
It Takes Two to Tango, Part II: Synthesis of A-Ring Functionalised Quinones Containing Two Redox-Active Centres with Antitumour Activities
 ,
,  , , and
, , and
Abstract
:1. Introduction

2. Results and Discussion
2.1. Synthesis of the Azide Units
2.2. Synthesis of the Aminoalkyne Units
2.3. Scopes Achieved
2.4. Anticancer Evaluation
2.4.1. Azide Substrates (4, 7 and 10)
2.4.2. Naphthoquinoidal Aminoalkyne Substrates (18a–e)
2.4.3. First Family of Triazoles (19a–e)
2.4.4. Second Family of Triazoles (20a–e)
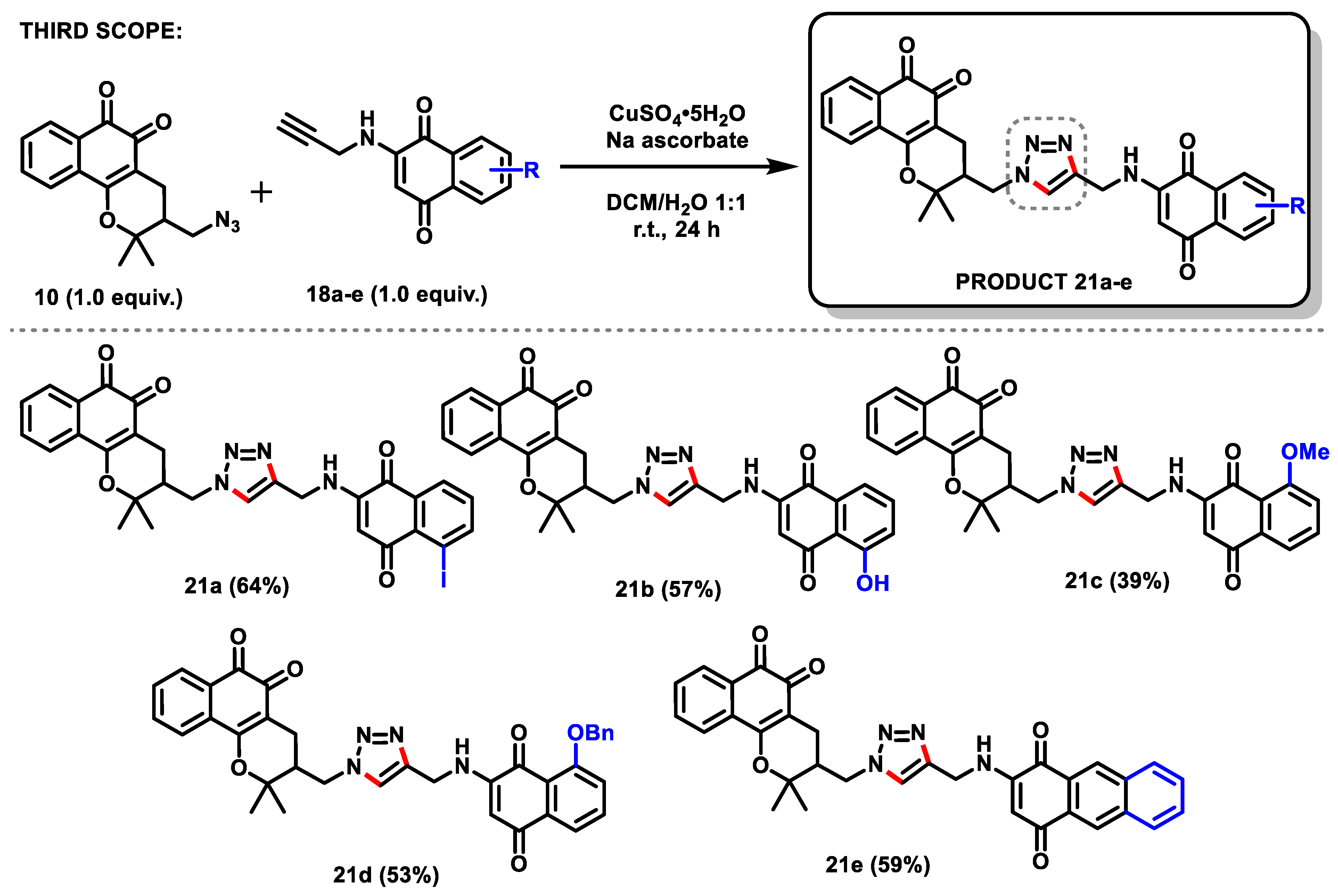
2.4.5. Third Family of Triazoles (21a–e)
3. Materials and Methods
3.1. General Remarks
3.2. Synthesis of Azide Precursors (4, 7, and 10)
3.3. Synthesis of A-Ring-Modified Quinoidal Substrates
3.4. General Procedure for the Synthesis of Amino-Alkynes (18a–e)
3.5. General Procedure for Triazole Synthesis via a Copper-Catalysed 1,3-Dipolar Cycloaddition (19a–21e)
3.6. Characterization Data of Products 19a–21e
3.7. Anti-Tumor Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Cancer—World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 18 November 2022).
- Zughaibi, T.A.; Suhail, M.; Tarique, M.; Tabrez, S. Targeting PI3K/Akt/mTOR pathway by different flavonoids: A cancer chemopreventive approach. Int. J. Mol. Sci. 2021, 22, 12455. [Google Scholar] [CrossRef] [PubMed]
- Islam, B.; Suhail, M.; Khan, M.S.; Ahmad, A.; Zughaibi, T.A.; Husain, F.M.; Rehman, M.T.; Tabrez, S. Flavonoids and PI3K/Akt/mTOR signaling cascade: A potential crosstalk in anticancer treatment. Curr. Med. Chem. 2021, 28, 8083–8097. [Google Scholar] [CrossRef] [PubMed]
- Hawash, M.; Jaradat, N.; Eid, A.M.; Abubaker, A.; Mufleh, O.; Al-Hroub, Q.; Sobuh, S. Synthesis of novel isoxazole–carboxamide derivatives as promising agents for melanoma and targeted nano-emulgel conjugate for improved cellular permeability. BMC Chem. 2022, 16, 47. [Google Scholar] [CrossRef] [PubMed]
- Kadi, I.; Şekerci, G.; Boulebd, H.; Zebbiche, Z.; Tekin, S.; Küçükbay, H.; Küçükbay, F.; Boumoud, T. Synthesis, in vitro, and in silico studies of novel poly-heterocyclic compounds bearing pyridine and furan moieties as potential anticancer agents. J. Mol. Struct. 2023, 1271, 134054. [Google Scholar] [CrossRef]
- Fnfoon, D.Y.; Al-Adilee, K.J. Synthesis and spectral characterization of some metal complexes with new heterocyclic azo imidazole dye ligand and study biological activity as anticancer. J. Mol. Struct. 2023, 1271, 134089. [Google Scholar] [CrossRef]
- Sallam, E.R.; Aboulnaga, S.F.; Samy, A.M.; Beltagy, D.M.; El Desouky, J.M.; Abdel-Hamid, H.; Fetouh, H.A. Synthesis, characterization of new heterocyclic compound: Pyrazolyl hydrazino quinoxaline derivative: 3-[5-(Hydroxy1methyl)-1-phenylpyrazol-3-yl]-2-[2, 4, 5-trimethoxybenzylidine] hydrazonyl-quinoxaline of potent antimicrobial, antioxidant, antiviral, and antitumor activity. J. Mol. Struct. 2023, 1271, 133983. [Google Scholar]
- Fujita, K. Cytochrome P450 and Anticancer Drugs. Curr. Drug Metabol. 2006, 7, 23–37. [Google Scholar] [CrossRef]
- Würth, R.; Thellung, S.; Bajetto, A.; Mazzanti, M.; Florio, T.; Barbieri, F. Drug-repositioning opportunities for cancer therapy: Novel molecular targets for known compounds. Drug Disc. Today 2016, 21, 190–199. [Google Scholar] [CrossRef]
- Smith, N.F.; Figg, W.D.; Sparreboom, A. Recent advances in pharmacogenetic approaches to anticancer drug development. Drug Develop. Res. 2004, 62, 233–253. [Google Scholar] [CrossRef]
- Mallavadhani, U.V.; Prasad, C.V.; Shrivastava, S.; Naidu, V.G.M. Synthesis and anticancer activity of some novel 5,6-fused hybrids of juglone based 1,4-naphthoquinones. Eur. J. Med. Chem. 2014, 83, 84–91. [Google Scholar] [CrossRef]
- Pingaew, R.; Prachayasittikul, V.; Worachartcheewan, A.; Nantasenamat, C.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Novel 1,4-naphthoquinone-based sulfonamides: Synthesis, QSAR, anticancer and antimalarial studies. Eur. J. Med. Chem. 2015, 103, 446–459. [Google Scholar] [CrossRef] [PubMed]
- Hussain, H.; Green, I.R. Lapachol and lapachone analogs: A journey of two decades of patent research (1997–2016). Exp. Op. Therap. Pat. 2017, 27, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Baiju, T.V.; Almeida, R.G.; Sivanandan, S.T.; de Simone, C.A.; Brito, L.M.; Cavalcanti, B.C.; Pessoa, C.; Namboothiri, I.N.N.; da Silva Júnior, E.N. Quinonoid compounds via reactions of lawsone and 2-aminonaphthoquinone with α-bromonitroalkenes and nitroallylic acetates: Structural diversity by C-ring modification and cytotoxic evaluation against cancer cells. Eur. J. Med. Chem. 2018, 151, 686–704. [Google Scholar] [CrossRef]
- Bolton, J.L.; Trush, M.A.; Penning, T.M.; Dryhurst, G.; Monks, T.J. Role of quinones in toxicology. Chem. Res. Toxicol. 2000, 13, 135–160. [Google Scholar] [CrossRef] [PubMed]
- da Silva Júnior, E.N.; Jardim, G.A.M.; Jacob, C.; Dhawa, U.; Ackermann, L.; de Castro, S.L. Synthesis of quinones with highlighted biological applications: A critical update on the strategies towards bioactive compounds with emphasis on lapachones. Eur. J. Org. Chem. 2019, 179, 863–915. [Google Scholar] [CrossRef] [PubMed]
- da Silva Júnior, E.N.; Jardim, G.A.M.; Menna-Barreto, R.F.S.; de Castro, S.L. Anti-Trypanosoma cruzi compounds: Our contribution for the evaluation and insights on the mode of action of naphthoquinones and derivatives. J. Braz. Chem. Soc. 2014, 25, 1780–1798. [Google Scholar]
- Patel, O.P.S.; Beteck, R.M.; Legoabe, L.J. Antimalarial application of quinones: A recent update. Eur. J. Med. Chem. 2021, 210, 113084. [Google Scholar] [CrossRef]
- Silva, R.L.; Demarque, D.P.; Dusi, R.G.; Sousa, J.P.B.; Albernaz, L.C.; Espindola, L.S. Residual larvicidal activity of quinones against Aedes aegypti. Molecules 2020, 25, 3978. [Google Scholar] [CrossRef]
- Carneiro, P.F.; Pinto, M.C.F.R.; Coelho, T.S.; Cavalcanti, B.C.; Pessoa, C.; de Simone, C.A.; Nunes, I.K.C.; de Oliveira, N.M.; de Almeida, R.G.; Pinto, A.V.; et al. Quinonoid and phenazine compounds: Synthesis and evaluation against H37Rv, rifampicin and isoniazid-resistance strains of Mycobacterium tuberculosis. Eur. J. Med. Chem. 2011, 46, 4521–4529. [Google Scholar] [CrossRef]
- da Silva Júnior, E.N.; de Deus, C.F.; Cavalcanti, B.C.; Pessoa, C.; Costa-Lotufo, L.V.; Montenegro, R.C.; de Moraes, M.O.; Pinto, M.C.F.R.; de Simone, C.A.; Ferreira, V.F.; et al. 3-Arylamino and 3-alkoxy-nor-β-lapachone derivatives: Synthesis and cytotoxicity against cancer cell lines. J. Med. Chem. 2010, 53, 504–508. [Google Scholar] [CrossRef]
- Lim, S.M.; Jeong, Y.; Lee, S.; Im, H.; Tae, H.S.; Kim, B.G.; Park, H.D.; Park, J.; Hong, S. Identification of β-lapachone analogs as novel MALT1 inhibitors to treat an aggressive subtype of diffuse large B-cell lymphoma. J. Med. Chem. 2015, 58, 8491–8502. [Google Scholar] [CrossRef] [PubMed]
- da Silva Júnior, E.N.; Cavalcanti, B.C.; Guimarães, T.T.; Pinto, M.C.F.R.; Cabral, I.O.; Pessoa, C.; Costa-Lotufo, L.V.; de Moraes, M.O.; de Andrade, C.K.Z.; dos Santos, M.R.; et al. Synthesis and evaluation of quinonoid compounds against tumor cell lines. Eur. J. Med. Chem. 2011, 46, 399–410. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Liu, A.; Li, Y.; Zhao, X.; Lv, S.; Zhu, W.; Jin, Y. Anticancer activity and mechanism of juglone on human cervical carcinoma HeLa cells. Canad. J. Physiol. Pharm. 2012, 90, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Gilloteaux, J.; Jamison, J.M.; Neal, D.; Summers, J.L. Synergistic antitumor cytotoxic actions of ascorbate and menadione on human prostate (DU145) cancer cells in vitro: Nucleus and other injuries preceding cell death by autoschizis. Ultrastruct. Pathol. 2014, 38, 116–140. [Google Scholar] [CrossRef] [PubMed]
- Lamson, D.W.; Plaza, S.M. The anticancer efects of Vitamin K. Altern. Med. Ver. 2003, 8, 303–318. [Google Scholar]
- Castellanos, J.R.G.; Prieto, J.M.; Heinrich, M. Red Lapacho (Tabebuia impetiginosa)—A global ethnopharmacological commodity? J. Ethnopharm. 2009, 121, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pink, J.J.; Planchon, S.M.; Tagliarino, C. Varnest, M.E.; Siegel, D.; Boothman, D.A. NAD(P)H:Quinone Oxidoreductase Activity Is the Principal Determinant of b-Lapachone Cytotoxicity. J. Biomol. Chem. 2000, 275, 5416–5424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Sun, X.; LaMont, J.T.; Li, C.J. Selective killing of cancer cells by β-lapachone: Direct checkpoint activation as a strategy against cancer. PNAS 2003, 100, 2674–2678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, D.E.; Beg, M.S.; Fattah, F.; Frankel, A.E.; Fatunde, O.; Arriaga, Y.; Dowell, J.E.; Bisen, A.; Leff, R.D.; Meek, C.C.; et al. Phase 1 study of ARQ 761, a β-lapachone analogue that promotes NQO1-mediated programmed cancer cell necrosis. Br. J. Cancer 2018, 119, 928–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araújo, A.J.; de Souza, A.A.; da Silva Júnior, E.N.; Marinho-Filho, J.D.B.; de Moura, M.A.B.F.; Rocha, D.D.; Vasconcellos, M.C.; Costa, C.O.; Pessoa, C.; de Moraes, M.O.; et al. Growth inhibitory effects of 3′-nitro-phenylamino nor-beta-lapachone against HL-60: A redox-dependant mechanism. Toxicol. Vitr. 2012, 26, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Lima, D.B.J.; Almeida, R.G.; Jardim, G.A.M.; Barbosa, B.P.A.; Santos, A.C.C.; Valença, W.O.; Scheide, M.R.; Gatto, C.C.; de Carvalho, G.G.C.; Costa, P.M.S.; et al. It takes two to tango: Synthesis of cytotoxic quinones containing two redox active centers with potential antitumor activity. RSC Med. Chem. 2021, 12, 1709–1721. [Google Scholar] [CrossRef] [PubMed]
- Jardim, G.A.M.; da Cruz, E.H.G.; Valença, W.O.; Lima, D.J.B.; Cavalcanti, B.C.; Pessoa, C.; Rafique, J.; Braga, A.L.; Jacob, C.; da Silva Júnior, E.N. Synthesis of selenium-quinone hybrid compounds with potential antitumor activity via Rh-catalyzed C–H bond activation and click chemistry. Molecules 2018, 23, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, G.G.; do Nascimento, T.A.; de Almeida, A.K.A.; Bombaça, A.C.S.; Menna-Barreto, R.F.S.; Jacob, C.; Warratz, S.; da Silva Júnior, E.N.; Ackermann, L. Ruthenium(II)-catalyzed C–H alkenylation of quinones: Diversity-oriented strategy for trypanocidal compounds. Eur. J. Org. Chem. 2019, 2019, 2344–2353. [Google Scholar] [CrossRef]
- Dias, G.G.; Rogge, T.; Kuniyil, R.; Jacob, C.; Menna-Barreto, R.F.S.; da Silva Júnior, E.N.; Ackermann, L. Ruthenium-catalyzed C–H oxygenation of quinones by weak O-coordination for potent trypanocidal agents. Chem. Commun. 2018, 54, 12840–12843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jardim, G.A.M.; Oliveira, W.X.C.; Freitas, R.P.; Menna-Barreto, R.F.S.; Silva, T.L.; Goulart, M.O.F.; da Silva Júnior, E.N. Direct sequential C–H iodination/organoyl-thiolation for the benzenoid A-ring modification of quinonoid deactivated systems: A new protocol for potent trypanocidal quinones. Org. Biomol. Chem. 2018, 16, 1686–1691. [Google Scholar] [CrossRef]
- Agalave, S.G.; Maujan, S.R.; Pore, V.S. Click chemistry: 1,2,3-triazoles as pharmacophores. Chem. As. J. 2011, 6, 2696–2718. [Google Scholar] [CrossRef] [PubMed]
- Xi, W.; Scott, T.F.; Kloxin, C.J.; Bowman, C.N. Click Chemistry in Materials Science. Adv. Funct. Mat. 2014, 24, 2572–2590. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Chan, T.R.; Hilgraf, R.; Fokin, V.V.; Sharpless, K.B.; Finn, M.G. Bioconjugation by copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition. J. Am. Chem. Soc. 2003, 125, 3192–3193. [Google Scholar] [CrossRef]
- Gomes, R.S.; Jardim, G.A.M.; de Carvalho, R.L.; Araújo, M.H.; da Silva Júnior, E.N. Beyond copper-catalyzed azide-alkyne 1,3-dipolar cycloaddition: Synthesis and mechanism insights. Tetrahedron 2019, 75, 3697–3712. [Google Scholar] [CrossRef]
- Valença, W.O.; Baiju, T.V.; Brito, F.G.; Araújo, M.H.; Pessoa, C.; Cavalcanti, B.C.; de Simone, C.A.; Jacob, C.; Namboothiri, I.N.N.; da Silva Júnior, E.N. Synthesis of quinone-based N-sulfonyl-1,2,3-triazoles: Chemical reactivity of Rh(II) azavinyl carbenes and antitumor activity. ChemistrySelect 2017, 2, 4301–4308. [Google Scholar] [CrossRef]
- Gontijo, T.B.; de Freitas, R.P.; Emery, F.S.; Pedrosa, L.F.; Vieira Neto, J.B.; Cavalcanti, B.C.; Pessoa, C.; King, A.; de Moliner, F.; Vendrell, M.; et al. On the synthesis of quinone-based BODIPY hybrids: New insights on antitumor activity and mechanism of action in cancer cells. Bioorg. Med. Chem. Lett. 2017, 27, 4446–4456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva Júnior, E.N.; Menna-Barreto, R.F.S.; Pinto, M.C.F.R.; Silva, R.S.F.; Teixeira, D.V.; de Souza, M.C.B.V.; de Simone, C.A.; de Castro, S.L.; Ferreira, V.F.; Pinto, A.V. Naphthoquinoidal [1,2,3]-triazole, a new structural moiety active against Trypanosoma cruzi. Eur. J. Med. Chem. 2008, 43, 1774–1780. [Google Scholar] [CrossRef]
- Jardim, G.A.M.; Cruz, E.H.G.; Valença, W.O.; Resende, J.M.; Rodrigues, B.L.; Ramos, D.F.; Oliveira, R.N.; Silva, P.E.A.; da Silva Júnior, E.N. On the search for potential antimycobacterial drugs: Synthesis of naphthoquinoidal, phenazinic and 1,2,3-triazolic compounds and evaluation against Mycobacterium tuberculosis. J. Braz. Chem. Soc. 2015, 26, 1013–1027. [Google Scholar] [CrossRef]
- Mezeiova, E.; Janockova, J.; Andrys, R.; Soukup, O.; Kobrlova, T.; Muckova, L.; Pejchal, J.; Simunkova, M.; Handl, J.; Micankova, P.; et al. 2-Propargylamino-naphthoquinone derivatives as multipotent agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2021, 211, 113112. [Google Scholar] [CrossRef] [PubMed]
- Jardim, G.A.M.; da Silva Júnior, E.N.; Bower, J.F. Overcoming naphthoquinone deactivation: Rhodium-catalyzed C-5 selective C–H iodination as a gateway to functionalized derivatives. Chem. Sci. 2016, 7, 3780–3784. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kongkathip, N.; Kongkathip, B.; Siripong, P.; Sangma, C.; Luangkamin, S.; Niyomdecha, M.; Pattanapa, S.; Piyaviriyagul, S.; Kongsaeree, P. Potent antitumor activity of synthetic 1,2-naphthoquinones and 1,4-naphthoquinones. Bioorg. Med. Chem. 2003, 11, 3179–3191. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.S.F.; Costa, E.M.; Trindade, U.L.T.; Teixeira, D.V.; Pinto, M.C.F.R.; Santos, G.L.; Malta, V.R.S.; de Simone, C.A.; Pinto, A.V.; de Castro, S.L. Synthesis of naphthofuranquinones with activity against Trypanosoma cruzi. Eur. J. Med. Chem. 2006, 41, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, R.C.; Araújo, A.J.; Molina, M.T.; Filho, J.D.B.M.; Rocha, D.D.; López-Montero, E.; Goulart, M.O.F.; Bento, E.S.; Alves, A.P.N.N.; Pessoa, C.; et al. Cytotoxic activity of naphthoquinones with special emphasis on juglone and its 5-O-methyl derivative. Chem.-Biol. Int. 2010, 184, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Chen, C.-C.; Yu, T.; Yang, J.; Hu, X. Enantioselective total synthesis of (−)-Spiroxins A, C, and D. Angew. Chem. Int. Ed. 2021, 60, 18514–18518. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Shen, X. Iron-catalyzed regioselective alkylation of 1,4-quinones and coumarins with functionalized alkyl bromides. Org Biomol. Chem. 2020, 18, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Nor, S.M.M.; Sukari, M.A.H.M.; Azziz, S.S.S.A.; Fah, W.C.; Alimon, H.; Juhan, S.F. Synthesis of new cytotoxic aminoanthraquinone derivatives via nucleophilic substitution reactions. Molecules 2013, 18, 8046–8062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd | HCT-116 | PC3 | SNB-19 | K-562 | HL60 | B16 | A549 | KG1 | RAJI | L929 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Azides | 4 | 1.96 (1.82–2.11) | 5.93 (5.11–6.91) | 2.09 (1.75–2.46) | 1.42 (1.18–1.69) | 1.08 (0.90–1.24) | 1.77 (1.65–1.89) | 3.65 (3.34–4.01) | 5.98 (5.04–7.08) | 2.46 (2.06–3.00) | 2.74 (2.33–3.24) |
| 7 | 1.80 (1.67–1.93) | 2.25 (1.86–2.70) | 1.43 (1.20–1.66) | 0.98 (0.84–1.15) | 0.59 (0.48–0.70) | 1.99 (1.86–2.14) | 2.63 (2.38–2.90) | 3.67 (2.97–4.52) | 0.53 (0.44–0.66) | 3.46 (2.71–4.51) | |
| 10 | 4.33 (3.39–5.56) | 7.05 (6.26–7.95) | 4.07 (3.73–4.45) | 4.37 (3.63–5.27) | 3.72 (2.87–4.61) | 3.51 (3.03–4.31) | 4.83 (3.75–6.29) | 8.03 (6.91–9.42) | 9.08 (7.80–10.61) | 7.22 (6.42–8.16) | |
| Alkynes | 18a | >100 | >100 | >100 | >100 | 38.33 (28.52–54.05) | 48.53 (43.85–53.93) | >100 | >100 | >100 | >100 |
| 18b | 12.73 (11.19–14.46) | >100 | 15.03 (13.35–16.96) | 24.98 (21.43–29.26) | >100 | 8.65 (7.48–10.07) | 15.04 (13.18–17.24) | >100 | 23.21 (19.70–27.52) | 23.95 (18.86–30.07) | |
| 18c | >100 | >100 | >100 | >100 | >100 | 43.82 (40.82–47.06) | >100 | >100 | 39.33 (34.02–45.60) | >100 | |
| 18d | >100 | >100 | >100 | >100 | 31.98 (24.53–41.04) | 35.47 (32.88–38.28) | >100 | >100 | >100 | >100 | |
| 18e | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | |
| First scope | 19a | 1.00 (0.93–1.06) | 3.65 (2.93–4.60) | 0.99 (0.90–1.06) | 1.21 (0.98–1.52) | 0.44 (0.39–0.50) | 1.87 (1.67–2.10) | 2.56 (2.20–3.07) | 1.37 (0.51–2.38) | 2.02 (1.74–2.36) | 1.21 (1.08–1.34) |
| 19b | 1.70 (1.53–1.89) | 3.16 (2.62–3.76) | 3.35 (3.11–3.63) | 3.16 (2.75–3.63) | 0.99 (0.91–1.08) | 3.55 (3.20–3.97) | 3.16 (2.75–3.63) | 5.24 (4.06–6.84) | 2.55 (2.24–2.93) | 5.97 (4.65–7.61) | |
| 19c | 1.60 (1.50–1.71) | 3.50 (3.21–3.82) | 8.19 (7.01–9.39) | 3.06 (2.51–3.70) | 1.07 (0.87–1.25) | 1.74 (1.55–1.96) | 3.06 (2.51–3.70) | 4.77 (4.00–5.80) | 0.93 (0.76–1.12) | 9.17 (8.15–10.35) | |
| 19d | 0.53 (0.47–0.60) | 1.23 (1.00–1.51) | 0.76 (0.68–0.85) | 1.04 (0.90–1.18) | 0.34 (0.32–0.36) | 0.93 (0.86–1.00) | 1.11 (1.01–1.24) | 2.94 (2.52–3.47) | 1.26 (1.06–1.50) | 2.29 (1.90–2.76) | |
| 19e | 2.63 (2.30–3.00) | 3.48 (3.04–4.00) | 3.52 (3.21–3.92) | 8.13 (7.58–8.69) | 0.86 (0.80–0.91) | 3.97 (3.47–4.54) | 6.14 (4.89–7.84) | 4.77 (3.77–6.13) | 1.69 (1.39–2.03) | 4.83 (3.86–6.07) | |
| Second scope | 20a | 5.47 (5.05–5.91) | 9.79 (8.21–11.55) | 8.50 (7.43–9.72) | 5.56 (5.22–5.90) | 2.84 (2.38–3.33) | 4.19 (3.87–4.53) | 15.32 (12.27–19.38) | 25.34 (19.55–33.35) | 2.80 (2.27–3.49) | 5.59 (4.64–6.90) |
| 20b | 7.22 (5.81–8.96) | 10.23 (8.67–12.02) | 9.47 (8.49–10.55) | 12.77 (10.40–15.43) | 5.00 (4.15–5.98) | 4.52 (3.94–5.20) | 18.92 (17.03–21.04) | 23.32 (19.56–28.29) | 2.49 (1.93–3.16) | 27.62 (22.96–33.39) | |
| 20c | >100 | >100 | 49.59 (38.93–64.11) | >100 | 38.29 (32.06–45.34) | 44.06 (40.86–47.26) | 24.54 (20.94–29.03) | 39.39 (28.22–59.67) | 37.04 (33.88–40.56) | >100 | |
| 20d | 6.16 (5.18–7.30) | 12.84 (10.63–15.51) | 10.22 (9.0–11.64) | 10.40 (8.95–12.06) | 3.74 (3.03–4.90) | 9.21 (8.48–9.98) | 27.37 (21.62–34.82) | 8.90 (6.78–11.74) | 4.32 (3.60–5.17) | 9.43 (8.32–10.73) | |
| 20e | 7.20 (6.30–8.20) | 17.60 (15.67–19.87) | 7.87 (5.67–11.04) | 7.58 (6.24–9.20) | 1.57 (1.31–1.86) | 9.57 (8.68–10.53) | 12.65 (10.10–16.17) | 37.03 (31.56–43.87) | 5.26 (4.28–6.44) | 15.88 (10.22–26.35) | |
| Third scope | 21a | 2.69 (2.34–3.13) | 6.19 (5.12–7.51) | 2.93 (2.48–3.50) | 5.03 (4.12–6.30) | 1.02 (0.89–1.17) | 0.96 (0.08–1.40) | 2.76 (2.40–3.16) | 4.20 (3.48–5.11) | 5.71 (4.43–7.50) | 1.86 (1.37–2.56) |
| 21b | 1.64 (1.39–1.94) | 3.85 (3.09–4.77) | 4.24 (3.92–4.59) | 4.13 (3.50–4.90) | 2.23 (0.74–1.06) | 3.94 (2.95–5.09) | 1.68 (1.41–2.13) | 3.01 (2.59–3.52) | 2.01 (1.68–2.43) | 2.07 (1.53–2.85) | |
| 21c | 4.85 (3.79–6.32) | 5.09 (3.92–6.72) | 4.92 (4.55–5.32) | 5.30 (4.71–6.00) | 2.28 (1.90–2.75) | 4.57 (3.97–5.31) | 4.78 (3.6–6.36) | 1.32 (1.07–1.65) | 1.22 (0.93–1.63) | 5.60 (5.06–6.21) | |
| 21d | 4.16 (3.42–5.10) | 9.76 (8.35–11.47) | 3.83 (3.30–4.44) | 10.06 (7.77–13.16) | 0.89 (0.78–1.01) | 5.12 (3.84–6.84) | 2.61 (2.15–3.15) | 11.52 (9.82–13.60) | 28.72 (21.64–40.62) | 5.27 (4.03–6.89) | |
| 21e | 1.74 (1.33–2.33) | 2.87 (2.44–3.40) | 1.33 (1.03–1.73) | 3.69 (3.07–4.46) | 0.26 (0.23–0.28) | 2.86 (1.76–3.03) | 0.88 (0.77–0.99) | 4.21 (3.47–5.17) | 6.83 (5.38–8.91) | 1.71 (1.51–2.06) | |
| Positive Control | DOXO | 0.21 * (0.16–0.29) | 0.76 * (0.59–0.93) | 1.20 * (1.03–1.39) | 0.91 (0.78–1.06) | 0.02 * (0.01–0.02) | 0.06 (0.05–0.07) | 0.57 (0.50–0.66) | 0.38 (0.33–0.44) | 2.33 (2.02–2.71) | 1.72 * (1.58–1.87) |
| Compound | HCT-116 | PC3 | SNB-19 | K-562 | HL60 | B16 | A549 | KG1 | RAJI |
|---|---|---|---|---|---|---|---|---|---|
| 4 | 1.4 | 0.5 | 1.3 | 1.9 | 2.5 | 1.5 | 0.8 | 0.5 | 1.1 |
| 7 | 1.9 | 1.5 | 2.4 | 3.5 | 5.9 | 1.7 | 1.3 | 0.9 | 6.5 |
| 10 | 1.7 | 1.0 | 1.8 | 1.7 | 1.9 | 2.1 | 1.5 | 0.9 | 0.8 |
| 18a | 1.0 | 1.0 | 1.0 | 1.0 | 2.6 | 2.1 | 1.0 | 1.0 | 1.0 |
| 18b | 1.9 | 0.2 | 1.6 | 1.0 | 0.2 | 2.8 | 1.6 | 0.2 | 1.0 |
| 18c | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 2.3 | 1.0 | 1.0 | 2.5 |
| 18d | 1.0 | 1.0 | 1.0 | 1.0 | 3.1 | 2.8 | 1.0 | 1.0 | 1.0 |
| 18e | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| 19a | 1.2 | 0.3 | 1.2 | 1.0 | 2.8 | 0.6 | 0.5 | 0.9 | 0.6 |
| 19b | 3.5 | 1.9 | 1.8 | 1.9 | 6.0 | 1.7 | 1.9 | 1.1 | 2.3 |
| 19c | 5.7 | 2.6 | 1.1 | 3.0 | 8.6 | 5.3 | 3.0 | 1.9 | 9.9 |
| 19d | 4.3 | 1.9 | 3.0 | 2.2 | 6.7 | 2.5 | 2.1 | 0.8 | 1.8 |
| 19e | 1.8 | 1.4 | 1.4 | 0.6 | 5.6 | 1.2 | 0.8 | 1.0 | 2.9 |
| 20a | 1.0 | 0.6 | 0.7 | 1.0 | 2.0 | 1.3 | 0.4 | 0.2 | 2.0 |
| 20b | 3.8 | 2.7 | 2.9 | 2.2 | 5.5 | 6.1 | 1.5 | 1.2 | 11.1 |
| 20c | 1.0 | 1.0 | 2.0 | 1.0 | 2.6 | 2.3 | 4.1 | 2.5 | 2.7 |
| 20d | 1.5 | 0.7 | 0.9 | 0.9 | 2.5 | 1.0 | 0.3 | 1.1 | 2.2 |
| 20e | 2.2 | 0.9 | 2.0 | 2.1 | 10.1 | 1.7 | 1.3 | 0.4 | 3.0 |
| 21a | 0.7 | 0.3 | 0.6 | 0.4 | 1.8 | 1.9 | 0.7 | 0.4 | 0.3 |
| 21b | 1.3 | 0.5 | 0.5 | 0.5 | 0.9 | 0.5 | 1.2 | 0.7 | 1.0 |
| 21c | 1.2 | 1.1 | 1.1 | 1.1 | 2.5 | 1.2 | 1.2 | 4.2 | 4.6 |
| 21d | 1.3 | 0.5 | 1.4 | 0.5 | 5.9 | 1.0 | 2.0 | 0.5 | 0.2 |
| 21e | 1.0 | 0.6 | 1.3 | 0.5 | 6.6 | 0.6 | 1.9 | 0.4 | 0.3 |
| Doxorubicin | 8.2 * | 2.3 * | 1.4 * | 1.9 | 86.0 * | 30.2 | 3.0 | 4.5 | 0.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, J.C.; de Carvalho, R.L.; Sampaio, H.G.S.; Honorato, J.; Ellena, J.A.; Martins, F.T.; Pereira, J.V.M.; Costa, P.M.S.; Pessoa, C.; Ferreira, R.S.; et al. It Takes Two to Tango, Part II: Synthesis of A-Ring Functionalised Quinones Containing Two Redox-Active Centres with Antitumour Activities. Molecules 2023, 28, 2222. https://doi.org/10.3390/molecules28052222
Oliveira JC, de Carvalho RL, Sampaio HGS, Honorato J, Ellena JA, Martins FT, Pereira JVM, Costa PMS, Pessoa C, Ferreira RS, et al. It Takes Two to Tango, Part II: Synthesis of A-Ring Functionalised Quinones Containing Two Redox-Active Centres with Antitumour Activities. Molecules. 2023; 28(5):2222. https://doi.org/10.3390/molecules28052222
Chicago/Turabian StyleOliveira, Joyce C., Renato L. de Carvalho, Hugo G. S. Sampaio, João Honorato, Javier A. Ellena, Felipe T. Martins, João V. M. Pereira, Pedro M. S. Costa, Claudia Pessoa, Rafaela S. Ferreira, and et al. 2023. "It Takes Two to Tango, Part II: Synthesis of A-Ring Functionalised Quinones Containing Two Redox-Active Centres with Antitumour Activities" Molecules 28, no. 5: 2222. https://doi.org/10.3390/molecules28052222