
Evaluation of Encapsulation Potential of Selected Star-Hyperbranched Polyglycidol Architectures: Predictive Molecular Dynamics Simulations and Experimental Validation

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
2.1. In Silico Studies
2.1.1. 3D Structure Generation and Surface Properties
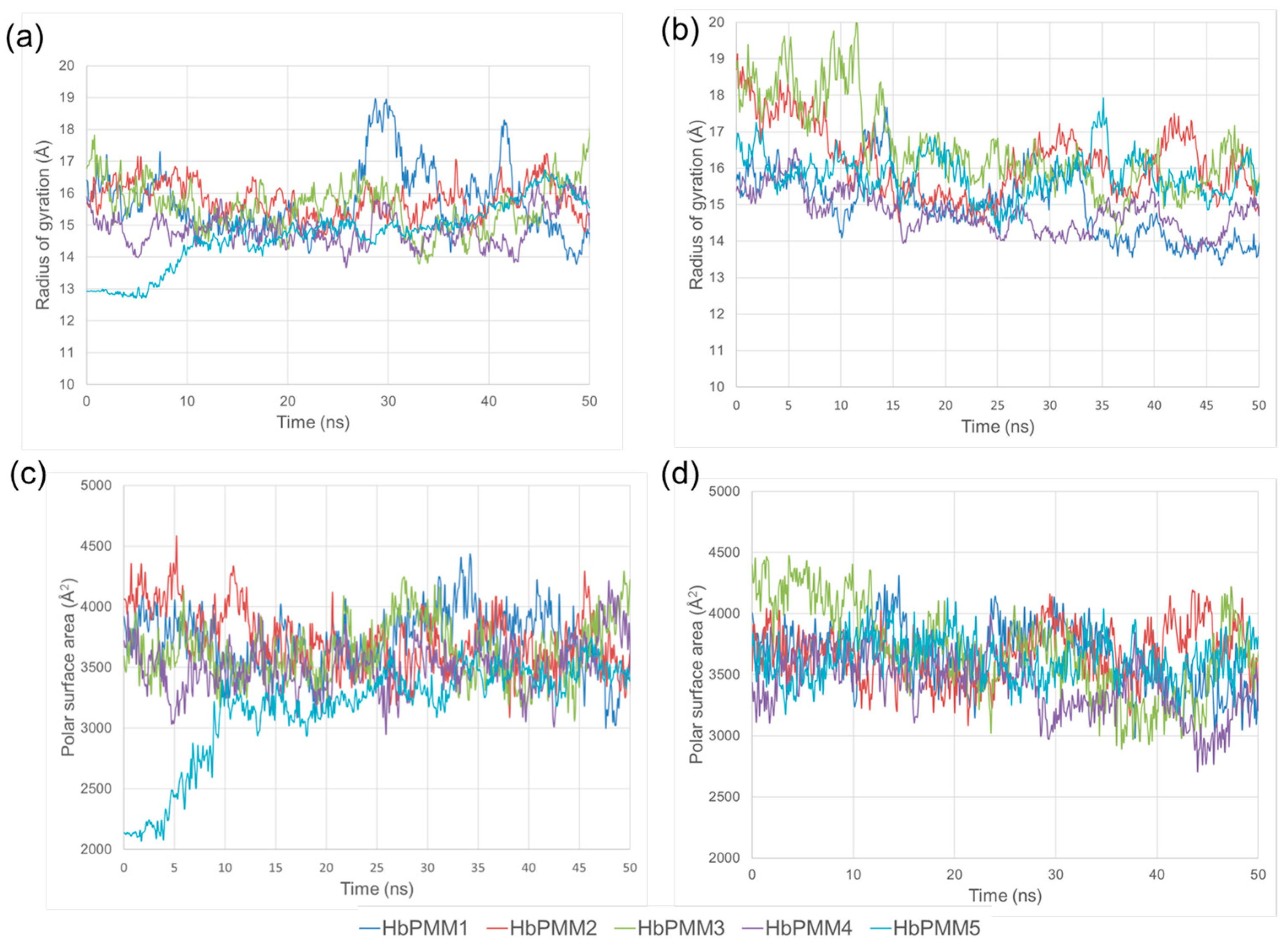
2.1.2. Molecular Dynamics Simulations of R17 and Tinidazole
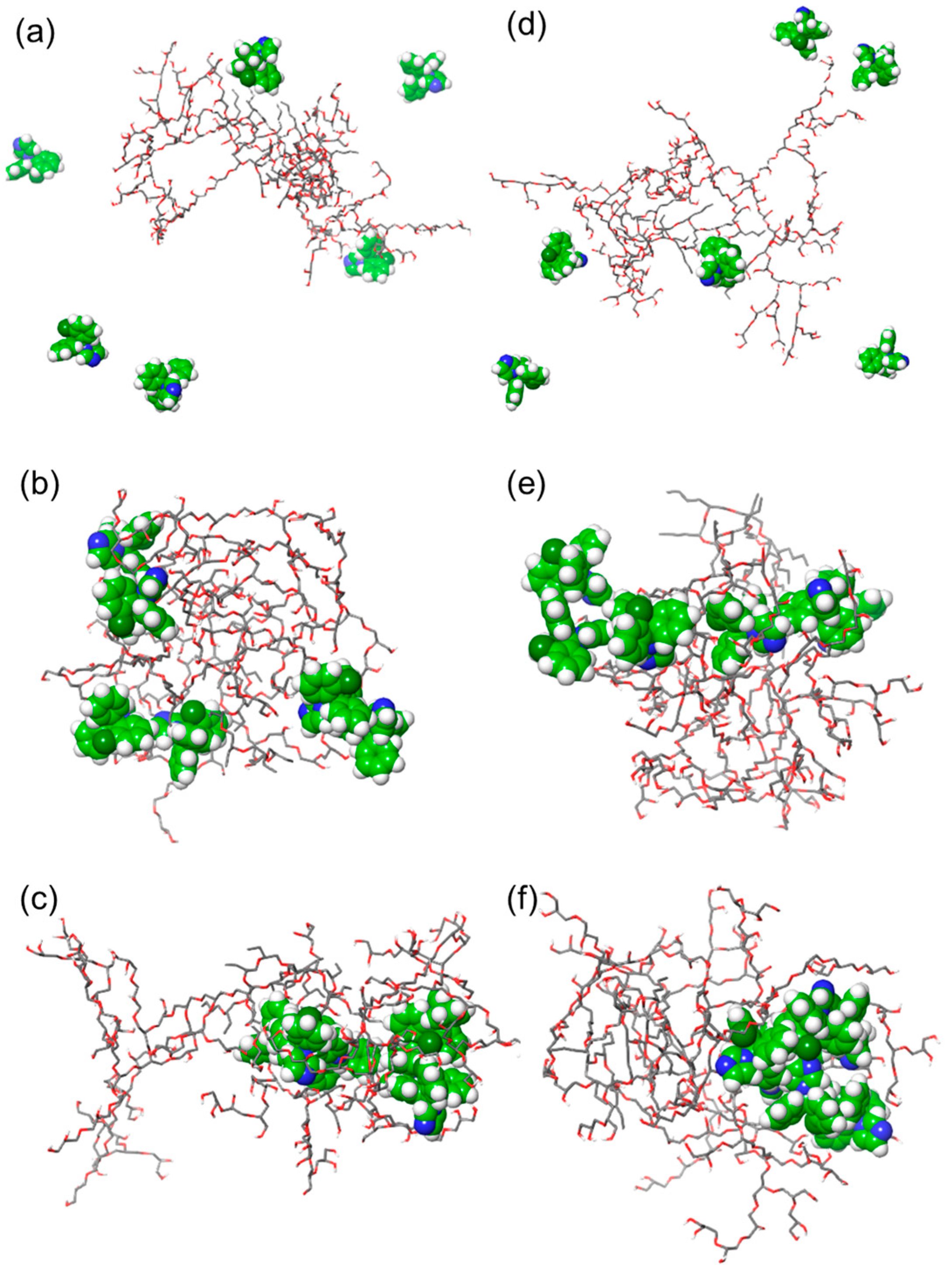
2.1.3. Molecular Dynamics Simulations of Clotrimazole Polymer Mixtures
2.2. Experimental Validation
2.2.1. R14 and R17 Synthesis and Characterization
2.2.2. Tinidazole and Clotrimazole Encapsulation Studies
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Poly(1,2-epoxybutane)-co-HbPGL (R14)
3.3. Synthesis of Poly(1,2-epoxyhexane)-co-HbPGL (R17)
3.4. Gel Permeation Chromatography, GPC
3.5. Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry, MALDI-TOF
3.6. Evalulation of Drug Loadings in Copolymers
3.7. Generation of 3D Structures
3.8. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jones, M.C.; Ranger, M.; Leroux, J.C. pH-sensitive unimolecular polymeric micelles: Synthesis of a novel drug carrier. Bioconjugate Chem. 2003, 14, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Sun, P.; Tong, G.S.; Zhu, X.Y. Star polymer-based unimolecular micelles and their application in bio-imaging and diagnosis. Biomaterials 2018, 178, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Ambade, A.V.; Savariar, E.N.; Thayumanavan, S. Dendrimeric micelles for controlled drug release and targeted delivery. Mol. Pharm. 2005, 2, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Gupta, U.; Agashe, H.B.; Asthana, A.; Jain, N.K. Dendrimers: Novel polymeric nanoarchitectures for solubility enhancement. Biomacromolecules 2006, 7, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Jose, J.; Charyulu, R.N. PAMAM Dendrimers: Novel polymeric nanoarchitectures for solubility enhancement of Ketoconazole. Optoelectron. Adv. Mat. 2016, 10, 604–608. [Google Scholar]
- Gosecki, M.; Kazmierski, S.; Gosecka, M. Diffusion-Controllable Biomineralization Conducted In Situ in Hydrogels Based on Reversibly Cross-Linked Hyperbranched Polyglycidol. Biomacromolecules 2017, 18, 3418–3431. [Google Scholar] [CrossRef] [PubMed]
- Gosecki, M.; Zgardzinska, B.; Gosecka, M. Temperature-Induced Changes in the Nanostructure of Hydrogels Based on Reversibly Cross-Linked Hyperbranched Polyglycidol with B(OH)(4)(circle minus) Ions. J. Phys. Chem. C 2016, 120, 18323–18332. [Google Scholar] [CrossRef]
- Ziemczonek, P.; Gosecka, M.; Gosecki, M.; Marcinkowska, M.; Janaszewska, A.; Klajnert-Maculewicz, B. Star-Shaped Poly(furfuryl glycidyl ether)-Block-Poly(glyceryl glycerol ether) as an Efficient Agent for the Enhancement of Nifuratel Solubility and for the Formation of Injectable and Self-Healable Hydrogel Platforms for the Gynaecological Therapies. Int. J. Mol. Sci. 2021, 22, 8386. [Google Scholar] [CrossRef]
- Gosecka, M.; Jaworska-Krych, D.; Gosecki, M.; Wielgus, E.; Marcinkowska, M.; Janaszewska, A.; Klajnert-Maculewicz, B. Self-Healable, Injectable Hydrogel with Enhanced Clotrimazole Solubilization as a Potential Therapeutic Platform for Gynecology. Biomacromolecules 2022, 23, 4203–4219. [Google Scholar] [CrossRef]
- Jafari, M.; Abolmaali, S.S.; Najafi, H.; Tamaddon, A.M. Hyperbranched polyglycerol nanostructures for anti-biofouling, multifunctional drug delivery, bioimaging and theranostic applications. Int. J. Pharm. 2020, 576, 118959. [Google Scholar] [CrossRef]
- Ying, H.; He, G.J.; Zhang, L.F.; Lei, Q.F.; Guo, Y.S.; Fang, W.J. Hyperbranched polyglycerol/poly(acrylic acid) hydrogel for the efficient removal of methyl violet from aqueous solutions. J. Appl. Polym. Sci. 2016, 133. [Google Scholar] [CrossRef]
- Wu, C.Z.; Strehmel, C.; Achazi, K.; Chiapisi, L.; Dernedde, J.; Lensen, M.C.; Gradzielski, M.; Ansorge-Schumacher, M.B.; Haag, R. Enzymatically Cross-Linked Hyperbranched Polyglycerol Hydrogels as Scaffolds for Living Cells. Biomacromolecules 2014, 15, 3881–3890. [Google Scholar] [CrossRef] [PubMed]
- Postnova, I.; Silant’ev, V.; Kim, M.H.; Song, G.Y.; Kim, I.; Ha, C.S.; Shchipunov, Y. Hyperbranched polyglycerol hydrogels prepared through biomimetic mineralization. Colloid Surf. B 2013, 103, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Haryanto; Singh, D.; Huh, P.H.; Kim, S.C. Hyperbranched poly(glycidol)/poly(ethylene oxide) crosslinked hydrogel for tissue engineering scaffold using e-beams. J. Biomed. Mater. Res. A 2016, 104, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Schomer, M.; Schull, C.; Frey, H. Hyperbranched aliphatic polyether polyols. J. Polym. Sci. Pol. Chem. 2013, 51, 995–1019. [Google Scholar] [CrossRef]
- Wilms, D.; Stiriba, S.E.; Frey, H. Hyperbranched Polyglycerols: From the Controlled Synthesis of Biocompatible Polyether Polyols to Multipurpose Applications. Acc. Chem. Res. 2010, 43, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Kurniasih, I.N.; Keilitz, J.; Haag, R. Dendritic nanocarriers based on hyperbranched polymers. Chem. Soc. Rev. 2015, 44, 4145–4164. [Google Scholar] [CrossRef] [PubMed]
- Javan Nikkhah, S.; Thompson, D. Molecular Modelling Guided Modulation of Molecular Shape and Charge for Design of Smart Self-Assembled Polymeric Drug Transporters. Pharmaceutics 2021, 13, 141. [Google Scholar] [CrossRef]
- Casalini, T. Not only in silico drug discovery: Molecular modeling towards in silico drug delivery formulations. J. Control. Release 2021, 332, 390–417. [Google Scholar] [CrossRef]
- Zloh, M.; Martinho, N. Modelling Approaches for Studies of Drug-Polymer Interactions in Drug Delivery Systems. In Computer Aided Pharmaceutics and Drug Delivery: An Application Guide for Students and Researchers of Pharmaceutical Sciences; Springer: Berlin/Heidelberg, Germany, 2022; pp. 561–591. [Google Scholar]
- Vu Kinh, L.; Nguyen Anh, D.; Romero Jovel, S.; Nguyen Hoang Khue, T. Tinidazole Delivery Improved by Nanosized Minicells Originated from Leuconostoc mesenteroides. J. Nanomater. 2019, 2019, 7684795. [Google Scholar] [CrossRef]
- Ibišević, M.; Pilipović, S.; Nešić, I.; Kerleta, V.; Husejnagić, D.; Kozarević, E.C.; Horozić, E.; Karić, E. Antimicrobial activity of liposomal and non-liposomal vaginal suppositories with Origanum compactum essential oil. Technol. Acta-Sci. Prof. J. Chem. Technol. 2020, 13, 5–10. [Google Scholar]
- Debnath, M.; Muzib, Y.I.; Kumar, S.A. Formulation development, optimization and in-vitro releas kinetic study on colon targeted tinidazole-guar gum microcapsules. Int. J. Pharm. Pharma. Sci. 2013, 5, 278–285. [Google Scholar]
- Catenacci, L.; Marrubini, G.; Sorrenti, M.; Rossi, S.; Sandri, G.; Ferrari, F.; Fagnani, V.; Valentino, C.; Bonferoni, M.C. Design of experiments-assisted development of clotrimazole-loaded ionic polymeric micelles based on hyaluronic acid. Nanomaterials 2020, 10, 635. [Google Scholar] [CrossRef] [PubMed]
- Gosecki, M.; Ziemczonek, P.; Gosecka, M.; Urbaniak, M.; Wielgus, E.; Marcinkowska, M.; Janaszewska, A.; Klajnert-Maculewicz, B. Cross-linkable star-hyperbranched unimolecular micelles for the enhancement of the anticancer activity of clotrimazole. J. Mater. Chem. B 2023, 11, 5552–5564. [Google Scholar] [CrossRef] [PubMed]
- Hosseinzadeh, F.; Tabesh, H.; Farzaneh, F. Nano drug delivery platform based on thermosensitive PEG-PCL hydrogel encapsulated in silver-bearing micelles and its antifungal activity investigation against vaginal candidiasis. Front. Mater. 2023, 10, 1210542. [Google Scholar] [CrossRef]
- Kareem, F.; Bhayo, A.M.; Imran, M.; Shah, M.R.; Khan, K.M.; Malik, M.I. Enhanced therapeutic efficacy of clotrimazole by delivery through poly (ethylene oxide)-block-poly (ε-caprolactone) copolymer-based micelles. J. Appl. Polym. Sci. 2019, 136, 47769. [Google Scholar] [CrossRef]
- Li, T.; Zhu, L.; Li, J.; Cao, Z.; Sha, J.; Li, Y.; Ren, B. Solubility, thermodynamic properties and molecular simulation of tinidazole in fourteen mono-solvents at different temperatures. J. Chem. Thermodyn. 2022, 170, 106767. [Google Scholar] [CrossRef]
- Wang, F.; Liu, Y.; Yan, H.; Wang, D.; Chu, Z.; Li, K.; Tong, L.; Chen, M.; Gong, J. Revealing dissolution behavior and thermodynamic properties of tinidazole in 12 mono-solvents based on experiments and molecular simulation. J. Mol. Liq. 2022, 366, 120081. [Google Scholar] [CrossRef]
- Shang, S.; Zhao, Q.; Zhang, D.; Sun, R.; Tang, Y. Molecular dynamics simulation of the adsorption behavior of two different drugs on hydroxyapatite and Zn-doped hydroxyapatite. Mater. Sci. Eng. C 2019, 105, 110017. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhang, D.; Sun, R.; Shang, S.; Wang, H.; Yang, Y.; Wang, L.; Liu, X.; Sun, T.; Chen, K. Adsorption behavior of drugs on hydroxyapatite with different morphologies: A combined experimental and molecular dynamics simulation study. Ceram. Int. 2019, 45, 19522–19527. [Google Scholar] [CrossRef]
- Gupta, K.M.; Das, S.; Wong, A.B.H.; Chow, P.S. Formulation and Skin Permeation of Active-Loaded Lipid Nanoparticles: Evaluation and Screening by Synergizing Molecular Dynamics Simulations and Experiments. Langmuir 2023, 39, 308–319. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; Association for Computing Machinery: New York, NY, USA, 2006. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Poly(1,2-epoxybutane)-co-HbPGL | Poly(1,2-epoxyhexane)-co-HbPGL | ||
|---|---|---|---|
| Tinidazole aimed (per copolymer molecule) | 15 | 25 | 15 |
| Tinidazole encapsulated | 14 | 22 | 14 |
| Encapsulation efficiency, % | 93 | 88 | 93 |
| Poly(1,2-epoxybutane)-co-HbPGL | Poly(1,2-epoxyhexane)-co-HbPGL | |
|---|---|---|
| Clotrimazole aimed (per copolymer molecule) | 10 | 6 |
| Clotrimazole encapsulated | 9 | 5 |
| Encapsulation efficiency, % | 90 | 83 |
| System 1 | System 2 | System 3 | System 4 | |
|---|---|---|---|---|
| Copolymer | R14 | R14 | R17 | R17 |
| Weight of copolymer (mg) | 42.5 | 30 | 46.5 | 30 |
| Drug | clotrimazole | tinidazole | clotrimazole | tinidazole |
| Volume of drug solution (mL) | 3 | from 2.25 to 3.70 | 2 | 2.25 |
| Stage | Stage Name | Solvent | Solvent Shell (Å) | Ensemble | Simulation (ns) |
|---|---|---|---|---|---|
| 1 | Dissolving | MeOH | NPT | 20 | |
| 2 | Drying stage 1 | MeOH | 25 | NVT | 10 |
| 3 | Drying stage 2 | MeOH | 20 | NVT | 10 |
| 4 | Drying stage 1 | MeOH | 15 | NVT | 10 |
| 5 | Drying stage 2 | MeOH | 10 | NVT | 10 |
| 6 | Drying stage 1 | MeOH | 5 | NVT | 10 |
| 7 | Drying stage 2 | MeOH | 2.5 | NVT | 10 |
| 8 | Redissolving | Water | NPT | 50 |
| Stage | Stage Name | Solvent | Solvent Shell (Å) | Ensemble | Simulation (ns) |
|---|---|---|---|---|---|
| 1 | Dissolving | MeOH | NPT | 20 | |
| 2 | Drying stage 1 | MeOH | 15 | NVT | 10 |
| 3 | Drying stage 2 | MeOH | 6 | NVT | 10 |
| 4 | Drying stage 1 | MeOH | 3 | NVT | 10 |
| 5 | Drying stage 2 | MeOH | 2 | NVT | 10 |
| 6 | Redissolving | Water | NPT | 50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gosecki, M.; Urbaniak, M.; Martinho, N.; Gosecka, M.; Zloh, M. Evaluation of Encapsulation Potential of Selected Star-Hyperbranched Polyglycidol Architectures: Predictive Molecular Dynamics Simulations and Experimental Validation. Molecules 2023, 28, 7308. https://doi.org/10.3390/molecules28217308
Gosecki M, Urbaniak M, Martinho N, Gosecka M, Zloh M. Evaluation of Encapsulation Potential of Selected Star-Hyperbranched Polyglycidol Architectures: Predictive Molecular Dynamics Simulations and Experimental Validation. Molecules. 2023; 28(21):7308. https://doi.org/10.3390/molecules28217308
Chicago/Turabian StyleGosecki, Mateusz, Malgorzata Urbaniak, Nuno Martinho, Monika Gosecka, and Mire Zloh. 2023. "Evaluation of Encapsulation Potential of Selected Star-Hyperbranched Polyglycidol Architectures: Predictive Molecular Dynamics Simulations and Experimental Validation" Molecules 28, no. 21: 7308. https://doi.org/10.3390/molecules28217308