
Multitargeting Histamine H3 Receptor Ligands among Acetyl- and Propionyl-Phenoxyalkyl Derivatives

 , , , , , and
, , , , , and
Abstract
:1. Introduction
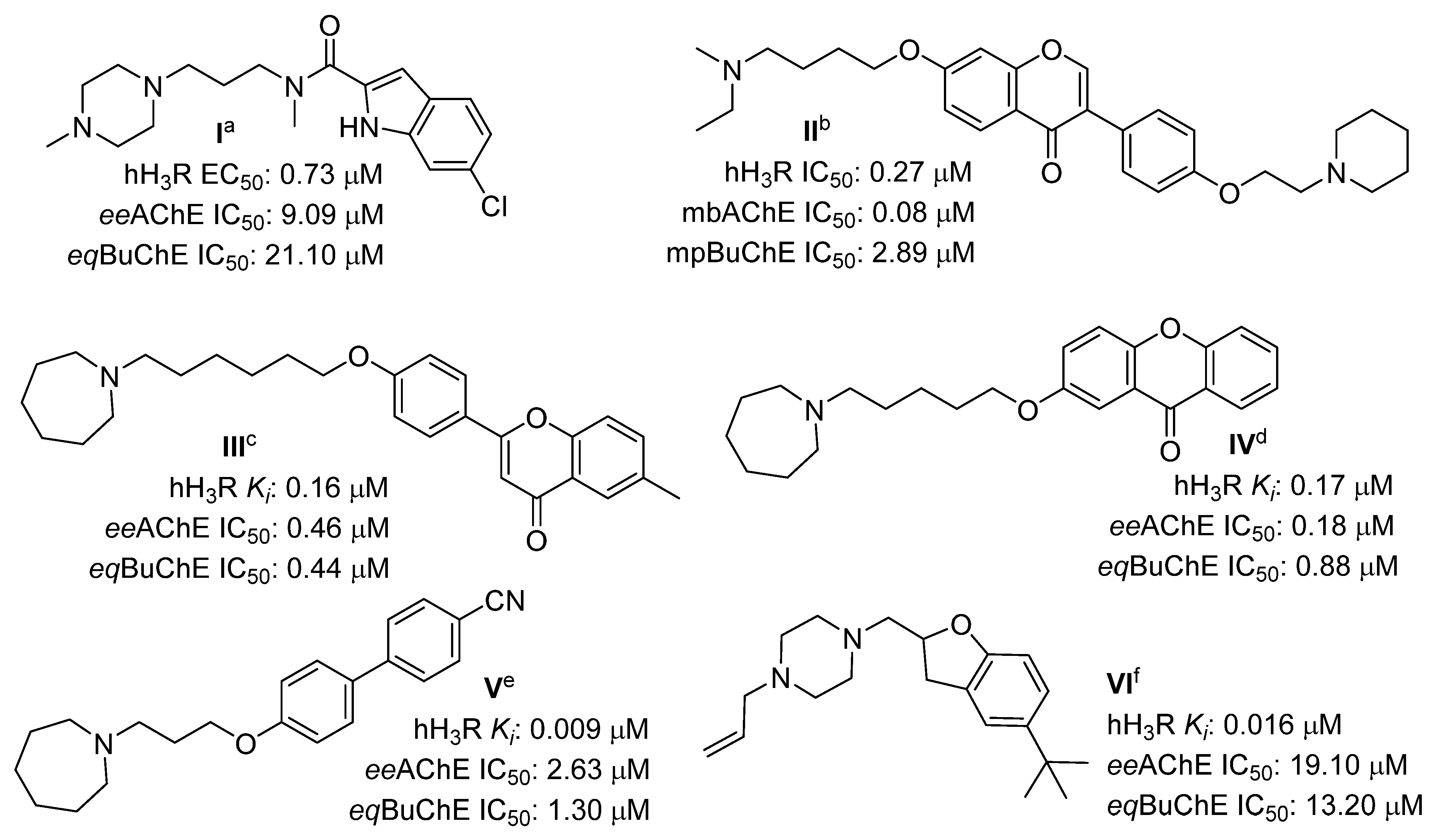
2. Results
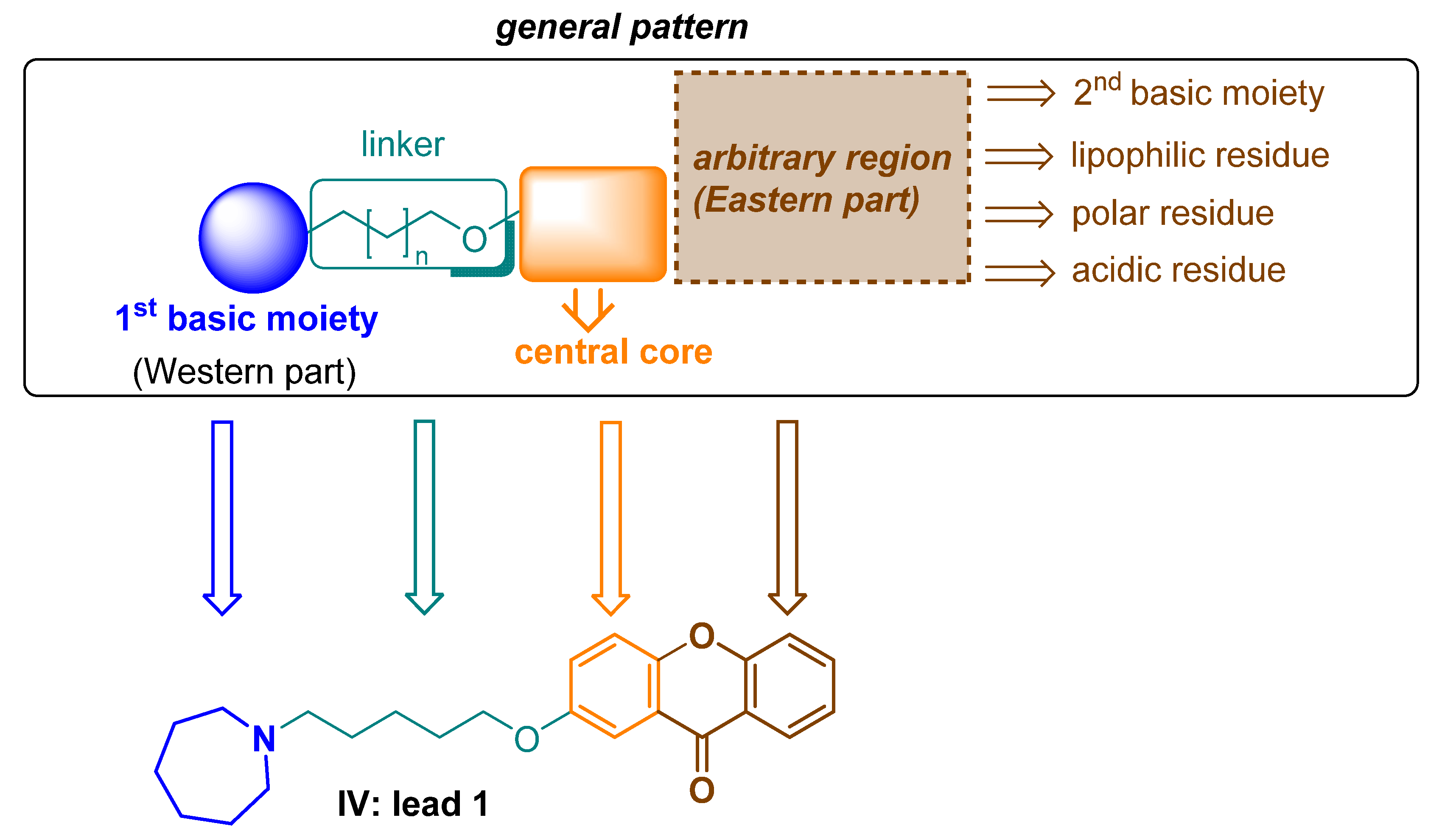
2.1. Design of Compounds
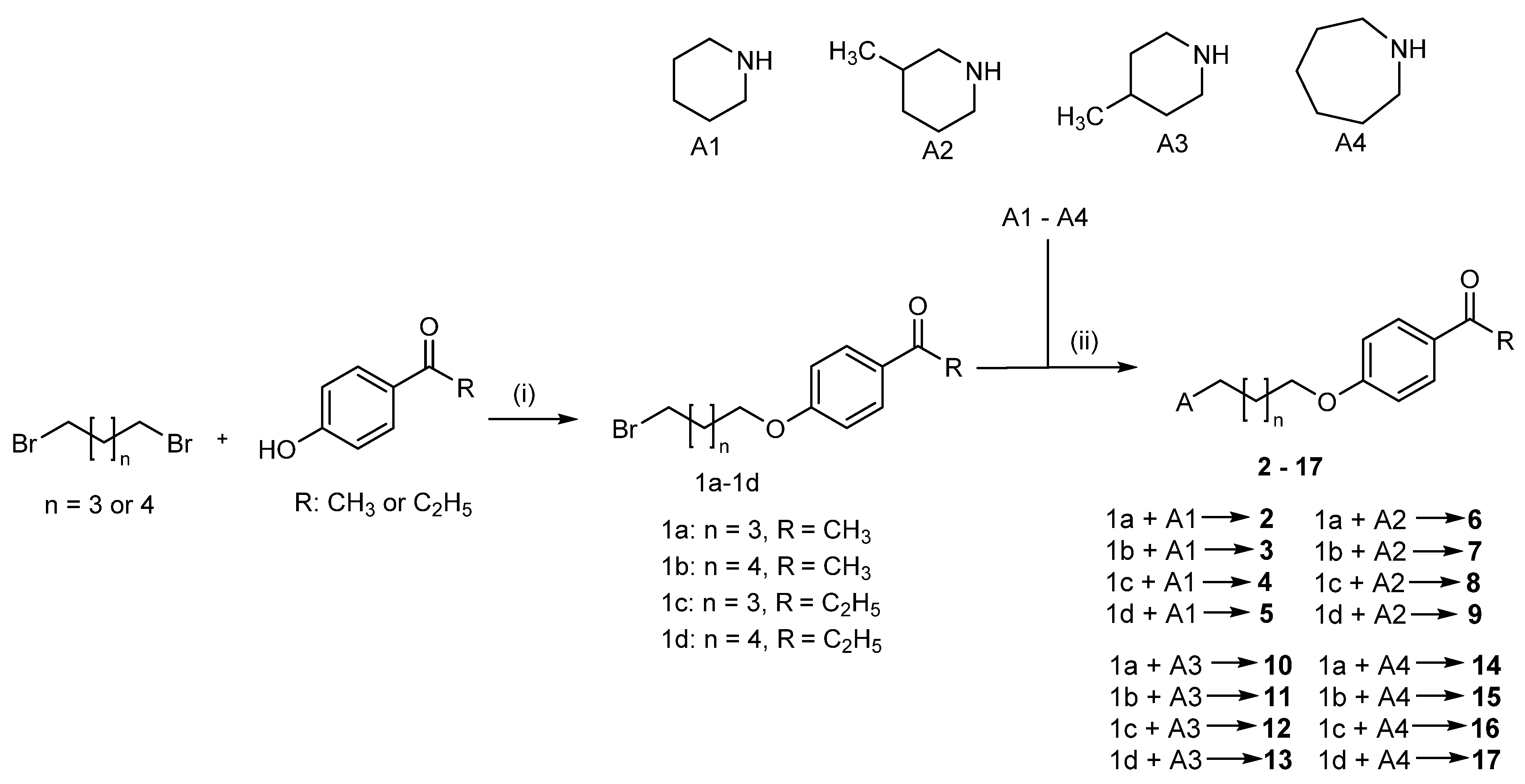
2.2. Synthesis of Compounds 2–17
2.3. Human Histamine H3 Receptor Affinity
2.4. Cholinesterase Inhibitory Activity
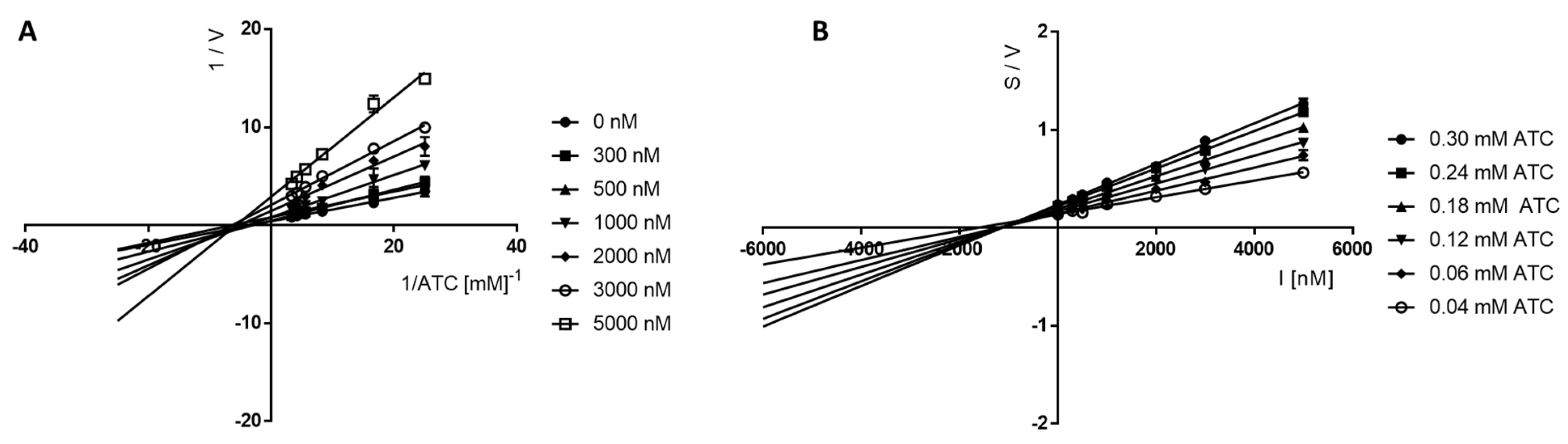
Kinetic Studies of the Inhibition of eeAChE and eqBuChE
2.5. Monoamine Oxidase B Inhibitory Activity
2.6. Cytotoxicity Studies of Selected Compounds
3. Discussion
4. Materials and Methods
4.1. Synthesis of Compounds
4.1.1. Synthesis of Compounds 1a–1d
- 1-(4-((5-bromopentyl)oxy)phenyl)ethan-1-one CAS 61270-21-1 (1a)
- 1-(4-((6-bromohexyl)oxy)phenyl)ethan-1-one CAS138107-19-4 (1b)
- 1-(4-((5-bromopentyl)oxy)phenyl)propan-1-one (1c)
- 1-(4-((6-bromohexyl)oxy)phenyl)propan-1-one (1d)
4.1.2. Synthesis of Compounds 2–17
4.2. Biological Studies
4.2.1. Histamine H3 Receptor Affinity
4.2.2. Cholinesterase Inhibitory Activity
4.2.3. Kinetic Studies of Cholinesterases
4.2.4. Monoamine Oxidase B Inhibitory Activity
4.2.5. Toxicity Evaluation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Samanta, S.; Ramesh, M.; Govindaraju, T. Chapter 1: Alzheimer’s is a Multifactorial Disease. In Alzheimer’s Disease: Recent Findings in Pathophysiology, Diagnostic and Therapeutic Modalities, 1st ed.; RCS: London, UK, 2022; pp. 1–34. [Google Scholar] [CrossRef]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef]
- Twohig, D.; Nielsen, H.M. α-synuclein in the pathophysiology of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, L.A.; Hong, N.S.; McDonald, R.J. Revisiting the cholinergic hypothesis in the development of Alzheimer’s disease. Neurosci. Biobehav. Rev. 2011, 35, 1397–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Wang, Y.; Wang, Y.; Li, X.; Wang, S.; Wang, Z. Recent advance on carbamate-based cholinesterase inhibitors as potential multifunctional agents against Alzheimer’s disease. Eur. J. Med. Chem. 2022, 240, 114606. [Google Scholar] [CrossRef]
- Pardo-Moreno, T.; González-Acedo, A.; Rivas-Domínguez, A.; García-Morales, V.; García-Cozar, F.J.; Ramos-Rodríguez, J.J.; Melguizo-Rodríguez, L. Therapeutic Approach to Alzheimer’s Disease: Current Treatments and New Perspectives. Pharmaceutics 2022, 14, 1117. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, I.; Sorrentino, L.; Paoletti, A.; Marra, R.; Arbitrio, M. The State of The Art on Acetylcholinesterase Inhibitors in the Treatment of Alzheimer’s Disease. J. Cent. Nerv. Syst. Dis. 2021, 13, 11795735211029113. [Google Scholar] [CrossRef] [PubMed]
- Cheong, S.L.; Tiew, J.K.; Fong, Y.H.; Leong, H.W.; Chan, Y.M.; Chan, Z.L.; Kong, E.W.J. Current Pharmacotherapy and Multi-Target Approaches for Alzheimer’s Disease. Pharmaceuticals 2022, 15, 1560. [Google Scholar] [CrossRef]
- Lopes, F.B.; Aranha, C.M.S.Q.; Fernandes, J.P.S. Histamine H3 receptor and cholinesterases as synergistic targets for cognitive decline: Strategies to the rational design of multitarget ligands. Chem. Biol. Drug Des. 2021, 98, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Arrang, J.M.; Garbarg, M.; Schwartz, J.C. Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature 1983, 302, 832–837. [Google Scholar] [CrossRef]
- Panula, P.; Chazot, P.L.; Cowart, M.; Gutzmer, R.; Leurs, R.; Liu, W.L.; Stark, H.; Thurmond, R.L.; Haas, H.L. International Union of Basic and Clinical Pharmacology. XCVIII. Histamine receptors. Pharmacol. Rev. 2015, 67, 601–655. [Google Scholar] [CrossRef] [Green Version]
- Nieto-Allamila, G.; Márquez-Gómez, R.; García-Gálvez, A.M.; Morales-Figueroa, G.E.; Arias-Montaño, J.A. The Histamine H3 Receptor: Structure, pharmacology, and Function. Mol. Pharmacol. 2016, 90, 649–673. [Google Scholar] [CrossRef] [Green Version]
- Ghamari, N.; Zarei, O.; Arias-Montaño, J.A.; Reiner, D.; Dastmalchi, S.; Stark, H.; Hamzeh-Mivehroud, M. Histamine H3 receptor antagonists/inverse agonists: Where do they go? Pharmacol. Ther. 2019, 200, 69–84. [Google Scholar] [CrossRef]
- Łażewska, D.; Kieć-Kononowicz, K. Progress in the development of histamine H3 receptor antagonists/inverse agonists: A patent review (2013–2017). Expert Opin. Ther. Pat. 2018, 28, 175–196. [Google Scholar] [CrossRef]
- Kubo, M.; Kishi, T.; Matsunaga, S.; Iwata, N. Histamine H3 receptor antagonists for Alzheimer’s Disease: A Systematic Review and Meta-Analysis of Randomized Placebo-Controlled Trials. J. Alzheimers Dis. 2015, 48, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Yang, L.; Liu, Z.; Lou, S.; Mei, S.; Li, M.; Chen, Z.; Zhang, H. Structural basis for recognition of antihistamine drug by human histamine receptor. Nat. Commun. 2022, 13, 6105. [Google Scholar] [CrossRef] [PubMed]
- Ghamari, N.; Dastmalchi, S.; Zarei, O.; Arias-Montaño, J.A.; Reiner, D.; Ustun-Alkan, F.; Stark, H.; Hamzeh-Mivehroud, M. In silico and in vitro studies of two non-imidazole multiple targeting agents at histamine H3 receptors and cholinesterase enzymes. Chem. Biol. Drug Des. 2020, 95, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hu, M.; Li, X.; Zhang, D.; Chen, C.; Fu, J.; Shao, S.; Shi, G.; Zhou, Y.; Wu, S.; et al. Design, synthesis, and evaluation of isoflavone analogs as multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 168, 207–220. [Google Scholar] [CrossRef]
- Bajda, M.; Łażewska, D.; Godyń, J.; Zaręba, P.; Kuder, K.; Hagenow, S.; Łątka, K.; Stawarska, E.; Stark, H.; Kieć-Kononowicz, K.; et al. Search for new multi-target compounds against Alzheimer’s disease among histamine H3 receptor ligands. Eur. J. Med. Chem. 2020, 185, 111785. [Google Scholar] [CrossRef] [PubMed]
- Łażewska, D.; Bajda, M.; Kaleta, M.; Zaręba, P.; Doroz-Płonka, A.; Siwek, A.; Alachkar, A.; Mogilski, S.; Saad, A.; Kuder, K.; et al. Rational design of new multitarget histamine H3 receptor ligands as potential candidates for treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2020, 207, 112743. [Google Scholar] [CrossRef]
- Godyń, J.; Zaręba, P.; Łażewska, D.; Stary, D.; Reiner-Link, D.; Frank, A.; Latacz, G.; Mogilski, S.; Kaleta, M.; Doroz-Płonka, A.; et al. Cyanobiphenyls: Novel H3 receptor ligands with cholinesterase and MAO B inhibitory activity as multitarget compounds for potential treatment of Alzheimer’s disease. Bioorg. Chem. 2021, 14, 105129. [Google Scholar] [CrossRef]
- Lopes, F.B.; Aranha, C.M.S.Q.; Corrêa, M.F.; Fernandes, G.A.B.; Okamoto, D.N.; Simões, L.P.M.; Junior, N.M.N.; Fernandes, J.P.S. Evaluation of the histamine H3 receptor antagonists from LINS01 series as cholinesterases inhibitors: Enzymatic and modeling studies. Chem. Biol. Drug Des. 2022, 100, 722–729. [Google Scholar] [CrossRef]
- Finberg, J.P.M. Inhibitors of MAO-B and COMT: Their Effects on Brain Dopamine Levels and Uses in Parkinson’s Disease. J. Neural. Transm. 2019, 126, 433–448. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Kaminga, A.C.; Wen, S.W.; Wu, X.; Acheampong, K.; Liu, A. Dopamine and Dopamine Receptors in Alzheimer’s Disease: A Systematic Review and Network Meta-Analysis. Front. Aging Neurosci. 2019, 11, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, E.; Derrick, J.S.; Lee, S.; Kang, J.; Han, J.; Lee, S.J.C.; Chung, S.W.; Lim, M.H. Regulatory Activities of Dopamine and Its Derivatives toward Metal-Free and Metal-Induced Amyloid-β Aggregation, Oxidative Stress, and Inflammation in Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 2655–2666. [Google Scholar] [CrossRef]
- Godyń, J.; Zaręba, P.; Stary, D.; Kaleta, M.; Kuder, K.J.; Latacz, G.; Mogilski, S.; Reiner-Link, D.; Frank, A.; Doroz-Płonka, A.; et al. Benzophenone Derivatives with Histamine H3 Receptor Affinity and Cholinesterase Inhibitory Potency as Multitarget-Directed Ligands for Possible Therapy of Alzheimer’s Disease. Molecules 2023, 28, 238. [Google Scholar] [CrossRef] [PubMed]
- Sadek, B.; Łażewska, D.; Hagenow, S.; Kieć-Kononowicz, K.; Stark, H. Histamine H3R Antagonists: From Scaffold Hopping to Clinical Candidates. In Histamine Receptors. Preclinical and Clinical Aspects; Blandina, P., Passani, M., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 109–155. [Google Scholar] [CrossRef]
- Chmiel, T.; Mieszkowska, A.; Kempińska-Kupczyk, D.; Kot-Wasik, A.; Namieśnik, J.; Mazerska, Z. The impact of lipophilicity on environmental processes, drug delivery and bioavailability of food components. Microchem. J. 2019, 146, 393–406. [Google Scholar] [CrossRef]
- Available online: http://www.swissadme.ch/ (accessed on 28 December 2022).
- Kottke, T.; Sander, K.; Weizel, L.; Schneider, E.H.; Seifert, R.; Stark, H. Receptor-specific functional efficacies of alkyl imidazoles as dual histamine H3/H4 receptor ligands. Eur. J. Pharmacol. 2011, 654, 200–208. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Anders, V., Jr.; Feather-Stone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Łażewska, D.; Olejarz-Maciej, A.; Kaleta, M.; Bajda, M.; Siwek, A.; Karcz, T.; Doroz-Płonka, A.; Cichoń, U.; Kuder, K.; Kieć-Kononowicz, K. 4-tert-Pentylphenoxyalkyl derivatives–Histamine H3 receptor ligands and monoamine oxidase B inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 3596–3600. [Google Scholar] [CrossRef]
- Łażewska, D.; Kaleta, M.; Schwed, J.S.; Karcz, T.; Mogilski, S.; Latacz, G.; Olejarz, A.; Siwek, A.; Kubacka, M.; Lubelska, A.; et al. Biphenyloxy-alkyl-piperidine and azepane derivatives as histamine H3 receptor ligands. Bioorg. Med. Chem. 2017, 25, 5341–5354. [Google Scholar] [CrossRef]
- Ramirez, T.; Strigun, A.; Verlohner, A.; Huener, H.A.; Peter, E.; Herold, M.; Bordag, N.; Mellert, W.; Walk, T.; Spitzer, M.; et al. Prediction of liver toxicity and mode of action using metabolomics in vitro in HepG2 cells. Arch Toxicol. 2018, 92, 893–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, M.; Zempel, H. SH-SY5Y-derived neurons: A human neuronal model system for investigating TAU sorting and neuronal subtype-specific TAU vulnerability. Rev. Neurosci. 2021, 33, 1–15. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
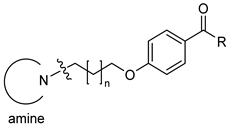 | |||||||
|---|---|---|---|---|---|---|---|
| Comp | Amine | n | R | hH3R aKi (nM) Mean [CI 95%] | eeAChE b IC50 (µM) Mean ± SEM (% inh) e | eqBuChE c IC50 (µM) Mean ± SEM (% inh) e | hMAO B d (% inh) f |
| 2 | 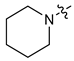 | 3 | CH3 | 34 [24;47] | (45%) | (25%) | (2%) |
| 3 | 4 | CH3 | 75 [34;169] | (67%) | (23%) | (3%) | |
| 4 | 3 | C2H5 | 24 [11;54] | (49%) | (32%) | (16%) | |
| 5 | 4 | C2H5 | 41 [27;64] | 4.79 ± 0.19 | 1.35 ± 0.04 | (22%) | |
| 6 | 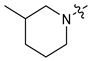 | 3 | CH3 | 26 [13;52] | 3.06 ± 0.10 | (14%) | (10%) |
| 7 | 4 | CH3 | 72 [41;127] | 1.75 ± 0.05 | (18%) | (0%) | |
| 8 | 3 | C2H5 | 12 [6;22] | 2.10 ± 0.06 | 1.75 ± 0.05 | (19%) | |
| 9 | 4 | C2H5 | 38 [20;72] | (68%) | (66%) | (12%) | |
| 10 | 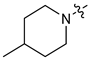 | 3 | CH3 | 120 [82;177] | (60%) | (43%) | (0%) |
| 11 | 4 | CH3 | 106 [76;149] | 2.65 ± 0.11 | (49%) | (0%) | |
| 12 | 3 | C2H5 | 57 [36;91] | (58%) | 1.29 ± 0.08 | (0%) | |
| 13 | 4 | C2H5 | 58 [35;96] | 2.56 ± 0.08 | (65%) | (1%) | |
| 14 | 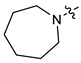 | 3 | CH3 | 52 [38;71] | (63%) | (63%) | (37%) |
| 15 | 4 | CH3 | 79 [41;150] | (67%) | (68%) | (6%) | |
| 16 | 3 | C2H5 | 30 [17;53] | 3.60 ± 0.12 | 0.55 ± 0.02 | (6%) | |
| 17 | 4 | C2H5 | 42 [20;85] | 1.06 ± 0.03 | 2.86 ± 0.03 | (5%) | |
| pitolisant | 12 ± 3 g | (3%) g | 8.42 ± 0.18 g | (2%) g | |||
| donepezil | nt h | 0.04 ± 0.01 g | 1.83 ± 0.02 | nt h | |||
| safinamide | nt h | nt h | nt h | (98%) 7.60 ± 1.0 i | |||
| Compound | HepG2 a IC50 [µM] | Cell Viability (%) b | SH-SY5Y a IC50 [µM] | Cell Viability (%) b |
|---|---|---|---|---|
| 5 | 158 | 69.55 ± 2.14 | - | No toxic effect |
| 6 | 120 | 74.32 ± 5.58 | 149 | 86.23 ± 2.20 |
| 7 | 148 | 72.38 ± 4.54 | - | No toxic effect |
| 8 | 142 | 70.89 ± 2.92 | 349 | 90.73 ± 1.50 |
| 11 | 223 | 77.52 ± 0.94 | - | No toxic effect |
| 12 | 149 | 77.20 ± 4.14 | 421 | 90.70 ± 12.88 |
| 13 | 105 | 64.66 ± 4.40 | - | No toxic effect |
| 16 | 92 | 67.69 ± 1.14 | 357 | 90.28 ± 5.32 |
| 17 | 60 | 58.02 ± 3.54 | 111 | 70.35 ± 4.46 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Łażewska, D.; Kaleta, M.; Zaręba, P.; Godyń, J.; Dubiel, M.; Honkisz-Orzechowska, E.; Doroz-Płonka, A.; Więckowska, A.; Stark, H.; Kieć-Kononowicz, K. Multitargeting Histamine H3 Receptor Ligands among Acetyl- and Propionyl-Phenoxyalkyl Derivatives. Molecules 2023, 28, 2349. https://doi.org/10.3390/molecules28052349
Łażewska D, Kaleta M, Zaręba P, Godyń J, Dubiel M, Honkisz-Orzechowska E, Doroz-Płonka A, Więckowska A, Stark H, Kieć-Kononowicz K. Multitargeting Histamine H3 Receptor Ligands among Acetyl- and Propionyl-Phenoxyalkyl Derivatives. Molecules. 2023; 28(5):2349. https://doi.org/10.3390/molecules28052349
Chicago/Turabian StyleŁażewska, Dorota, Maria Kaleta, Paula Zaręba, Justyna Godyń, Mariam Dubiel, Ewelina Honkisz-Orzechowska, Agata Doroz-Płonka, Anna Więckowska, Holger Stark, and Katarzyna Kieć-Kononowicz. 2023. "Multitargeting Histamine H3 Receptor Ligands among Acetyl- and Propionyl-Phenoxyalkyl Derivatives" Molecules 28, no. 5: 2349. https://doi.org/10.3390/molecules28052349