
Triphenylborane in Metal-Free Catalysis

1
Department of Natural Sciences, The University of Virginia’s College, Wise, VA 24293-4400, USA
2
Department of Chemistry and Biochemistry, Texas Tech University, Lubbock, TX 79409-1061, USA
*
Authors to whom correspondence should be addressed.
Molecules 2023, 28(3), 1340; https://doi.org/10.3390/molecules28031340
Submission received: 1 January 2023
/
Revised: 23 January 2023
/
Accepted: 24 January 2023
/
Published: 31 January 2023
(This article belongs to the Special Issue Featured Reviews in Organometallic Chemistry)
Abstract
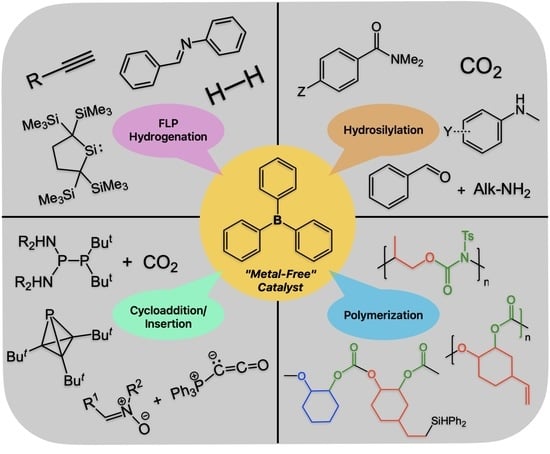
:The development and application of new organoboron reagents as Lewis acids in synthesis and metal-free catalysis have dramatically expanded over the past 20 years. In this context, we will show the recent uses of the simple and relatively weak Lewis acid BPh3—discovered 100 years ago—as a metal-free catalyst for various organic transformations. The first part will highlight catalytic applications in polymer synthesis such as the copolymerization of epoxides with CO2, isocyanate, and organic anhydrides to various polycarbonate copolymers and controlled diblock copolymers as well as alternating polyurethanes. This is followed by a discussion of BPh3 as a Lewis acid component in the frustrated Lewis pair (FLP) mediated cleavage of hydrogen and hydrogenation catalysis. In addition, BPh3-catalyzed reductive N-methylations and C-methylations with CO2 and silane to value-added organic products will be covered as well along with BPh3-catalyzed cycloadditions and insertion reactions. Collectively, this mini-review showcases the underexplored potential of commercially available BPh3 in metal-free catalysis.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Last year marked the 100th anniversary of the discovery of triphenyl borane, BPh3, the first isolated triaryl borane by E. Krause and R. Nitsche [1]. The authors obtained BPh3 as a crystalline solid through the treatment of BF3 with excess phenyl magnesium bromide followed by distillation under vacuum. About 50 years later, its solid-state structure was determined by single crystal X-ray crystallography [2]. The results revealed a trigonal planar coordination environment for boron with the three phenyl rings being tilted by about 30° toward the plane.
BPh3 has found widespread application as a promoter in the hydrocyanation of olefins in the presence of Ni complexes and is used industrially by Du Pont for its hydrocyanation of butadiene to adiponitrile, a nylon intermediate [3]. In addition, BPh3 has been employed extensively as a diphenyl boryl transfer agent in the synthesis of boron-containing heterocyclic materials with remarkable photophysical properties, such as electro- and photoluminescence and aggregation-induced emission (AIE) [4,5,6,7,8,9,10,11,12,13,14,15,16,17]. BPh3 is a relatively weak Lewis acid that forms Lewis acid–base adducts with pyridine and a wide variety of aliphatic amines [18]. These stable adducts have found use as catalysts for the polymerization of acrylic esters [3], as antifouling reagents in marine environments [19,20,21] and as agrochemical fungicides [22,23,24].
Despite its early discovery and structural simplicity, BPh3 has not risen to the same prominence amongst scientists in academia as has its ingenious perfluorinated and highly Lewis acidic counterpart, B(C6F5)3, first reported in 1963 [25]. Perhaps one major factor that has caused BPh3 to lag behind B(C6F5)3, particularly in the emerging field of metal-free Lewis acid catalysis, is its comparably low Lewis acidity [26]. As high Lewis acidity is often key to the activation and rapid catalytic transformation of organic substrates, it is not a surprise that B(C6F5)3 has been at the forefront of current research in metal-free catalysis [27,28,29,30,31,32,33,34,35,36]. However, as the development and application of new boron Lewis acids have expanded at an amazing pace over the past 20 years, it has also become apparent that tuning the Lewis acidity of the catalyst can be an important factor in achieving new modes of substrate activation and selectivity. A prime example of this development has been the application of frustrated Lewis pairs (FLPs) [37,38,39] in catalytic hydrogenations of unsaturated organic molecules. Work from various groups has shown that weaker Lewis acid components may exhibit better performances, improved functional group tolerances, or different selectivities [40,41,42,43,44,45].
In this context, this mini-review aims to showcase the underexplored potential of commercially available and weakly Lewis-acidic BPh3 in metal-free catalysis. Particular emphasis is given to the role of BPh3 as a catalyst in polymer synthesis, in frustrated Lewis pair (FLP) mediated hydrogen cleavage, and hydrogenation catalysis as well as transformations of CO2 to value-added organic products and Lewis acid catalyzed cycloadditions and insertion reactions.
2. BPh3 in Polymerization Catalysis
In developing effective “latent catalysts” for the curing of epoxy resins that overcome issues associated with previously utilized systems, such as poor solubility and hygroscopicity, Endo and co-workers studied Lewis pairs of general formula Ph3PCHRBX3, where X = H, Ph, F (Scheme 1) [46,47]. Most of these air- and moisture-stable zwitterionic phosphonium borates 1–6, derived from reactions of the respective boranes with phosphonium ylides, proved to be active pre-catalysts in the co-polymerization of bisphenol A and bisphenol A diglycidyl ether at high temperatures (Scheme 1). The pre-catalyst activity at 120 °C was found to be in the following order: 4 > 2 > 1 > 3 > 5 > 6. The fact that BPh3-adduct 4 and BH3-adduct 2 converted the bisphenol A diglycidyl ether within 3 h to ca. 90% and 70%, respectively, whereas BF3-adduct 6 was essentially inactive, impressively shows the importance of steric and electronic parameters in the design of Lewis pairs as active polymerization catalysts.
The key to the catalytic activity of the phosphonium borate catalysts 1-6 was assumed to be their thermally induced dissociation to BX3 and Ph3P=CHR, with the degree of dissociation being a sensitive function of temperature as well as the strength of the C-B bond (Scheme 2). The significantly better performances of the BPh3-ylide and BH3-ylide adducts 4 and 2 compared to the BF3-ylides 5 and 6 have been attributed to their weaker B-C bonds due to the lower Lewis acidity of BPh3 and BH3. Upon dissociation of the adduct, the resulting phosphonium ylide serves as an initiator deprotonating the phenolic OH group of bisphenol A to generate a phenoxide anion. BX3 activates, as a Lewis acid, the epoxide functionality, facilitating its ring-opening via nucleophilic attack of the phenoxide anion. Whether BX3 remains to be intact as a catalyst during the course of the reaction is questionable given the harsh conditions and the Bronsted acidity of bisphenol A.
The past decade has witnessed increasing interest in the metal-free catalyzed formation of cyclic organic carbonates and polycarbonates from epoxide monomers and CO2 as an alternative to metal-based catalysts [48,49,50,51]. Recent reports from Feng [50] and Darensbourg [51] demonstrated that metal-free Lewis pairs can be utilized as effective catalysts in the copolymerizing of propylene oxide (PO) with CO2 or carbonyl sulfide (COS). The catalyst systems comprised triethyl borane as a Lewis acid, and amines, onium salts, or alkoxides as Lewis bases.
The Kerton group investigated the activity of BPh3 and B(C6F5)3 as catalysts and bis(triphenylphosphine)iminium chloride (PPNCl) as a co-catalyst for the reaction of propylene oxide (PO) and CO2 at 100 °C (Scheme 3) [52]. Both were active catalysts in generating propylene carbonate under identical conditions. These findings were in stark contrast with previous work using triethyl borane as the catalyst, which produced poly(propylene carbonate) [50]. It is worthwhile noting that based on initial rate measurements, the catalyst system BPh3/PPNCl was more than five times faster than B(C6F5)3/PPNCl. This is most likely due to the significantly higher Lewis acidity of B(C6F5)3, which binds much more strongly to the Cl- anion of co-catalyst PPNCl than BPh3 does. Consistent with kinetic studies revealing that lowering the CO2 pressure increases the reaction rate, BPh3 catalyzed the reaction of epichlorohydrin with CO2, even under atmospheric CO2 pressure, to give the respective cyclic carbonate as the sole product.
In addition, BPh3 was found to catalyze the copolymerization of CO2 with cyclohexene oxide (CHO) and vinyl cyclohexene oxide (VCHO), respectively, at low catalyst loadings. Polycarbonates with high numbers of averaged molecular weights and excellent polydispersities were obtained. In the absence of CO2, BPh3 catalyzed the ring opening of CHO to give the epoxide homopolymer (Scheme 3).
In a follow-up study, Kerton and co-workers investigated the ability of BPh3 to catalyze the addition of Si–H groups onto a vinyl-substituted polycarbonate [53]. In a ‘one-pot’ sequence, BPh3 catalyzed both reactions: the copolymerization of VCHO and CO2 to polycarbonate followed by the hydrosilylation of the vinyl groups in the polymer with phenyl dimethyl silane to give side-chain silylated polycarbonate (Scheme 4). Perhaps for steric reasons, the degree of hydrosilylation of polycarbonate was only 10%. However, when using a polycarbonate terpolymer derived from CHO, VCHO, and CO2, the degree of hydrosilylation could be increased to 36%, in this case with H2SiPh2 as the hydrosilylation reagent.
In 2021, Kerton et al. disclosed the BPh3/PPNCl catalyzed copolymerization of organic anhydrides and epoxides [54]. Cyclohexene oxide (CHO), vinyl cyclohexene oxide (VCHO) and also limonene oxide (LO) could be polymerized with phthalic anhydride (PAH) and cis-4-cyclohexene-1,2-dicarboxylic anhydride (CDA), respectively, to give perfectly alternating copolymers with excellent polydispersities (Figure 1).
In addition, the authors discovered that these perfectly alternating co-polymers can be further polymerized to form controlled diblock copolymers using BPh3/PPNCl as the catalyst system. For example, PAH was first allowed to react with excess CHO to generate copolymer I. Once the full conversion of PAH was achieved, the second anhydride, CDA, was added and allowed to react to completion, resulting in the selective formation of controlled diblock copolymer II (Scheme 5). Similarly, by sequentially adding CO2 to the in situ generated copolymer I, controlled diblock copolymer III could be obtained again with high selectivity and excellent polydispersities. It is worthwhile noting that the markedly stronger Lewis acid B(C6F5)3 was not an active catalyst either for epoxide/anhydride copolymerization or for epoxide/anhydride/CO2 block copolymerizations. However, it was found that when B(C6F5)3 was added to diblock copolymer III, the carbonate block of the polymer was depolymerized to give the respective cyclic carbonate.
Gnanou, Feng, and co-workers investigated triethyl borane (BEt3) catalyzed copolymerizations of epoxides with organic isocyanates to polyurethanes [55], challenging transformations due to the propensity of most isocyanates to undergo side-reactions such as homopolymerization, cyclotrimerization and [2 + 3] cycloaddition with epoxides. Strongly electron-withdrawing p-tosyl isocyanate (TSI) was found to be the most suitable substrate selectively undergoing copolymerization with a range of epoxides to form almost perfectly alternating polyurethanes (less than 1% ether linkages in the polymer) in high yields and purities. Interestingly, when investigating the impact of the Lewis acidity of the borane catalyst on the rate of propylene oxide/TSI copolymerization, it was found that with BPh3, a stronger Lewis acid than BEt3, the reaction occurred explosively with the release of large amounts of heat. Astonishingly, with very low catalyst loadings of 0.05 mol% BPh3 and 0.1 mol% PPNCl, complete polymerization was accomplished within 10 min with turnover frequencies of over 10,000 h−1, and a remarkably high number of averaged molecular weight (Mn = 225,000 g/mol), exclusively alternating polyurethane structure and a polydispersity of 1.51. Unfortunately, a substrate scope with this highly active and selective catalyst system was not investigated (Scheme 6).
3. BPh3 as an FLP Component in Hydrogenation Catalysis
Stephan and co-workers’ discovery that Lewis acidic boranes can actively participate in the heterolytic cleavage of H2 via a frustrated Lewis pair (FLP) approach [37,38,39] opened the door to the design of metal-free catalysts for the hydrogenation of unsaturated organic substrates [56,57,58,59,60]. Briefly, FLPs comprise sterically encumbered Lewis acid–base pairs, unable to form Lewis acid–base adducts due to unfavorable steric interactions. As a result, Lewis acidity and basicity of the individual FLP components remain unquenched, thus enabling the heterolytic cleavage of H2, a key step in catalytic hydrogenation reactions [41,42,43,44,45]. Typically, active FLP catalysts are comprised of strongly Lewis acidic, often highly fluorinated or chlorinated aryl boranes coupled with relatively weak N- or P-containing Lewis bases. Theoretical studies concerning the thermodynamic feasibility of the FLP-mediated H2 cleavage, however, suggested that weak Lewis acids could be active as well, provided that a sufficiently strong base is present [61]. In this context, it is worthwhile noting that Stephan, in one of his seminal papers [38], disclosed the stochiometric H2 cleavage utilizing the FLPs B(C6F5)3/P(But)3 and BPh3/P(But)3. The former FLP with B(C6F5)3 (ΔHHA = −112 kcal/mol) quickly and quantitatively generated the phosphonium borate salt [HB(C6F5)3][HP(But)3] at room temperature, whereas with less acidic BPh3 (ΔHHA = −74.4 kcal/mol) salt [HBPh3][HP(But)3] was formed in only 33% yields after 24 h. However, theoretical calculations by Papai and co-workers appear to contradict the effectiveness of BPh3/P(But)3 to cleave H2 as the reaction was calculated to be endergonic (ΔGR = +18.2 kcal/mol), while for B(C6F5)3/P(But)3 the reaction with H2 was exergonic (ΔGR = −14.7 kcal/mol) [61].
Building on these and other results [62,63], Krempner et al. employed the bulky organosuperbases Verkade base (pKa = ~33 in CH3CN) and phosphazene (pKa ~28 in CH3CN) in combination with BPh3 (Scheme 7) [64]. Upon exposure to H2, both FLPs instantly generated the corresponding borate salts 7 and 8 in yields of 71% and 85%, resp. Solutions of 8 appeared to be thermally stable, while 7 in solution quickly released H2 when heated to 60 °C, indicating reversibility of heterolytic hydrogen cleavage, which is key to the development of effective hydrogenation catalysts. In fact, BPh3/phosphazene was demonstrated to be an active FLP catalyst system in the quantitative hydrogenation of the N-benzylidene aniline to N-benzyl aniline in THF as solvent.
An interesting extension of this concept was recently introduced by Hu and co-workers for the FLP-catalyzed hydrogenation of alkynes [65]. Because the molecular FLP BPh3/pyridine thermally degraded during hydrogenation of phenylacetylene (conversion 12% after 12 h at 120 °C and 50 bar H2), the group developed a polymeric Lewis acid based on BPh3. The synthesis is illustrated in Scheme 8 and involves a classical Friedel–Crafts reaction of BPh3 with 1,2-dichloroethane in the presence of AlCl3.
This polymeric Lewis acid, Poly-BPh3, combined with pyridine as the Lewis base was shown to be active in the semi-hydrogenation of a variety of aliphatic and aromatic terminal alkynes at 120 °C to preferentially give the corresponding alkenes. The Lewis acid component of this FLP, Poly-BPh3, is reusable without loss of catalytic activity after being recovered from the reaction mixture, and appears to be thermally and hydrolytically remarkably stable.
Recent developments concerning iridium- and ruthenium-catalyzed hydrogenations of organosilicon compounds [66,67,68,69,70] motivated the Cantat group to design FLP catalysts for the metal-free hydrogenation of various organosilanes with Si-X bonds (X = OTf, I, Br, Cl) [71]. After extensive screening, B(2,6-F2-C6H3)3 as the Lewis acid combined with stochiometric amounts of 2,2,6,6-tetramethylpiperidine (TMP) as the base was identified as the most effective FLP catalyst for the hydrogenation of Me3SiX (X = OTf, I, Br) to Me3SiH (Scheme 9). Employing the same FLP catalyst, Et3SiOTf, Ph3SiOTf, and (Pri)2Si(OTf)2 could be hydrogenated to the corresponding hydrosilanes in good yields as well. However, irrespective of the base used, B(2,6-F2-C6H3)3 was not able to convert the more challenging substrate Me3SiCl to Me3SiH, primarily because of the comparably stronger Si-Cl bond and lower hydricity of the respective borohydride anion, HB(2,6-F2-C6H3)3–, formed upon H2-splitting. Building on the idea of favoring the thermodynamics by increasing the hydricity of the borohydride, less Lewis acidic BPh3 was tested in combination with stronger base phosphazene to ensure H2-cleavage. With this FLP in hand, 28% of Me3SiCl were converted after 72 h to Me3SiH in yields of 26%. Moreover, upon adding the chloride abstracting additive NaOTf, the yields of Me3SiH were further increased to a respectable 52% (Scheme 9).
Silylenes, divalent silicon species of general formula R1R2Si, are known to exhibit amphoteric behavior, capable of serving either as Lewis acids or Lewis bases. In this respect, Kira, Mueller, and co-workers investigated the potential of sterically overcrowded silylene 9 to be an active component in the heterolytic cleavage of H2 via an FLP approach (Scheme 10) [72]. It was found that 9 not only actively engaged in H2 splitting, it was also fully hydrogenated to hydrosilane 13 in isolated yields ranging from 75–84% with catalytic amounts of either Lewis base (PPh3, NPh3, PEt3, and NEt3) or Lewis acids such as BEt3 and BPh3. NMR spectroscopic investigations in solution showed that neither PPh3 nor BPh3 forms a classical Lewis acid–base adduct, indicative of FLP formation in both cases. Nonetheless, when H2 was introduced into the solution, rapid formation of hydrosilane 13 occurred. Based on DFT calculations, a mechanism was proposed in which BPh3 initially forms the weak complex 10 with the silylene, which is stabilized by ca. −7 kcal/mol compared to the starting materials. Subsequent heterolytic cleavage of H2 leads to the formation of silylium borate 12; its formation is slightly endothermic (+14 kcal/mol). Finally, the hydride is transferred from the borohydride to the silylium cation to give the final product 2, a strongly exothermic process (−37 kcal/mol). It should be noted that strong Lewis acid B(C6F5)3 was not capable of hydrogenating silylene 9, instead, degradation occurred.
4. Hydrosilylation Catalysis
Arguably, one of the most challenging metal-free catalyzed reductive transformations represents the reduction of organic amides to amines [73,74,75]. In 2016, Okuda and co-workers reported the highly selective hydrosilylation of a variety of aromatic and aliphatic tertiary amides to the corresponding amines with 10 mol% BPh3 as the catalyst system and 2 eq. of PhMeSiH2 as the reducing agent (Scheme 11) [76].
Aromatic and aliphatic tertiary amines were obtained in good to excellent isolated yields. In contrast, α,β-unsaturated amides underwent hydrosilylation of the olefinic groups (C=C) to give the respective α-silylated amides. In addition, this process showed a good functional group tolerance with additives such as acetophenone, ethyl acetate, N-benzylidene aniline, tert-butyl isocyanate, and thiophene not inhibiting the activity of the catalyst. Only in the case of benzaldehyde and ethanol, hydrosilylation and dehydrosilylation, respectively, occurred prior to amide reduction. The addition of pyridine deactivated the catalyst due to irreversible Lewis acid–base adduct formation with BPh3. The BPh3-catalyzed amide hydrosilylations described by Okuda’s group appear to be superior in terms of both functional group tolerance and chemo-selectivity when compared with other metal-free catalysts reported in the literature [74,75]. For example, B(C6F5)3 catalyzes the reduction of both functional groups of 14, the ester as well as the amide, to give benzylamine 15. In contrast, BPh3 reduces the amide group while keeping the ester group intact leading to the selective formation of the ester-functionalized benzylamine 16 (Scheme 11) [74].
The same group reported the BPh3-catalyzed hydrosilylation of CO2 (Scheme 12) [77]. This approach enabled the highly selective formation of silyl formates with various hydro silanes such as PhSiH3, PhMeSiH2 or Et3SiH in polar solvents such as acetonitrile, nitromethane and propylene carbonate. No turnover was observed in less polar and non-polar solvents such as benzene, toluene, tetrahydrofuran (THF) and CH2Cl2. Mechanistically the BPh3-catalyzed hydrosilylation of CO2 is suggested to proceed via the dual activation of CO2 and organosilane by BPh3, where polar solvents with high dielectric constants stabilize the partially charged transient species (Scheme 11). Note that strongly Lewis acidic B(C6F5)3 itself was not capable of catalyzing the hydrosilylation of CO2, while as the FLP component in combination with tetramethyl piperidine (TMP) as a Lewis base and with an excess of triethylsilane CO2 was quantitatively reduced to give CH4 [78].
More recently, Ema and co-workers reported the utilization of CO2 as C1-feedstock for the reductive methylation of secondary and tertiary aromatic amines derivatives, employing PhSiH3 as a reducing agent and BPh3 as the catalyst (Scheme 13) [79,80]. The N-methylation of sec-amines was achieved with 1 atm of CO2, 4.0 equivalents of PhSiH3 and 5 mol% of BPh3 under solvent-free conditions at 30 °C. The process tolerates various functional groups such as halides, nitrile, nitro, ester and alkoxy groups. On the other hand, C-methylenation was observed with tertiary aromatic amines to give the corresponding diarylmethanes in moderate yields at 40 °C. Note that B(C6F5)3 was inactive even at higher temperatures in any of these transformations, probably due to irreversible Lewis acid–base adduct formation. Similar to what was seen for the reaction of tertiary aromatic amines, 1-methylindole at 30°C converted to 3,3′-methylenebis(1-methylindole) 18 in good yields. On the contrary, B(C6F5)3 catalyzed the formal hydrogenation of the olefinic bond of N-methyl indole to selectively give N-methyl dihydroindole 19.
Motivated by the moderate water tolerance of B(C6F5)3 in catalytic hydrogenation reactions [81], Ingleson and co-workers disclosed the triaryl borane catalyzed reductive aminations of aldehydes to aromatic and aliphatic amines using Me2PhSiH as the reducing agent [82,83]. Notably, the employed catalysts BPh3 and B(C6F5)3 showed strikingly different selectivities for aliphatic and aromatic amine substrates (Scheme 14). With B(C6F5)3 as the catalyst, the amine substrates were limited to aryl amines (pKa of the conjugate acid < 12), while with weaker Lewis acidic BPh3, the more basic alkyl amines (pKa of the conjugate acid > 16) performed well with excellent conversions and yields. These findings were attributed to the different interactions of the amine base with the intermediately formed water adducts H2O→BPh3, and H2O→B(C6F5)3. Thus, with more basic alkyl amines, irreversible deprotonation of the highly acidic adduct H2O→B(C6F5)3 (pKa = 8.4 in CH3CN) occurred, resulting in the degradation of the catalyst. BPh3, on the other hand, undergoes rapid protodeboronation in the presence of the more acidic aryl amines. It is worth noting that the in situ generated Lewis acid B(3,5-Cl2-C6H3)3, whose Lewis acidity is in between B(C6F5)3 and BPh3, was capable of catalyzing the reductive amination with both aryl and alkyl amines. However, a detailed substrate scope was not explored with B(3,5-Cl2-C6H3)3 as the catalyst.
5. Miscellaneous
Krempner and co-worker reported the Lewis acid catalyzed [2 + 3] cycloaddition of Bestmann’s ylide, Ph3P=C=C=O, with nitrones to produce a variety of previously unknown 5-isoxazolidinones with exocyclic phosphonium ylide functionality in excellent isolated yields [84]. Subsequent quenching with reactive aldehydes via a classical Wittig reaction gave access to 5-isoxazolidinones with exocyclic double bonds (Scheme 15).
Of the boron-containing Lewis acid catalysts tested, BPh3 proved to be most effective in providing the cycloaddition product in almost quantitative yields within 8 h at room temperature. Weaker Lewis acids such as BEt3, BMes3 and B(OMe)3 were inactive even at elevated temperatures, while B(C6F5)3, the strongest amongst the Lewis acids studied, required 80 °C and 16 h to quantitatively produce the cycloaddition product. The authors proposed a mechanism (Scheme 16) in which the borane catalyst activates the nitrone via Lewis acid–base interactions, which facilitates the nucleophilic attack of the ylidic carbon of Ph3PCCO resulting in the cyclized borane-5-isoxazolidinone adduct from which the 5-isoxazolidinone is liberated via borane–nitrone adduct formation. The higher activity of the BPh3 over its more acidic counterpart B(C6F5)3 is attributed to the latter forming stable adducts with both Ph3PCCO and the nitrone, while BPh3 does not [85].
Recently, Cummins and co-workers disclosed the synthesis and reaction behavior of highly strained tri-tert-butylphosphatetrahedrane 20 (Scheme 17) [86]. Thus, upon the addition of BPh3 (20 mol%), 20 underwent rapid dimerization to form the bicyclic structure 22 in 72% yield. To trap potential intermediates, tetrahedrane 20 was treated with a 20-fold excess of styrene, which in the presence of BPh3 (20 mol%) furnished the cycloaddition product 23 in a yield of 88%. Similarly, BPh3 catalyzed the cycloaddition of 20 with 1 atm of ethylene to give the corresponding bicyclic structure 24 in yields of 74%. Quantum chemical calculations revealed BPh3 to mediate C−P bond cleavage to give intermediately tri-tert-butyl-phospacyclobutadiene 21, which subsequently either dimerizes to 22 or undergoes a formal [2 + 4] cycloaddition with styrene and ethylene to yield 23 and 24, respectively.
Finally, Grubba et al. demonstrated the ability of BPh3 to act as an efficient Lewis acid catalyst in the diphosphination of CO2 and CS2 (Scheme 18) [87]. BPh3-catalyzed insertion of CO2 and CS2 into the P-P bond of unsymmetrical diphosphines 25 led to the selective formation of products 26 of the general formula (R2N)2P–E–C(=E)–P(But)2, where the central carbon of CE2 binds to the more nucleophilic P(But)2 moiety. In elucidating the reaction mechanism, it was found that neither CO2 nor the bulky diphosphines reacted with BPh3 individually to form the Lewis acid–base adducts, respectively, suggesting FLP-type behavior. It was proposed that the FLP diphosphine/BPh3 synergistically interacts with CO2 resulting in rapid P-P bond insertion.
Author Contributions
Writing—review and editing, S.M. and C.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Catalysis Science Program, under Award DE-SC0019094.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Krause, E.; Nitsche, R. Darstellung von Organischen Bor-Verbindungen Mit Hilfe von Borfluorid, II.: Bortriphenyl Und Phenyl-borsäure. Ber. Dtsch. Chem. Ges. A/B 1922, 55, 1261–1265. [Google Scholar] [CrossRef] [Green Version]
- Zettler, F.; Hausen, H.D.; Hess, H. Crystal and Molecular Structure of Triphenylborane. J. Organomet. Chem. 1974, 72, 157–162. [Google Scholar] [CrossRef]
- Guibert, C.R.; Little, J.L. Alkyl- and Arylboranes. In Ullmanns’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag: Weinheim, Germany, 2005. [Google Scholar] [CrossRef]
- Wu, Q.; Esteghamatian, M.; Hu, N.-X.; Popovic, Z.; Enright, G.; Tao, Y.; D’Iorio, M.; Wang, S. Synthesis, Structure, and Electroluminescence of BR2q (R = Et, Ph, 2-Naphthyl and q = 8-Hydroxyquinolato). Chem. Mater. 2000, 12, 79–83. [Google Scholar] [CrossRef]
- Liu, Q.-D.; Mudadu, M.S.; Thummel, R.; Tao, Y.; Wang, S. From blue to red: Syntheses, structures, electronic, and electroluminescent properties of tunable luminescent N,N chelate boron complexes. Adv. Funct. Mater. 2005, 15, 143–154. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Chi, Y.; Liu, C.-S.; Yu, J.-K.; Cheng, Y.-M.; Chen, K.-S.; Chou, P.-T.; Peng, S.-M.; Lee, G.-H.; Carty, A.J.; et al. Rational color tuning and luminescent properties of functionalized boron-containing 2-pyridylpyrrolide complexes. Adv. Funct. Mater. 2005, 15, 567–574. [Google Scholar] [CrossRef]
- Liddle, B.J.; Silva, R.M.; Morin, T.J.; Macedo, F.P.; Shukla, R.; Lindeman, S.V.; Gardinier, J.R. BORAZANs: Tunable fluorophores based on 2-(pyrazolyl)aniline chelates of diphenylboron. J. Org. Chem. 2007, 72, 5637–5646. [Google Scholar] [CrossRef] [Green Version]
- Tokoro, Y.; Nagai, A.; Kokado, K.; Chujo, Y. Synthesis of Organoboron Quinoline-8-thiolate and Quinoline-8-selenolate Complexes and Their Incorporation into the π-Conjugated Polymer Main-Chain. Macromolecules 2009, 42, 2988–2993. [Google Scholar] [CrossRef]
- Li, D.; Wang, K.; Huang, S.; Qu, S.; Liu, X.; Zhu, Q.; Zhang, H.; Wang, Y. Brightly fluorescent red organic solids bearing boron-bridged π-conjugated skeletons. J. Mater. Chem. 2011, 21, 15298–15304. [Google Scholar] [CrossRef]
- Frath, D.; Azizi, S.; Ulrich, G.; Retailleau, P.; Ziessel, R. Facile Synthesis of Highly Fluorescent Boranil Complexes. Org. Lett. 2011, 13, 3414–3417. [Google Scholar] [CrossRef]
- Kubota, Y.; Hara, H.; Tanaka, S.; Funabiki, K.; Matsui, M. Synthesis and Fluorescence Properties of Novel Pyrazine-Boron Complexes Bearing a β-Iminoketone Ligand. Org. Lett. 2011, 13, 6544–6547. [Google Scholar] [CrossRef]
- Li, D.; Zhang, H.; Wang, C.; Huang, S.; Guo, J.; Wang, Y. Construction of full-color-tunable and strongly emissive materials by functionalizing a boron-chelate four-ring-fused π-conjugated core. J. Mater. Chem. 2012, 22, 4319–4328. [Google Scholar] [CrossRef]
- Shimizu, S.; Murayama, A.; Haruyama, T.; Iino, T.; Mori, S.; Furuta, H.; Kobayashi, N. Benzo[c,d]indole-Containing Aza-BODIPY Dyes: Asymmetrization-Induced Solid-State Emission and Aggregation-Induced Emission Enhancement as New Properties of a Well-Known Chromophore. Chem. Eur. J. 2015, 21, 12996–13003. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, Y.; Liu, Q.; Li, Z.; Yan, H.; Ji, C.; Duan, J.; Liu, Z. Aggregation-induced emission (AIE) of pyridyl-enamido-based organoboron luminophores. Chem. Commun. 2015, 51, 784–787. [Google Scholar] [CrossRef] [PubMed]
- Dou, C.; Ding, Z.; Zhang, Z.; Xie, Z.; Liu, J.; Wang, L. Developing Conjugated Polymers with High Electron Affinity by Replacing a C-C Unit with a B-N Unit. Angew. Chem., Int. Ed. 2015, 54, 3648–3652. [Google Scholar] [CrossRef]
- Liu, F.; Ding, Z.; Liu, J.; Wang, L. An organoboron compound with a wide absorption spectrum for solar cell applications. Chem. Commun. 2017, 53, 12213–12216. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Nakamuro, T.; Yamashita, K.; Yanagisawa, H.; Nureki, O.; Kikkawa, M.; Gao, H.; Tian, J.; Shang, R.; Nakamura, E. B/N-Doped p-Arylenevinylene Chromophores: Synthesis, Properties, and Microcrystal Electron Crystallographic Study. J. Am. Chem. Soc. 2020, 142, 18990–18996. [Google Scholar] [CrossRef]
- Krause, E. Valence problem of boron: A series of strikingly stable secondary valence compounds of boron triphenyl. Ber. Dtsch. Chem. Ges. 1924, 57B, 813–818. [Google Scholar] [CrossRef]
- Nakamura, T.; Umeno, M. Triarylborane-Amine Compounds, and Their (co)Polymers, and Antifouling Agents. Japan Patent JP2000143673 A, 26 May 2000. [Google Scholar]
- Shimada, A.; Kohara, M.; Shibuya, Y. Triphenylboron-Containing Polymers and Their Use as Marine Antifouling Agents. WO9833829 A1, 6 August 1998. [Google Scholar]
- Saeki, Y.; Kumagaya, H. Synergistic Marine Antifouling Agents Containing Triphenylboranes and Higher Aliphatic Polyamines. Japan Patent JP10182322 A, 7 July 1998. [Google Scholar]
- Lawson, K.R.; Mound, W.R.; Whittingham, W.G. Preparation of Pyrazolylborane Derivatives as Fungicides in Plants. World Intellectual Property Organization. WO9711952 A1, 3 April 1997. [Google Scholar]
- Lawson, K.R.; Mound, W.R.; Whittingham, W.G. Preparation of Pyrimidinylborane Derivatives as Fungicides. World Intellectual Property Organization. WO9711951 A1, 3 April 1997. [Google Scholar]
- Tsang, T.H.; Strutzel, J.L. Preparation of Bis(amineborane)alkanes as Agrochemical Fungicides. U.S. Patent US5075292 A, 24 December 1991. [Google Scholar]
- Massey, A.G.; Park, A.J.; Stone, F.G.A. Tris(pentafluorophenyl)boron. Proc. Chem. Soc. 1963, 2, 212. [Google Scholar]
- Mayer, R.J.; Hampel, N.; Ofial, A.R. Lewis Acidic Boranes, Lewis Bases, and Equilibrium Constants: A Reliable Scaffold for a Quantitative Lewis Acidity/Basicity Scale. Chem. Eur. J. 2021, 27, 4070–4080. [Google Scholar] [CrossRef]
- Gao, H.; Battley, A.; Leitao, E.M. The Ultimate Lewis Acid Catalyst: Using Tris(Pentafluorophenyl) Borane to Create Bespoke Siloxane Architectures. Chem. Commun. 2022, 58, 7451–7465. [Google Scholar] [CrossRef] [PubMed]
- Nori, V.; Pesciaioli, F.; Sinibaldi, A.; Giorgianni, G.; Carlone, A. Boron-Based Lewis Acid Catalysis: Challenges and Perspectives. Catalysts 2022, 12, 5. [Google Scholar] [CrossRef]
- Kumar, G.; Roy, S.; Chatterjee, I. Tris(Pentafluorophenyl)Borane Catalyzed C–C and C–Heteroatom Bond Formation. Org. Biomol. Chem. 2021, 19, 1230–1267. [Google Scholar] [CrossRef] [PubMed]
- Hackel, T.; McGrath, N.A. Tris(Pentafluorophenyl)Borane-Catalyzed Reactions Using Silanes. Molecules 2019, 24, 432. [Google Scholar] [CrossRef] [Green Version]
- Park, S. B(C6F5)3-Catalyzed Sp3 C—Si Bond Forming Consecutive Reactions. Chin. J. Chem. 2019, 37, 1057–1071. [Google Scholar] [CrossRef]
- Lawson, J.R.; Melen, R.L. Tris(Pentafluorophenyl)Borane and Beyond: Modern Advances in Borylation Chemistry. Inorg. Chem. 2017, 56, 8627–8643. [Google Scholar] [CrossRef] [Green Version]
- Oestreich, M.; Hermeke, J.; Mohr, J. A Unified Survey of Si–H and H–H Bond Activation Catalysed by Electron-Deficient Boranes. Chem. Soc. Rev. 2015, 44, 2202–2220. [Google Scholar] [CrossRef] [Green Version]
- Brook, M.A.; Grande, J.B.; Ganachaud, F. New Synthetic Strategies for Structured Silicones Using B(C6F5)3. In Silicon Polymers; Muzafarov, A.M., Ed.; Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 2010; Volume 235, pp. 161–183. ISBN 978-3-642-16047-9. [Google Scholar]
- Erker, G. Tris(Pentafluorophenyl)Borane: A Special Boron Lewis Acid for Special Reactions. Dalton Trans. 2005, 36, 1883–1890. [Google Scholar] [CrossRef]
- Piers, W.E.; Chivers, T. Pentafluorophenylboranes: From Obscurity to Applications. Chem. Soc. Rev. 1997, 26, 345. [Google Scholar] [CrossRef]
- Welch, G.C.; Juan, R.R.S.; Masuda, J.D.; Stephan, D.W. Reversible, Metal-Free Hydrogen Activation. Science 2006, 314, 1124–1126. [Google Scholar] [CrossRef] [Green Version]
- Welch, G.C.; Stephan, D.W. Facile Heterolytic Cleavage of Dihydrogen by Phosphines and Boranes. J. Am. Chem. Soc. 2007, 129, 1880–1881. [Google Scholar] [CrossRef]
- Spies, P.; Erker, G.; Kehr, G.; Bergander, K.; Fröhlich, R.; Grimme, S.; Stephan, D.W. Rapid Intramolecular Heterolytic Dihydrogen Activation by a Four-Membered Heterocyclic Phosphane–Borane Adduct. Chem. Commun. 2007, 5072–5074. [Google Scholar] [CrossRef]
- Mummadi, S.; Brar, A.; Wang, G.; Kenefake, D.; Diaz, R.; Unruh, D.K.; Li, S.; Krempner, C. “Inverse” Frustrated Lewis Pairs: An Inverse FLP Approach to the Catalytic Metal Free Hydrogenation of Ketones. Chem. Eur. J. 2018, 24, 16526–16531. [Google Scholar] [CrossRef]
- Paradies, J. Metal-Free Hydrogenation of Unsaturated Hydrocarbons Employing Molecular Hydrogen. Angew. Chem. Int. Ed. 2014, 53, 3552–3557. [Google Scholar] [CrossRef] [PubMed]
- Farrell, J.M.; Posaratnanathan, R.T.; Stephan, D.W. A Family of N-Heterocyclic Carbene-Stabilized Borenium Ions for Metal-Free Imine Hydrogenation Catalysis. Chem. Sci. 2015, 6, 2010–2015. [Google Scholar] [CrossRef] [Green Version]
- Erős, G.; Mehdi, H.; Pápai, I.; Rokob, T.A.; Király, P.; Tárkányi, G.; Soós, T. Expanding the Scope of Metal-Free Catalytic Hydrogenation through Frustrated Lewis Pair Design. Angew. Chem. Int. Ed. 2010, 49, 6559–6563. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.-H.; Kehr, G.; Fröhlich, R.; Wibbeling, B.; Schirmer, B.; Grimme, S.; Erker, G. Reaction of Frustrated Lewis Pairs with Conjugated Ynones-Selective Hydrogenation of the Carbon-Carbon Triple Bond. Angew. Chem. Int. Ed. 2011, 50, 7183–7186. [Google Scholar] [CrossRef]
- Chernichenko, K.; Madarász, Á.; Pápai, I.; Nieger, M.; Leskelä, M.; Repo, T. A Frustrated-Lewis-Pair Approach to Catalytic Reduction of Alkynes to Cis-Alkenes. Nature Chem. 2013, 5, 718–723. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Sanda, F.; Endo, T. Application of (Triphenylphosphinemethyl-Ene)Boranes to Thermally Latent Catalysts for Polyaddition of Bisphenol A Diglycidyl Ether with Bisphenol A: Model System of Epoxy−Novolac Resin. Macromolecules 2001, 34, 1134–1136. [Google Scholar] [CrossRef]
- Kobayashi, M.; Sanda, F.; Endo, T. Substituent Effect of (Triphenylphosphinemethylene)Boranes on Latent Catalytic Activity for Polyaddition of Bisphenol A Diglycidyl Ether with Bisphenol A: Model System of Epoxy−Novolac Resin. Macromolecules 2002, 35, 346–348. [Google Scholar] [CrossRef]
- Wang, L.; Kodamaa, K.; Hirose, T. DBU/benzyl bromide: An efficient catalytic system for the chemical fixation of CO2 into cyclic carbonates under metal- and solvent-free conditions. Catal. Sci. Technol. 2016, 6, 3872–3877. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, G.; Kodamaa, K.; Hirose, T. An efficient metal- and solvent-free organocatalytic system for chemical fixation of CO2 into cyclic carbonates under mild conditions. Green Chem. 2016, 18, 1229–1233. [Google Scholar] [CrossRef]
- Zhang, D.; Boopathi, S.K.; Hadjichristidis, N.; Gnanou, Y.; Feng, X. Metal-Free Alternating Copolymerization of CO2 with Epoxides: Fulfilling “Green” Synthesis and Activity. J. Am. Chem. Soc. 2016, 138, 11117–11120. [Google Scholar] [CrossRef]
- Yang, J.-L.; Wu, H.-L.; Li, Y.; Zhang, X.-H.; Darensbourg, D.J. Perfectly Alternating and Regioselective Copolymerization of Carbonyl Sulfide and Epoxides by Metal-Free Lewis Pairs. Angew. Chem. Int. Ed. 2017, 56, 5774–5779. [Google Scholar] [CrossRef] [PubMed]
- Andrea, K.A.; Kerton, F.M. Triarylborane-Catalyzed Formation of Cyclic Organic Carbonates and Polycarbonates. ACS Catal. 2019, 9, 1799–1809. [Google Scholar] [CrossRef]
- Andrea, K.A.; Kerton, F.M. Functionalized Polycarbonates via Triphenylborane Catalyzed Polymerization-Hydrosilylation. RSC Adv. 2019, 9, 26542–26546. [Google Scholar] [CrossRef] [Green Version]
- Andrea, K.A.; Wheeler, M.D.; Kerton, F.M. Borane Catalyzed Polymerization and Depolymerization Reactions Controlled by Lewis Acidic Strength. Chem. Commun. 2021, 57, 7320–7322. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Hadjichristidis, N.; Gnanou, Y.; Feng, X. Polyurethanes from Direct Organocatalytic Copolymerization of p -Tosyl Isocyanate with Epoxides. Angew. Chem. Int. Ed. 2021, 60, 1593–1598. [Google Scholar] [CrossRef]
- Stephan, D.W. Catalysis without Precious Metals; Bullock, R.M., Ed.; Wiley-VCH: Weinheim, Germany, 2010; pp. 261–275. ISBN 978-3-527-32354-32358. [Google Scholar]
- Stephan, D.W.; Greenberg, S.; Graham, T.W.; Chase, P.; Hastie, J.J.; Geier, S.J.; Farrell, J.M.; Brown, C.C.; Heiden, Z.M.; Welch, G.C.; et al. Metal-Free Catalytic Hydrogenation of Polar Substrates by Frustrated Lewis Pairs. Inorg. Chem. 2011, 50, 12338–12348. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.W. “Frustrated Lewis Pair” Hydrogenations. Org. Biomol. Chem. 2012, 10, 5740. [Google Scholar] [CrossRef]
- Erker, G.; Stephan, D.W. Frustrated Lewis Pairs I: Uncovering and Understanding; Topics in current chemistry; Springer: Heidelberg, Germany; New York, NY, USA, 2013; ISBN 978-3-642-36697-0. [Google Scholar]
- Hounjet, L.J.; Stephan, D.W. Hydrogenation by Frustrated Lewis Pairs: Main Group Alternatives to Transition Metal Catalysts? Org. Process Res. Dev. 2014, 18, 385–391. [Google Scholar] [CrossRef]
- Rokob, T.A.; Hamza, A.; Pápai, I. Rationalizing the Reactivity of Frustrated Lewis Pairs: Thermodynamics of H 2 Activation and the Role of Acid−Base Properties. J. Am. Chem. Soc. 2009, 131, 10701–10710. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Aquino, A.J.A.; Cordes, D.B.; Hung-Low, F.; Hase, W.L.; Krempner, C. A Zwitterionic Carbanion Frustrated by Boranes—Dihydrogen Cleavage with Weak Lewis Acids via an “Inverse” Frustrated Lewis Pair Approach. J. Am. Chem. Soc. 2013, 135, 16066–16069. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.J.M.; Labinger, J.A.; Bercaw, J.E. Homogeneous CO Hydrogenation: Dihydrogen Activation Involves a Frustrated Lewis Pair Instead of a Platinum Complex. J. Am. Chem. Soc. 2010, 132, 3301–3303. [Google Scholar] [CrossRef] [Green Version]
- Mummadi, S.; Unruh, D.K.; Zhao, J.; Li, S.; Krempner, C. “Inverse” Frustrated Lewis Pairs—Activation of Dihydrogen with Organosuperbases and Moderate to Weak Lewis Acids. J. Am. Chem. Soc. 2016, 138, 3286–3289. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Wu, Y.; Hu, X. Controlling the Lewis Acidity and Polymerizing Effectively Prevent Frustrated Lewis Pairs from Deactivation in the Hydrogenation of Terminal Alkynes. Org. Lett. 2021, 23, 3685–3690. [Google Scholar] [CrossRef]
- Tsushima, D.; Igarashi, M.; Sato, K.; Shimada, S. Ir-Catalyzed Hydrogenolysis Reaction of Silyl Triflates and Halides with H2. Chem. Lett. 2017, 46, 1532–1534. [Google Scholar] [CrossRef]
- Beppu, T.; Sakamoto, K.; Nakajima, Y.; Matsumoto, K.; Sato, K.; Shimada, S. Hydrosilane Synthesis via Catalytic Hydrogenolysis of Halosilanes Using a Metal-Ligand Bifunctional Iridium Catalyst. J. Organomet. Chem. 2018, 869, 75–80. [Google Scholar] [CrossRef]
- Glüer, A.; Schweizer, J.I.; Karaca, U.S.; Würtele, C.; Diefenbach, M.; Holthausen, M.C.; Schneider, S. Hydrosilane Synthesis by Catalytic Hydrogenolysis of Chlorosilanes and Silyl Triflates. Inorg. Chem. 2018, 57, 13822–13828. [Google Scholar] [CrossRef]
- Durin, G.; Berthet, J.-C.; Nicolas, E.; Thuéry, P.; Cantat, T. The Role of (tBuPOCOP)Ir(I) and Iridium(III) Pincer Complexes in the Catalytic Hydrogenolysis of Silyl Triflates into Hydrosilanes. Organometallics 2022, 41, 1786–1796. [Google Scholar] [CrossRef]
- Durin, G.; Berthet, J.-C.; Nicolas, E.; Cantat, T. Unlocking the Catalytic Hydrogenolysis of Chlorosilanes into Hydrosilanes with Superbases. ACS Catal. 2021, 11, 10855–10861. [Google Scholar] [CrossRef]
- Durin, G.; Fontaine, A.; Berthet, J.; Nicolas, E.; Thuéry, P.; Cantat, T. Metal-Free Catalytic Hydrogenolysis of Silyl Triflates and Halides into Hydrosilanes. Angew. Chem. Int. Ed. 2022, 61, e202200911. [Google Scholar] [CrossRef]
- Kira, M.; Müller, T. Dihydrogen Splitting Using Dialkylsilylene-Based Frustrated Lewis Pairs. Chem. Asian J. 2017, 12, 1204–1207. [Google Scholar] [CrossRef]
- Li, Y.; Molina de La Torre, J.A.; Grabow, K.; Bentrup, U.; Junge, K.; Zhou, S.; Brückner, A.; Beller, M. Selective Reduction of Amides to Amines by Boronic Acid Catalyzed Hydrosilylation. Angew. Chem. Int. Ed. 2013, 52, 11577–11580. [Google Scholar] [CrossRef]
- Chadwick, R.C.; Kardelis, V.; Lim, P.; Adronov, A. Metal-Free Reduction of Secondary and Tertiary N -Phenyl Amides by Tris(Pentafluorophenyl)Boron-Catalyzed Hydrosilylation. J. Org. Chem. 2014, 79, 7728–7733. [Google Scholar] [CrossRef]
- Blondiaux, E.; Cantat, T. Efficient Metal-Free Hydrosilylation of Tertiary, Secondary and Primary Amides to Amines. Chem. Commun. 2014, 50, 9349–9352. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, D.; Shirase, S.; Mashima, K.; Okuda, J. Chemoselective Reduction of Tertiary Amides to Amines Catalyzed by Triphenylborane. Angew. Chem. Int. Ed. 2016, 55, 13326–13329. [Google Scholar] [CrossRef]
- Mukherjee, D.; Sauer, D.F.; Zanardi, A.; Okuda, J. Selective Metal-Free Hydrosilylation of CO2 Catalyzed by Triphenylborane in Highly Polar, Aprotic Solvents. Chem. Eur. J. 2016, 22, 7730–7733. [Google Scholar] [CrossRef] [PubMed]
- Berkefeld, A.; Piers, W.E.; Parvez, M. Tandem Frustrated Lewis Pair/Tris(Pentafluorophenyl)Borane-Catalyzed Deoxygenative Hydrosilylation of Carbon Dioxide. J. Am. Chem. Soc. 2010, 132, 10660–10661. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Hiyoshi, M.; Maekawa, S.; Saiki, Y.; Ratanasak, M.; Hasegawa, J.; Ema, T. Deoxygenative CO2 Conversions with Triphenylborane and Phenylsilane in the Presence of Secondary Amines or Nitrogen-Containing Aromatics. Green Chem. 2022, 24, 2385–2390. [Google Scholar] [CrossRef]
- Ratanasak, M.; Murata, T.; Adachi, T.; Hasegawa, J.; Ema, T. Mechanism of BPh3-Catalyzed N-Methylation of Amines with CO2 and Phenylsilane: Cooperative Activation of Hydrosilane. Chem. A Eur. J. 2022, 28, e202202210. [Google Scholar] [CrossRef]
- Scott, D.J.; Simmons, T.R.; Lawrence, E.J.; Wildgoose, G.G.; Fuchter, M.J.; Ashley, A.E. Facile Protocol for Water-Tolerant “Frustrated Lewis Pair”-Catalyzed Hydrogenation. ACS Catal. 2015, 5, 5540–5544. [Google Scholar] [CrossRef] [Green Version]
- Fasano, V.; Ingleson, M.J. Expanding Water/Base Tolerant Frustrated Lewis Pair Chemistry to Alkylamines Enables Broad Scope Reductive Aminations. Chem. Eur. J. 2017, 23, 2217–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasano, V.; Radcliffe, J.E.; Ingleson, M.J. B(C6F5)3-Catalyzed Reductive Amination Using Hydrosilanes. ACS Catal. 2016, 6, 1793–1798. [Google Scholar] [CrossRef] [Green Version]
- Brar, A.; Unruh, D.K.; Ling, N.; Krempner, C. BPh3-Catalyzed [2+3] Cycloaddition of Ph3PCCO with Aldonitrones: Access to 5-Isoxazolidinones with Exocyclic Phosphonium Ylide Moieties. Org. Lett. 2019, 21, 6305–6309. [Google Scholar] [CrossRef]
- Brar, A.; Unruh, D.K.; Aquino, A.J.; Krempner, C. Lewis Acid Base Chemistry of Bestmann’s Ylide, Ph3PCCO, and Its Bulkier Analogue, (Cyclohexyl)3PCCO. Chem. Commun. 2019, 55, 3513–3516. [Google Scholar] [CrossRef]
- Riu, M.-L.Y.; Eckhardt, A.K.; Cummins, C.C. Dimerization and Cycloaddition Reactions of Transient Tri- Tert -Butylphosphacyclobutadiene Generated by Lewis Acid Induced Isomerization of Tri- Tert -Butylphosphatetrahedrane. J. Am. Chem. Soc. 2021, 143, 13005–13009. [Google Scholar] [CrossRef] [PubMed]
- Szynkiewicz, N.; Ponikiewski, Ł.; Grubba, R. Diphosphination of CO2 and CS2 Mediated by Frustrated Lewis Pairs—Catalytic Route to Phosphanyl Derivatives of Formic and Dithioformic Acid. Chem. Commun. 2019, 55, 2928–2931. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Catalytic copolymerization of bisphenol A and bisphenol A diglycidyl ether.

Scheme 2.
Proposed mechanistic steps of the catalytic copolymerization of bisphenol A and bisphenol A diglycidyl ether.
Scheme 2.
Proposed mechanistic steps of the catalytic copolymerization of bisphenol A and bisphenol A diglycidyl ether.

Scheme 3.
BPh3-catalyzed formation of cyclic carbonates and polycarbonates.

Scheme 4.
Sequential BPh3-catalyzed copolymerization and hydrosilylation.

Figure 1.
BPh3-catalyzed copolymerization of organic anhydrides and epoxides.

Scheme 5.
BPh3-catalyzed copolymerization to alternating copolymers and controlled diblock copolymers.
Scheme 5.
BPh3-catalyzed copolymerization to alternating copolymers and controlled diblock copolymers.

Scheme 6.
BPh3-catalyzed copolymerization of propylene oxide (PO) and p-tosyl isocyanate (TSI).

Scheme 7.
FLP-mediated stochiometric H2-cleavage and catalytic hydrogenation of N-benzylidene aniline.
Scheme 7.
FLP-mediated stochiometric H2-cleavage and catalytic hydrogenation of N-benzylidene aniline.

Scheme 8.
Synthesis and catalytic activity of Poly-BPh3 in the FLP-mediated semi-hydrogenation of alkynes to alkenes.
Scheme 8.
Synthesis and catalytic activity of Poly-BPh3 in the FLP-mediated semi-hydrogenation of alkynes to alkenes.

Scheme 9.
FLP-catalyzed hydrogenation of Me3SiX to Me3SiH.

Scheme 10.
BPh3-catalyzed hydrogenation of a silylene and proposed mechanism of formation.

Scheme 11.
Catalytic hydrosilylation of organic amides to amines.

Scheme 12.
Lewis acid catalyzed hydrosilylation of CO2 and proposed mechanism of silyl formate formation (TMP = 2,2,6,6-tetramethylpiperidine).
Scheme 12.
Lewis acid catalyzed hydrosilylation of CO2 and proposed mechanism of silyl formate formation (TMP = 2,2,6,6-tetramethylpiperidine).

Scheme 13.
BPh3-catalyzed N-methylation and C-methylenation of secondary and tertiary aromatic amines.
Scheme 13.
BPh3-catalyzed N-methylation and C-methylenation of secondary and tertiary aromatic amines.

Scheme 14.
Triaryl borane catalyzed reductive amination of aldehydes.

Scheme 15.
BPh3-catalyzed formal [2 + 3] cycloaddition of Ph3PCCO with nitrones to 5-isoxazolidinone with exocyclic phosphonium ylide moiety and their Wittig reaction.
Scheme 15.
BPh3-catalyzed formal [2 + 3] cycloaddition of Ph3PCCO with nitrones to 5-isoxazolidinone with exocyclic phosphonium ylide moiety and their Wittig reaction.

Scheme 16.
Proposed mechanism of the BPh3-catalyzed formal [2 + 3] cycloaddition of Ph3PCCO with nitrones (LA = Lewis acid).
Scheme 16.
Proposed mechanism of the BPh3-catalyzed formal [2 + 3] cycloaddition of Ph3PCCO with nitrones (LA = Lewis acid).

Scheme 17.
BPh3-catalyzed reactions of tri-tert-butylphosphatetrahedrane.

Scheme 18.
BPh3-catalyzed insertion of CE2 (E = O, S) into diphosphines.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mummadi, S.; Krempner, C. Triphenylborane in Metal-Free Catalysis. Molecules 2023, 28, 1340. https://doi.org/10.3390/molecules28031340
AMA Style
Mummadi S, Krempner C. Triphenylborane in Metal-Free Catalysis. Molecules. 2023; 28(3):1340. https://doi.org/10.3390/molecules28031340
Chicago/Turabian StyleMummadi, Suresh, and Clemens Krempner. 2023. "Triphenylborane in Metal-Free Catalysis" Molecules 28, no. 3: 1340. https://doi.org/10.3390/molecules28031340