

The Impact of Fluorination on the Design of Histone Deacetylase Inhibitors

1
Department of Pharmaceutical Chemistry, Hanoi University of Pharmacy, 13-15 Le Thanh Tong, Hanoi 10000, Vietnam
2
Immunobiology and Stem Cell Laboratory, Department of Internal Medicine V (Hematology and Oncology), Medical University Innsbruck, Anichstraße 35, 6020 Innsbruck, Austria
3
Tyrolean Cancer Research Institute, Innrain 66, 6020 Innsbruck, Austria
4
Department of Pharmaceutical and Medicinal Chemistry, Institute of Pharmacy, University of Greifswald, Friedrich-Ludwig-Jahn-Straße 17, 17489 Greifswald, Germany
*
Authors to whom correspondence should be addressed.
Molecules 2023, 28(4), 1973; https://doi.org/10.3390/molecules28041973
Submission received: 8 January 2023
/
Revised: 13 February 2023
/
Accepted: 16 February 2023
/
Published: 19 February 2023
(This article belongs to the Special Issue Bioorganic Chemistry: Current and Future Perspectives)
Abstract
:In recent years, histone deacetylases (HDACs) have emerged as promising targets in the treatment of cancer. The approach is to inhibit HDACs with drugs known as HDAC inhibitors (HDACis). Such HDACis are broadly classified according to their chemical structure, e.g., hydroxamic acids, benzamides, thiols, short-chain fatty acids, and cyclic peptides. Fluorination plays an important role in the medicinal–chemical design of new active representatives. As a result of the introduction of fluorine into the chemical structure, parameters such as potency or selectivity towards isoforms of HDACs can be increased. However, the impact of fluorination cannot always be clearly deduced. Nevertheless, a change in lipophilicity and, hence, solubility, as well as permeability, can influence the potency. The selectivity towards certain HDACs isoforms can be explained by special interactions of fluorinated compounds with the structure of the slightly different enzymes. Another aspect is that for a more detailed investigation of newly synthesized fluorine-containing active compounds, fluorination is often used for the purpose of labeling. Aside from the isotope 19F, which can be detected by nuclear magnetic resonance spectroscopy, the positron emission tomography of 18F plays a major role. However, to our best knowledge, a survey of the general effects of fluorination on HDACis development is lacking in the literature to date. Therefore, the aim of this review is to highlight the introduction of fluorine in the course of chemical synthesis and the impact on biological activity, using selected examples of recently developed fluorinated HDACis.

1. Introduction
In recent decades, progress has been made towards the faster diagnosis of cancer, for example, by multimodal imaging [1], focusing on particular biomarkers and subsequent analysis using omics technologies [2,3]. Distinct treatment approaches—apart from classical chemotherapy using anticancer drugs—such as through sophisticated stem cell transplantation [4,5] are resulting in novel opportunities in cancer treatment. Despite these achievements, cancer remains one of the leading causes of death worldwide [6].
Intense research into the pathogenesis of cancer generally improves our knowledge of the disease. However, the current advances in understanding the pathogenesis of cancer are only a small step forward. Indeed, many aspects are still completely unclear and a comprehensive understanding is needed for successful prevention and targeted treatment [7].
Further challenges include circumventing the development of resistance and achieving the best possible selectivity against cancer cells. Therefore, it is important to make smart structural modifications to existing anticancer drugs, or even better, to find auspicious targets that can be addressed by novel classes of bioactive compounds. In this context, various cancer-associated enzymes are coming into focus to serve as potential targets for antitumor therapeutics [8]. One promising representative among them are histone deacetylases (HDACs) [9].
Histones are the substrates of HDACs. After the deacetylation of acetylated lysine, the amino acid located at the N-terminal end of the histones is again positively charged. This is accompanied by an increased affinity for the negatively charged phosphate backbone of the DNA, ultimately resulting in a downregulation of its transcription. The dysregulation of gene expression, which is actually mediated by HDACs, is known to occur in a variety of cancers [10]. The abnormal activation of HDACs can be targeted by HDAC inhibitors (HDACis).
Probably the first HDACi to be discovered is trichostatin A (Figure 1), which is currently undergoing preclinical studies. However, some HDACis have already been approved by the United States Food and Drug Administration (FDA) for the treatment of various types of cancer. Some examples include the cyclic peptide romidepsin, and the hydroxamates vorinostat (also known as SAHA—suberoylanilide hydroxamic acid), belinostat, and panobinostat (Figure 1) [11]. In addition, some interesting synergistic properties were also discovered when HDACis were combined with established anticancer drugs [12]. All these encouraging features ensure that new HDACis are constantly being developed.
Our previous research on the design of HDACis involved derivatives bearing different heterocyclic cores, such as compounds based on quinazoline [13,14,15,16,17], indirubin [18], as well as 1,3-oxazole and 1,3-thiazole [19]. The use of heterocycles plays an important role in medicinal chemistry [20]. Within the concept of bioisosterism, heterocycles can be used to optimize drugs, for example, regarding pharmacokinetic and toxicological properties, efficacy, and selectivity [21].
Fluorination is another very common method of fine-tuning drug molecules [22,23,24]. This concerns, for instance, the development of glycomimetic drugs [25], metal complexes [26], and nucleosides [27], with all of these exerting also anticancer activity. The introduction of fluorine substituents in antitumor active compounds is performed for various reasons, such as bioanalytical labeling, in order to increase selectivity [28] or regulate the mechanism of cell death [29]. Fluorination is also a common method in the design of HDACis. However, to the best of our knowledge, it has no explicit focus in the literature thus far. Therefore, the aim of this review is to gain insight into the purpose and influence of fluorination in the recent development of HDACis.
2. Introduction of the Chemical Modification Generating Fluorination in HDACis
In the last decade, many HDACis with different core structures have been discovered, including short-chain fatty acids (e.g., phenylbutyric acid, valproic acid), hydroxamic acids (e.g., SAHA or suberoylanilide hydroxamic acid), benzamides, and acyclic peptides (e.g., depsipeptide). Nevertheless, they are subject to a very similar structural setup. The established pharmacophore comprises a cap and a zinc-binding group (ZBG), both of which are connected by a linker that occupies the binding channel (Figure 1) [30]. The cap interacts with the pockets of zinc-dependent HDAC, while the zinc-binding domain chelates zinc [31].
Several representatives of HDACis have undergone different phases of clinical trials for numerous types of cancer [32]. With the efforts to find new anticancer drugs, many studies have been carried out to search for novel potential HDACis, while fluorination plays a significant role. The different processes of introducing fluoro groups into these HDACis vary regarding the synthetic pathways, methods, and reaction conditions. This comprehensive work aims to summarize the progress that has been made in recent years on the fluorination of HDACis as anticancer agents.
2.1. Hydroxamic Acids
Hydroxamic acids are the best studied class of HDACis. To date, three of the four agents approved by the FDA with a confirmed HDAC-mediated mechanism of action for treatment in the clinic are hydroxamic acids. The representatives of this subclass are also the most studied group of fluorine-containing HDACis.
Generally, the simplest way to introduce fluorine into the chemical structure of compounds is to use precursors that already contain the fluorine substituent. Derivatives synthesized by this approach mostly use basic coupling reactions such as alkylation, reductive amination, or an amide-coupling reaction. The advantage of these methods is that they are straightforward, easy to apply, and can thus also be performed in small research laboratories.
Aboukhatwa and co-workers [33] designed, synthesized, and evaluated the bioactivity of a series of novel chemically diverse photoreactive probes (PRPs). The general strategy for the synthesis of PRPs shows that the process of introducing fluoride-containing subunits into the designed compounds is by reductive amination. This approach included two steps (Scheme 1). The first was the reaction between the amine derivative and perfluorobenzaldehyde in dichloromethane (DCM). Then, the deprotonation of imines was carried out with sodium borohydride (NaBH4) in methanol (MeOH) to obtain the product containing a fluorine group.
Furthermore, in this study by Aboukhatwa [33], several novel indol-containing hydroxamic acids were synthesized using the N-alkylation of an indole moiety or secondary amine. The conditions of this step depended on the structure of the starting materials (Scheme 2). This reaction in the synthesis pathway of the final products bearing an indole ring was carried out in dimethylformamide (DMF) in the presence of sodium hydride (NaH). The process of introducing fluorine into a secondary amine, however, was performed under alkaline conditions (K2CO3), and acetonitrile (MeCN) proved to be an optimal solvent.
In another study by Wang et al. [34], a series of hydroxamic acids bearing an indole ring were designed and synthesized. Fluorine-containing derivatives were obtained based on an amide-coupling reaction. This amide reaction was carried out in 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU) and trimethylamine (Me3N) in DCM (Scheme 3).
Using the same method, Goehringer, Peng, and co-authors [35] published a scheme for the synthesis of a novel pentafluorothio-substituted vorinostat-type HDACi (Scheme 4). This study described how the ethyl ester precursor SF5-SAHEt was obtained via amide-coupling reaction between ethyl hydrogen suberate and 4-pentafluorothioaniline in a moderate yield (63%). The catalysts used in this reaction were 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI), (dimethylamino)pyridine (DMAP), and triethylamine (Et3N). DCM was reported as a suitable solvent for this reaction.
Meyners et al. [36] investigated a series of novel perfluorinated hydroxamic acids. These final compounds were synthesized by an amide-coupling reaction following a two-step procedure (Scheme 5). The first was the conversion from fluoro-containing carboxylic acids to the respective acyl chlorides using thionyl chloride (SOCl2). Secondly, the reaction between the acyl chlorides and the particular aromatic amines (RNH2) or heteroaromatic amines produced intermediate amides. The strategy pursued for the synthesis of these compounds was to use already fluorinated starting materials.
Toutah et al. [37] reported the synthetic pathways used to prepare hydroxamic acids containing fluorine substituents (Scheme 6). Fluorine was introduced into these compounds by amine sulfonylation employing polyhalogenated benzenesulfonyl chloride to yield sulfonamide precursors. This reaction was conducted smoothly in DCM and Et3N (yield: 61–91%).
A chemical reaction regularly used for the synthesis of fluorinated hydroxamic acids is represented by esterification. Walton et al. [38] reported several series of novel target compounds incorporating perfluorinated chains linked by a ester group. The ester bond plays an important role in connecting perfluorinated alkyl chains to a SAHA phenyl head group. Steglich esterification proceeded with the relevant alcohols in the presence of EDCI and DMAP, which produced intermediate compounds (Scheme 7).
Vu et al. [39] reported a synthetic pathway of several novel fluorinated N-hydroxyheptanamides containing quinazolin-4(3H)-ones. The fluorine substituent was introduced into these compounds by a nucleophilic substitution reaction between the 3-butyl-6-hydroxy-2-methylquinazolin-4(3H)-one and fluorine-containing aldehydes in acetic acid to afford intermediates in 61–82% yields (Scheme 8).
Another strategy for the synthesis of novel fluorinated analogues covers the synthesis of heterocycles from the starting materials, which already contain the fluorine substituent. New phthalazino[1,2-b]-quinazolinone derivatives, which are multitarget HDAC inhibitors, were synthesized by Liu and co-authors [40]. The quinazolinone moieties were obtained from the reaction of 2-amino-5-fluorobenzoic acids and triphosgene (BTC, bis(trichloromethyl) carbonate). This reaction occurred smoothly in tetrahydrofurane (THF) (Scheme 9).
Fluorine-containing hydroxamic acids can also be synthesized by direct, catalytic vicinal difluorination. In the current research by Erdeljac et al. [41], novel fluorinated analogues of the HDACi vorinostat were synthesized from commercial sources or prepared from readily available starting materials (Scheme 10). The authors reported that 4-iodotoluene and Selectfluor® were employed as an inexpensive organo catalyst and terminal oxidant, respectively. This reaction was carried in a solution mixture of amine:HF (1:5) in the presence of Et3N·3HF; Olah’s reagent, which served as fluoride source; and a Brønsted acid activator.
In more detail, a Teflon® reaction vessel was loaded with the starting material, i.e., 4-iodotoluene, dichloroethan (DCE), an amine:HF (1:5) solution mixture of Olah’s reagent, Et3N·3HF, and Selectfluor®. The reaction vessel was sealed with a Teflon® screw cap and the mixture was stirred at room temperature for 25 h. The yields of the intermediate products in this reaction were acceptable (39–94%).
A potential strategy for attaching fluorine atoms to the linker chain of difluorinated SAHA analogues was published by Ariawan et al. [42]. Several methods exist to introduce two fluorine atoms into hydroxamic acids (Scheme 11). The monofluorinated compounds were synthesized through a four-step sequence procedure via a cyclic sulfate intermediate. These four steps included: 1. SOCl2, pyridine, DCM; 2. NaIO4, RuCl3, H2O, DCM, MeCN; 3. TBAF, THF, MeCN; 4. p-TsOH, dioxane. The second hydroxyl group was activated as the triflate with (CF3SO2)2O, pyridine in DCM, which, when displaced with fluoride produced the difluorinated product in a moderate yield. This reaction was conducted in TBAF, THF, and DCM. Nevertheless, a naphthalimide-containing compound was achieved through chlorination (SOCl2, pyridine, DCM), cyclic sulfate formation (1. NaIO4, RuCl3, H2O, DCM, MeCN; 2. H2SO4, H2O, THF), followed by ring-opening with TBAF in THF and MeCN, to obtain the fluorohydrin. After that, the fluorohydrin was treated with DeoxoFluor to yield the difluorinated products.
One of the main goals to fluorinate the derivatives of known HDACis is to study their pharmacokinetic properties (see below). This is often accomplished by labeling with radioactive isotopes of fluorine such as 18F.
Therefore, Strebl et al. [43] reported the radiochemical synthesis of [18F]Bavarostat. While previous studies showed that the radiolabeling of hydroxamic acids such as Bavarostat were difficult to achieve because of protracted, inefficient, low-yielding methods, the authors followed a novel approach by using ruthenium-mediated radiofluorination. This pathway has various advantages, including high efficiency, yield, and specific radioactivity. Bavarostat can be radiolabeled with 18F by deoxyfluorination through the in situ formation of an air-stable ruthenium π-complex of the intermediate phenol precursor (Scheme 12). The labeling proceeding had high conversion (more than 70% radiochemical yield, as estimated by TLC). After HPLC (high-performance liquid chromatography) purification, the overall non-decay corrected radiochemical yield of [18F]Bavarostat was 8.1% (n = 2).
In the study of Hendricks, Keliher, et al. [44] a synthetic strategy towards 18F-suberoylanilide hydroxamic acid (18F-SAHA) was developed. 18F (without carrier) was transferred to the starting material by the direct fluorination of 1,4-dinitrobenzene under optimized conditions for radiochemical synthesis and microwave heating at 120 ℃, in order to give 18F-labeled 1-fluoro-4-nitrobenzene (Scheme 13).
Recently, Strebl et al. [45] reported three novel fluorine-18 analogues of [11C]Martinostat ([18F]MGS1–3) via fluorination of the aromatic ring. The radiosynthesis of these radiotracers is illustrated in Scheme 14. In the synthetic routes of [18F]MGS1 and [18F]MGS2, the nitro group was replaced by 18F by using [18F]CsF, and DMSO was found as a suitable solvent. Meanwhile, in the synthetic pathway of [18F]MGS3, this reaction was carried out in DMSO with the presence of [18F]KF.
2.2. Benzamides
Benzamide-containing HDACis are the second most studied class of derivatives along with hydroxamic acids. Inhibitors with 2-aminobenzamide as ZBG show potential activity against HDAC class I, such as HDAC3 and HDAC8 [46]. There are several methods for the synthesis of fluorinated benzamides. The most common way to introduce fluorine into the intended compounds is again to use starting materials that are already fluorinated. In this context, simple reactions such as amide-coupling are used.
Jayathilaka and co-workers [47] investigated the synthetic pathway to give the fluorinated analogue NKL54 (Scheme 15). The trifluoromethyl group was introduced into the designed compounds using fluorinated aniline. The amide product is generally formed from the carboxylic acid and the aromatic amine in a 68% yield.
In the research of Bonomi et al. [48], the synthesis of novel histone deacetylase radio-tracers for positron-emission tomography (PET) imaging, including DFAHA and TFAHA precursors, was reported. The amide bonds of DFAHA and TFAHA were formed using particular anhydrides in DCM at room temperature (Scheme 16). The TFAHA precursor was synthesized in pyridine and acetyl chloride, while the reaction of the amine with bromofluoroacetyl chloride and triethylamine in DCM obtained the DFAHA precursor in a modest yield (15%).
In the current research of La and co-authors [49], the synthesis of novel fluorinated benzamide compounds as potential HDACis was described. The final products with the cap containing a fluorine moiety were obtained via substitution of the hydroxyl functional group to a fluorine substituent applying tosylate compounds (Scheme 17). DCM and tert-amyl alcohol were found as suitable solvents for this reaction.
In the study by Ibrahim et al. [50], a series of fluorine-containing compounds was prepared through Suzuki coupling reaction between the Boc-protected 4-bromo-2-nitroaniline and the corresponding fluorinated boronic acid compounds (Scheme 18). Tetrakis P(Ph)3Pd served as an efficient catalyst, and 1,2-dimethoxyethane was reported to be the optimal solvent. Sodium carbonate was added to the reaction mixture, which was needed for the sequestration of acid generated.
Also in this research [50], the authors reported another method for the synthesis of fluorinated 2-aminobenzamides using amide coupling. The amide intermediates were prepared by the reactions between the carboxylic acid derivatives and 1,2-phenylenediamines employing the coupling reagent HATU (hexafluorophosphate azabenzotriazole tetramethyl uronium) and N,N-diisopropylethylamine (DIPEA) as the base (Scheme 19). DMF was described to be the optimal solvent for these amide-forming reactions.
Schäker-Hübner and co-workers [51] also synthesized a series of novel benzamides. The Boc-protected intermediates containing fluorine were obtained via the Suzuki reaction of tert-butyl-(4-bromo-2-nitrophenyl)carbamate with alkyl boronic acids (Scheme 20). Tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4) was reported as a satisfactory catalyst for these coupling reactions. The mixture of toluene and ethanol was adduced as solvent.
In the research work of Liu and co-workers [40], as mentioned above among the hydroxamic acid derivatives (Scheme 9), the authors used the same strategy as for the synthesis of fluorinated hydroxamic acids to obtain new fluorinated benzamides. The quinazolinone ring was synthesized from the reaction between the initial 2-aminobenzoic acids with a fluorine substituent and triphosgene (Scheme 21).
Similar to hydroxamic acids (see above), some studies have also been performed to synthesize fluorinated benzamide derivatives containing the radiolabel 18F. Moreover, in the study of Bonomi et al. [48], the authors investigated a series of novel histone deacetylase radiotracers for PET imaging, including [18F]DFAHA and [18F]TFAHA (Scheme 22). The target benzamides were synthesized in radiochemical yields of 25% and 22%, respectively. The chemical structures of the radioactive products were confirmed by nuclear magnetic resonance (NMR) spectroscopy, and the purity was more than 95%, which was determined by analytical radio-HPLC.
Li and co-authors [52] investigated the synthesis of [18F] Fluoroethyl-INER1577 (i.e., INER1577-3, 92, Scheme 23). The first step was the fluorination of ethane-1,2-diyl bis(4-methylbenzenesulfonate) (TsOCH2CH2OTs) with K[18F] and kryptofix 2.2.2 (K2.2.2). This reaction was conducted smoothly in MeCN at 90 °C for 10 min. The radiochemical yield of TsOCH2CH2O18F was approximately 70%. Then, the intermediate reacted with INER1577 (91) in DMSO at 100 °C to produce [18F]Fluoroethyl-INER1577 (92). After 60 min, the radiochemical yield of [18F]Fluoroethyl-INER1577 was approximately 5–10%, and both chemical and radiochemical purities were more than 99%.
In another work by Li et al. [53], [18F] INER1577-3 was synthesized by the one-step method (Scheme 24). This reaction was carried out with [18F]fluoride without carrier addition in the presence of K2CO3 and K2.2.2 in 85% MeCN/water. The optimum temperature of this step was approximately 100 °C. The radiochemical purity of the final product was more than 95% (determined by the analytical RP-HPLC method).
2.3. Thiols
In recent years, besides the research on HDACis based on hydroxamic acids or benzamides, thiol derivatives have also been attracting much attention. This may be related to the ability of thiols to chelate zinc well [54]. Several research studies on fluorinated thiol derivatives have also been published.
For example, Chuman et al. [55] described methods for the synthesis of fluoroalkene-containing HDACis. As outlined in Scheme 25, the designed thiols were obtained through the reactions between the intermediate aldehyde and triethyl 2-fluoro-2-phosphonoacetate (EtO)2P(O)CHFCOOEt to obtain fluoroalkene in the presence of n-butyllithium (n-BuLi) in THF. Interestingly, the advantage of this method is that the fluoroalkene was formed with E-selectivity.
In another study of Wen et al. [56], a series of novel thiol-based HDACis was synthesized. Fluorine was introduced into the designed compounds by condensation reactions. These reactions occurred between 4-fluoroacetophenone or 4-fluoropropiophenone and dimethyl oxalate (Scheme 26). The intermediates were transferred to phenylpyrazole esters via the treatment of these compounds with hydrazine hydrate in acetic acid, followed by hydrolysis in the presence of sodium hydroxide (NaOH) in MeOH, to produce carboxylic acids.
2.4. Short-Chain Fatty Acids
Several natural short-chain fatty acids have shown promise to serve as potential HDACis [57,58]. There were also studies on fluorine-containing fatty acids comparing the HDAC inhibitory activities of natural amino acids and fluorinated amino acid derivatives.
A series of fluorinated amino acid esters were reported in the research study of Lübke et al. [59]. These potent and selective HDACis were synthesized from several unnatural amino acids bearing monofluoroalkyl side chains (Scheme 27). The process of introducing fluorine-containing moieties into the designed compounds is accomplished through alkylation.
A second method described by Lübke et al. [59] to introduce fluorine into the chemical structure of amino acid esters was to use an addition reaction (Scheme 28). The fluorinated intermediates were achieved by bromofluorination with 1,3-dibromohydantoine (DBH) and Olah’s reagent (Py·9HF) or N-bromosuccinimide (NBS), Et3N·3HF. After this step, a mixture of bromofluoro ester and dibromides was obtained. Following separation gave the desired products.
The authors reported that fluorine substituents played an important role in the interaction of HDACis with the active site of the enzymes. This research study also showed that a fluoro vinyl moiety had a similar role to an apolar amide group in terms of physical or chemical properties, which can lead to similar biological properties [59].
2.5. Cyclic Peptides
There are also fluorinated cyclic peptides with potential activity as HDACis. Such derivatives were recently designed and synthesized by Zhang, Liu, Gao, and co-workers [60,61]. All compounds reported in these studies were analogues of largazole (Figure 2). This is a cyclic depsipeptide derived from a cyanobacterium of the genus Symploca. It effectively inhibits the HDACs of class I [62].
The fluorine-containing side chain fragments were provided via several types of reactions, which are all described in Scheme 29 below. The first step to introduce the fluorine for the design of cyclic peptides was the alkylation with ethyl 2-bromo-2,2-difluoroacetate in the presence of triphenylphosphine and diethylzinc in THF. An alternative method for fluorination was through a Reformatsky reaction with ethyl 2-bromo-2,2-difluoroacetate, zinc in THF. Another pathway involved a two-step process. The first step was the preparation of 1-alkynyl(phenyl)iodonium tetrafluoroborate from alkyne. Then, the intermediate was fluorinated by the reaction with 20% HF aqueous solution in CHCl3. Moreover, the use of starting materials that already contain the fluorine substituent is also feasible.
3. Biological Significance and Purpose of Fluorination in HDACis
3.1. Potency
The potency of a drug is often a very important parameter in the medicinal chemistry design of novel compounds [63]. Potency generally refers to the effectivity of an active ingredient. This is expressed by the concentration required to achieve a certain pharmacological effect, e.g., enzyme inhibition or cytotoxicity [64]. Increasing the potency can ensure both engagement with the target and the avoidance of off-target activity. If it is possible to use a drug at a lower dose due to its high potency, this may be associated with less toxicity. The particular target plays a role in each case, in order to prove which transformations of the chemical structure ultimately also lead to an increase in potency. Nevertheless, some modifications occur with high frequency in different drug classes and contribute to an improvement in potency [65]. These structural alterations include, among others, fluorination. Recent examples where fluorination has had an impact on the efficacy of HDACis are described below.
Erdeljac and co-workers [41] investigated the influence of the introduction of the chiral 1,2-difluoroethylene unit as a hybrid bioisostere of the trifluoromethyl and ethyl (BITE) group in a series of eight vorinostat derivatives (Scheme 10). Compared to pure vorinostat, the latter additionally possess an ester moiety with different alkyl lengths of the alcohol component. The potency of the fluorine-containing BITE compounds with respect to the in vitro inhibition of HDAC1 and HDAC6 was compared with that of the non-fluorinated ethyl congeners.
Against HDAC1, all eight fluorinated homologs were more potent than the corresponding ethyl derivatives. Four of the fluorinated compounds (n = 1–4) even exceeded the potency of the clinically approved vorinostat. Targeting the isoenzyme HDAC6, six of the 1,2-difluoroethylene-bearing compounds were more active than the respective ethyl derivatives and vorinostat. However, the potency against HDAC1 and HDAC6 generally decreased with the elongation of the methylene spacer in both series.
In a related investigation of the trifluoromethyl group as a bioisostere to the ethyl substituent, the authors recognized a low lipophilicity in this particular compound as a result of fluorination. Furthermore, a relationship between lipophilicity and passive permeability was deduced. The introduction of fluorine also had an effect on the solubility of the compounds. However, no consistent correlation with an increasing degree of fluorination could be established, but a heterogeneous order [66].
Based on these results, the authors judged the BITE group to be a promising bioisostere for the development of potent HDACis. Moreover, they suggested that the concept of 1,2-difluoroethylene derivatives can be successfully applied to other small molecules in the future [41].
A similar approach was already taken by Walton et al. [38] in designing a series of six hydroxamic acids derived from vorinostat with perfluorinated alkyl side chains of different lengths (Scheme 7). The antiproliferative effect of the compounds was investigated against ovarian cancer cells A2780. Five of the six perfluorinated compounds showed cytotoxicity in the single-digit micromolar range, two of which were minimally better and another two of which were significantly more potent than vorinostat. This proves a positive effect of the introduction of perfluorinated alkyl side chains. In this context, the authors suggested that compounds with a longer perfluorinated chain have lower uptake in cancer cells due to their decreased solubility. This in turn leads to less toxicity.
Also in the work of Goehringer, Peng, and colleagues [35], the structure of the clinically approved drug vorinostat was altered. In particular, the modification is based on the introduction of a 4-pentafluorosulfanyl substituent leading to a vorinostat derivative with a para-SF5 aryl cap (Scheme 4). The authors justified their decision for the SF5-group with the fact that it is a stable imitator of negatively charged biomolecules, due to its lipophilicity and electron-withdrawing properties [67]. The growth inhibitory activity of SF5-vorinostat against six human cell lines was compared with that of vorinostat and revealed a beneficial effect on increasing potency.
While SF5-vorinostat exhibited approximately the same cytotoxicity as the parent compound against the human prostate cell line DU145, it had approximately two-fold higher potency in the other cell lines tested (hepatocellular carcinoma Hep-G2, and T-cell leukemia/lymphoma cell lines Jurkat, Hut78, SupT11, and SMZ1) due to an IC50 value half as high. In all six cell lines, the IC50 value of SF5-vorinostat was in the low single-digit micromolar range.
Similar effects were detected for the respective trifluoromethyl analogue (Figure 3) in the study by Salmi-Smail et al. [68]. This showed approximately equal antiproliferative activity as vorinostat in the breast carcinoma SKBR3, chronic myelogenous leukemia K562, and acute myelogenous leukemia HL60 cell lines. More importantly, CF3-vorinostat was superior by a factor of approximately 1.4–1.8 in colon carcinoma HT29, histiocytic lymphoma U937, and Jurkat JA16 cell lines. The 4F-vorinostat derivative (Figure 3) was only tested in the cell lines SKBR3, HT29, and U937. Its IC50 value was not better than that of vorinostat; however, it was still in the low single-digit micromolar range. In contrast, 3F-vorinostat (Figure 3) was equivalent or slightly superior to vorinostat in these three cell lines.
Zhang, Liu, Gao et al. [60,61] adduced the HDACi largazole (Figure 2) and performed a fluoroscan to extend the structural diversity. This resulted in largazole thiol derivatives with different fluorination in the thiolate side chain (Scheme 29). Derivatives of 116 exhibiting single fluorination at positions 18 and 19, respectively, or the compound which was double fluorinated at position 20, were investigated. The efficacy of the compounds to inhibit human HDACs 1–3, 6, 8 was investigated. In comparison with largazole, these three fluorinated derivatives were more potent. It should be mentioned that largazole thiol exerted stronger effects than largazole (Figure 2) itself. However, the derivative fluorosubstituted in the C19 position even exceeded its activity.
The research of Luckhurst and co-workers [69] was focused on the development of tetrasubstituted cyclopropane hydroxamic acids as HDACis (Figure 4). Formally starting with the trisubstituted compounds, they took the approach of introducing a fluorine substituent leading to the tetrasubstituted congeners. This was based on the assumption that the fluorine at the Cα adjacent to the hydroxamic acid would lead to enhanced acidity, and thus, increase activity. The authors reported on six pairs of compounds, each with and without the fluorine substituent. The potency of these compounds to inhibit the HDAC1 enzyme was investigated. The working hypothesis was confirmed, as all fluorinated derivatives were two to nine times more active than the unfluorinated analogues. In the series of compounds, the pyrimidine capping group was also modified. In this context, fluorination in the form of the introduction of a single fluorine substituent, a difluoromethoxy or a trifluoromethyl group, was considered. However, only minimal effects on activity were observed in this regard.
The study by Yao, Li, and coworkers [70] reported on HDACis, in which structural modifications were made to the surface recognition cap, including the introduction of a 4,4-difluoropiperidin-1-yl residue (Figure 5). However, the fluorinated compounds only showed moderate antiproliferative activity against MDA-MB-231 cells.
Gawel, Shouksmith, Raouf, Nawar, and co-workers [71] described some HDACis belonging to hydroxamic acids with different fluorination patterns in the phenyl cap group. The cap moiety serves to anchor the inhibitor to the outside of the active center of the HDAC. In their work, the authors also introduced derivatives with a pentafluorophenyl group (Figure 6). The cytotoxicity was investigated using the leukemia cell line MV4-11.
The p-fluorinated compound showed cytotoxicity with an IC50 value of 3.64 μM. Interestingly, the compound with a trifluoromethyl substituent in the para-position was even more potent (0.88 μM). The compound with a five-fold fluorination of the aromatic residue (pentafluorobenzene derivative, 1.51 μM) was also stronger than the monofluorinated derivative. This trend could indicate a positive effect on potency upon increasing fluorination. A comparison of the other pentafluoro compounds in their study (i.e., compounds 60 and 84 in the original work: 1.85 μM and 7.37 μM, respectively) with the corresponding p-tert-butyl-substituted compounds (i.e., compounds 68 and 83 in the original work: 0.57 μM and 8.57 μM, respectively) only partially confirms this. Here, the different spacers between the cap group and the hydroxamic acid moiety seem to have an influence. This can also be seen in the pentafluoro compounds (i.e., compounds 100 and 101 in the original work: 0.32 μM and 0.76 μM, respectively), which induced even stronger cytotoxicity against MV4-11 cells than citarinostat (1.24 μM).
A HDACi also fluorinated in the cap group was described in the work of Kim, La and co-workers [72]—in particular, a fluorinated aminophenyl-benzamide-based drug (Figure 7). In addition, in the chemical structure of the HDACi, the zinc-binding group was substituted by a “foot pocket” unit, that is, a thiophen-2-yl substituent. The compound was designed to specifically inhibit HDAC1. It exhibited promising cytotoxicity against the colorectal cancer cell lines HCT116 (IC50 = 5.59 μM) and DLD-1 (IC50 = 4.05 μM). Remarkably, the activity against normal intestinal epithelial cells (ICEs, IC50 = 18.38 μM) was approximately four times less pronounced, demonstrating some kind of selectivity towards colon cancer cells. Based on these results, the authors concluded that the biarylbenzamide structure, as well as fluorination within the cap moiety, represent a promising strategy for the development of potent HDAC inhibitors.
Schäker-Hübner et al. [51] also studied the effect of fluorination of the “foot pocket” unit attached to the zinc-binding domain (Scheme 20). They observed ambivalent results due to fluorination when the inhibitory potential of the newly synthesized fluorine-containing compounds 85 was investigated. The introduction of a 4-fluorophenyl substituent resulted in a slightly more potent inhibition of the isoenzymes HDAC1-3 compared to the phenyl parent compound. However, when a fluorine substituent was additionally introduced at positions 2 or 6 starting from the pyridin-3-yl substituted compound, the inhibition of HDAC1-3 was lower. The authors investigated that fluorination on the aromatic system increases lipophilicity, and thus, decreases solubility, which could in turn explain a poorer inhibitory effect. For the 2-fluoropyridin-4-yl derivative, compared with the non-fluorinated pyridin-4-yl compound, inhibition was reduced towards HDAC1-2 and enhanced towards HDAC3. Depending on the position where the fluorine substituent is introduced, not only the potency can be increased, but also the selectivity can be fine-tuned.
3.2. Selectivity
In addition to potency, another parameter that plays an important role in any drug design is selectivity [73]. Selectivity generally describes the extent to which a drug binds to targets that are different than the desired target (e.g., enzymes, receptors, ion channels) or acts on different tissues (e.g., cancerous vs. non-cancerous). High selectivity means that few additional targets are bound. Ideally, binding occurs only to the target through which the pharmacological effect is exerted and not to other targets that may be associated with undesirable side effects.
In the development of HDACis as antitumor compounds, it is important that the compounds induce their effects on cancer cells and not on healthy cells. On the other hand, there are also efforts to create selectivity towards the different HDAC isoenzymes [74]. This is based on the fact that not all HDAC isoforms are overexpressed in all types of cancers [75].
For the vorinostat derivative 26 bearing perfluorinated alkyl side chains (Scheme 7), as reported in the study of Walton et al. [38], approximately five-fold selectivity over ovarian carcinoma cells A2780 can be seen for the compound with the polyfluorinated C10 chain compared to non-cancerous HEK cells. Since for the unfluorinated analogue the selectivity was approximately two and a half times, fluorination could lead to the doubling of the selectivity. With the parent compound vorinostat, there was almost no selectivity for the observed tumor cells. For the sake of completeness, however, it must be mentioned that for the other derivatives with C8 and C12 chains, the selectivity with respect to tumor cells could not be increased by fluorination. Ultimately, selectivity towards tumor cells can be achieved if there is selectivity with respect to tumor-specific targets [76]. In the development of HDACis, it takes an important role to specifically address certain isoforms of HDAC with the newly developed compounds.
Gryder, Wu, and co-workers [77] used the example of the benzamide-based compound “Merck60” (Figure 8) in their study. They demonstrated that an exchange of the thieno ring in the para-position of the benzamide core against a fluorine substituent in the meta-position can positively influence the selectivity for the isoenzyme HDAC3 compared to HDAC1/2. The reason for this can be explained in terms of the different sizes of the side pockets. It is smaller for HDAC3 than for HDAC1/2 and can be well targeted by the fluorine-substituted derivative, whereas the bulkier thieno ring does not favor this. Addressing smaller and hydrophobic binding pockets as a result of fluorine substitution is a common tool in medicinal chemistry [78]. The strategy has also been successfully applied, for example, in the design of fluorinated ligands, which exert their antitumor effect primarily through a more selective inhibition of the cyclooxygenase-2 (COX-2) enzyme, but not through influence on COX-1, unlike the corresponding methylated analogues [28,79].
In the series of triazolylphenyl-based hydroxamic acid derivatives (Figure 9) described as HDACis in the work of Chen et al. [80,81], fluorination of the phenyl ring in the para-position resulted in a doubling or tripling of the selectivity for the isoenzyme HDAC6 over HDAC1. Moreover, the introduction of a pentafluorophenyl cap in the study by Gawel, Shouksmith, Raouf, Nawar, and co-workers [71] was found to increase selectivity over HDAC6.
The HDAC6 isoenzyme is of particular interest because its structure is somewhat different from the other isoenzymes [82]. Furthermore, the research work of Sandrone et al. [83] investigated the influence of fluorination on the selectivity towards the isoenzyme HDAC6. In a series of benzohydroxamate-based compounds (Figure 10), the linker in the form of the phenyl ring was single- or double-fluorinated. The effect on HDAC6 was not much improved, but the activity towards HDAC1 was decreased. This is ultimately accompanied by an increase in selectivity for HDAC6. Compared to the unfluorinated linkers, there is approximately a six-fold increase in selectivity for HDAC6 over HDAC1 after the introduction of two fluorine substituents. Specific interactions between the fluorine-containing linker and residues responsible in the binding pocket (i.e., Gly582, Ser531, and His614) of HDAC6 (HDAC class IIb) are suggested as the reason for this. In contrast, this is hindered in class I HDAC (e.g., HDAC1) because the Ser531 is exchanged by an Arg. These results of Sandrone et al. [83] were similarly confirmed in the study of Reßing, Schliehe-Diecks et al., where an increased inhibition of HDAC4 (HDAC class IIa) was detected due to compounds with a fluorinated linker [84].
Apparently opposed results were obtained from the fluorine scan undertaken by Zhang, Liu, Gao et al. [60,61] (Scheme 29). Fluorination on the thiolate residue of largazole derivatives revealed that the fluorinated compounds shifted in selectivity in favor of the isoenzyme HDAC1 relative to HDAC6.
The situation is again different for the 4-pentafluorosulfanyl-substituted vorinostat derivative. Compared to the parent compound, the activity towards HDAC6 is minimally increased. The introduction of the fluorine-containing substituent (Scheme 4) did not affect the inhibition profile of HDAC1 and HDAC2 [35]. This trend was also apparent with the trifluoromethylated congener [68].
Chuman et al. developed derivatives of vorinostat [55]. The hydroxamic acid residue, which serves as the zinc-binding site, was exchanged for a fluoroalkene containing a sulfanylmethyl group (Scheme 25). Additionally, the effects of the E- and Z-configurations on selectivity towards HDAC isozymes were investigated. In relation to the parent compound SAHA, there was no real change in the HDAC1/HDAC6 selectivity ratio. Nevertheless, the affinity of the Z-isomer towards the enzymes HDAC1, HDAC8, HDAC4, and HDAC6 was two to three times greater than with SAHA. The E-isomer had lower affinity, particularly for the HDAC1 and HDAC4 enzymes. However, these results do not necessarily indicate a shift in selectivity based on replacement by fluorine-containing substituents. The effects are more likely due to the E/Z-configuration. The authors considered this as a potential novel approach to designing isoform-selective HDACis.
The work of Keuler, König, Bückreiß, and colleagues [85] reports the development of compounds with a difluoromethyl-1,3,4-oxadiazole scaffold that serves to bind zinc instead of a hydroxamate (Figure 11). These non-hydroxamate compounds were found to function as proteolysis-targeting chimeras (PROTACs) for the intended degradation of a specific protein, e.g., HDAC. The compounds bearing the aminomethyl linker in the meta-position instead of the para-position, and with a spacer with oxyethylene subunits instead of pure alkyl groups, were found to be selective HDAC6 degraders compared to HDAC1 and HDAC4. Although the agents carry terminal conspicuous difluoromethyl groups, the influence of selectivity due to fluorination cannot be clearly deduced from this study.
In the paper by Ariawan et al. [42], the effect of vicinal difluorination in the chemical structure of the hydroxamate-based HDACis vorinostat and scriptaid was investigated (Scheme 11). The authors found that the introduction of fluorine substituents does not have a positive effect on the potency to inhibit HDAC. Among the vorinostat derivatives, there was little change in overall selectivity towards the eleven HDAC isoenzymes, as a result of fluorination. However, subtle effects on the selectivity of the class I HDACs could be detected. Finally, the authors concluded that it is rather challenging to engineer selective HDACis, especially when the binding pockets of the substrates (i.e., HDAC1, HDAC2, HDAC3) are narrow. This shows that fluorination alone is not necessarily crucial for bringing the substrate into the appropriate binding pocket of any isoenzyme. However, once it has reached the binding pocket, hydrophobic interactions formed by fluorine substituents can, of course, contribute to selectivity.
3.3. Labeling
The focus in medicinal chemistry is on designing new therapeutic agents through chemical synthesis. This development also includes the optimization of properties such as potency and selectivity. Furthermore, a major goal is understanding the influence of modifications in the chemical structure. Aside from the impact on the effect, there is interest in biodistribution, metabolism, or interaction with a target. In order to analyze the effectivity, it must be possible to sensitively detect the active agent in complex biological matrices. Fluorination is a good tracer for this purpose because fluorinated compounds rarely occur in nature. In addition, based on the isotopes 19F and 18F, it is possible to perform detection by spectroscopy and positron emission tomography (PET), respectively.
3.3.1. Labeling Using 19F
Sankaranarayanapillai and co-workers [86,87] developed a method based on NMR of 19F to monitor the inhibition of HDAC, since the fluorinated lysine derivative Boc-Lys-(Tfa)-OH (BLT) is a substrate for cleavage mediated by HDAC. Of note, treatment with BLT had no effect on the viability of PC3 cancer cells or on the activity of HDAC. Therefore, the amount of the 19F-labeled imaging agent BLT (Figure 12) detected by NMR spectroscopy allows conclusions to be drawn about the activity of HDAC. This method is non-invasive but does not rely on the fluorination of HDACis. Nevertheless, it has a distinct advantage. Since the treatment of a tumor with HDACis often leads to tumor stasis and not necessarily to its shrinkage, standard imaging methods would not necessarily be suitable to monitor this behavior. However, one drawback is that the different trifluoroacetyl-lysine-based substrates have different specificity with respect to HDAC isoenzymes; therefore, such substrates have limited applications [44].
In the study by Jayathilaka et al. [47], the introduction of a 19F tracer in the form of a trifluoromethyl group (Scheme 15) was used to gain insight into the mechanism of action of the known HDACi N-(2-aminophenyl)-N’-phenyloctanol diamine (BML-210). The activity of class IIa HDACs such as HDAC4 depends on the interaction with the Myocyte Enhancer Factor-2 (MEF2). Therefore, interference in this context represents a promising strategy to indirectly inhibit class IIa HDACs. The 19F-containing derivatives of BML-210 were used to study the interaction of the compound to bind MEF2 using NMR analysis. By comparing the 19F NMR spectrum of the free fluorinated compound with an approach in which the MEF2 protein was present in excess, it was possible to conclude that the drug interacts with MEF2.
Although these two examples represent the sophisticated use of 19F to characterize HDACis, NMR spectroscopy is inferior to PET in its prevalence and application.
3.3.2. Labeling Using 18F
Labeling drugs with the radioisotope 18F is the most common modality of radiolabeling in both preclinical and clinical drug development [88]. There are a variety of chemical reactions, e.g., electrophilic or nucleophilic substitutions, to yield in radiofluorine-labeled drugs. Its importance is related to its accessibility by PET, as the isotope 18F undergoes β+ decay with a half-life of 109.7 min. By employing PET, it is possible to allow molecular imaging.
Probably the first HDAC imaging agent based on 18F-PET was developed by Hendricks, Keliher, and colleagues [44]. Compound 50 is a derivative of the clinically important HDACi vorinostat. The novel compound was obtained by introducing a radiofluorine substituent at position 4 of the aromatic ring of vorinostat (Scheme 13). With its low nanomolar potency, the compound showed a very similar biological profile to its parent compound. Using an ovarian cancer model in mice, the binding affinity to HDAC was investigated by 18F-PET. The authors found that quantification of the binding was possible up to 24 h after application. This paved the way for real-time in vivo imaging of HDACs, which the authors suggested will make a useful contribution to the characterization of HDACis.
Further studies include, for example, the development of radiofluorinated bioisosteres of the HDACi santacruzamate A as potential PET tracers [89]. Li and colleagues [52] introduced a [18F]fluoroethyl tracer into a biphenyl-based benzamide HDAC (i.e., INER-1577) to enable imaging (Scheme 23). They studied the distribution in the body and brain of rats using a micro-PET scanner. The compound was also able to inhibit the growth of breast cancer cell lines 4T1 and MCF-55. The authors continued synthesizing further benzamide-based HDACis with 18F tracers as imaging agents for mice [90]. The review by Dasko et al. [91] summarized further examples in which HDACis were fluorinated by the introduction of the 18F radiotracer, in order to study, among other things, the biodistribution and target binding of the compounds.
In their first study, Strebl et al. [45] used 18F labeling to assess the penetrance of the hydroxamic acid-based and non-fluorinated HDACi martinostat and the fluorinated derivative [18F]martinostat, respectively (Scheme 14). The radiofluorinated compounds showed promising properties as a radiotracer, as brain accumulation and distribution were similar to those of the 11C-labeled but non-fluorinated martinostat. However, the use of the 19F radioisotope instead of an 11C-labeled derivative (half-life 11C: 20.3 min) was associated with a longer half-life. In their following study [43], the authors prepared a radiofluorine version of the drug, i.e., [18F]bavarostat 47 (Scheme 12). Bavarostat belongs to the class of hydroxamic acid derivatives as HDAC inhibitors. The chemical structure of bavarostat also contains a fluorine substituent. However, it is assumed that the fluorine substituent does not affect the binding to HDAC [45]. Bavarostat itself is characterized by high selectivity to HDAC6. Since HDAC6 appears to play a role not only in cancer, but also in central nervous system diseases (e.g., Alzheimer’s disease), penetration into the brain is important, albeit difficult to reach with HDACis. The authors used the radiolabeled compound to determine the uptake into the brains of rodents and non-human primates. Bavarostat showed high penetration. Therefore, it was suggested that bavarostat is suitable for future investigations to target HDAC6 in living human brains.
The study by Chen et al. [92] also picked up four fluorinated HDACis designed for PET imaging due to their corresponding labeling with 18F (coded as INER-1577 #1 to INER-1577 #4, Figure 13). In particular, the compounds were again designed for Alzheimer’s disease detection, as HDACis are considered to play an important role in this context. The radiofluorinated HDACis serving as PET imaging compounds were investigated for their metabolism in biological systems, including a mass spectrometric approach. The authors concluded from their work that imaging studies should be performed promptly after application.
4. Issues of Fluorination
Even if fluorination can be beneficial for biological effects in terms of potency and selectivity or can be used in the context of labeling for bioanalytical studies, there are also some disadvantages.
The introduction of fluorine substituents into molecules is not always as trivial as it might seem and presents challenges to the medicinal chemist [93]. This may relate, for example, to the high toxicity or poor practical handling of fluorination reagents, as well as to low yields.
Among the HDACis mentioned by Luckhurst et al. [69], fluorine was also introduced in the form of difluoromethoxylation. The synthesis of difluoromethoxy derivatives is very commonly performed using chlorodifluoromethane and a base, following the elimination of HCl from chlorodifluoromethane. Chlorodifluoromethane is gaseous, and this synthetic procedure requires an ozone depleting reagent; these are major drawbacks of this approach [94]. On the other hand, the direct trifluoromethoxylation of aromatic compounds can be carried out under radical reactions, as described, for example, by Venturini et al. [95]. The gas trifluoromethyl hypofluorite is used. This particular reagent suffers from high toxicity [96]. Moreover, gases are generally more difficult to handle compared to solids. Even though liquids are easier to handle than gases, it can be considered a disadvantage to work with liquid reagents compared to solids as an alternative. The DexoFluor used for the synthesis of HDACis in the work of Ariawan et al. [42] is subject to this limitation to a certain extent. Fortunately, crystalline solids have now also been described as an improvement on DeoxoFluor for deoxyfluorination [97]. A different aspect is that if a C–F bond is to be introduced stereoselectively in the course of the synthesis, chiral auxiliaries can be used. This has the disadvantage that the auxiliary reagent must first be attached and then removed [98]. However, this is a general restriction and not limited to fluorine.
Examination of the series of HDACis by Meyners et al. [36] and Walton et al. [38] seems to reveal another issue. These are compounds that have a perfluorinated alkyl core. For compounds in which carbon atoms are completely substituted with halogens, photodissociation can occur under the release of the halogen into the stratosphere and the depletion of the ozone layer [97]. However, this effect seems to mainly apply to chlorine and bromine atoms rather than fluorine [99], but it would come into play when mixed halogenated alkanes are present. The formation of greenhouse gases is also mentioned in the review by Caron [97] as a disadvantage of the synthesis of fluorinated compounds. This environmental aspect is extended by the poor profile of fluorinating reagents with respect to green chemistry. In addition, the costs of the fluorinated reagents are very high in some cases, and there are safety concerns [97].
However, such concerns are not limited to the reagents used in the synthesis, but also affect the safety of drug use. Chemical instability and enzymes that metabolize active agents could lead to a loss of fluorination and the formation of metabolites that could probably affect the safety of therapy [100].
One of the main reasons why fluorination is used in medicinal chemistry drug design is its influence on metabolism. Since the dissociation energy of a C–F bond is very high, it seems that the generation of active metabolites can be avoided and the metabolic clearance can be decreased [100]. Nevertheless, a resealing of fluoride can occur. This can be seen by metabolizing enzymes or mediation by a nucleophile. If an agent undergoes a high cleavage of the C–F bond with the subsequent occurrence of relevant amounts of fluoride, skeletal fluorosis such as exostoses or periostitis may develop as adverse drug effects [101].
As comprehensively described by Pan [100], defluorination from monofluorinated alkyls bearing an intramolecular nucleophile can occur by an SN2 reaction. The nucleophile does not necessarily have to be present intramolecularly, but also in the form of biologically relevant compounds, such as glutathione. Furthermore, in the course of such an SN2 reaction, not only can fluoride be released, but also, biological targets can be alkylated, which could result in toxicity. Apart from the SN2 reaction, the heterolytic cleavage of a C–F bond yielding to the release of fluoride may also be due to a lone pair of electrons at the β-position of the fluorine [100]. The cleavage of fluoride can also be mediated by cytochrome P450 enzymes that catalyze the hydroxylation of fluorinated alkanes [100]. However, it is not only fluoride that leads to toxicological concerns, but also fluoroacetate [102]. This can be formed, for example, during the metabolism of molecules containing 1,3-difluoro-2-propyl or O-/N-2-fluoroethyl substituents [100].
5. Conclusions
Currently, HDACs increasingly prove to play an important role in cancer via chromatin-modifying complexes and modulating gene expression [10]. Therefore, these enzymes become potential and attractive targets in the research and development of new anticancer drugs [9]. As a result, many HDACis have been discovered and studied in recent years. One of the strategies for the design of novel pharmaceuticals is to introduce fluorine into the chemical structures [103]. This approach has also been performed with HDACis such as hydroxamic acids, benzamides, thiols, or cyclic peptides. However, no overview is available on this aspect so far in the literature to the best of our knowledge.
Fluorination has been shown to have many advantages, as described above in this review. These include partly higher potency, increased binding affinity, and expanded selectivity towards the different HDAC isoenzymes [44]. Moreover, altered physicochemical properties and improved metabolic stability can also be attributed to the introduction of fluorine 92. Yet, it should also be noted that chemical instability and enzymes that metabolize active agents could lead to a loss of fluorination and the formation of metabolites that could probably affect the safety of drug use [104]. Nevertheless, chemical instability and metabolizing enzymes (e.g., CYP enzymes) could cause a loss of fluorination and the generation of likely toxic metabolites that compromise drug safety [100].
In the field of radiation oncology, fluorine-18 (18F) represents a radioisotope which is one of the most common isotopes used for PET imaging [105]. It has a half-life of approximately 110 min, more than five times longer compared to the half-life of carbon-11 (approximately 20 min) [44].
This review summarized several HDACis-related studies focusing on the synthesis, as well as the purposes and influences, of fluorination developed in recent years. The development of synthetic methods for the formation of fluorinated derivatives and the advantages of incorporating fluorine into the chemical scaffold of new HDACis will certainly open new opportunities in the research and development of anticancer agents in the future.
Author Contributions
Conceptualization, D.T.A. and D.B.; formal analysis, D.T.A. and D.B.; writing—original draft preparation, D.T.A. and D.B.; writing—review and editing, N.H.N. and B.K.; visualization, D.T.A.; supervision, N.H.N. and B.K.; project administration, D.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tempany, C.M.C.; Jayender, J.; Kapur, T.; Bueno, R.; Golby, A.; Agar, N.; Jolesz, F.A. Multimodal imaging for improved diagnosis and treatment of cancers. Cancer 2015, 121, 817–827. [Google Scholar] [CrossRef] [Green Version]
- Bünger, S.; Laubert, T.; Roblick, U.J.; Habermann, J.K. Serum biomarkers for improved diagnostic of pancreatic cancer: A current overview. J. Cancer Res. Clin. Oncol. 2011, 137, 375–389. [Google Scholar] [CrossRef]
- Dalal, N.; Jalandra, R.; Sharma, M.; Prakash, H.; Makharia, G.K.; Solanki, P.R.; Singh, R.; Kumar, A. Omics technologies for improved diagnosis and treatment of colorectal cancer: Technical advancement and major perspectives. Biomed. Pharmacother. 2020, 131, 110648. [Google Scholar] [CrossRef]
- Steiner, N.; Schober, P.; Willenbacher, W.; Kircher, B.; Gunsilius, E.; Wolf, D.; Nachbaur, D. Autologous and Allogeneic Stem Cell Transplantation as Salvage Treatment Options for Relapsed/Refractory Multiple Myeloma: A Single-center Experience over 20 Years. Anticancer Res. 2022, 42, 5825–5832. [Google Scholar] [CrossRef]
- Steiner, N.; Göbel, G.; Mauser, L.; Mühlnikel, L.; Fischinger, M.; Künz, T.; Willenbacher, W.; Hetzenauer, G.; Rudzki, J.; Nussbaumer, W.; et al. Poor Mobilizers in Lymphoma but Not Myeloma Patients Had Significantly Poorer Progression-Free Survival after Autologous Stem Cell Transplantation: Results of a Large Retrospective, Single-Center Observational Study. Cancers 2023, 15, 608. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Feng, Y.; Spezia, M.; Huang, S.; Yuan, C.; Zeng, Z.; Zhang, L.; Ji, X.; Liu, W.; Huang, B.; Luo, W.; et al. Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis. 2018, 5, 77–106. [Google Scholar] [CrossRef]
- Baig, M.H.; Adil, M.; Khan, R.; Dhadi, S.; Ahmad, K.; Rabbani, G.; Bashir, T.; Imran, M.A.; Husain, F.M.; Lee, E.J.; et al. Enzyme targeting strategies for prevention and treatment of cancer: Implications for cancer therapy. Semin. Cancer Biol. 2019, 56, 1–11. [Google Scholar] [CrossRef]
- Li, G.; Tian, Y.; Zhu, W.-G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 576946. [Google Scholar] [CrossRef]
- Ruzic, D.; Djoković, N.; Srdić-Rajić, T.; Echeverria, C.; Nikolic, K.; Santibanez, J.F. Targeting Histone Deacetylases: Opportunities for Cancer Treatment and Chemoprevention. Pharmaceutics 2022, 14, 209. [Google Scholar] [CrossRef]
- Jin, Y.; Liu, T.; Luo, H.; Liu, Y.; Liu, D. Targeting Epigenetic Regulatory Enzymes for Cancer Therapeutics: Novel Small-Molecule Epidrug Development. Front. Oncol. 2022, 12, 848221. [Google Scholar] [CrossRef]
- Behnisch-Cornwell, S.; Grathwol, C.W.; Schulig, L.; Voigt, A.; Baecker, D.; Link, A.; Bednarski, P.J. Correlation Analysis of Protein Expression of 10 HDAC/Sirtuin Isoenzymes with Sensitivities of 23 Anticancer Drugs in 17 Cancer Cell Lines and Potentiation of Drug Activity by Co-Treatment with HDAC Inhibitors. Cancers 2022, 14, 187. [Google Scholar] [CrossRef]
- Anh, D.T.; Hai, P.-T.; Huy, L.D.; Ngoc, H.B.; Ngoc, T.T.M.; Dung, D.T.M.; Park, E.J.; Song, I.K.; Kang, J.S.; Kwon, J.-H.; et al. Novel 4-Oxoquinazoline-Based N-Hydroxypropenamides as Histone Deacetylase Inhibitors: Design, Synthesis, and Biological Evaluation. ACS Omega 2021, 6, 4907–4920. [Google Scholar] [CrossRef]
- Hieu, D.T.; Anh, D.T.; Hai, P.-T.; Thuan, N.T.; Huong, L.-T.-T.; Park, E.J.; Young Ji, A.; Soon Kang, J.; Phuong Dung, P.T.; Han, S.-B.; et al. Quinazolin-4(3H)-one-Based Hydroxamic Acids: Design, Synthesis and Evaluation of Histone Deacetylase Inhibitory Effects and Cytotoxicity. Chem. Biodivers. 2019, 16, e1800502. [Google Scholar] [CrossRef]
- Minh, V.N.; Thanh, T.N.; Lien, T.H.; Anh, T.P.D.; Cuong, D.H.; Nam, H.N.; Hai, T.P.; Minh-Ngoc, L.; Le-Thi-Thu, H.; Chinh, V.L.; et al. Design, Synthesis and Biological Evaluation of Novel N-hydroxyheptanamides Incorporating 6-hydroxy-2-methylquinazolin-4(3H)-ones as Histone Deacetylase Inhibitors and Cytotoxic Agents. Anti-Cancer Agents Med. Chem. 2019, 19, 1543–1557. [Google Scholar] [CrossRef]
- Hieu, D.T.; Anh, D.T.; Hai, P.-T.; Huong, L.-T.-T.; Park, E.J.; Choi, J.E.; Kang, J.S.; Dung, P.T.P.; Han, S.-B.; Nam, N.-H. Quinazoline-Based Hydroxamic Acids: Design, Synthesis, and Evaluation of Histone Deacetylase Inhibitory Effects and Cytotoxicity. Chem. Biodivers. 2018, 15, e1800027. [Google Scholar] [CrossRef]
- Hieu, D.T.; Anh, D.T.; Tuan, N.M.; Hai, P.-T.; Huong, L.-T.-T.; Kim, J.; Kang, J.S.; Vu, T.K.; Dung, P.T.P.; Han, S.-B.; et al. Design, synthesis and evaluation of novel N-hydroxybenzamides/N-hydroxypropenamides incorporating quinazolin-4(3H)-ones as histone deacetylase inhibitors and antitumor agents. Bioorganic Chem. 2018, 76, 258–267. [Google Scholar] [CrossRef]
- Anh, D.T.; Hai, P.-T.; Dung, D.T.M.; Dung, P.T.P.; Huong, L.-T.-T.; Park, E.J.; Jun, H.W.; Kang, J.S.; Kwon, J.-H.; Tung, T.T.; et al. Design, synthesis and evaluation of novel indirubin-based N-hydroxybenzamides, N-hydroxypropenamides and N-hydroxyheptanamides as histone deacetylase inhibitors and antitumor agents. Bioorganic Med. Chem. Lett. 2020, 30, 127537. [Google Scholar] [CrossRef]
- Anh, D.T.; Hai, P.-T.; Huong, L.-T.-T.; Park, E.J.; Jun, H.W.; Kang, J.S.; Kwon, J.-H.; Dung, D.T.M.; Anh, V.T.; Hue, V.T.M.; et al. Exploration of certain 1,3-oxazole- and 1,3-thiazole-based hydroxamic acids as histone deacetylase inhibitors and antitumor agents. Bioorganic Chem. 2020, 101, 103988. [Google Scholar] [CrossRef]
- Jampilek, J. Heterocycles in Medicinal Chemistry. Molecules 2019, 24, 3839. [Google Scholar] [CrossRef] [Green Version]
- Gomtsyan, A. Heterocycles in drugs and drug discovery. Chem. Heterocycl. Compd. 2012, 48, 7–10. [Google Scholar] [CrossRef]
- Gupta, P.S. Roles of Fluorine in Drug Design and Drug Action. Lett. Drug Des. Discov. 2019, 16, 1089–1109. [Google Scholar] [CrossRef]
- Grygorenko, O.O.; Melnykov, K.P.; Holovach, S.; Demchuk, O. Fluorinated Cycloalkyl Building Blocks for Drug Discovery. ChemMedChem 2022, 17, e202200365. [Google Scholar] [CrossRef]
- Richardson, P. Applications of fluorine to the construction of bioisosteric elements for the purposes of novel drug discovery. Expert Opin. Drug Discov. 2021, 16, 1261–1286. [Google Scholar] [CrossRef]
- Hevey, R. The Role of Fluorine in Glycomimetic Drug Design. Chem. Eur. J. 2021, 27, 2240–2253. [Google Scholar] [CrossRef]
- Páez-Franco, J.C.; Zermeño-Ortega, M.R.; de la O.-Contreras, C.M.; Canseco-González, D.; Parra-Unda, J.R.; Avila-Sorrosa, A.; Enríquez, R.G.; Germán-Acacio, J.M.; Morales-Morales, D. Relevance of Fluorinated Ligands to the Design of Metallodrugs for Their Potential Use in Cancer Treatment. Pharmaceutics 2022, 14, 402. [Google Scholar] [CrossRef]
- Pal, S.; Chandra, G.; Patel, S.; Singh, S. Fluorinated Nucleosides: Synthesis, Modulation in Conformation and Therapeutic Application. Chem. Rec. 2022, 22, e202100335. [Google Scholar] [CrossRef]
- Baecker, D.; Obermoser, V.; Kirchner, E.A.; Hupfauf, A.; Kircher, B.; Gust, R. Fluorination as tool to improve bioanalytical sensitivity and COX-2-selective antitumor activity of cobalt alkyne complexes. Dalton Trans. 2019, 48, 15856–15868. [Google Scholar] [CrossRef]
- Sagasser, J.; Ma, B.N.; Baecker, D.; Salcher, S.; Hermann, M.; Lamprecht, J.; Angerer, S.; Obexer, P.; Kircher, B.; Gust, R. A New Approach in Cancer Treatment: Discovery of Chlorido[N,N′-disalicylidene-1,2-phenylenediamine]iron(III) Complexes as Ferroptosis Inducers. J. Med. Chem. 2019, 62, 8053–8061. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, J.; Jiang, Q.; Zhang, L.; Song, W. Zinc binding groups for histone deacetylase inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef] [Green Version]
- Gong, W.; Wu, R.; Zhang, Y. Thiol versus hydroxamate as zinc binding group in HDAC inhibition: An ab initio QM/MM molecular dynamics study. J. Comput. Chem. 2015, 36, 2228–2235. [Google Scholar] [CrossRef] [Green Version]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Br. J. Clin. Pharmacol. 2021, 87, 4577–4597. [Google Scholar] [CrossRef]
- Aboukhatwa, S.M.; Hanigan, T.W.; Taha, T.Y.; Neerasa, J.; Ranjan, R.; El-Bastawissy, E.E.; Elkersh, M.A.; El-Moselhy, T.F.; Frasor, J.; Mahmud, N.; et al. Structurally Diverse Histone Deacetylase Photoreactive Probes: Design, Synthesis, and Photolabeling Studies in Live Cells and Tissue. ChemMedChem 2019, 14, 1096–1107. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, X.; Li, J.; Hou, J.; Qu, Y.; Yu, C.; He, F.; Xu, W.; Wu, J. Design, synthesis, and preliminary bioactivity evaluation of N1-hydroxyterephthalamide derivatives with indole cap as novel histone deacetylase inhibitors. Chem. Biol. Drug Des. 2017, 89, 38–46. [Google Scholar] [CrossRef]
- Goehringer, N.; Peng, Y.; Nitzsche, B.; Biermann, H.; Pradhan, R.; Schobert, R.; Herling, M.; Höpfner, M.; Biersack, B. Improved Anticancer Activities of a New Pentafluorothio-Substituted Vorinostat-Type Histone Deacetylase Inhibitor. Pharmaceuticals 2021, 14, 1319. [Google Scholar] [CrossRef]
- Meyners, C.; Wolff, B.; Kleinschek, A.; Krämer, A.; Meyer-Almes, F.-J. Perfluorinated hydroxamic acids are potent and selective inhibitors of HDAC-like enzymes from Pseudomonas aeruginosa. Bioorganic Med. Chem. Lett. 2017, 27, 1508–1512. [Google Scholar] [CrossRef]
- Toutah, K.; Nawar, N.; Timonen, S.; Sorger, H.; Raouf, Y.S.; Bukhari, S.; von Jan, J.; Ianevski, A.; Gawel, J.M.; Olaoye, O.O.; et al. Development of HDAC Inhibitors Exhibiting Therapeutic Potential in T-Cell Prolymphocytic Leukemia. J. Med. Chem. 2021, 64, 8486–8509. [Google Scholar] [CrossRef]
- Walton, J.W.; Cross, J.M.; Riedel, T.; Dyson, P.J. Perfluorinated HDAC inhibitors as selective anticancer agents. Org. Biomol. Chem. 2017, 15, 9186–9190. [Google Scholar] [CrossRef] [Green Version]
- Vu, K.T.; Thanh, T.N.; Minh, V.N.; Linh, H.N.; Thao, T.P.N.; Nguyen, T.B.T.; Hien, T.D.; Chinh, V.L.; Duc, H.T.; Anh, D.L.; et al. Novel Conjugated Quinazolinone-Based Hydroxamic Acids: Design, Synthesis and Biological Evaluation. Med. Chem. 2021, 17, 732–749. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, B.; Wang, Y.; Wang, X.; Gou, S. Discovery of phthalazino[1,2-b]-quinazolinone derivatives as multi-target HDAC inhibitors for the treatment of hepatocellular carcinoma via activating the p53 signal pathway. Eur. J. Med. Chem. 2022, 229, 114058. [Google Scholar] [CrossRef]
- Erdeljac, N.; Bussmann, K.; Schöler, A.; Hansen, F.K.; Gilmour, R. Fluorinated Analogues of the Histone Deacetylase Inhibitor Vorinostat (Zolinza): Validation of a Chiral Hybrid Bioisostere, BITE. ACS Med. Chem. Lett. 2019, 10, 1336–1340. [Google Scholar] [CrossRef]
- Ariawan, A.D.; Mansour, F.; Richardson, N.; Bhadbhade, M.; Ho, J.; Hunter, L. The Effect of Vicinal Difluorination on the Conformation and Potency of Histone Deacetylase Inhibitors. Molecules 2021, 26, 3974. [Google Scholar] [CrossRef]
- Strebl, M.G.; Campbell, A.J.; Zhao, W.-N.; Schroeder, F.A.; Riley, M.M.; Chindavong, P.S.; Morin, T.M.; Haggarty, S.J.; Wagner, F.F.; Ritter, T.; et al. HDAC6 Brain Mapping with [18F]Bavarostat Enabled by a Ru-Mediated Deoxyfluorination. ACS Cent. Sci. 2017, 3, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Hendricks, J.A.; Keliher, E.J.; Marinelli, B.; Reiner, T.; Weissleder, R.; Mazitschek, R. In Vivo PET Imaging of Histone Deacetylases by 18F-Suberoylanilide Hydroxamic Acid (18F-SAHA). J. Med. Chem. 2011, 54, 5576–5582. [Google Scholar] [CrossRef] [Green Version]
- Strebl, M.G.; Wang, C.; Schroeder, F.A.; Placzek, M.S.; Wey, H.-Y.; Van de Bittner, G.C.; Neelamegam, R.; Hooker, J.M. Development of a Fluorinated Class-I HDAC Radiotracer Reveals Key Chemical Determinants of Brain Penetrance. ACS Chem. Neurosci. 2016, 7, 528–533. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.J.; Herman, D.; Gottesfeld, J.M. Pimelic Diphenylamide 106 Is a Slow, Tight-binding Inhibitor of Class I Histone Deacetylases. J. Biol. Chem. 2008, 283, 35402–35409. [Google Scholar] [CrossRef] [Green Version]
- Jayathilaka, N.; Han, A.; Gaffney, K.J.; Dey, R.; Jarusiewicz, J.A.; Noridomi, K.; Philips, M.A.; Lei, X.; He, J.; Ye, J.; et al. Inhibition of the function of class IIa HDACs by blocking their interaction with MEF2. Nucleic Acids Res. 2012, 40, 5378–5388. [Google Scholar] [CrossRef] [Green Version]
- Bonomi, R.; Mukhopadhyay, U.; Shavrin, A.; Yeh, H.-H.; Majhi, A.; Dewage, S.W.; Najjar, A.; Lu, X.; Cisneros, G.A.; Tong, W.P.; et al. Novel Histone Deacetylase Class IIa Selective Substrate Radiotracers for PET Imaging of Epigenetic Regulation in the Brain. PLoS ONE 2015, 10, e0133512. [Google Scholar] [CrossRef] [Green Version]
- La, M.T.; Jeong, B.-H.; Kim, H.-K. Design and Synthesis of Novel N-(2-aminophenyl)benzamide Derivatives as Histone Deacetylase Inhibitors and Their Antitumor Activity Study. Bull. Korean Chem. Soc. 2021, 42, 740–743. [Google Scholar] [CrossRef]
- Ibrahim, H.S.; Abdelsalam, M.; Zeyn, Y.; Zessin, M.; Mustafa, A.-H.M.; Fischer, M.A.; Zeyen, P.; Sun, P.; Bülbül, E.F.; Vecchio, A.; et al. Synthesis, Molecular Docking and Biological Characterization of Pyrazine Linked 2-Aminobenzamides as New Class I Selective Histone Deacetylase (HDAC) Inhibitors with Anti-Leukemic Activity. Int. J. Mol. Sci. 2022, 23, 369. [Google Scholar] [CrossRef]
- Schäker-Hübner, L.; Haschemi, R.; Büch, T.; Kraft, F.B.; Brumme, B.; Schöler, A.; Jenke, R.; Meiler, J.; Aigner, A.; Bendas, G.; et al. Balancing Histone Deacetylase (HDAC) Inhibition and Drug-likeness: Biological and Physicochemical Evaluation of Class I Selective HDAC Inhibitors. ChemMedChem 2022, 17, e202100755. [Google Scholar] [CrossRef]
- Li, M.H.; Shiue, C.Y.; Chang, H.-C.; Chu, H.H. Synthesis of [18F]benzamide ([18F]INER-1577) as Histone Deacetylase (HDACs) Imaging Agent. J. Nucl. Med. 2016, 57, 2664. [Google Scholar]
- Li, M.-H.; Chang, H.-C.; Feng, C.-F.; Yu, H.-W.; Shiue, C.-Y. Synthesis and Evaluation of <sup>18</sup>F-INER-1577-3 as a Central Nervous System (CNS) Histone Deacetylase Imaging Agent. Curr. Med. Imaging 2020, 16, 978–990. [Google Scholar] [CrossRef]
- Tavares, M.T.; Kozikowski, A.P.; Shen, S. Mercaptoacetamide: A promising zinc-binding group for the discovery of selective histone deacetylase 6 inhibitors. Eur. J. Med. Chem. 2021, 209, 112887. [Google Scholar] [CrossRef]
- Chuman, Y.; Ueyama, M.; Sano, S.; Wu, F.; Kiyota, Y.; Higashi, T.; Osada, S.; Sakaguchi, K. Effects of E/Z Configuration of Fluoroalkene-containing HDAC Inhibitors on Selectivity for HDAC Isoforms. Chem. Lett. 2013, 42, 833–835. [Google Scholar] [CrossRef]
- Wen, J.; Niu, Q.; Liu, J.; Bao, Y.; Yang, J.; Luan, S.; Fan, Y.; Liu, D.; Zhao, L. Novel thiol-based histone deacetylase inhibitors bearing 3-phenyl-1H-pyrazole-5-carboxamide scaffold as surface recognition motif: Design, synthesis and SAR study. Bioorganic Med. Chem. Lett. 2016, 26, 375–379. [Google Scholar] [CrossRef]
- Waldecker, M.; Kautenburger, T.; Daumann, H.; Busch, C.; Schrenk, D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J. Nutr. Biochem. 2008, 19, 587–593. [Google Scholar] [CrossRef]
- Lu, Q.; Yang, Y.-T.; Chen, C.-S.; Davis, M.; Byrd, J.C.; Etherton, M.R.; Umar, A.; Chen, C.-S. Zn2+-Chelating Motif-Tethered Short-Chain Fatty Acids as a Novel Class of Histone Deacetylase Inhibitors. J. Med. Chem. 2004, 47, 467–474. [Google Scholar] [CrossRef]
- Lübke, M.; Jung, M.; Haufe, G. New histone deacetylase inhibitors based on 4-fluoro-2-amino acid esters: Synthesis and activity. J. Fluor. Chem. 2013, 152, 144–156. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, J.; Gao, D.; Yu, X.; Wang, J.; Lei, X. A fluorine scan on the Zn2+-binding thiolate side chain of HDAC inhibitor largazole: Synthesis, biological evaluation, and molecular modeling. Eur. J. Med. Chem. 2019, 182, 111672. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, J.; Gao, D.; Yu, X.; Wang, J.; Lei, X. Corrigendum to “A fluorine scan on the Zn2+-binding thiolate side chain of HDAC inhibitor largazole: Synthesis, biological evaluation, and molecular modeling”. Eur. J. Med. Chem. 2019, 182, 111672, Erratum in Eur. J. Med. Chem. 2020, 198, 112340. [Google Scholar] [CrossRef]
- Hong, J.; Luesch, H. Largazole: From discovery to broad-spectrum therapy. Nat. Prod. Rep. 2012, 29, 449–456. [Google Scholar] [CrossRef] [Green Version]
- Schwaid, A.G.; Cornella-Taracido, I. Causes and Significance of Increased Compound Potency in Cellular or Physiological Contexts. J. Med. Chem. 2018, 61, 1767–1773. [Google Scholar] [CrossRef]
- Neubig, R.R.; Spedding, M.; Kenakin, T.; Christopoulos, A. International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on Terms and Symbols in Quantitative Pharmacology. Pharmacol. Rev. 2003, 55, 597–606. [Google Scholar] [CrossRef] [Green Version]
- Dimova, D.; Stumpfe, D.; Bajorath, J. Specific chemical changes leading to consistent potency increases in structurally diverse active compounds. MedChemComm 2014, 5, 742–749. [Google Scholar] [CrossRef]
- Erdeljac, N.; Kehr, G.; Ahlqvist, M.; Knerr, L.; Gilmour, R. Exploring physicochemical space via a bioisostere of the trifluoromethyl and ethyl groups (BITE): Attenuating lipophilicity in fluorinated analogues of Gilenya® for multiple sclerosis. Chem. Commun. 2018, 54, 12002–12005. [Google Scholar] [CrossRef]
- Altomonte, S.; Zanda, M. Synthetic chemistry and biological activity of pentafluorosulphanyl (SF5) organic molecules. J. Fluor. Chem. 2012, 143, 57–93. [Google Scholar] [CrossRef] [Green Version]
- Salmi-Smail, C.; Fabre, A.; Dequiedt, F.; Restouin, A.; Castellano, R.; Garbit, S.; Roche, P.; Morelli, X.; Brunel, J.M.; Collette, Y. Modified Cap Group Suberoylanilide Hydroxamic Acid Histone Deacetylase Inhibitor Derivatives Reveal Improved Selective Antileukemic Activity. J. Med. Chem. 2010, 53, 3038–3047. [Google Scholar] [CrossRef]
- Luckhurst, C.A.; Breccia, P.; Stott, A.J.; Aziz, O.; Birch, H.L.; Bürli, R.W.; Hughes, S.J.; Jarvis, R.E.; Lamers, M.; Leonard, P.M.; et al. Potent, Selective, and CNS-Penetrant Tetrasubstituted Cyclopropane Class IIa Histone Deacetylase (HDAC) Inhibitors. ACS Med. Chem. Lett. 2016, 7, 34–39. [Google Scholar] [CrossRef] [Green Version]
- Yao, D.; Li, C.; Jiang, J.; Huang, J.; Wang, J.; He, Z.; Zhang, J. Design, synthesis and biological evaluation of novel HDAC inhibitors with improved pharmacokinetic profile in breast cancer. Eur. J. Med. Chem. 2020, 205, 112648. [Google Scholar] [CrossRef]
- Gawel, J.M.; Shouksmith, A.E.; Raouf, Y.S.; Nawar, N.; Toutah, K.; Bukhari, S.; Manaswiyoungkul, P.; Olaoye, O.O.; Israelian, J.; Radu, T.B.; et al. PTG-0861: A novel HDAC6-selective inhibitor as a therapeutic strategy in acute myeloid leukaemia. Eur. J. Med. Chem. 2020, 201, 112411. [Google Scholar] [CrossRef]
- Kim, S.L.; La, M.T.; Shin, M.W.; Kim, S.W.; Kim, H.K. A novel HDAC1 inhibitor, CBUD-1001, exerts anticancer effects by modulating the apoptosis and EMT of colorectal cancer cells. Int. J. Oncol. 2020, 57, 1027–1038. [Google Scholar] [CrossRef]
- Fischer, T.; Gazzola, S.; Riedl, R. Approaching Target Selectivity by De Novo Drug Design. Expert Opin. Drug Discov. 2019, 14, 791–803. [Google Scholar] [CrossRef]
- Yang, F.; Zhao, N.; Ge, D.; Chen, Y. Next-generation of selective histone deacetylase inhibitors. RSC Adv. 2019, 9, 19571–19583. [Google Scholar] [CrossRef] [Green Version]
- Shetty, M.G.; Pai, P.; Deaver, R.E.; Satyamoorthy, K.; Babitha, K.S. Histone deacetylase 2 selective inhibitors: A versatile therapeutic strategy as next generation drug target in cancer therapy. Pharmacol. Res. 2021, 170, 105695. [Google Scholar] [CrossRef]
- Suzuki-Karasaki, Y.; Suzuki-Karasaki, M.; Uchida, M.; Ochiai, T. Depolarization Controls TRAIL-Sensitization and Tumor-Selective Killing of Cancer Cells: Crosstalk with ROS. Front. Oncol. 2014, 4, 128. [Google Scholar] [CrossRef] [Green Version]
- Gryder, B.E.; Wu, L.; Woldemichael, G.M.; Pomella, S.; Quinn, T.R.; Park, P.M.C.; Cleveland, A.; Stanton, B.Z.; Song, Y.; Rota, R.; et al. Chemical genomics reveals histone deacetylases are required for core regulatory transcription. Nat. Commun. 2019, 10, 3004. [Google Scholar] [CrossRef] [Green Version]
- Shah, P.; Westwell, A.D. The role of fluorine in medicinal chemistry. J. Enzym. Inhib. Med. Chem. 2007, 22, 527–540. [Google Scholar] [CrossRef] [Green Version]
- Baecker, D.; Sagasser, J.; Karaman, S.; Hörmann, A.A.; Gust, R. Development of methylated cobalt–alkyne complexes with selective cytotoxicity against COX-positive cancer cell lines. Arch. Pharm. 2022, 355, 2100408. [Google Scholar] [CrossRef]
- Chen, Y.; Lopez-Sanchez, M.; Savoy, D.N.; Billadeau, D.D.; Dow, G.S.; Kozikowski, A.P. A Series of Potent and Selective, Triazolylphenyl-Based Histone Deacetylases Inhibitors with Activity against Pancreatic Cancer Cells and Plasmodium falciparum. J. Med. Chem. 2008, 51, 3437–3448. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhang, X. Recent advances in small molecular modulators targeting histone deacetylase 6. Future Drug Discov. 2020, 2, FDD53. [Google Scholar] [CrossRef]
- Butler, K.V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A.P. Rational Design and Simple Chemistry Yield a Superior, Neuroprotective HDAC6 Inhibitor, Tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846. [Google Scholar] [CrossRef] [Green Version]
- Sandrone, G.; Cukier, C.D.; Zrubek, K.; Marchini, M.; Vergani, B.; Caprini, G.; Fossati, G.; Steinkühler, C.; Stevenazzi, A. Role of Fluorination in the Histone Deacetylase 6 (HDAC6) Selectivity of Benzohydroxamate-Based Inhibitors. ACS Med. Chem. Lett. 2021, 12, 1810–1817. [Google Scholar] [CrossRef]
- Reßing, N.; Schliehe-Diecks, J.; Watson, P.R.; Sönnichsen, M.; Cragin, A.D.; Schöler, A.; Yang, J.; Schäker-Hübner, L.; Borkhardt, A.; Christianson, D.W.; et al. Development of Fluorinated Peptoid-Based Histone Deacetylase (HDAC) Inhibitors for Therapy-Resistant Acute Leukemia. J. Med. Chem. 2022, 65, 15457–15472. [Google Scholar] [CrossRef]
- Keuler, T.; König, B.; Bückreiß, N.; Kraft, F.B.; König, P.; Schäker-Hübner, L.; Steinebach, C.; Bendas, G.; Gütschow, M.; Hansen, F.K. Development of the first non-hydroxamate selective HDAC6 degraders. Chem. Commun. 2022, 58, 11087–11090. [Google Scholar] [CrossRef]
- Sankaranarayanapillai, M.; Tong, W.P.; Maxwell, D.S.; Pal, A.; Pang, J.; Bornmann, W.G.; Gelovani, J.G.; Ronen, S.M. Detection of histone deacetylase inhibition by noninvasive magnetic resonance spectroscopy. Mol. Cancer Ther. 2006, 5, 1325–1334. [Google Scholar] [CrossRef] [Green Version]
- Sankaranarayanapillai, M.; Tong, W.P.; Yuan, Q.; Bankson, J.A.; Dafni, H.; Bornmann, W.G.; Soghomonyan, S.; Pal, A.; Ramirez, M.S.; Webb, D.; et al. Monitoring Histone Deacetylase Inhibition In Vivo: Noninvasive Magnetic Resonance Spectroscopy Method. Mol. Imaging 2008, 7, 7290.2008.0011. [Google Scholar] [CrossRef]
- Jacobson, O.; Kiesewetter, D.O.; Chen, X. Fluorine-18 Radiochemistry, Labeling Strategies and Synthetic Routes. Bioconjugate Chem. 2015, 26, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Ahamed, M.; Vermeulen, K.; Schnekenburger, M.; Moltzau, R.L.; Levy, O.F.; Marton, J.; Froeyen, M.; Olberg, E.D.; Diederich, M.; Bormans, G. Synthesis, Enzyme Assays and Molecular Docking Studies of Fluorinated Bioisosteres of Santacruzamate A as Potential HDAC Tracers. Lett. Drug Des. Discov. 2017, 14, 787–797. [Google Scholar] [CrossRef]
- Li, M.-H.; Shiue, C.-Y.; Chang, H.-C.; Feng, C.-F.; Chang, C.-H. Synthesis and evaluation of F-18-INER-1577 as HDACi Imaging Agent in Mice. J. Nucl. Med. 2017, 58, 1101. [Google Scholar]
- Daśko, M.; de Pascual-Teresa, B.; Ortín, I.; Ramos, A. HDAC Inhibitors: Innovative Strategies for Their Design and Applications. Molecules 2022, 27, 715. [Google Scholar] [CrossRef]
- Chen, W.; Hsiao, Y.; Li, M.; Tsai, M.; Feng, C.; Chang, H.; Yu, H.; Shiue, C. Determination of Fragmentation Schemes and Metabolites of Fluorinated Histone Deacetylase Inhibitors for Use as Positron Emission Tomography Imaging Agents Using HPLC-MS/MS. Int. J. Anal. Mass Spectrom. Chromatogr. 2018, 6, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Halder, R.; Ritter, T. 18F-Fluorination: Challenge and Opportunity for Organic Chemists. J. Org. Chem. 2021, 86, 13873–13884. [Google Scholar] [CrossRef]
- Xing, L.; Blakemore, D.C.; Narayanan, A.; Unwalla, R.; Lovering, F.; Denny, R.A.; Zhou, H.; Bunnage, M.E. Fluorine in Drug Design: A Case Study with Fluoroanisoles. ChemMedChem 2015, 10, 715–726. [Google Scholar] [CrossRef]
- Venturini, F.; Navarrini, W.; Famulari, A.; Sansotera, M.; Dardani, P.; Tortelli, V. Direct trifluoro-methoxylation of aromatics with perfluoro-methyl-hypofluorite. J. Fluor. Chem. 2012, 140, 43–48. [Google Scholar] [CrossRef]
- Landelle, G.; Panossian, A.; Leroux, F.R. Trifluoromethyl ethers and -thioethers as tools for medicinal chemistry and drug discovery. Curr. Top. Med. Chem. 2014, 14, 941–951. [Google Scholar] [CrossRef]
- Caron, S. Where Does the Fluorine Come From? A Review on the Challenges Associated with the Synthesis of Organofluorine Compounds. Org. Process Res. Dev. 2020, 24, 470–480. [Google Scholar] [CrossRef]
- Kirk, K.L. Fluorination in Medicinal Chemistry: Methods, Strategies, and Recent Developments. Org. Process Res. Dev. 2008, 12, 305–321. [Google Scholar] [CrossRef]
- Ricaud, P.; Lefèvre, F. Chapter 1: Fluorine in the Atmosphere. In Advances in Fluorine Science; Tressaud, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 1, pp. 1–32. [Google Scholar]
- Pan, Y. The Dark Side of Fluorine. ACS Med. Chem. Lett. 2019, 10, 1016–1019. [Google Scholar] [CrossRef] [Green Version]
- Wermers, R.A.; Cooper, K.; Razonable, R.R.; Deziel, P.J.; Whitford, G.M.; Kremers, W.K.; Moyer, T.P. Fluoride Excess and Periostitis in Transplant Patients Receiving Long-Term Voriconazole Therapy. Clin. Infect. Dis. 2011, 52, 604–611. [Google Scholar] [CrossRef]
- Goncharov, N.V.; Jenkins, R.O.; Radilov, A.S. Toxicology of fluoroacetate: A review, with possible directions for therapy research. J. Appl. Toxicol. 2006, 26, 148–161. [Google Scholar] [CrossRef]
- Inoue, M.; Sumii, Y.; Shibata, N. Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 2020, 5, 10633–10640. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Alauddin, M.M. Positron emission tomography (PET) imaging with (18)F-based radiotracers. Am. J. Nucl. Med. Mol. Imaging 2012, 2, 55–76. [Google Scholar]
Figure 1.
Chemical structures of some HDACis considering the pharmacophore consisting of cap, linker, and zinc-binding group (ZBG).
Figure 1.
Chemical structures of some HDACis considering the pharmacophore consisting of cap, linker, and zinc-binding group (ZBG).

Scheme 1.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids via reductive amination by Aboukhatwa et al. [33].
Scheme 1.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids via reductive amination by Aboukhatwa et al. [33].

Scheme 2.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids via alkylation by Aboukhatwa et al. [33].
Scheme 2.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids via alkylation by Aboukhatwa et al. [33].

Scheme 3.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids by Wang et al. [34].
Scheme 3.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids by Wang et al. [34].

Scheme 4.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids by Goehringer, Peng, et al. [35].
Scheme 4.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids by Goehringer, Peng, et al. [35].

Scheme 5.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids by Meyners et al. [36].
Scheme 5.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids by Meyners et al. [36].

Scheme 6.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids by Toutah et al. [37].
Scheme 6.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids by Toutah et al. [37].

Scheme 7.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids by Walton et al. [38].
Scheme 7.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids by Walton et al. [38].

Scheme 8.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids by Vu et al. [39].
Scheme 8.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids by Vu et al. [39].

Scheme 9.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids by Liu et al. [40].
Scheme 9.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids by Liu et al. [40].

Scheme 10.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids by Erdeljac et al. [41].
Scheme 10.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids by Erdeljac et al. [41].

Scheme 11.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids by Ariawan et al. [42].
Scheme 11.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids by Ariawan et al. [42].

Scheme 12.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids by Strebl et al. [43].
Scheme 12.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids by Strebl et al. [43].

Scheme 13.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids by Hendricks, Keliher, et al. [44].
Scheme 13.
Fluorination in the synthetic pathway of fluorinated hydroxamic acids by Hendricks, Keliher, et al. [44].

Scheme 14.
Fluorination in the two synthetic pathways (A,B) of fluorinated hydroxamic acids by Strebl et al. [45]. Radiochemical procedures for MGS1–2 (A) and MGS3 (B).
Scheme 14.
Fluorination in the two synthetic pathways (A,B) of fluorinated hydroxamic acids by Strebl et al. [45]. Radiochemical procedures for MGS1–2 (A) and MGS3 (B).

Scheme 15.
Fluorination in the synthetic pathway of fluorinated benzamides by Jayathilaka et al. [47].
Scheme 15.
Fluorination in the synthetic pathway of fluorinated benzamides by Jayathilaka et al. [47].

Scheme 16.
Fluorination in the synthetic pathway of fluorinated benzamides by Bonomi et al. [48].
Scheme 16.
Fluorination in the synthetic pathway of fluorinated benzamides by Bonomi et al. [48].

Scheme 17.
Fluorination in the synthetic pathway of fluorinated benzamides by La et al. [49].
Scheme 17.
Fluorination in the synthetic pathway of fluorinated benzamides by La et al. [49].

Scheme 18.
Fluorination in the synthetic pathway of fluorinated benzamides via Suzuki reaction by Ibrahim et al. [50].
Scheme 18.
Fluorination in the synthetic pathway of fluorinated benzamides via Suzuki reaction by Ibrahim et al. [50].

Scheme 19.
Fluorination in the synthetic pathway of fluorinated benzamides via amide-coupling reactions by Ibrahim et al. [50].
Scheme 19.
Fluorination in the synthetic pathway of fluorinated benzamides via amide-coupling reactions by Ibrahim et al. [50].

Scheme 20.
Fluorination in the synthetic pathway of fluorinated benzamides by Schäker-Hübner et al. [51].
Scheme 20.
Fluorination in the synthetic pathway of fluorinated benzamides by Schäker-Hübner et al. [51].

Scheme 21.
Fluorination in the synthetic pathway of fluorinated benzamides by Liu et al. [40].
Scheme 21.
Fluorination in the synthetic pathway of fluorinated benzamides by Liu et al. [40].

Scheme 22.
Fluorination in the radiosynthetic pathway of fluorinated benzamides by Bonomi et al. [48].
Scheme 22.
Fluorination in the radiosynthetic pathway of fluorinated benzamides by Bonomi et al. [48].

Scheme 23.
Fluorination in the radiosynthetic pathway of [18F] Fluoroethyl-INER1577 by Li et al. [52].
Scheme 23.
Fluorination in the radiosynthetic pathway of [18F] Fluoroethyl-INER1577 by Li et al. [52].

Scheme 24.
Fluorination in the radiosynthetic pathway of [18F] Fluoroethyl-INER1577 by Li et al. [53].
Scheme 24.
Fluorination in the radiosynthetic pathway of [18F] Fluoroethyl-INER1577 by Li et al. [53].

Scheme 25.
Fluorination in the synthetic pathway of fluorinated thiols by Chuman et al. [55].
Scheme 25.
Fluorination in the synthetic pathway of fluorinated thiols by Chuman et al. [55].

Scheme 26.
Fluorination in the synthetic pathway of fluorinated thiols by Wen et al. [56].
Scheme 26.
Fluorination in the synthetic pathway of fluorinated thiols by Wen et al. [56].

Scheme 27.
Fluorination in the synthetic pathway of fluorinated short-chain fatty acids via alkylation by Lübke et al. [59].
Scheme 27.
Fluorination in the synthetic pathway of fluorinated short-chain fatty acids via alkylation by Lübke et al. [59].

Scheme 28.
Fluorination in the synthetic pathway of fluorinated short-chain fatty acids via bromofluorination by Lübke et al. [59].
Scheme 28.
Fluorination in the synthetic pathway of fluorinated short-chain fatty acids via bromofluorination by Lübke et al. [59].

Figure 2.
Chemical structure of largazole.

Scheme 29.
Fluorination in the synthetic pathway of fluorinated cyclic peptides by Zhang, Liu, Gao, et al. [60,61].

Figure 3.
Chemical structures of fluorinated hydroxamic acids by Salmi-Smail et al. [68].
Figure 3.
Chemical structures of fluorinated hydroxamic acids by Salmi-Smail et al. [68].

Figure 4.
Chemical structures of fluorinated hydroxamic acids by Luckhurst [69].
Figure 4.
Chemical structures of fluorinated hydroxamic acids by Luckhurst [69].

Figure 5.
Chemical structure of fluorinated hydroxamic acid by Yao, Li et al. [70].
Figure 5.
Chemical structure of fluorinated hydroxamic acid by Yao, Li et al. [70].

Figure 6.
Chemical structure of fluorinated hydroxamic acid (PTG-0861) by Gawel, Shouksmith, Raouf, Nawar et al. [71].
Figure 6.
Chemical structure of fluorinated hydroxamic acid (PTG-0861) by Gawel, Shouksmith, Raouf, Nawar et al. [71].

Figure 7.
Chemical structure of fluorinated benzamide (CBUD-1001) by Kim, La et al. [72].
Figure 7.
Chemical structure of fluorinated benzamide (CBUD-1001) by Kim, La et al. [72].

Figure 8.
Chemical structure of Merck60.


Figure 10.
Chemical structure of fluorinated hydroxamic acids by Sandrone et al. [83].
Figure 10.
Chemical structure of fluorinated hydroxamic acids by Sandrone et al. [83].

Figure 11.
Chemical structures of fluorinated HDAC6-addressing PROTACs by Keuler, König, Bückreiß et al. [85].
Figure 11.
Chemical structures of fluorinated HDAC6-addressing PROTACs by Keuler, König, Bückreiß et al. [85].


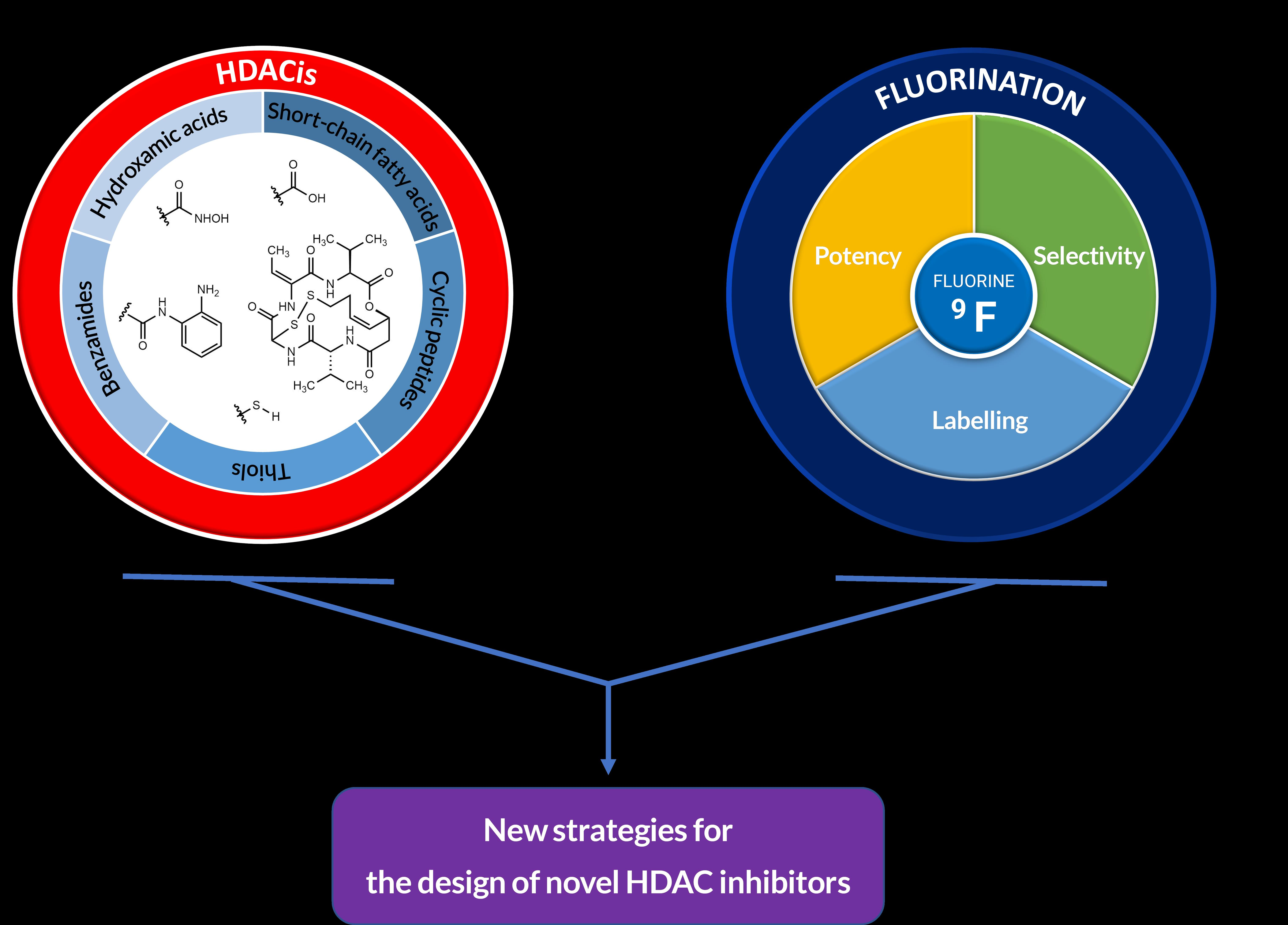{kind=link}
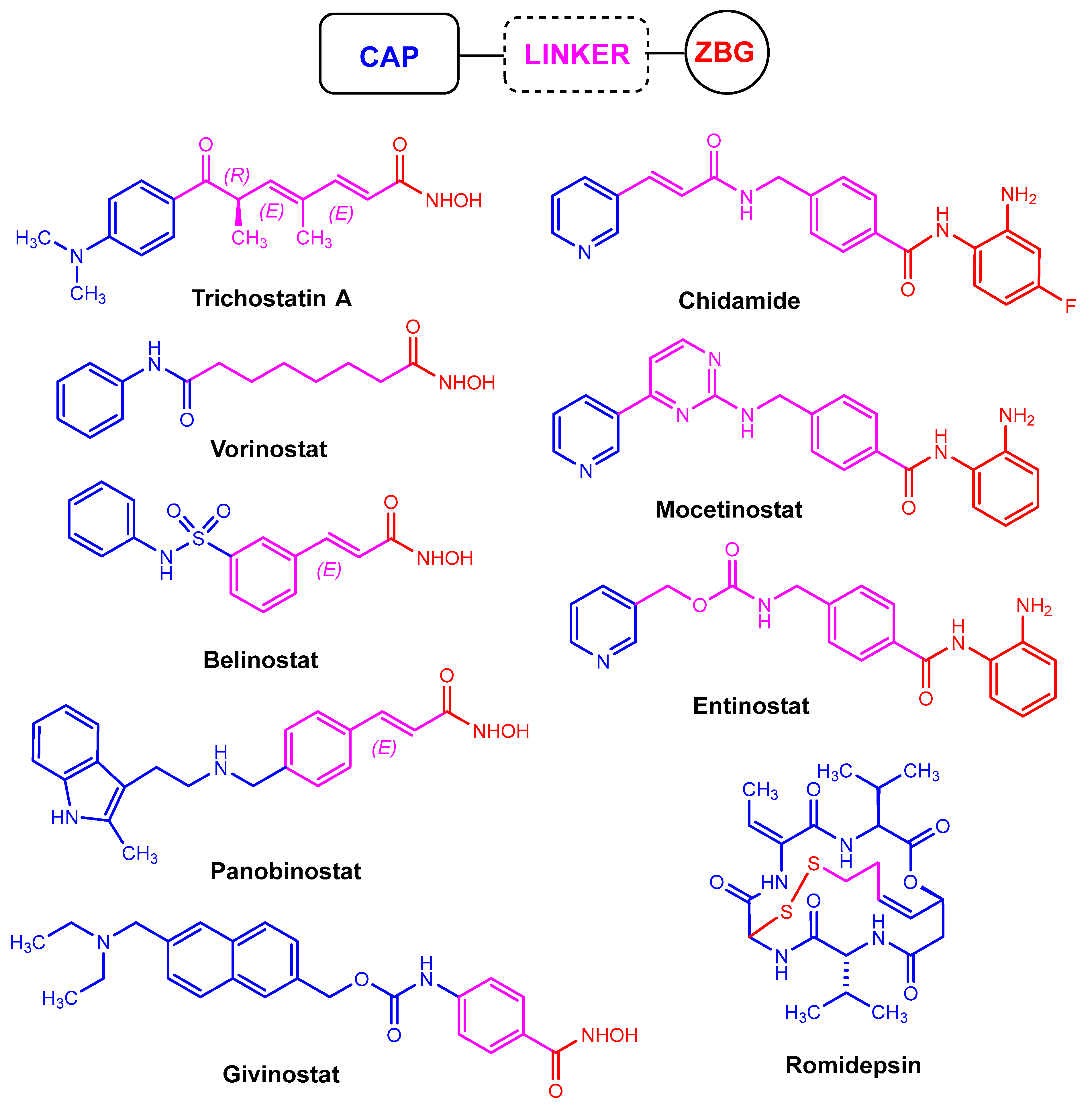{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
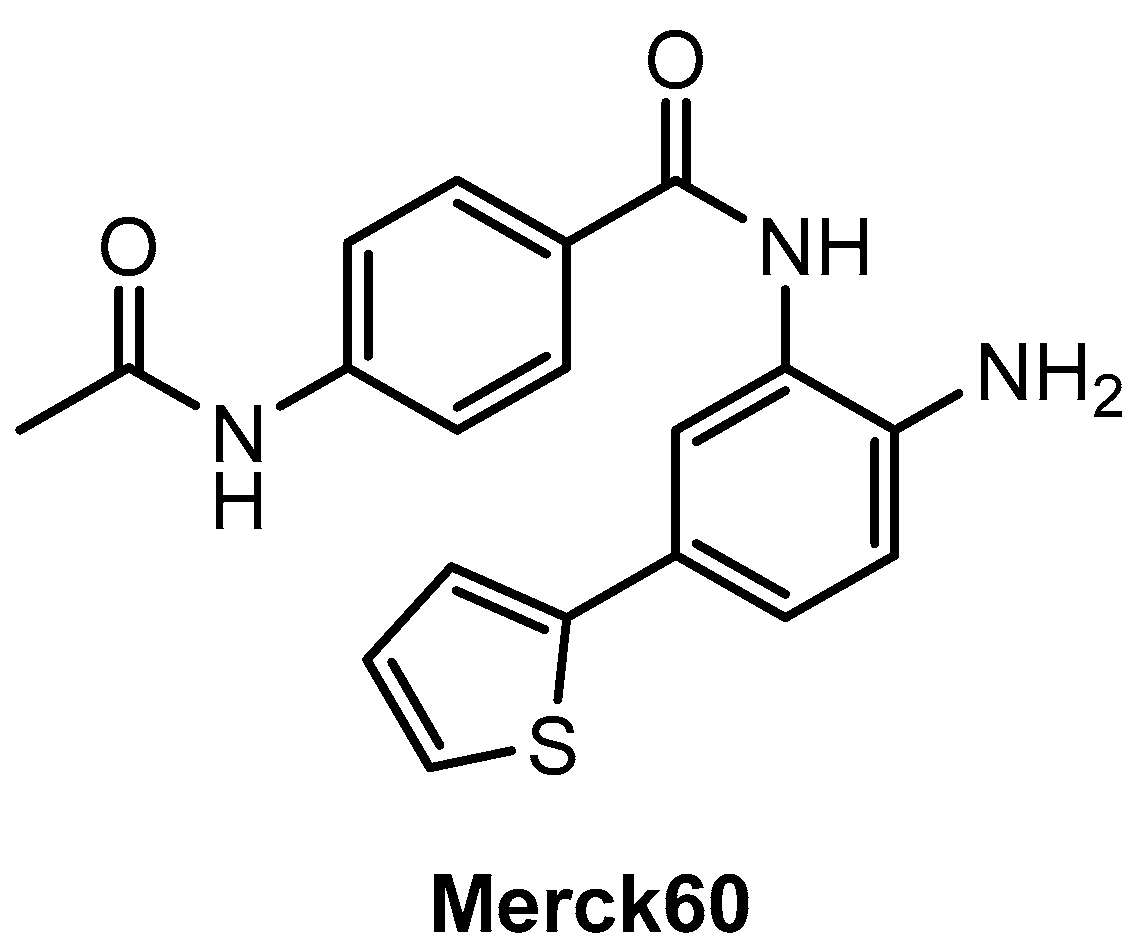{kind=link}
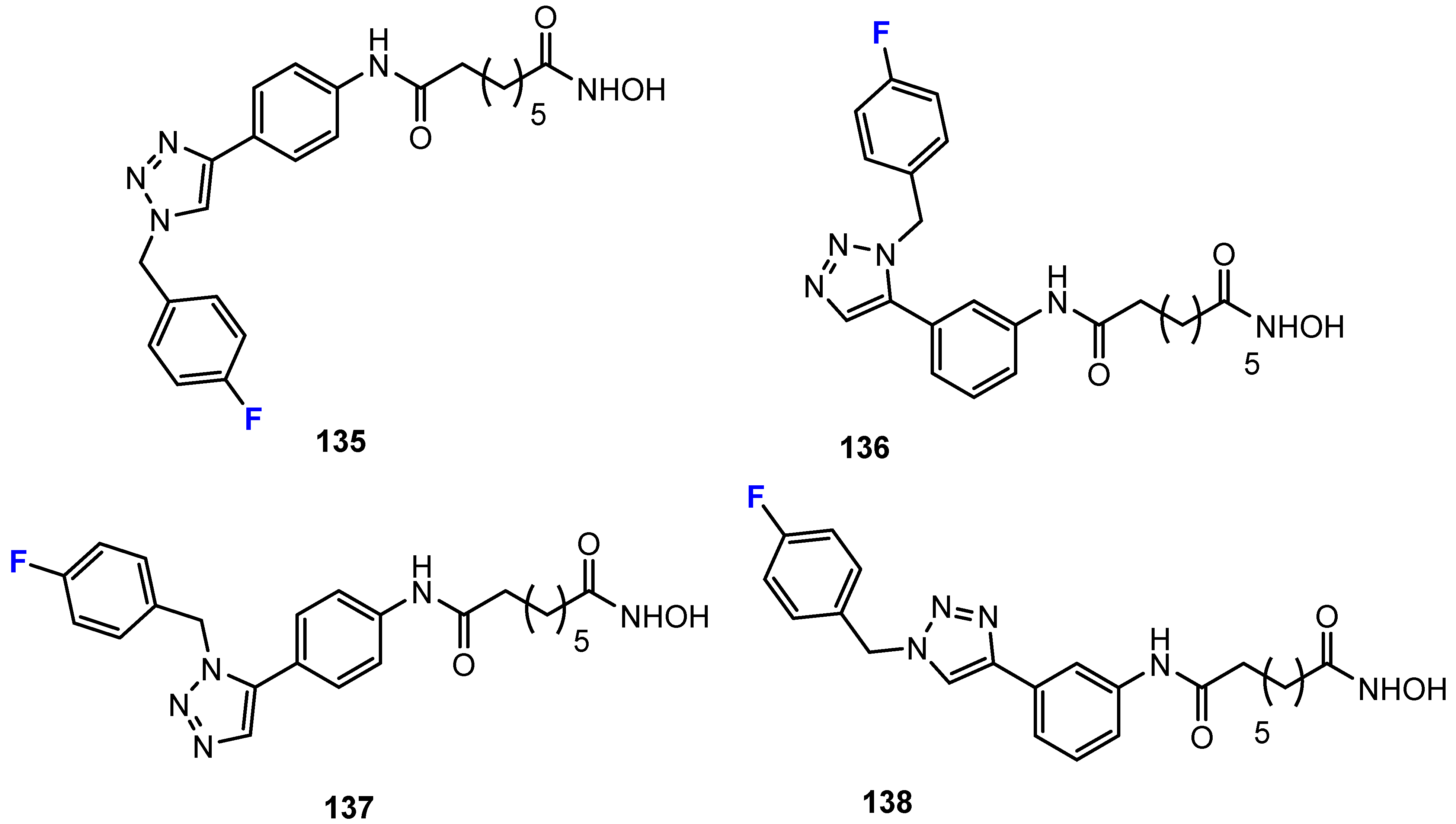{kind=link}
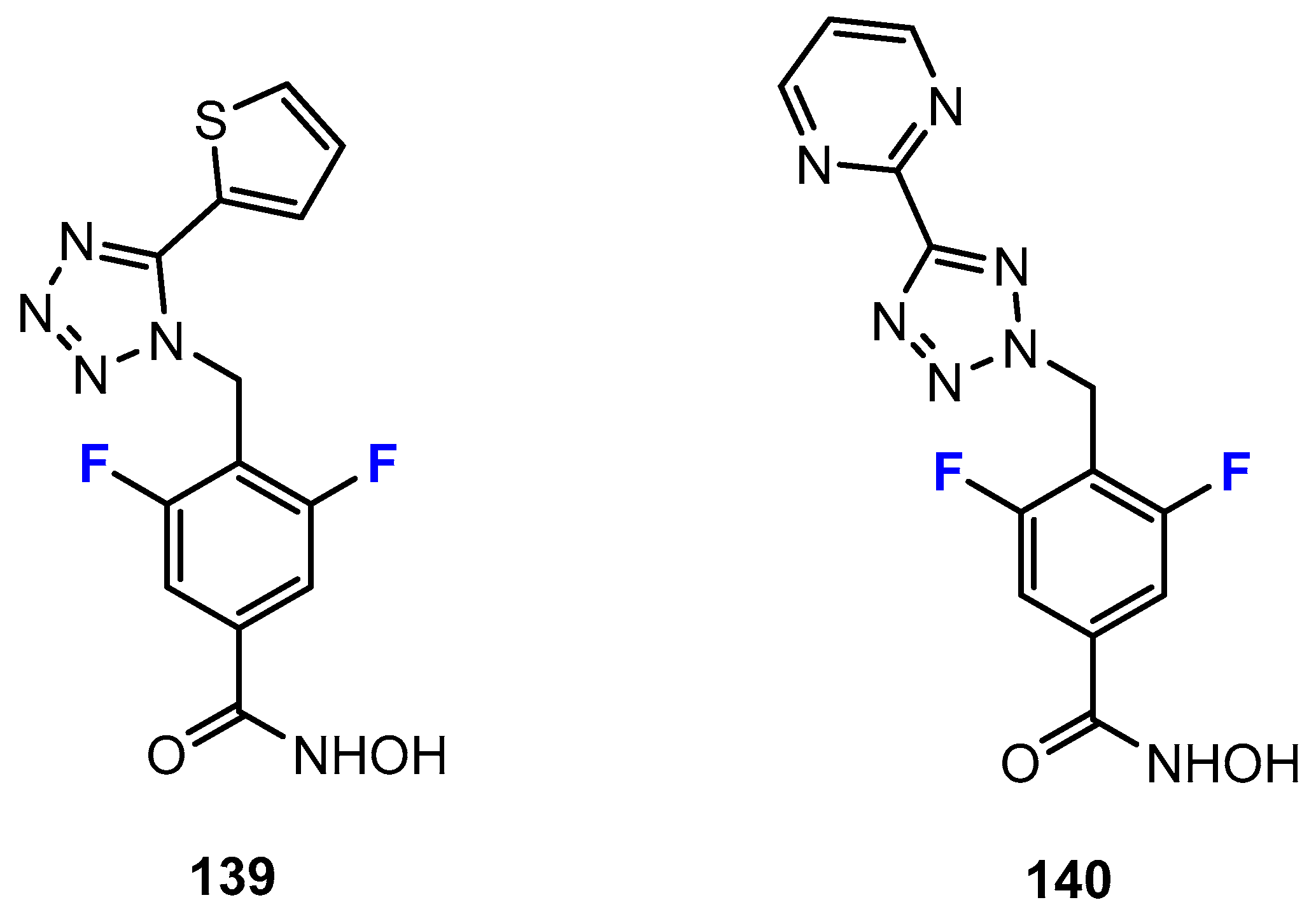{kind=link}
{kind=link}
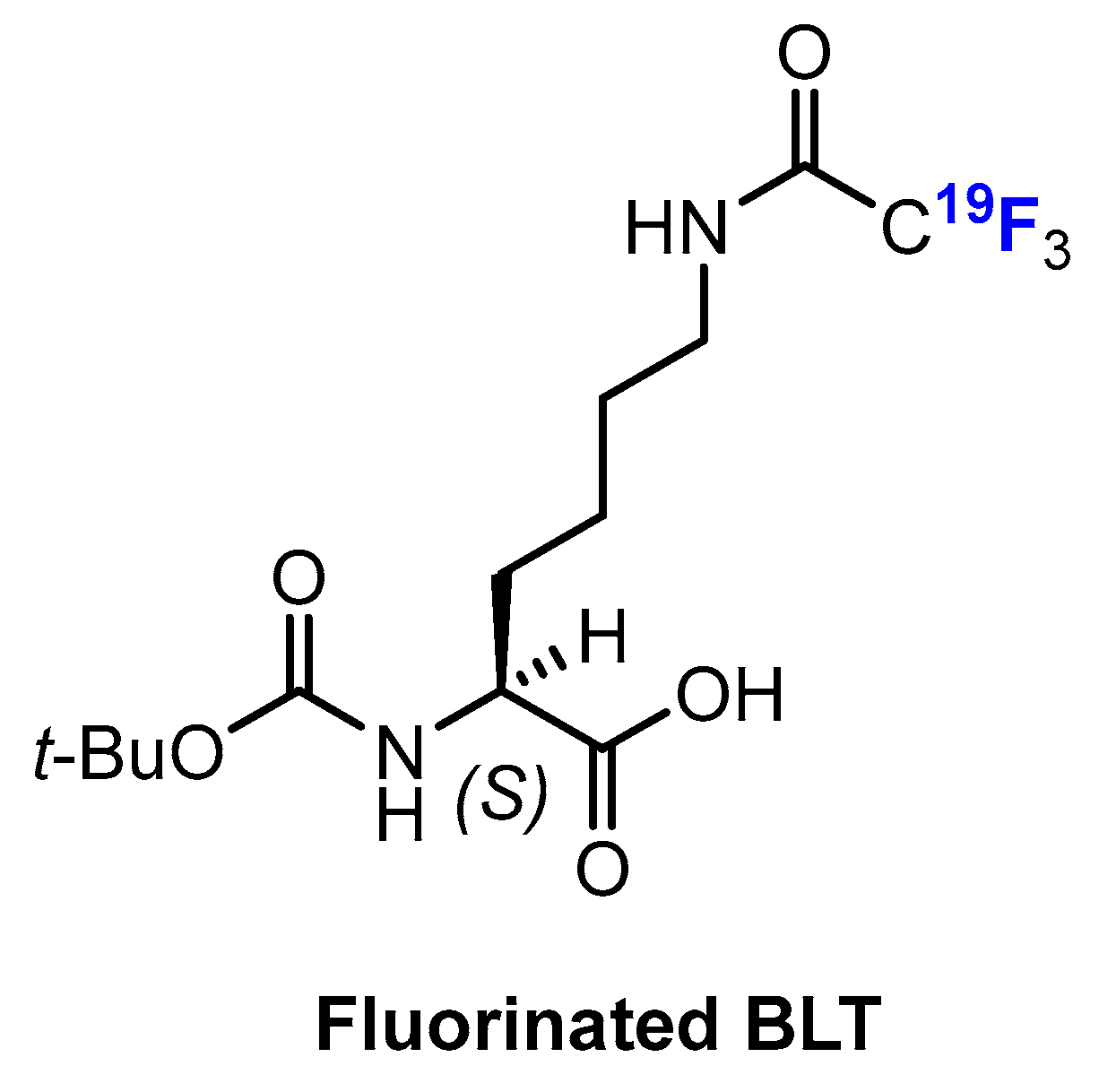{kind=link}
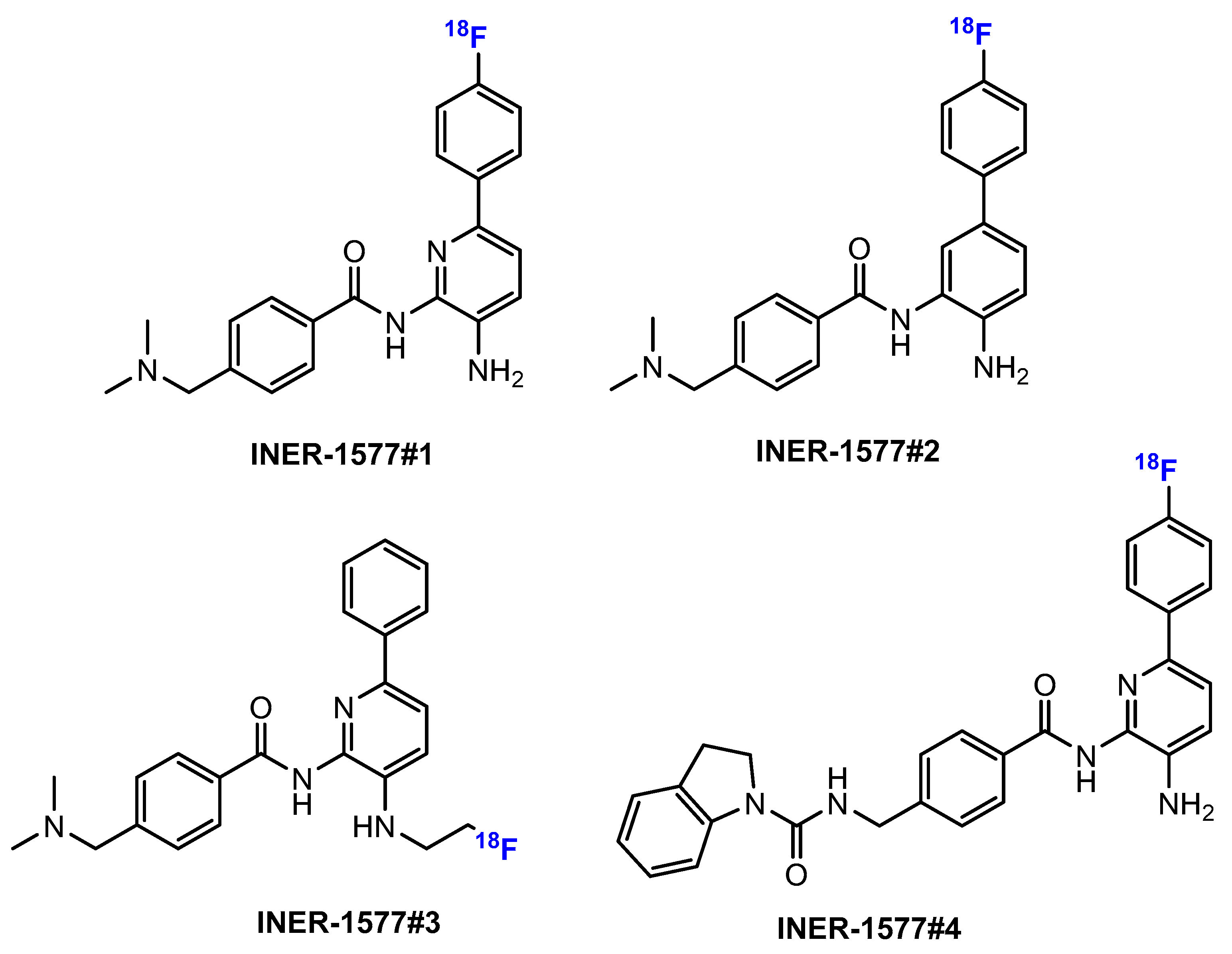{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
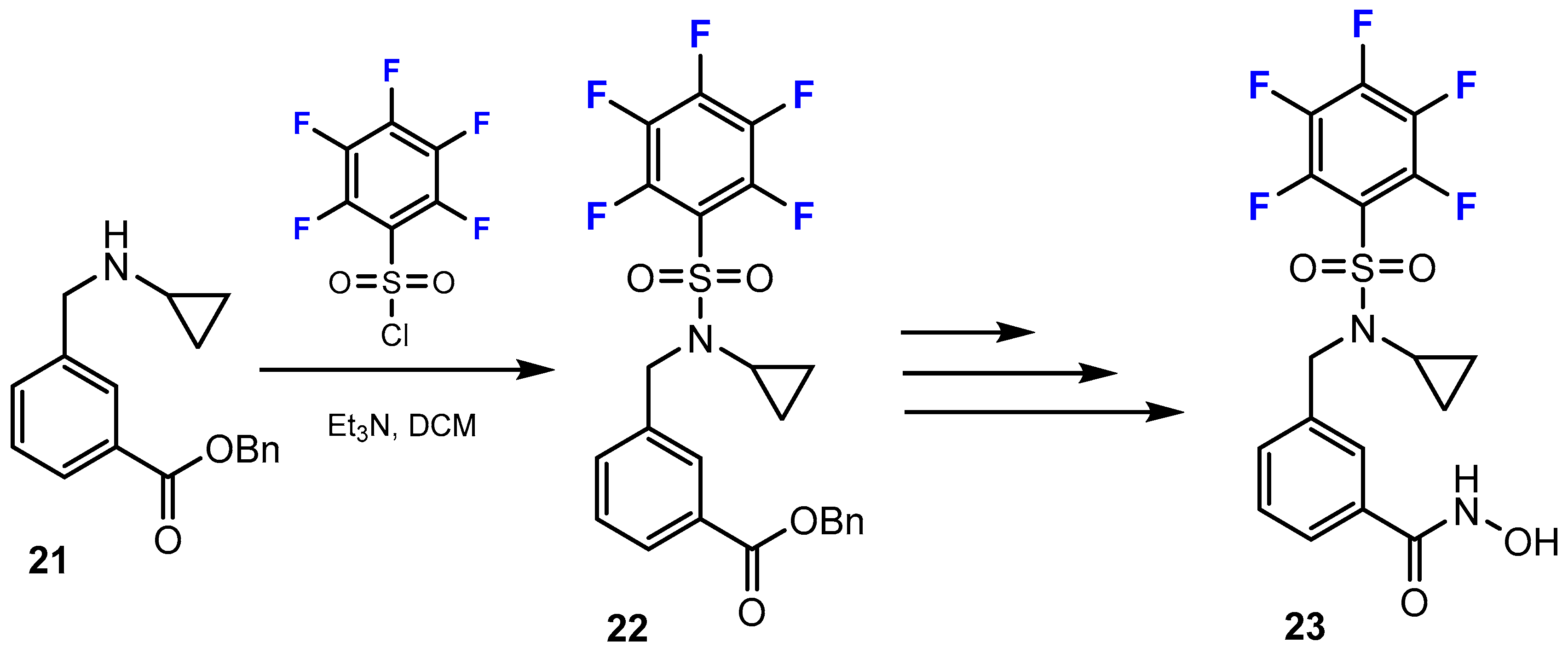{kind=link}
{kind=link}
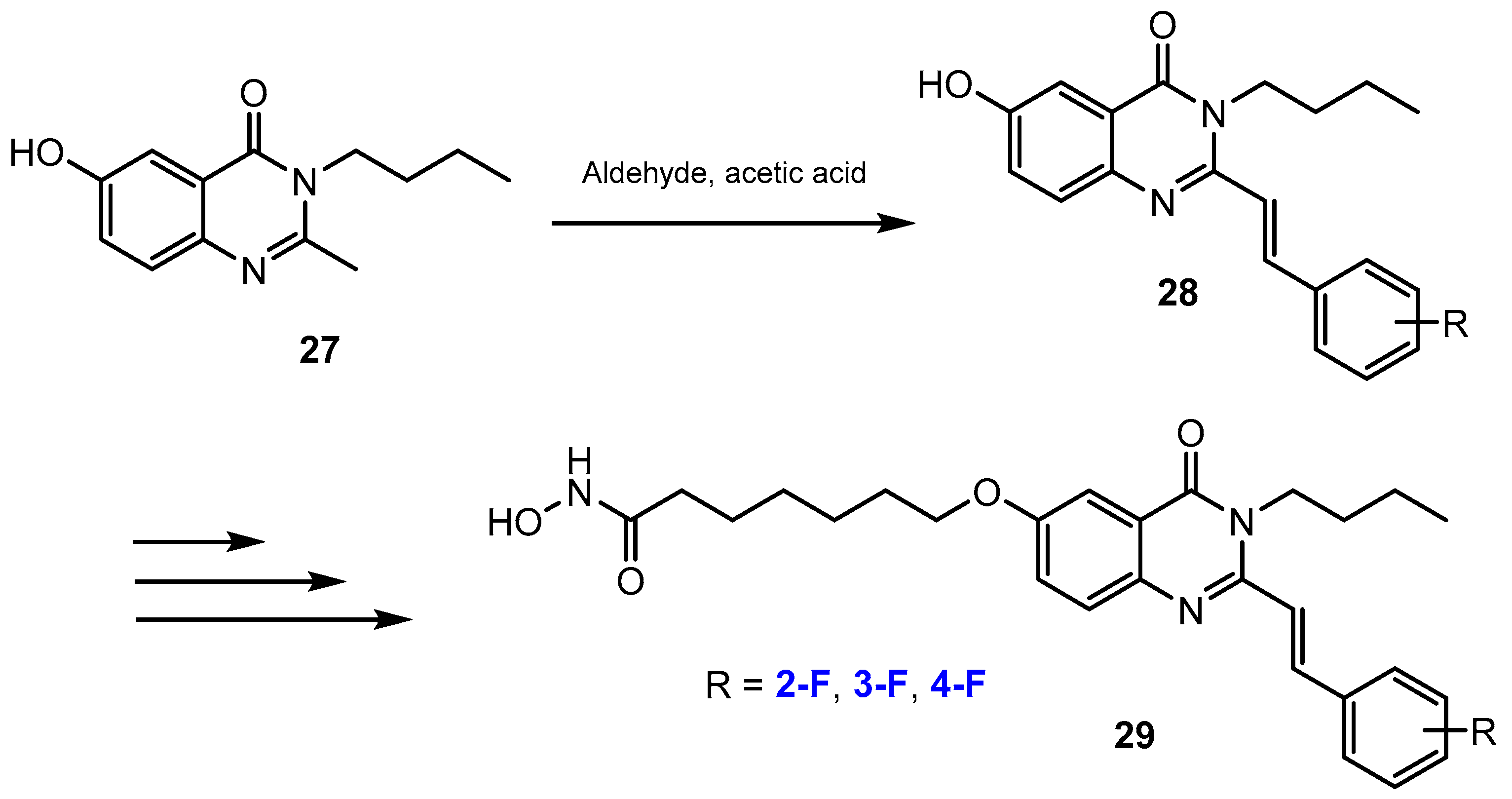{kind=link}
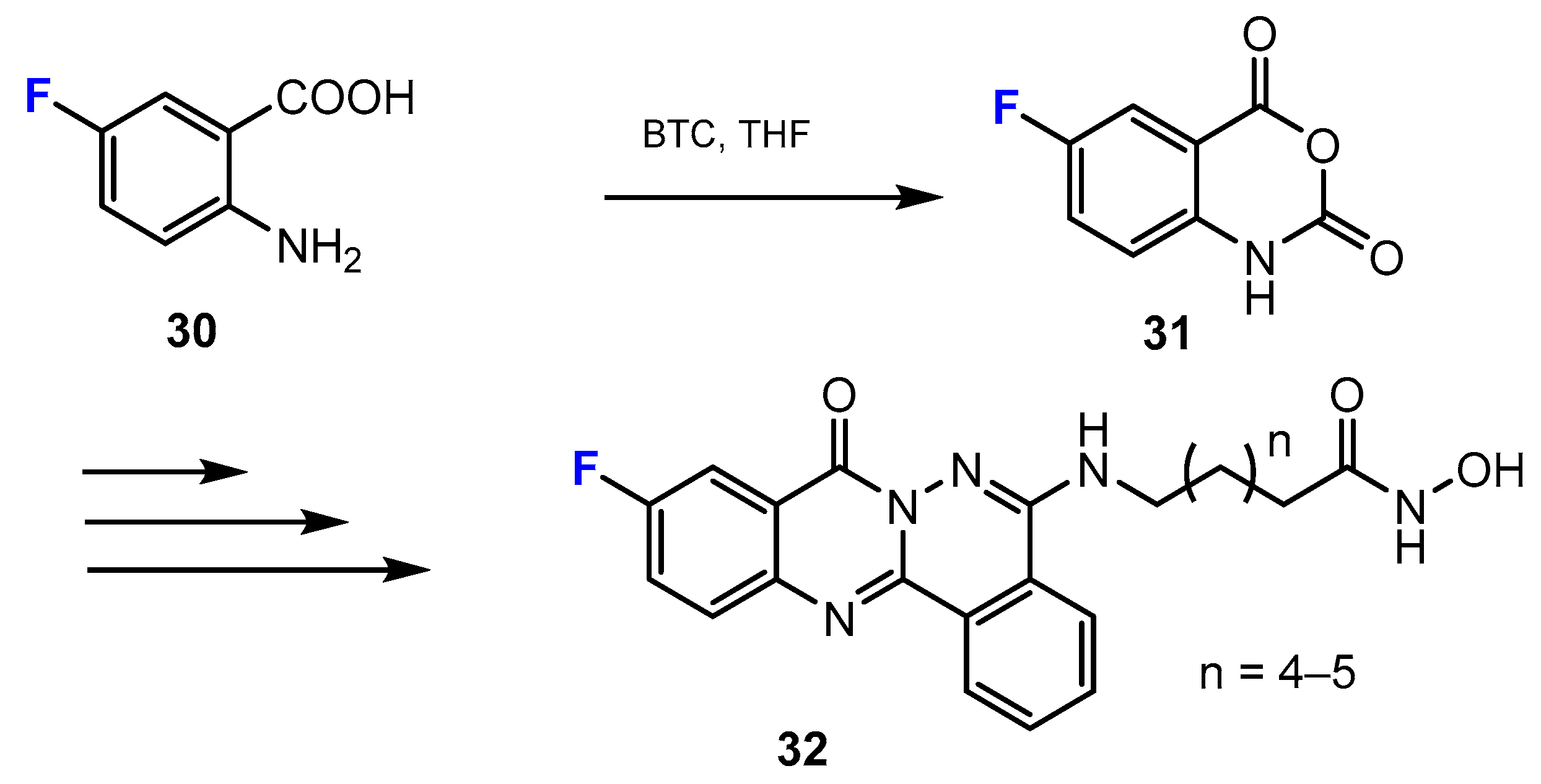{kind=link}
{kind=link}
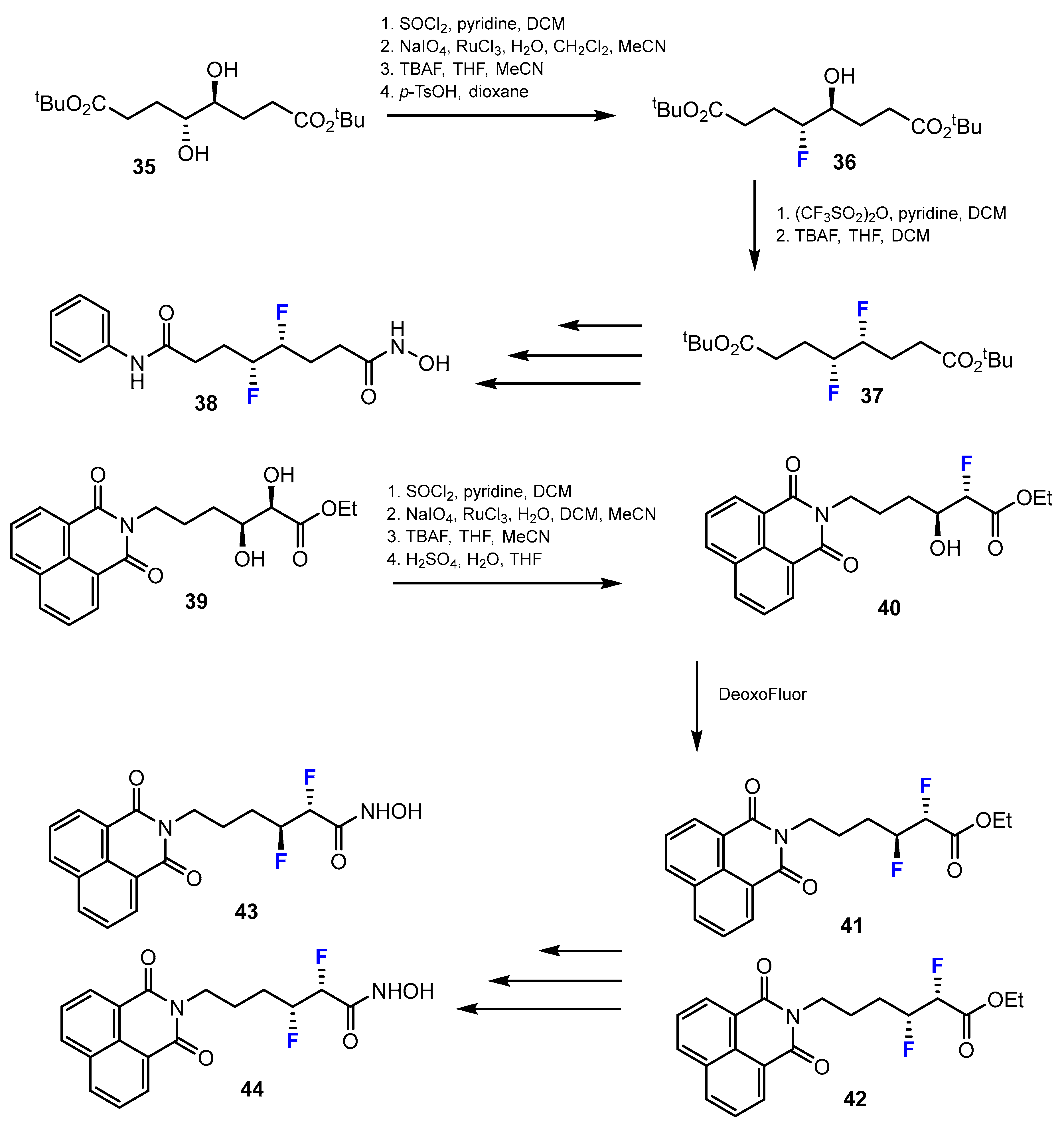{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
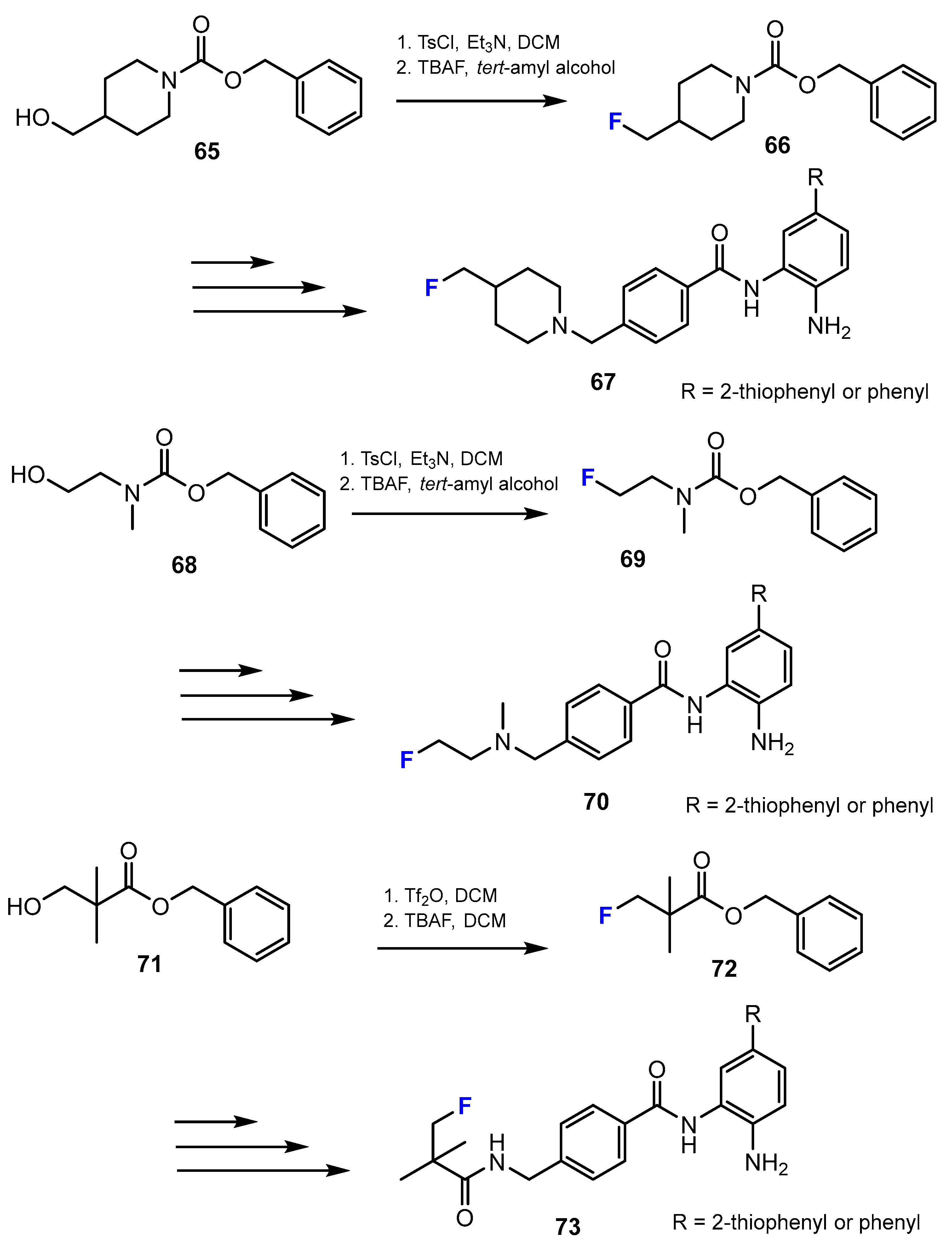{kind=link}
{kind=link}
{kind=link}
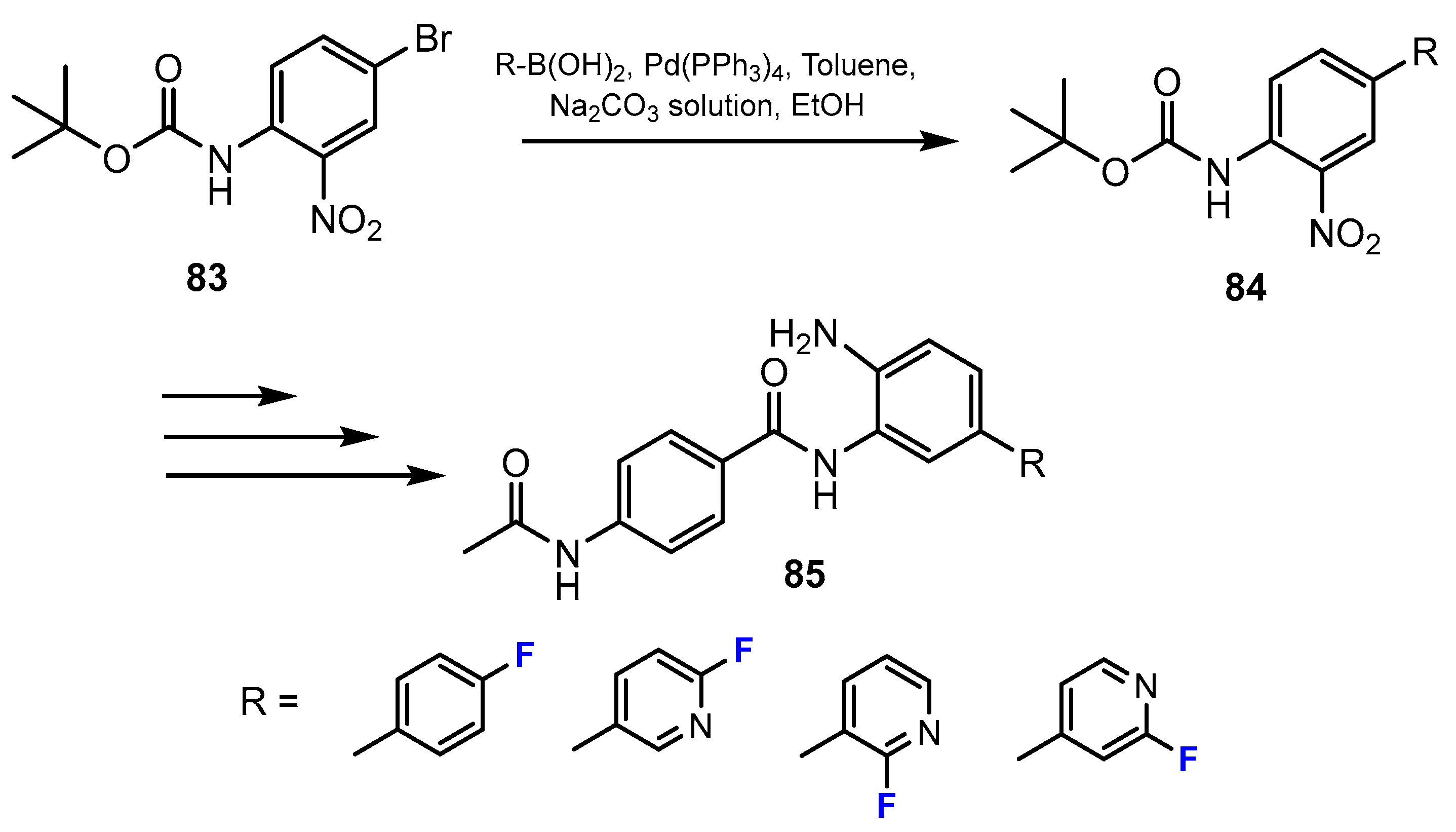{kind=link}
{kind=link}
{kind=link}
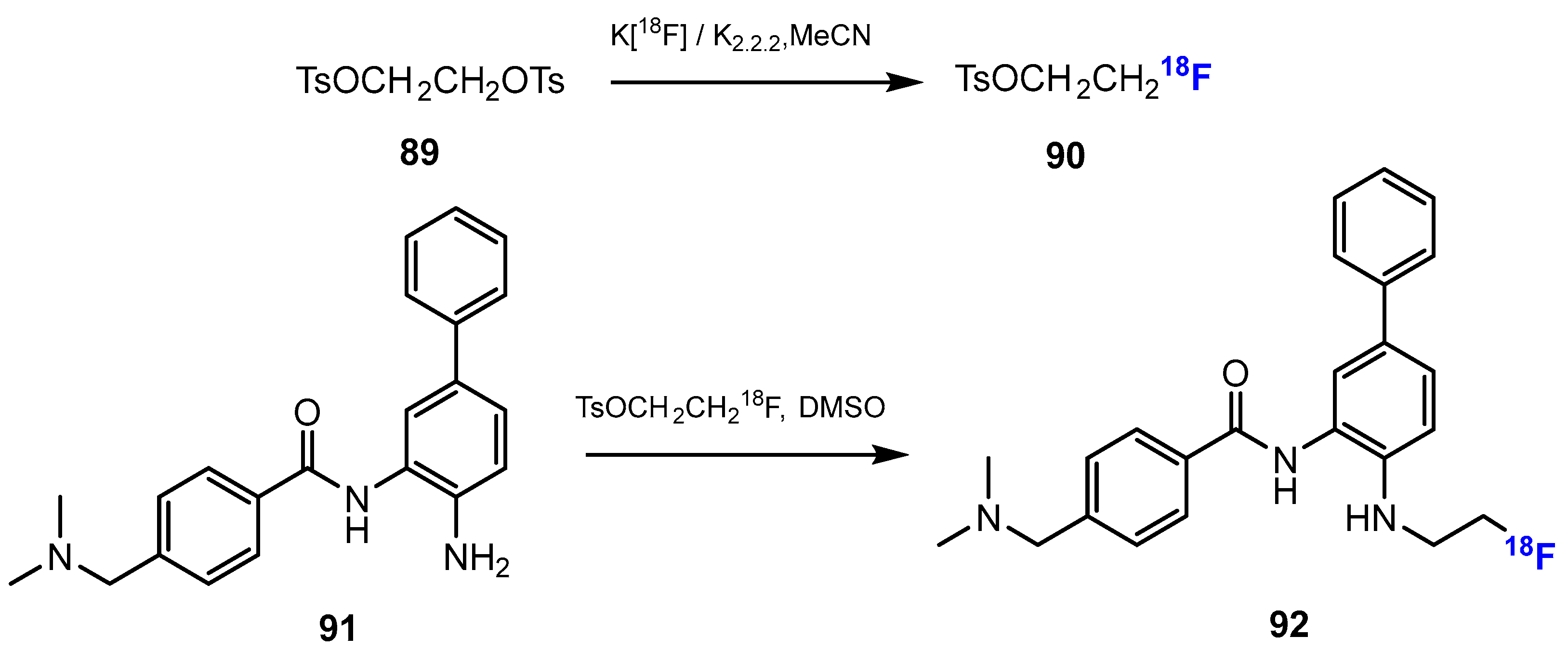{kind=link}
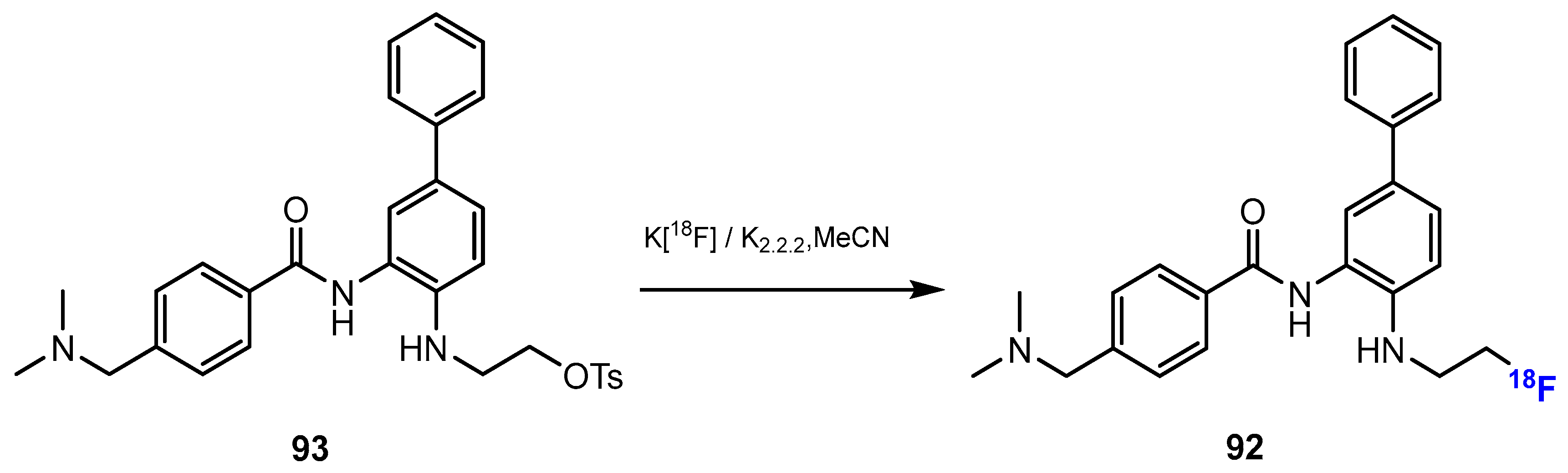{kind=link}
{kind=link}
{kind=link}
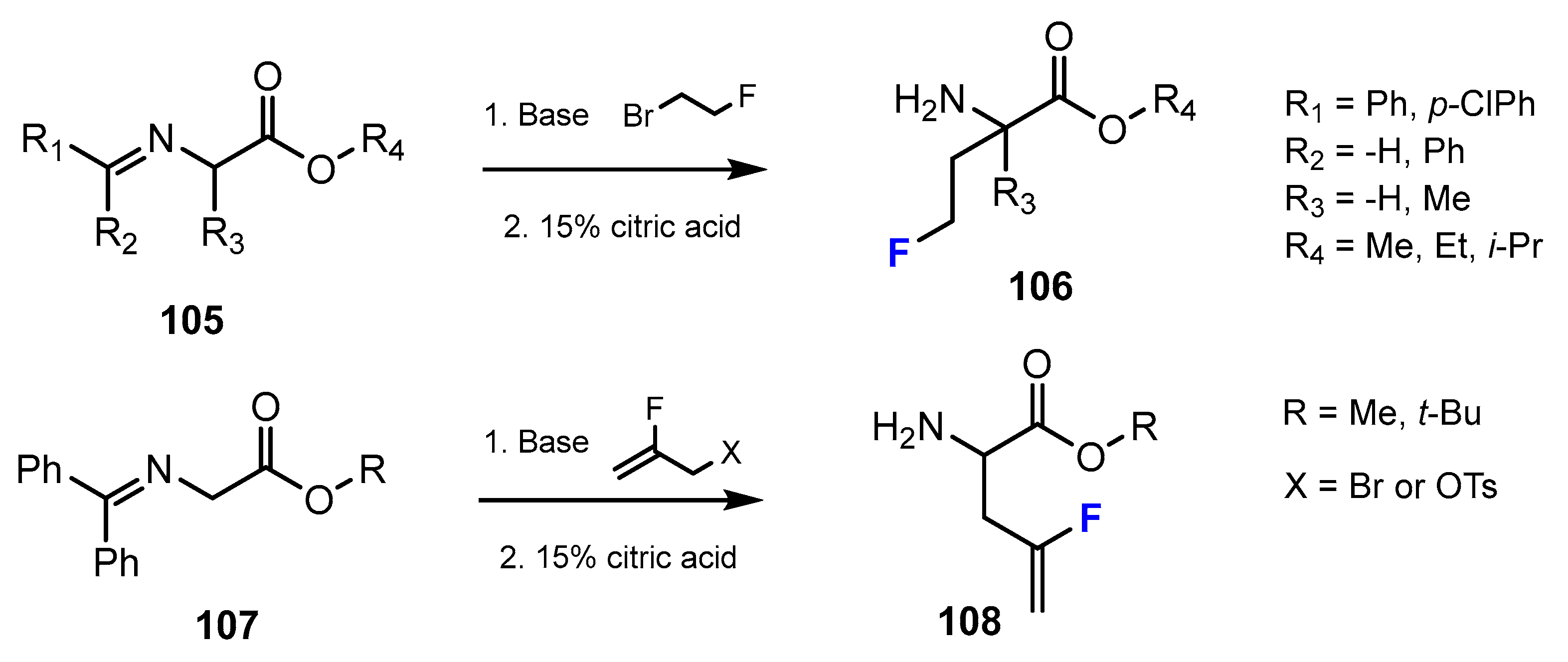{kind=link}
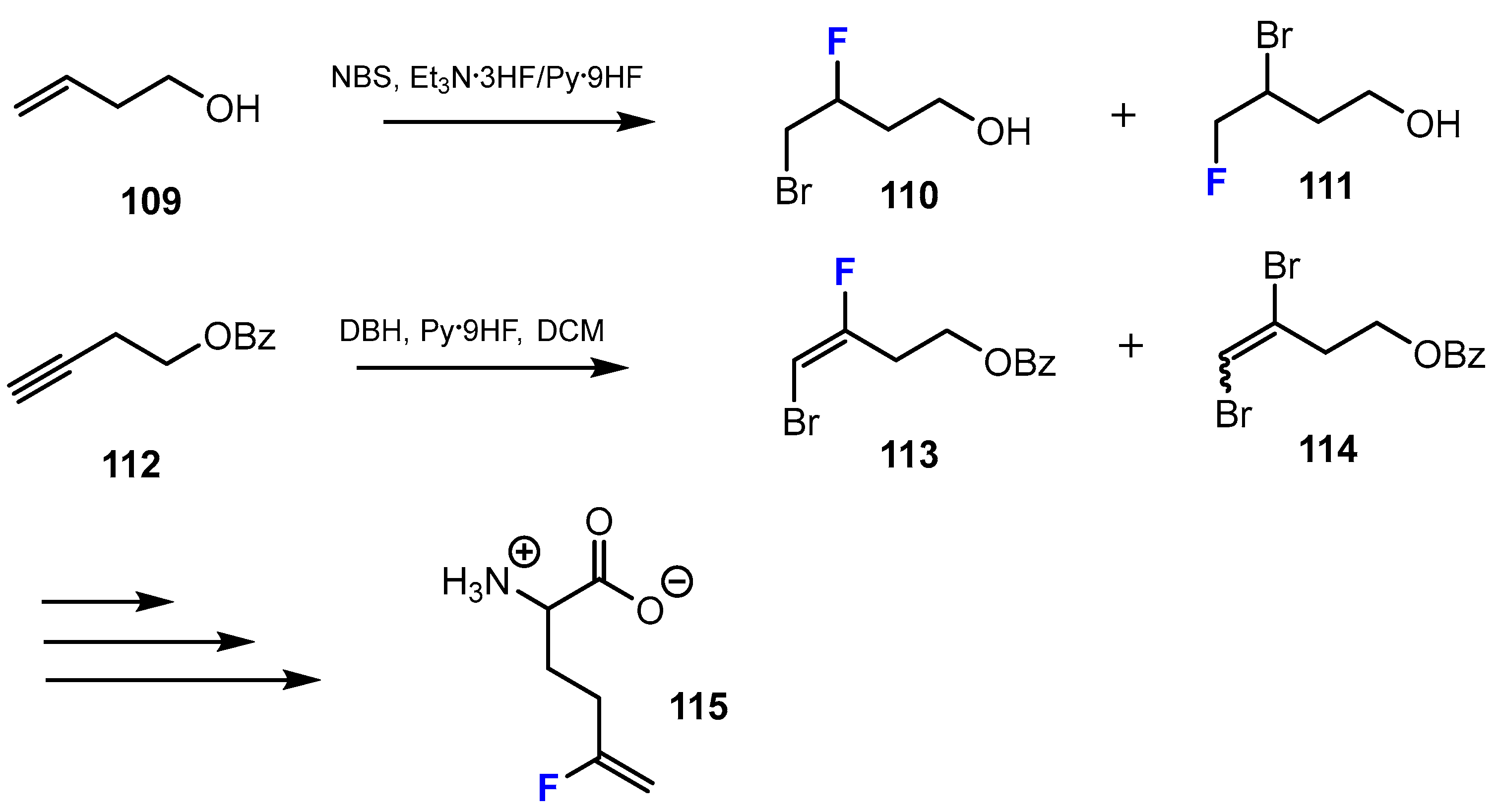{kind=link}
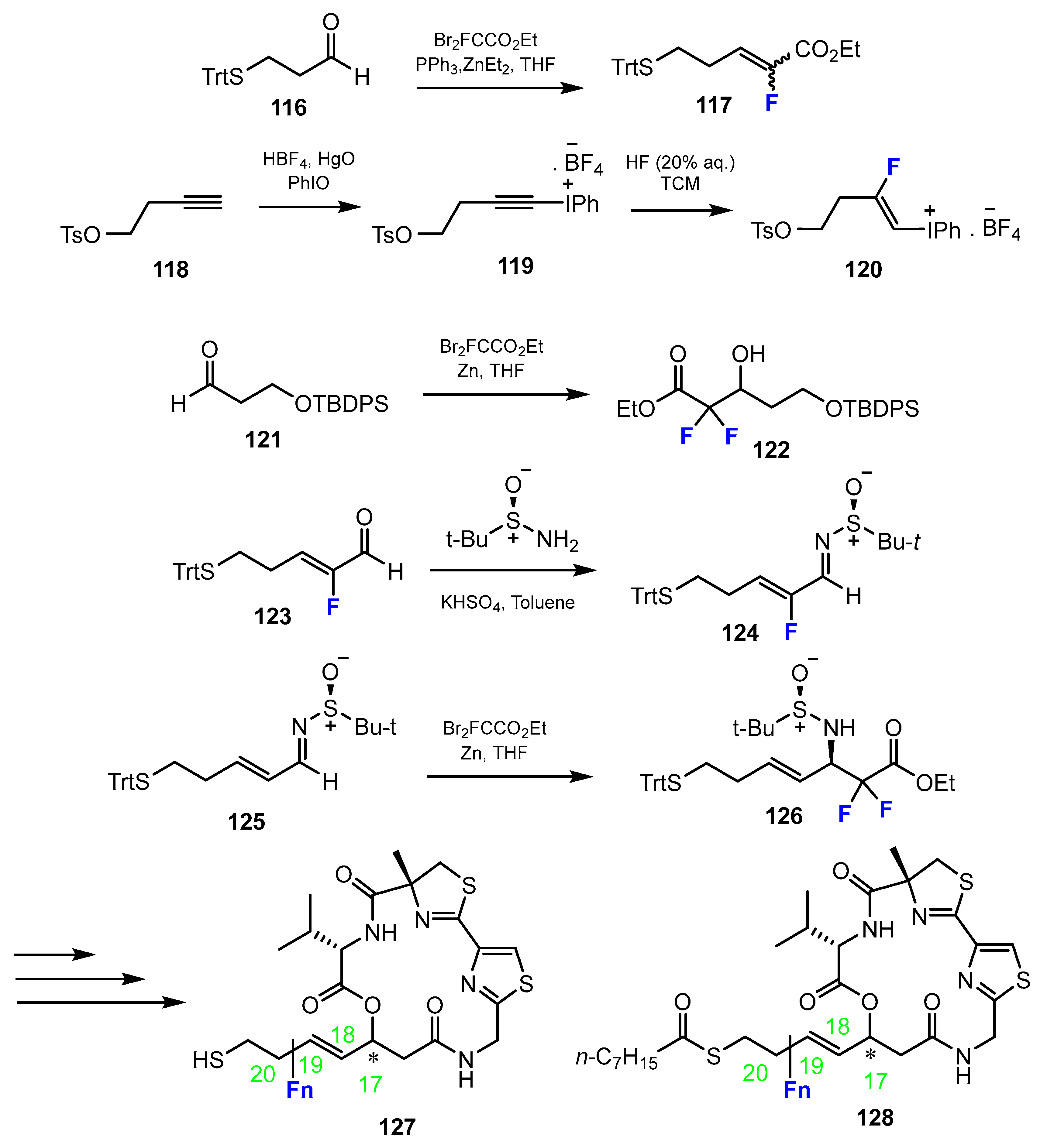{kind=link}
Figure 13.
Chemical structures of four INER-1577 derivatives by Chen et al. [92].
Figure 13.
Chemical structures of four INER-1577 derivatives by Chen et al. [92].

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tien Anh, D.; Hai Nam, N.; Kircher, B.; Baecker, D. The Impact of Fluorination on the Design of Histone Deacetylase Inhibitors. Molecules 2023, 28, 1973. https://doi.org/10.3390/molecules28041973
AMA Style
Tien Anh D, Hai Nam N, Kircher B, Baecker D. The Impact of Fluorination on the Design of Histone Deacetylase Inhibitors. Molecules. 2023; 28(4):1973. https://doi.org/10.3390/molecules28041973
Chicago/Turabian StyleTien Anh, Duong, Nguyen Hai Nam, Brigitte Kircher, and Daniel Baecker. 2023. "The Impact of Fluorination on the Design of Histone Deacetylase Inhibitors" Molecules 28, no. 4: 1973. https://doi.org/10.3390/molecules28041973