner, C.; et al. Replacing DMF in Solid-Phase Peptide Synthesis: Varying the Composition of Green Binary Solvent Mixtures as a Tool to Mitigate Common Side-Reactions. Green Chem. 2021, 23, 3312–3321. [Google Scholar] [CrossRef]
- El-Faham, A.; Albericio, F. COMU: A Third Generation of Uronium-Type Coupling Reagents. J. Pept. Sci. 2010, 16, 6–9. [Google Scholar] [CrossRef]
- Subirós-Funosas, R.; Prohens, R.; Barbas, R.; El-Faham, A.; Albericio, F. Oxyma: An Efficient Additive for Peptide Synthesis to Replace the Benzotriazole-Based HOBt and HOAt with a Lower Risk of Explosion. Chem. Weinh. Bergstr. Ger. 2009, 15, 9394–9403. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, M.T.; Boulton, L.T.; Sneddon, H.F.; Sheppard, T.D. A Green Chemistry Perspective on Catalytic Amide Bond Formation. Nat. Catal. 2019, 2, 10–17. [Google Scholar] [CrossRef]
- Todorovic, M.; Perrin, D.M. Recent Developments in Catalytic Amide Bond Formation. Pept. Sci. 2020, 112, e24210. [Google Scholar] [CrossRef]
- Tosi, E.; de Figueiredo, R.M.; Campagne, J.-M. Enantioselective Catalytic C-H Amidations: An Highlight. Catalysts 2021, 11, 471. [Google Scholar] [CrossRef]
- Nagahara, S.; Okada, Y.; Kitano, Y.; Chiba, K. Biphasic Electrochemical Peptide Synthesis. Chem. Sci. 2021, 12, 12911–12917. [Google Scholar] [CrossRef]
- Kapuśniak, Ł.; Plessow, P.N.; Trzybiński, D.; Woźniak, K.; Hofmann, P.; Jolly, P.I. A Mild One-Pot Reduction of Phosphine(V) Oxides Affording Phosphines(III) and Their Metal Catalysts. Organometallics 2021, 40, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Manabe, S.; Wong, C.M.; Sevov, C.S. Direct and Scalable Electroreduction of Triphenylphosphine Oxide to Triphenylphosphine. J. Am. Chem. Soc. 2020, 142, 3024–3031. [Google Scholar] [CrossRef] [PubMed]
- Elias, J.S.; Costentin, C.; Nocera, D.G. Direct Electrochemical P(V) to P(III) Reduction of Phosphine Oxide Facilitated by Triaryl Borates. J. Am. Chem. Soc. 2018, 140, 13711–13718. [Google Scholar] [CrossRef]
- Collina, S.; Volpe, S.D. 18. The Use of Microwaves in Drug Discovery; De Gruyter: Berlin, Germany, 2017; pp. 334–357. ISBN 978-3-11-047993-5. [Google Scholar]
- Sharma, N.; Sharma, U.K.; Van der Eycken, E.V. Microwave-Assisted Organic Synthesis: Overview of Recent Applications. In Green Techniques for Organic Synthesis and Medicinal Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2018; pp. 441–468. ISBN 978-1-119-28815-2. [Google Scholar]
- Singh, S.K.; Collins, J.M. New Developments in Microwave-Assisted Solid Phase Peptide Synthesis. Methods Mol. Biol. 2020, 2103, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.M.; Porter, K.A.; Singh, S.K.; Vanier, G.S. High-Efficiency Solid Phase Peptide Synthesis (HE-SPPS). Org. Lett. 2014, 16, 940–943. [Google Scholar] [CrossRef] [PubMed]
- Varela, Y.F.; Vanegas Murcia, M.; Patarroyo, M.E. Synthetic Evaluation of Standard and Microwave-Assisted Solid Phase Peptide Synthesis of a Long Chimeric Peptide Derived from Four Plasmodium Falciparum Proteins. Molecules 2018, 23, 2877. [Google Scholar] [CrossRef]
- Galanis, A.S.; Albericio, F.; Grøtli, M. Solid-Phase Peptide Synthesis in Water Using Microwave-Assisted Heating. Org. Lett. 2009, 11, 4488–4491. [Google Scholar] [CrossRef]
- Mahindra, A.; Nooney, K.; Uraon, S.; Sharma, K.K.; Jain, R. Microwave-Assisted Solution Phase Peptide Synthesis in Neat Water. RSC Adv. 2013, 3, 16810–16816. [Google Scholar] [CrossRef]
- Přibylka, A.; Pastorek, M.; Grepl, M.; Schütznerová, E.P. The Application of Anisole in Greener Solid-Phase Peptide Synthesis Protocols—Compatibility with Green Bases in Fmoc Removal and New Green Binary Mixture for Coupling. Tetrahedron 2021, 99, 132452. [Google Scholar] [CrossRef]
- Sharma, A.; Kumar, A.; de la Torre, B.G.; Albericio, F. Liquid-Phase Peptide Synthesis (LPPS): A Third Wave for the Preparation of Peptides. Chem. Rev. 2022, 122, 13516–13546. [Google Scholar] [CrossRef]
- Martin, V.; Egelund, P.H.G.; Johansson, H.; Quement, S.T.L.; Wojcik, F.; Pedersen, D.S. Greening the Synthesis of Peptide Therapeutics: An Industrial Perspective. RSC Adv. 2020, 10, 42457–42492. [Google Scholar] [CrossRef]
- Takahashi, D.; Inomata, T.; Fukui, T. AJIPHASE®: A Highly Efficient Synthetic Method for One-Pot Peptide Elongation in the Solution Phase by an Fmoc Strategy. Angew. Chem. 2017, 129, 7911–7915. [Google Scholar] [CrossRef]
- Okada, Y.; Suzuki, H.; Nakae, T.; Fujita, S.; Abe, H.; Nagano, K.; Yamada, T.; Ebata, N.; Kim, S.; Chiba, K. Tag-Assisted Liquid-Phase Peptide Synthesis Using Hydrophobic Benzyl Alcohols as Supports. J. Org. Chem. 2013, 78, 320–327. [Google Scholar] [CrossRef]
- Okada, Y.; Takasawa, R.; Kubo, D.; Iwanaga, N.; Fujita, S.; Suzuki, K.; Suzuki, H.; Kamiya, H.; Chiba, K. Improved Tag-Assisted Liquid-Phase Peptide Synthesis: Application to the Synthesis of the Bradykinin Receptor Antagonist Icatibant Acetate. Org. Process Res. Dev. 2019, 23, 2576–2581. [Google Scholar] [CrossRef]
- Yeo, J.; Peeva, L.; Chung, S.; Gaffney, P.; Kim, D.; Luciani, C.; Tsukanov, S.; Seibert, K.; Kopach, M.; Albericio, F.; et al. Liquid Phase Peptide Synthesis via One-Pot Nanostar Sieving (PEPSTAR). Angew. Chem. Int. Ed. 2021, 60, 7786–7795. [Google Scholar] [CrossRef]
- Fryszkowska, A.; Devine, P.N. Biocatalysis in Drug Discovery and Development. Curr. Opin. Chem. Biol. 2020, 55, 151–160. [Google Scholar] [CrossRef]
- Kinner, A.; Nerke, P.; Siedentop, R.; Steinmetz, T.; Classen, T.; Rosenthal, K.; Nett, M.; Pietruszka, J.; Lütz, S. Recent Advances in Biocatalysis for Drug Synthesis. Biomedicines 2022, 10, 964. [Google Scholar] [CrossRef] [PubMed]
- Rossino, G.; Robescu, M.S.; Licastro, E.; Tedesco, C.; Martello, I.; Maffei, L.; Vincenti, G.; Bavaro, T.; Collina, S. Biocatalysis: A Smart and Green Tool for the Preparation of Chiral Drugs. Chirality 2022, 34, 1403–1418. [Google Scholar] [CrossRef] [PubMed]
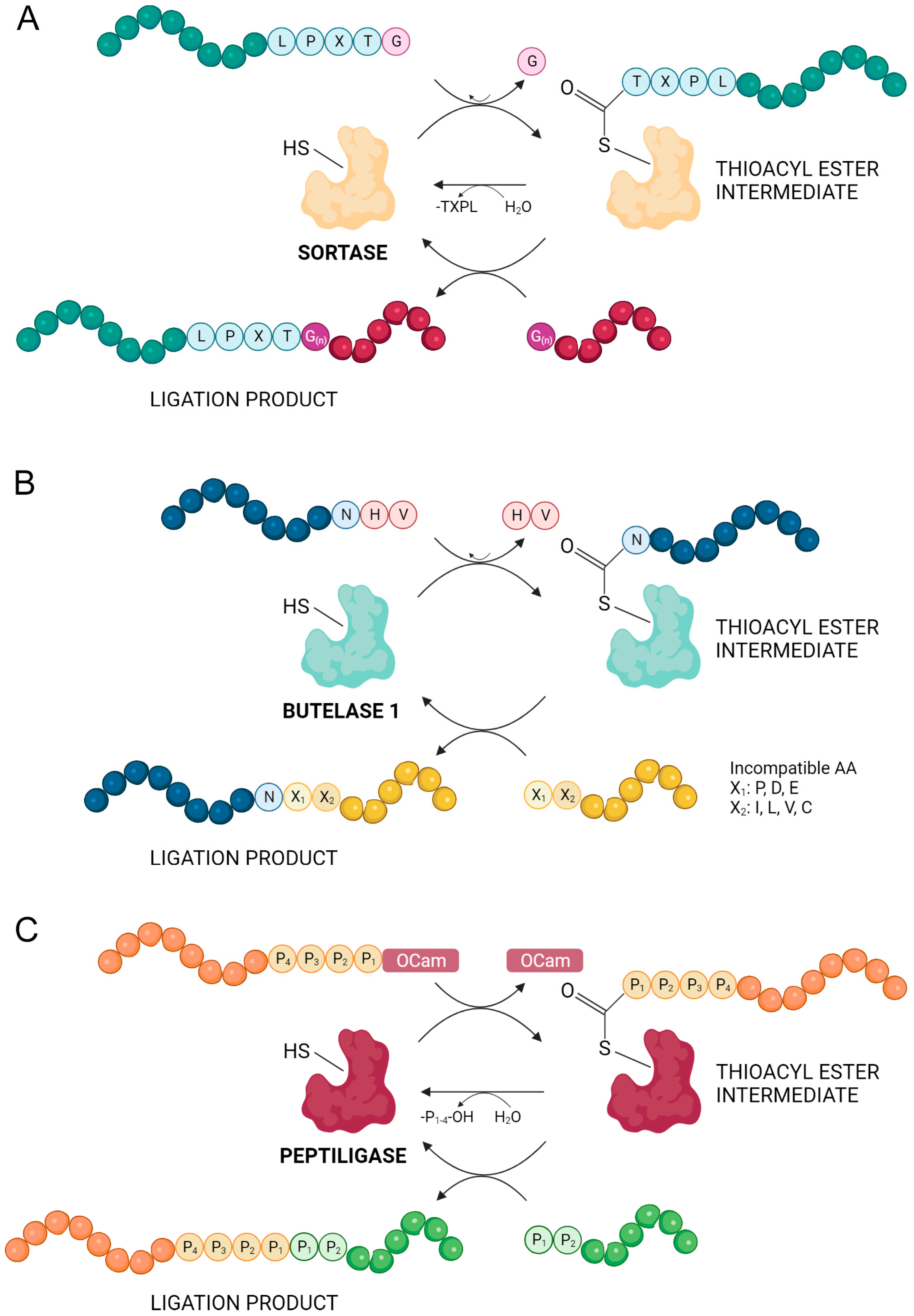
- Nuijens, T.; Toplak, A.; Schmidt, M.; Ricci, A.; Cabri, W. Natural Occurring and Engineered Enzymes for Peptide Ligation and Cyclization. Front. Chem. 2019, 7, 829. [Google Scholar] [CrossRef] [PubMed]
- Nuijens, T.; Toplak, A.; Quaedflieg, P.J.L.M.; Drenth, J.; Wu, B.; Janssen, D.B. Engineering a Diverse Ligase Toolbox for Peptide Segment Condensation. Adv. Synth. Catal. 2016, 358, 4041–4048. [Google Scholar] [CrossRef]
- Nuijens, T.; Toplak, A.; Van de Meulenreek, M.B.A.C.; Schmidt, M.; Goldbach, M.; Janssen, D.B.; Quaedflieg, P.J.L.M. Chemo-Enzymatic Peptide Synthesis (CEPS) Using Omniligases and Selective Peptiligases Efficient Biocatalysts for Assembling Linear and Cyclic Peptides and Protein Conjugates. Chim. Oggi-Chem. Today 2016, 34, 16–19. [Google Scholar]
- Pawlas, J.; Nuijens, T.; Persson, J.; Svensson, T.; Schmidt, M.; Toplak, A.; Nilsson, M.; Rasmussen, J.H. Sustainable, Cost-Efficient Manufacturing of Therapeutic Peptides Using Chemo-Enzymatic Peptide Synthesis (CEPS). Green Chem. 2019, 21, 6451–6467. [Google Scholar] [CrossRef]

























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
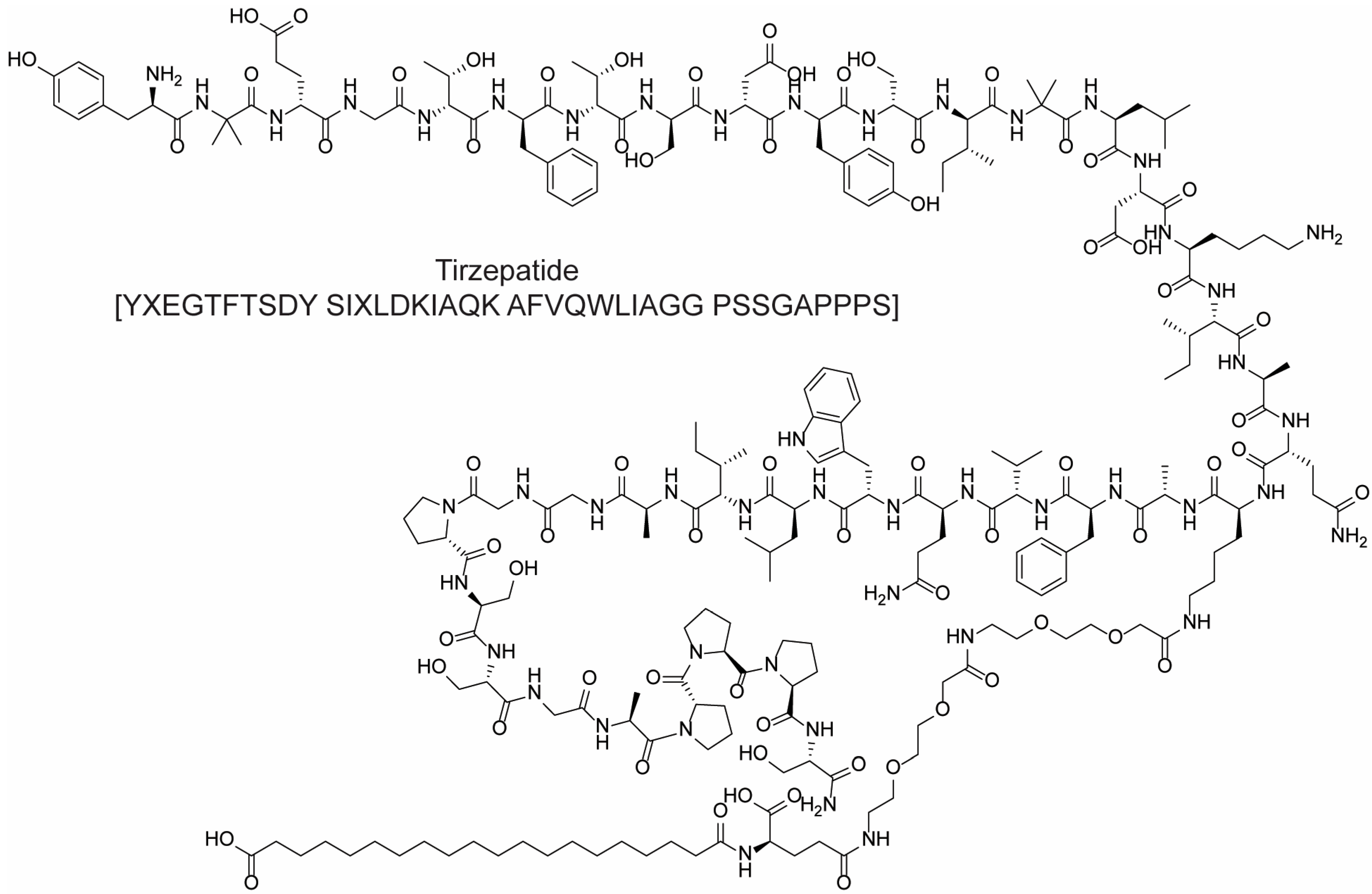{kind=link}
{kind=link}
{kind=link}
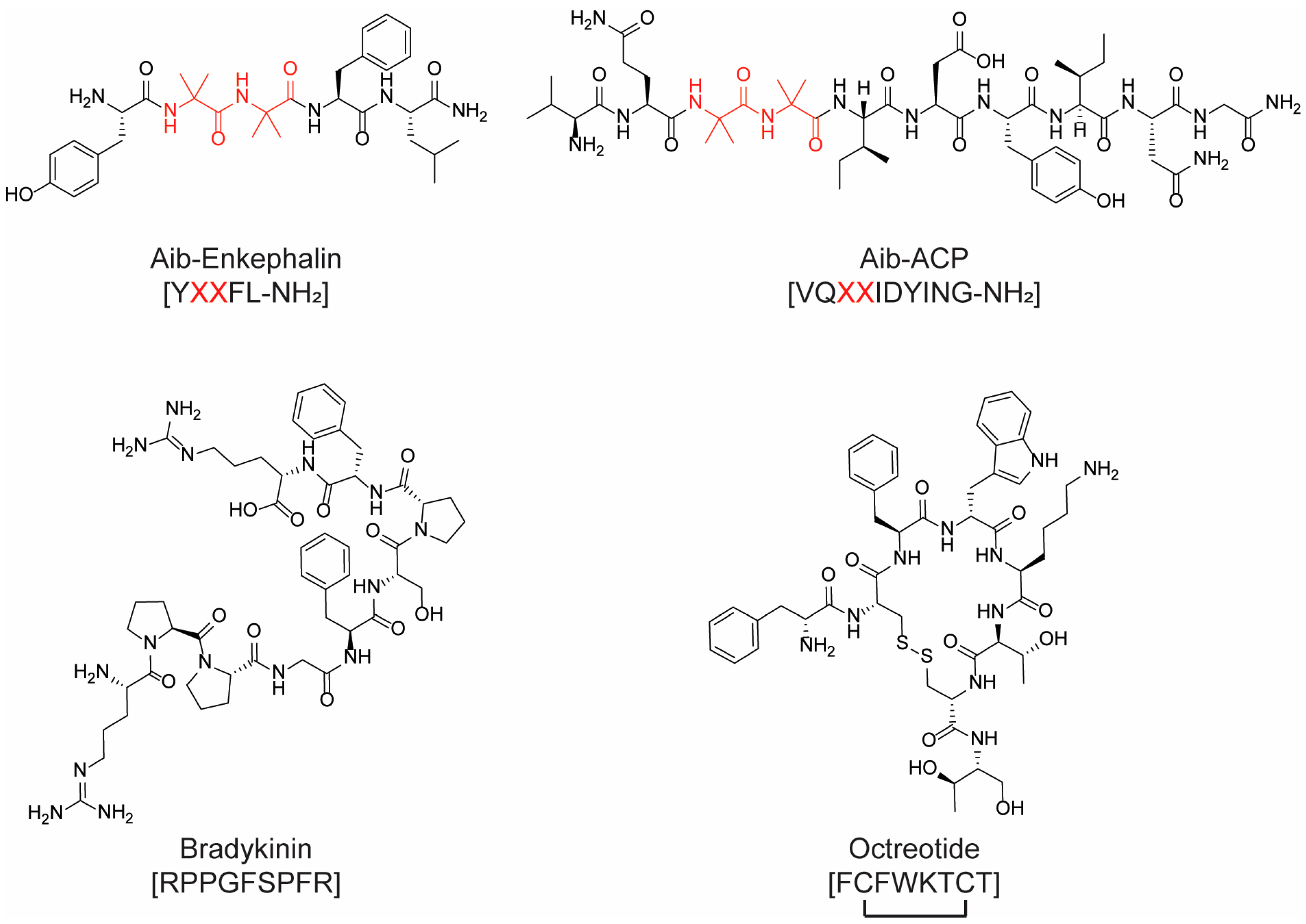{kind=link}
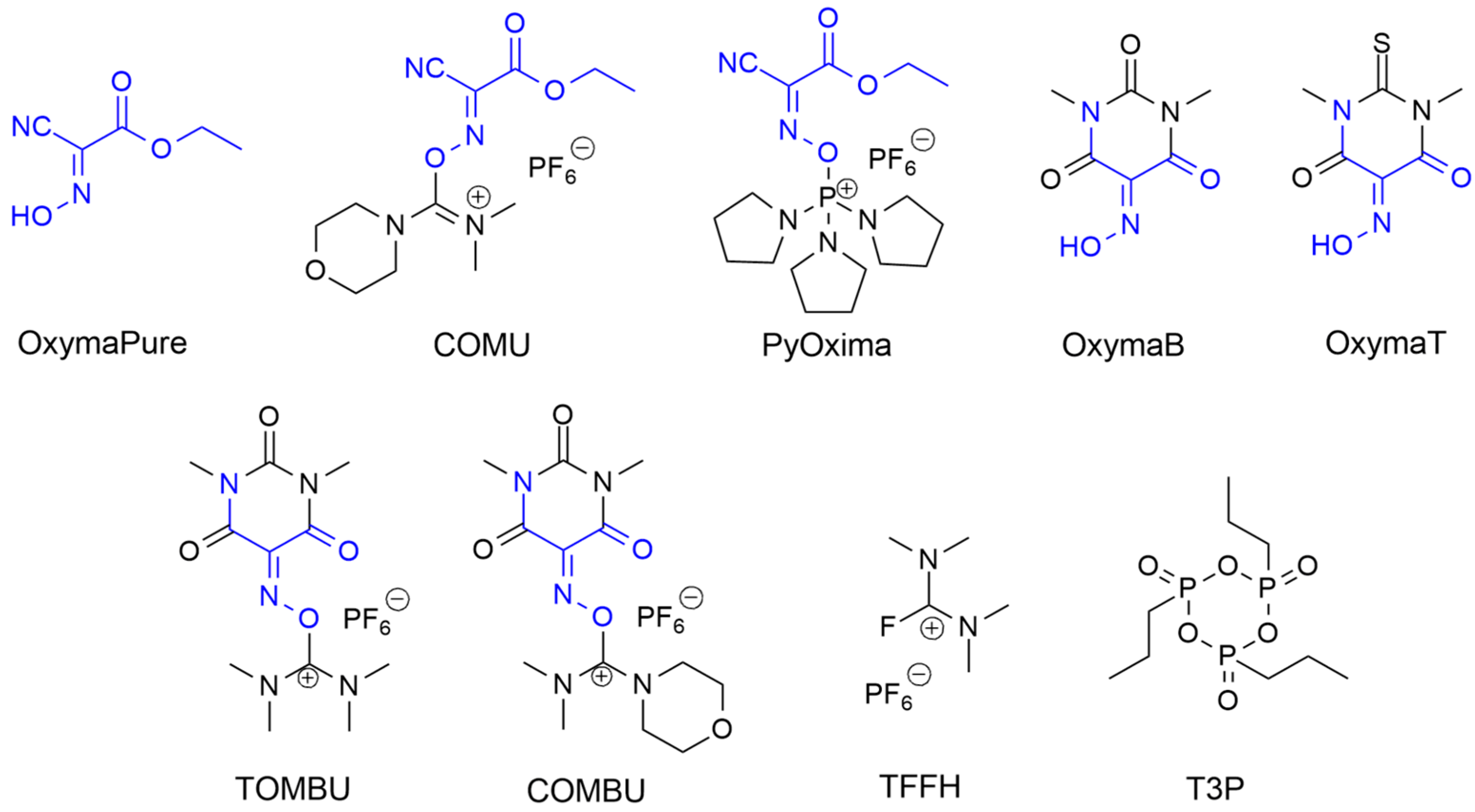{kind=link}
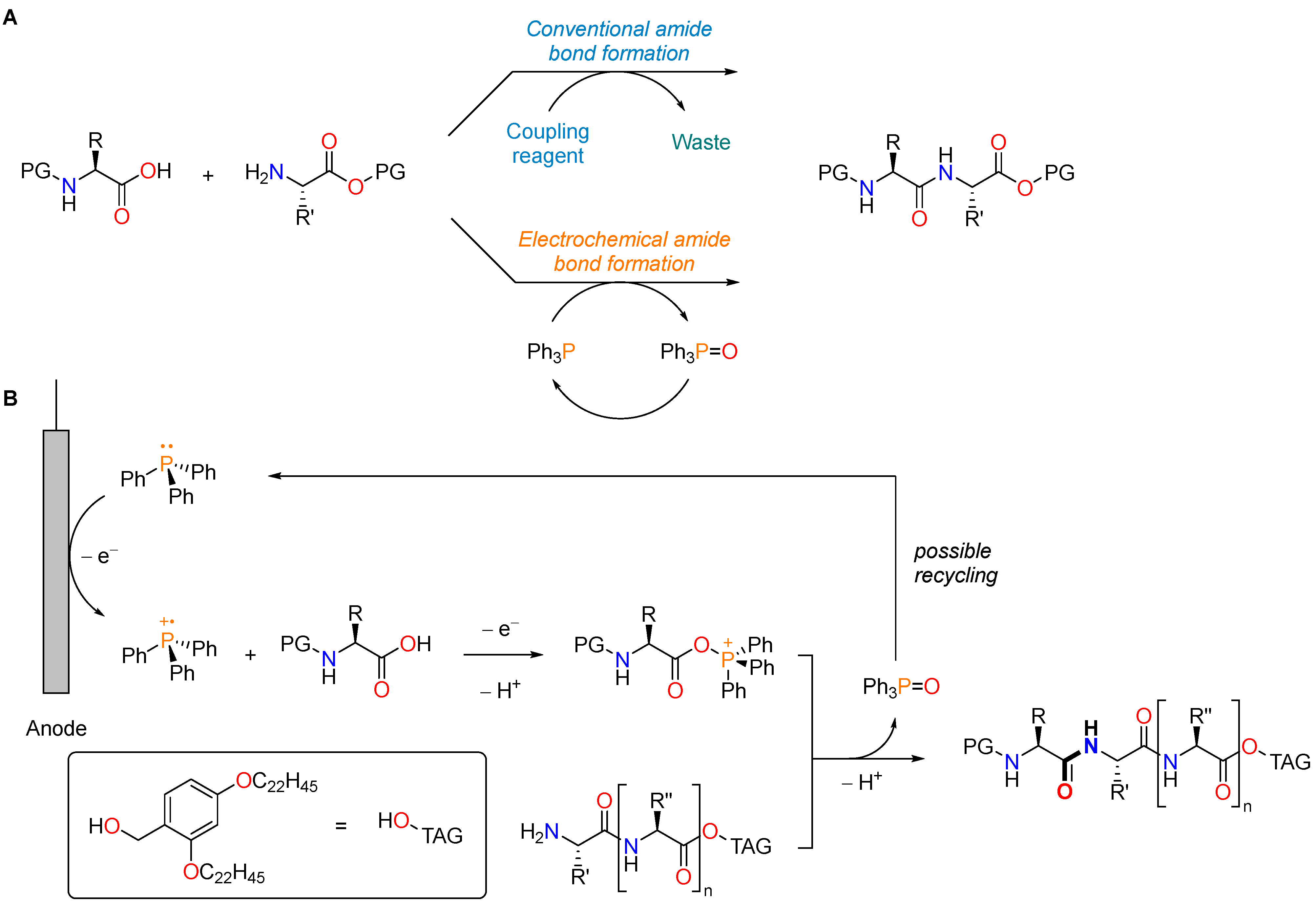{kind=link}
{kind=link}
{kind=link}
| Liquid-Phase Peptide Synthesis (LPPS) | Solid-Phase Peptide Synthesis (SPPS) | |
|---|---|---|
| Medium | Solution | Insoluble polymer |
| Batch size | Any (usually large scale) | Any (usually small scale) |
| Timing | Slow | Fast |
| Automation | Difficult | Semi- or fully-automated |
| Synthetic strategy | Generally convergent | Stepwise |
| Protecting groups employed | Typically Boc or Z | Typically Fmoc |
| Side-chain protection | Minimum | Maximum |
| Consumption of AA | Moderate | High |
| In-process analysis | Direct monitoring (e.g., HPLC) | Indirect monitoring |
| Purification of intermediates | Possible (usually by precipitation) | Not possible |
| Final purification | Relatively simple | Laborious |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossino, G.; Marchese, E.; Galli, G.; Verde, F.; Finizio, M.; Serra, M.; Linciano, P.; Collina, S. Peptides as Therapeutic Agents: Challenges and Opportunities in the Green Transition Era. Molecules 2023, 28, 7165. https://doi.org/10.3390/molecules28207165
Rossino G, Marchese E, Galli G, Verde F, Finizio M, Serra M, Linciano P, Collina S. Peptides as Therapeutic Agents: Challenges and Opportunities in the Green Transition Era. Molecules. 2023; 28(20):7165. https://doi.org/10.3390/molecules28207165
Chicago/Turabian StyleRossino, Giacomo, Emanuela Marchese, Giovanni Galli, Francesca Verde, Matteo Finizio, Massimo Serra, Pasquale Linciano, and Simona Collina. 2023. "Peptides as Therapeutic Agents: Challenges and Opportunities in the Green Transition Era" Molecules 28, no. 20: 7165. https://doi.org/10.3390/molecules28207165