
Synthesis and Biological Evaluation of New Dihydrofuro[3,2-b]piperidine Derivatives as Potent α-Glucosidase Inhibitors
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Structure–Activity Relationship
2.3. Drug Likeness
3. Materials and Methods
3.1. Molecular Docking
3.2. General Methods
3.3. General Procedure for α-Glucosidase Inhibition Assay
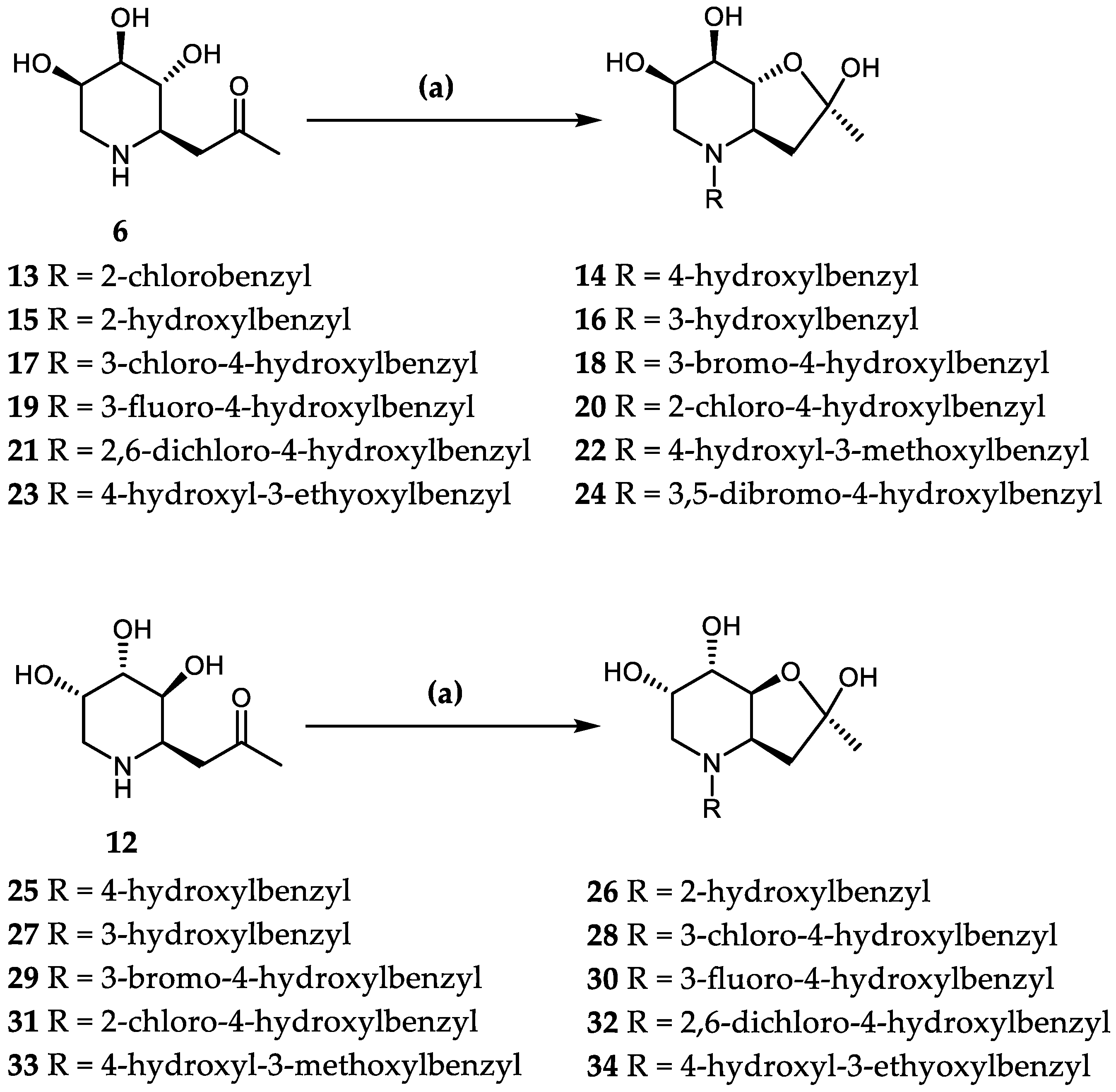
3.4. Synthesis Compounds 13–34
- (2R, 3aR, 6R, 7R, 7aR)-2,6,7-trihydroxy-2-methyl-4-(2-chlorobenzyl)-3a,7a-Dihydrofuro[3,2-b]piperidine (13) (Figures S21 and S22): A suspension of compound 6 (40 mg, 0.2 mmol) containing activated 4Å molecular sieves in anhydrous methanol (3 mL) was stirred at room temperature for 10 min. After cooling to −20 °C, the corresponding aldehydes (0.6 mmol, 3eq) and NaBH(OAc)3 (0.6 mmol, 3eq) were added, and the solution was stirred for 3 h under an argon atmosphere. The reaction solution was concentrated in vacuo and purified by silica gel flash column chromatography (dichloromethane/methanol, 25:1 → 5:1) to afford colorless, syrupy products. Colorless syrup, yield 49%, [α]25D + 35.5 (c 0.11, MeOH). 1H NMR (600 MHz, DMSO-d6) δ 7.39 (dd, J = 7.6, 1.6 Hz, 1H, HAr), 7.36–7.29 (m, 2H, 2 × HAr), 7.28–7.21 (m, 1H, HAr), 3.99 (d, J = 13.6 Hz, 1H, O-CH), 3.91 (s, 1H, O-CH), 3.80–3.70 (m, 3H, O-CH, ArCH2), 2.46 (dd, J = 10.6, 4.1 Hz, 1H, NCH), 2.38–2.26 (m, 2H, NCH2), 2.08 (d, J = 5.5 Hz, 1H, CH2), 1.98 (d, J = 7.0 Hz, 1H, CH2), 1.31 (s, 3H, CH3). 13C NMR (151 MHz, Acetone) δ 135.2 (CArCl), 134.2 (CAr), 131.5 (CAr), 129.6 (CAr), 129.0 (CAr), 127.1 (CAr), 104.5 (O-C-OH), 82.1(O-CH), 67.7(HO-CH), 65.9(HO-CH), 60.8(NCH), 55.9 (ArCH2), 49.9 (NCH2), 42.4 (CH2), 25.2 (CH3). ESI-HRMS: m/z calcd for C15H20O4NClNa [M + Na]+: 336.0973; found: 336.0957.
- (2R, 3aR, 6R, 7R, 7aR)-2,6,7-trihydroxy-2-methyl-4-(4-hydroxylbenzyl)-3a,7a-Dihydrofuro[3,2-b]piperidine (14) (Figures S23 and S24): The synthesized procedure was the same as compound 13. Colorless syrup, yield 42%, [α]25D +50.0 (c 0.13, MeOH). 1H NMR (600 MHz, CD3OD) δ 7.09 (m, 2H, 2 × HAr), 6.74 (m, 2 × HAr), 4.05–4.00 (m, 2H, 2 × O-CH), 3.89(s, 1H, O-CH), 3.88–3.70 (m, 2H, ArCH2), 2.58 (dd, J = 10.8, 4.1 Hz, 1H, NCH), 2.47 (d, J = 13.5 Hz, 1H, NCH2), 2.32 (t, J = 10.9 Hz, 1H, NCH2), 2.18 (d, J = 12.9 Hz, 1H, CH2), 2.00 (d, J = 13.1 Hz, 1H, CH2), 1.44 (s, 3H, CH3). 13C NMR (151 MHz, CD3OD) δ 156.7 (CArOH), 130.2 (CAr), 127.6 (CAr), 127.6 (CAr), 114.9 (CAr), 114.6 (CAr), 105.3 (O-C-OH), 82.6(O-CH), 67.3(HO-CH), 65.8(HO-CH), 59.7 (NCH), 57.8 (ArCH2), 48.4 (NCH2), 41.5 (CH2), 24.0 (CH3). ESI-HRMS: m/z calcd for C15H21O5N [M + H]+: 296.1407; found: 296.1481.
- (2R, 3aR, 6R, 7R, 7aR)-2,6,7-trihydroxy-2-methyl-4-(2-hydroxylbenzyl)-3a,7a-Dihydrofuro[3,2-b]piperidine (15): The synthesized procedure was the same as compound 13. Colorless syrup, yield 41%, [α]25D +42.6 (c 0.46, MeOH). 1H NMR (600 MHz, CD3OD) δ 7.11 (m, 2H, 2 × HAr), 6.80–6.70 (m, 2H, 2 × HAr), 4.23 (s, 1H, O-CH), 4.02 (s, 1H, O-CH), 3.94 (d, J = 12.6 Hz, 2H, O-CH, ArCH2), 3.52 (s, 1H, ArCH2), 3.20 (d, J = 12.7 Hz, 1H, NCH), 2.61 (dd, J = 29.8, 7.9 Hz, 1H, NCH2), 2.50 (d, J = 13.0 Hz, 1H, NCH2), 2.44 (t, J = 10.5 Hz, 1H, CH2), 2.34 (t, J = 10.8 Hz, 1H, CH2), 1.44 (s, 3H, CH3). 13C NMR (151 MHz, CD3OD) δ 154.6 (CArOH), 129.3 (CAr), 127.2 (CAr), 127.0 (CAr), 117.6 (CAr), 113.4 (CAr), 103.0 (O-C-OH), 80.9 (O-CH), 78.3 (HO-CH), 66.0 (HO-CH), 64.5 (NCH), 58.6 (ArCH2), 53.2 (NCH2), 47.5 (CH2), 23.6 (CH3). ESI-HRMS: m/z calcd for C15H21O5NNa [M + Na]+: 318.1312; found: 318.1322.
- (2R, 3aR, 6R, 7R, 7aR)-2,6,7-trihydroxy-2-methyl-4-(3-hydroxylbenzyl)-3a,7a-Dihydrofuro[3,2-b]piperidine (16) (Figures S25 and S26): The synthesized procedure was the same as compound 13. Colorless syrup, yield 45%, [α]25D +61.5 (c 0.16, MeOH). 1H NMR (600 MHz, CD3OD) δ 7.14–7.10 (m, 1H, HAr), 6.78–6.66 (m, 3H, 3×HAr), 4.20–3.80 (m, 2H, ArCH2), 4.07 (d, J = 13.1 Hz, 1H, O-CH), 3.91–3.80 (m, 2H, 2 × O-CH), 2.60 (d, J = 7.4 Hz, 1H, NCH), 2.46 (d, J = 13.4 Hz, 1H, NCH2), 2.33 (t, J = 11.0 Hz, 1H, NCH2), 2.16 (d, J = 9.0 Hz, 1H, CH2), 2.01 (d, J = 11.3 Hz, 1H, CH2), 1.44 (s, 3H, CH3). 13C NMR (151 MHz, CD3OD) δ 155.9 (CArOH), 137.0 (CAr), 127.7 (CAr), 118.5 (CAr), 114.2 (CAr), 112.7 (CAr), 103.8 (O-C-OH), 81.1(O-CH), 65.8(HO-CH), 64.3(HO-CH), 58.4 (NCH), 56.9 (ArCH2), 47.4 (NCH2), 40.0 (CH2), 22.6 (CH3). ESI-HRMS: m/z calcd for C15H21O5NNa [M + Na]+: 318.1312; found: 318.1317.
- (2R, 3aR, 6R, 7R, 7aR)-2,6,7-trihydroxy-2-methyl-4-(3-chloro-4-hydroxylbenzyl)-3a,7a-Dihydrofuro[3,2-b]piperidine (17) (Figures S27 and S28): The synthesized procedure was the same as compound 13. Colorless syrup, yield 35%, [α]25D +38.0 (c 0.10, MeOH). 1H NMR (600 MHz, CD3OD) δ 7.06–7.01 (m, 1H, HAr), 6.88 (d, J = 8.3 Hz, 1H, HAr), 6.84 (d, J = 8.2 Hz, 1H, HAr), 4.19–3.96 (m, 3H, 3 × O-CH), 3.92–3.82 (m, 2H, ArCH2), 2.56 (dd, J = 10.8, 4.1 Hz, 1H, NCH), 2.47 (d, J = 13.5 Hz, 1H, NCH2), 2.35 (t, J = 10.9 Hz, 1H, NCH2), 2.16 (d, J = 8.1 Hz, 1H, CH2), 2.03–1.98 (m, 1H, CH2), 1.44 (s, 3H, CH3). 13C NMR (151 MHz, CD3OD) δ 152.5 (CArOH), 130.4 (CAr), 129.1 (CAr), 128.5 (CAr), 120.3 (CArCl), 116.2 (CAr), 105.3 (O-C-OH), 82.6(O-CH), 67.3(HO-CH), 65.8(HO-CH), 59.7 (NCH), 57.2 (ArCH2), 48.7 (NCH2), 41.5 (CH2), 24.1 (CH3). ESI-HRMS: m/z calcd for C15H20O5NClNa [M + Na]+: 352.0922; found: 352.0917.
- (2R, 3aR, 6R, 7R, 7aR)-2,6,7-trihydroxy-2-methyl-4-(3-bromo-4-hydroxylbenzyl)-3a,7a-Dihydrofuro[3,2-b]piperidine (18) (Figures S29 and S30): The synthesized procedure was the same as compound 13. Colorless syrup, yield 46%, [α]25D +37.3 (c 0.26, MeOH). 1H NMR (600 MHz, CD3OD) δ 7.08–7.04 (m, 1H, HAr), 6.86 (d, J = 8.2 Hz, 1H, HAr), 6.84 (d, J = 8.2 Hz, 1H, HAr), 4.05–4.01 (m, 2H, 2 × O-CH), 3.96 (s, 1H, O-CH), 3.90–3.75 (m, 2H, ArCH2), 2.56 (dd, J = 10.7, 4.1 Hz, 1H, NCH), 2.47 (d, J = 13.5 Hz, 1H, NCH2), 2.31 (t, J = 10.9 Hz, 1H, NCH2), 2.16 (d, J = 9.2 Hz, 1H, CH2), 2.00 (dd, J = 13.5, 3.8 Hz, 1H, CH2), 1.44 (s, 3H, CH3). 13C NMR (151 MHz, CD3OD) δ 153.6 (CArOH), 133.5 (CAr), 129.4 (CAr), 129.3 (CAr), 115.9 (CAr), 109.4 (CArBr), 105.3 (O-C-OH), 82.6(O-CH), 67.3(HO-CH), 65.8(HO-CH), 59.7 (NCH), 57.1 (ArCH2), 48.7 (NCH2), 41.5 (CH2), 24.1 (CH3). ESI-HRMS: m/z calcd for C15H20O5NBrNa [M + Na]+: 396.0417; found: 396.0415.
- (2R, 3aR, 6R, 7R, 7aR)-2,6,7-trihydroxy-2-methyl-4-(3-fluoro-4-hydroxylbenzyl)-3a,7a-Dihydrofuro[3,2-b]piperidine (19) (Figures S31 and S32): The synthesized procedure was the same as compound 13. Colorless syrup, yield 40%, [α]25D +43.9 (c 0.19, MeOH). 1H NMR (600 MHz, CD3OD) δ 7.10–6.75 (m, 3H, 3×HAr), 4.03 (t, J = 3.2 Hz, 1H, OH), 3.90–3.85 (m, 2H, 2 × O-CH), 3.75–3.60(m, 2H, ArCH2), 2.56 (dd, J = 10.7, 4.0 Hz, 1H, NCH), 2.47 (d, J = 13.5 Hz, 1H, NCH2), 2.34 (t, J = 10.9 Hz, 1H, NCH2), 2.15 (d, J = 9.3 Hz, 1H, CH2), 2.01 (dd, J = 13.5, 3.9 Hz, 1H, CH2), 1.44 (s, 3H, CH3). 13C NMR (151 MHz, CD3OD) δ 151.4 (CArF), 144.3 (CArOH), 128.8 (CAr), 125.1 (CAr), 117.3 (CAr), 116.3 (CAr), 105.3 (O-C-OH), 82.6(O-CH), 67.3(HO-CH), 65.8(HO-CH), 59.7 (NCH), 57.4 (ArCH2), 48.6 (NCH2), 41.5 (CH2), 24.1 (CH3). ESI-HRMS: m/z calcd for C15H20O5NFNa [M + Na]+: 336.1218; found: 336.1211.
- (2R, 3aR, 6R, 7R, 7aR)-2,6,7-trihydroxy-2-methyl-4-(2-chloro-4-hydroxylbenzyl)-3a,7a-Dihydrofuro[3,2-b]piperidine (20) (Figures S33 and S34): The synthesized procedure was the same as compound 13. Colorless syrup, yield 44%, [α]25D +30.6 (c 0.11, MeOH). 1H NMR (600 MHz, CD3OD) δ 7.16 (d, J = 8.4 Hz, 1H, HAr), 6.83 (d, J = 2.4 Hz, 1H, HAr), 6.72 (dd, J = 8.5, 2.4 Hz, 1H, HAr), 4.08–4.01 (m, 2H, 2 × O-CH), 3.96 (s, 1H, O-CH), 3.91–3.70 (m, 2H, ArCH2), 2.53 (dd, J = 10.7, 3.9 Hz, 1H, NCH), 2.48 (d, J = 13.5 Hz, 1H, NCH2), 2.37 (t, J = 10.9 Hz, 1H, NCH2), 2.16 (d, J = 12.8 Hz, 1H, CH2), 2.01 (d, J = 4.0 Hz, 1H, CH2), 1.42 (s, 3H, CH3). 13C NMR (151 MHz, CD3OD) δ 157.9 (CArOH), 134.7 (CArCl), 132.5 (CAr), 124.7 (CAr), 115.9 (CAr), 114.0 (CAr), 105.0 (O-C-OH), 82.6(O-CH), 67.3(HO-CH), 65.8(HO-CH), 60.6 (ArCH2), 55.5 (NCH2), 48.7 (NCH), 41.5 (CH2), 24.2 (CH3). ESI-HRMS: m/z calcd for C15H21O5NCl [M + H]+: 330.1103; found: 330.1104.
- (2R, 3aR, 6R, 7R, 7aR)-2,6,7-trihydroxy-2-methyl-4-(2,6-dichloro-4-hydroxylbenzyl)-3a,7a-Dihydrofuro[3,2-b]piperidine (21) (Figures S35 and S36): The synthesized procedure was the same as compound 13. Colorless syrup, yield 31%, [α]25D +6.78 (c 0.12, MeOH). 1H NMR (600 MHz, CD3OD) δ 6.84 (s, 1H, HAr), 6.80 (d, J = 3.6 Hz, 1H, HAr), 4.11 (d, J = 12.7 Hz, 1H, O-CH), 4.04–4.01 (m, 1H, O-CH), 3.95 (s, 1H, O-CH), 3.85–3.40 (m, 2H, ArCH2), 2.62 (t, J = 10.8 Hz, 1H, NCH), 2.50 (d, J = 5.7 Hz, 1H, NCH2), 2.43 (dd, J = 10.6, 4.1 Hz, 1H, NCH2), 2.21 (d, J = 3.1 Hz, 1H, CH2), 1.98(m, 1H, CH2), 1.39 (s, 3H, CH3). 13C NMR (151 MHz, CD3OD) δ 157.8 (CArOH), 136.9 (CArCl), 136.9 (CArCl), 122.7 (CAr),115.5 (CAr), 115.4 (CAr), 104.7 (O-C-OH), 82.6(O-CH), 67.3(HO-CH), 66.0(HO-CH), 61.6 (NCH), 53.7 (ArCH2), 48.7 (NCH2), 41.3 (CH2), 20.1 (CH3). ESI-HRMS: m/z calcd for C15H21O5NCl2 [M + H]+: 364.0719; found: 364.0713.
- (2R, 3aR, 6R, 7R, 7aR)-2,6,7-trihydroxy-2-methyl-4-(4-hydroxyl-methoxylbenzyl)-3a,7a-Dihydrofuro[3,2-b]piperidine (22) (Figures S37 and S38): The synthesized procedure was the same as compound 13. Colorless syrup, yield 47%, [α]25D +36.4 (c 0.06, MeOH). 1H NMR (600 MHz, CD3OD) δ 6.83–6.70 (m, 3H, 3×HAr), 4.05 (d, J = 11.9 Hz, 2H, 2 × O-CH), 3.97 (s, 1H, O-CH), 3.85–3.75 (m, 2H, ArCH2), 3.83 (s, 3H, OCH3), 2.61 (dd, J = 10.8, 4.0 Hz, 1H, NCH), 2.50 (d, J = 13.5 Hz, 1H, NCH2), 2.34 (t, J = 11.0 Hz, 1H, NCH2), 2.21 (d, J = 6.6 Hz, 1H, CH2), 2.01 (dd, J = 13.5, 3.8 Hz, 1H, CH2), 1.45 (s, 3H, CH3). 13C NMR (151 MHz, CD3OD) δ 147.8 (CArOCH3), 145.9 (CArOH), 128.3 (CAr), 121.8(CAr), 114.7(CAr), 112.3(CAr), 105.8 (O-C-OH), 82.6 (O-CH), 67.3 (HO-CH), 66.5(HO-CH), 65.7 (NCH), 58.2 (ArCH2), 54.9 (OCH3), 48.7 (NCH2), 24.0 (CH2), 20.1 (CCH3). ESI-HRMS: m/z calcd for C16H23O6NNa [M + Na]+: 348.1418; found: 348.1427.
- (2R, 3aR, 6R, 7R, 7aR)-2,6,7-trihydroxy-2-methyl-4-(4-hydroxyl-3-ethyoxylbenzyl)-3a,7a-Dihydrofuro[3,2-b]piperidine (23) (Figures S39 and S40): The synthesized procedure was the same as compound 13. Colorless syrup, yield 50%, [α]25D +40.6 (c 0.11, MeOH). 1H NMR (600 MHz, CD3OD) δ 6.80 (s, 1H, phOHAr), 6.77–6.65 (m, 3H, 3×HAr), 4.11–3.99 (m, 5H, OCH2, 3 × O-CH), 3.90–3.75 (m, 2H, ArCH2), 2.60 (dd, J = 10.6, 4.0 Hz, 1H, NCH), 2.31 (m, 1H, NCH2), 2.22–2.16 (m, 1H, NCH2), 2.02–1.95 (m, 2H, CCH2), 1.45 (s, 3H, CCH3), 1.41 (t, J = 9.0 Hz, 3H, CH2CH3). 13C NMR (151 MHz, CD3OD) δ 146.8 (CArOEt), 146.1 (CArOH), 128.3 (CAr), 121.8 (CAr), 114.6 (CAr), 113.6 (CAr), 105.3 (O-C-OH), 82.6 (O-CH), 67.3 (HO-CH), 65.8 (HO-CH), 64.2 (OCH2), 59.7(NCH), 58.2 (ArCH2), 48.7 (NCH2), 48.2 (CCH2), 24.1 (CCH3), 13.7 (CH2CH3). ESI-HRMS: m/z calcd for C17H25O6NNa [M + Na]+: 362.1574; found: 362.1579.
- (2R, 3aR, 6R, 7R, 7aR)-2,6,7-trihydroxy-2-methyl-4-(3,5-dibromo-4-hydroxyl-benzyl)-3a,7a-Dihydrofuro[3,2-b]piperidine (24) (Figures S41 and S42): The synthesized procedure was the same as compound 13. Colorless syrup, yield 37%, [α]25D +18.6 (c 0.10, MeOH). 1H NMR (600 MHz, CD3OD) δ 7.44 (s, 1H, HAr), 7.40 (s, 1H, HAr), 4.02 (dd, J = 12.0, 8.5 Hz, 2H, 2 × O-CH), 3.96 (s, 1H, O-CH), 3.90–3.80 (m, 2H, ArCH2), 2.55 (d, J = 10.7 Hz, 1H, NCH), 2.46 (m, 1H, NCH2), 2.36 (t, J = 10.8 Hz, 1H, NCH2), 2.18 (s, 1H, CH2), 2.00 (d, J = 4.0 Hz, 1H, CH2), 1.45 (s, 3H, CH3). 13C NMR (151 MHz, CD3OD) δ 150.7(CArOH), 132.8 (CAr), 132.1(CAr), 131.4(CAr), 111.0 (CArBr), 110.8 (CArBr), 105.3 (O-C-OH), 82.5 (O-CH), 67.3 (HO-CH), 65.8 (HO-CH), 59.6(NCH), 56.6 (ArCH2), 48.6(NCH2), 41.4 (CH2), 24.1(CH3). ESI-HRMS: m/z calcd for C15H20O5NBr2[M + H]+: 451.9703; found: 451.9698.
- (2R, 3aR, 6S, 7S, 7aS)-2,6,7-trihydroxy-2-methyl-4-(4-hydroxylbenzyl)-3a,7a-di-hydrofuro[3,2-b]piperidine (25) (Figures S43 and S44): A suspension of compound 12 (40 mg, 0.2 mmol) containing activated 4Å molecular sieves in anhydrous methanol (3 mL) was stirred at room temperature for 10 min. After cooling to −20 °C, the corresponding aldehydes (0.6 mmol, 3eq) and NaBH(OAc)3 (0.6 mmol, 3eq) were added, and the solution was stirred for 3 h under an argon atmosphere. The reaction solution was concentrated in vacuo and purified by silica gel flash column chromatography (dichloromethane/methanol, 25:1 → 5:1) to afford 25. Colorless syrup, yield 45%, [α]25D−16.0 (c 0.10, MeOH). 1H NMR (600 MHz, CD3OD) δ 7.08 (d, J = 8.3 Hz, 2H, 2 × HAr), 6.74 (dd, J = 11.3, 2.9 Hz, 2H, 2 × HAr), 4.06–4.00 (m, 2H, 2 × O-CH), 3.96 (s, 1H, O-CH), 3.89–3.75 (m, 2H, ArCH2), 2.59 (dd, J = 10.9, 4.2 Hz, 1H, NCH), 2.49 (d, J = 13.5 Hz, 1H, NCH2), 2.31 (t, J = 8.4 Hz, 1H, NCH2), 2.20 (d, J = 8.5 Hz, 1H, CH2), 2.04–1.98 (m, 1H, CH2), 1.44 (s, 3H, CH3). 13C NMR (151 MHz, CD3OD) δ 156.7 (CArOH), 130.2 (CAr), 129.9 (CAr), 127.5 (CAr), 114.9 (CAr), 114.6 (CAr), 105.2 (O-C-OH), 82.6 (O-CH), 67.3 (HO-CH), 65.8 (HO-CH), 59.6 (NCH), 57.8 (ArCH2), 48.6 (NCH2), 42.5 (CH2), 24.0 (CH3). ESI-HRMS: m/z calcd for C15H22O5NNa [M + Na]+: 296.1492; found: 296.1501.
- (2R, 3aR, 6S, 7S, 7aS)-2,6,7-trihydroxy-2-methyl-4-(2-hydroxylbenzyl)-3a,7a-di-hydrofuro[3,2-b]piperidine (26) (Figures S45 and S46): The synthesized procedure was the same as compound 25. Colorless syrup, yield 47%, [α]25D−35.0 (c 0.10, MeOH). 1H NMR (600 MHz, CD3OD) δ 7.12–7.09 (m, 1H, HAr), 6.78–6.75 (m, 2H, 2 × HAr), 6.72 (dd, J = 8.1, 4.3 Hz, 1H, HAr), 4.01 (d, J = 3.3 Hz, 1H, O-CH), 3.98–3.93 (m, 1H, O-CH), 3.92–3.80 (m, 3H, O-CH, ArCH2), 2.61 (dd, J = 11.3, 3.8 Hz, 1H, NCH), 2.57 (t, J = 6 Hz, 1H, NCH2), 2.51 (d, J = 4.8 Hz, 1H, NCH2), 2.45 (dd, J = 17.2, 6.9 Hz, 1H, CH2), 2.32 (t, J = 10.8 Hz, 1H, CH2), 1.44 (s, 3H, CH3). 13C NMR (151 MHz, CD3OD) δ 156.1 (CArOH), 130.9 (CAr), 128.6 (CAr), 123.0 (CAr), 118.7 (CAr), 114.7 (CAr), 104.5 (O-C-OH), 82.5 (O-CH), 67.7 (HO-CH), 66.5 (HO-CH), 60.3 (NCH), 54.7 (ArCH2), 49.0 (NCH2), 41.5 (CH2), 25.1 (CH3). ESI-HRMS: m/z calcd for C15H22O5N [M + H]+: 296.1492; found: 296.1493.
- (2R, 3aR, 6S, 7S, 7aS)-2,6,7-trihydroxy-2-methyl-4-(3-hydroxylbenzyl)-3a,7a-di-hydrofuro[3,2-b]piperidine (27) (Figures S47 and S48): The synthesized procedure was the same as compound 25. Colorless syrup, yield 48%, [α]25D−32.8 (c 0.08, MeOH). 1H NMR (600 MHz, CD3OD) δ 7.15–7.10 (m, 1H, HAr), 6.80–6.30 (m, 3H, HAr), 4.08–4.02 (m, 2H, 2 × O-CH), 3.97 (s, 1H, O-CH), 3.90–3.75 (m, 2H, ArCH2), 2.59 (d, J = 4.1 Hz, 1H, NCH), 2.47 (d, J = 13.5 Hz, 1H, NCH2), 2.32 (t, J = 10.9 Hz, 1H, NCH2), 2.14 (d, J = 9.2 Hz, 1H, CH2), 2.01 (d, J = 9.6 Hz, 1H, CH2), 1.44 (s, 3H, CH3). 13C NMR (151 MHz, CD3OD) δ 157.4 (CArOH), 138.5 (CAr), 129.2 (CAr), 120.0 (CAr), 115.6 (CAr), 114.1 (CAr), 105.3 (O-C-OH), 82.6 (O-CH), 67.3 (HO-CH), 65.8 (HO-CH), 59.9 (NCH), 58.4 (ArCH2), 48.9 (NCH2), 41.5 (CH2), 24.1 (CH3). ESI-HRMS: m/z calcd for C15H22O5N [M + H]+: 296.1492; found: 296.1493.
- (2R, 3aR, 6S, 7S, 7aS)-2,6,7-trihydroxy-2-methyl-4-(3-chloro-4-hydroxylbenzyl)-3a,7a-Dihydrofuro[3,2-b]piperidine (28) (Figures S49 and S50): The synthesized procedure was the same as compound 25. Colorless syrup, yield 37%, [α]25D−23.7 (c 0.19, MeOH). 1H NMR (600 MHz, CD3OD) δ 7.25–7.15 (m, 1H, HAr), 7.10–7.00 (m, 1H, HAr), 6.90–6.80 (m, 1H, HAr), 4.04–4.00 (m, 1H, O-CH), 4.00 (s, 1H, O-CH), 3.96 (s, 1H, O-CH), 3.86–3.65 (m, 2H, ArCH2), 2.55 (dd, J = 10.7, 4.1 Hz, 1H, NCH), 2.47 (d, J = 13.5 Hz, 1H, NCH2), 2.30 (t, J = 10.9 Hz, 1H, NCH2), 2.15 (d, J = 11.1 Hz, 1H, CH2), 2.00 (dd, J = 13.5, 3.9 Hz, 1H, CH2), 1.44 (s, 3H, CH3). 13C NMR (151 MHz, CD3OD) δ 152.5 (CArOH), 130.4 (CAr), 128.5 (CAr), 128.5 (CAr), 120.3 (CArCl), 116.2 (CAr), 105.3 (O-C-OH), 82.6 (O-CH), 67.3 (HO-CH), 65.8 (HO-CH), 59.7 (NCH), 57.2 (ArCH2), 48.6 (NCH2), 41.5 (CH2), 24.1 (CH3). ESI-HRMS: m/z calcd for C15H21O5NCl [M + H]+: 330.1103; found: 330.1102.
- (2R, 3aR, 6S, 7S, 7aS)-2,6,7-trihydroxy-2-methyl-4-(3-bromo-4-hydroxylbenzyl)-3a,7a-Dihydrofuro[3,2-b]piperidine (29) (Figures S51 and S52): The synthesized procedure was the same as compound 25. Colorless syrup, yield 40%, [α]25D−47.5 (c 0.04, MeOH). 1H NMR (600 MHz, CD3OD) δ 7.37 (d, J = 2.0 Hz, 1H, HAr), 7.06 (dd, J = 8.3, 2.0 Hz, 1H, HAr), 6.86 (d, J = 7.6 Hz, 1H, HAr), 4.10–4.00 (m, 2H, O-CH), 3.96 (s, 1H, O-CH), 3.90–3.75 (m, 2H, ArCH2), 2.55 (dd, J = 10.7, 4.0 Hz, 1H, NCH), 2.48 (d, J = 13.5 Hz, 1H, NCH2), 2.30 (t, J = 10.9 Hz, 1H, NCH2), 2.15 (d, J = 10.8 Hz, 1H, CH2), 2.01 (dd, J = 13.5, 3.8 Hz, 1H, CH2), 1.44 (s, 3H, CH3). 13C NMR (151 MHz, CD3OD) δ 153.6 (CArOH), 133.5 (CAr), 129.2 (CAr), 115.8 (CAr), 115.6 (CAr), 109.4 (CArBr), 105.3 (O-C-OH), 82.6 (O-CH), 67.3 (HO-CH), 65.8 (HO-CH), 59.7 (NCH), 57.1 (ArCH2), 48.7 (NCH2), 41.5 (CH2), 24.1 (CH3). ESI-HRMS: m/z calcd for C15H21O5NBr [M + H]+: 374.0598; found: 374.0597.
- (2R, 3aR, 6S, 7S, 7aS)-2,6,7-trihydroxy-2-methyl-4-(3-fluoro-4-hydroxylbenzyl)-3a,7a-Dihydrofuro[3,2-b]piperidine (30) (Figures S53 and S54): The synthesized procedure was the same as compound 25. Colorless syrup, yield 35%, [α]25D−50.0 (c 0.08, MeOH). 1H NMR (600 MHz, CD3OD) δ 6.96–6.88 (m, 3H, 2 × HAr), 4.03 (s, 1H, O-CH), 4.00 (s, 1H, O-CH), 3.96 (s, 1H, O-CH), 3.90–3.40 (m, 2H, ArCH2), 2.56 (dd, J = 10.7, 4.1 Hz, 1H, NCH), 2.48 (d, J = 13.5 Hz, 1H, NCH2), 2.34 (t, J = 10.9 Hz, 1H, NCH2), 2.20–2.14 (m, 1H, CH2), 2.01 (dd, J = 13.5, 3.9 Hz, 1H, CH2), 1.44 (s, 3H, CH3). 13C NMR (151 MHz, CD3OD) δ 152.2 (CArF), 150.6 (CArOH), 128.8 (CAr), 125.1 (CAr), 117.3 (CAr), 116.3 (CAr), 105.3 (O-C-OH), 82.6 (O-CH), 67.3 (HO-CH), 65.8 (HO-CH), 59.7 (NCH), 57.4 (ArCH2), 48.7 (NCH2), 41.5 (CH2), 24.1 (CH3). ESI-HRMS: m/z calcd for C15H21O5NF [M + H]+: 314.1398; found: 314.1404.
- (2R, 3aR, 6S, 7S, 7aS)-2,6,7-trihydroxy-2-methyl-4-(2-chloro-4-hydroxylbenzyl)-3a,7a-Dihydrofuro[3,2-b]piperidine (31) (Figures S55 and S56): The synthesized procedure was the same as compound 25. Colorless syrup, yield 35%, [α]25D−37.5 (c 0.16, MeOH). 1H NMR (600 MHz, CD3OD) δ 7.23 (d, J = 8.3 Hz, 1H, HAr), 6.80 (d, J = 2.3 Hz, 1H, HAr), 6.70 (dd, J = 8.4, 2.5 Hz, 1H, HAr), 4.06 (t, J = 3.9 Hz, 1H, O-CH), 3.91 (s, 1H, O-CH), 3.80–3.78 (m, 1H, O-CH), 3.74 (d, J = 13.8, 1H, ArCH2), 3.26 (d, J = 15.5, 1H, ArCH2), 2.55 (dd, J = 11.0, 4.3 Hz, 1H, NCH), 2.31 (t, J = 10.9 Hz, 1H, NCH2), 2.19 (d, J = 13.6 Hz, 1H, NCH2), 2.15–2.09 (m, 1H, CH2), 2.00–1.90 (m, 1H, CH2), 1.49 (s, 3H, CH3). 13C NMR (151 MHz, CD3OD) δ 157.2 (CArOH), 134.3 (CArCl), 131.7 (CAr), 126.1 (CAr), 115.7 (CAr), 113.7 (CAr), 107.3 (O-C-OH), 81.0 (O-CH), 67.9 (HO-CH), 66.7 (HO-CH), 60.8 (NCH), 55.0 (ArCH2), 50.1 (NCH2), 42.6 (CH2), 22.4 (CH3). ESI-HRMS: m/z calcd for C15H21O5NCl [M + H]+: 330.1103; found: 330.1104.
- (2R, 3aR, 6S, 7S, 7aS)-2,6,7-trihydroxy-2-methyl-4-(2,6-dichloro-4-hydroxylbenzyl)-3a,7a-Dihydrofuro[3,2-b]piperidine (32) (Figures S57 and S58): The synthesized procedure was the same as compound 25. Colorless syrup, yield 30%, [α]25D−11.4 (c 0.06, MeOH). 1H NMR (600 MHz, CD3OD) δ 6.84 (s, 2H, 2 × HAr), 4.10 (d, J = 12.7 Hz, 1H, O-CH), 4.03 (d, J = 3.4 Hz, 1H, O-CH), 3.95 (s, 1H, O-CH), 3.85–3.65 (m, 2H, ArCH2), 2.62 (t, J = 10.8 Hz, 1H, NCH2), 2.50 (d, J = 6.9 Hz, 1H, NCH), 2.43 (dd, J = 10.7, 4.2 Hz, 1H, NCH2), 2.38 (d, J = 13.5 Hz, 1H, CH2), 1.98 (dd, J = 13.5, 4.1 Hz, 1H, CH2), 1.39 (s, 3H, CH3). 13C NMR (151 MHz, CD3OD) δ 158.1 (CArOH), 136.9 (CArCl), 136.9 (CArCl), 122.7 (CAr), 115.6 (CAr), 115.4 (CAr), 104.7 (O-C-OH), 82.6 (O-CH), 67.3 (HO-CH), 66.0 (HO-CH), 61.6 (NCH), 53.7 (ArCH2), 48.7 (NCH2), 41.3 (CH2), 24.2 (CH3). ESI-HRMS: m/z calcd for C15H20O5NCl2 [M + H]+: 364.0713; found: 364.0703.
- (2R, 3aR, 6S, 7S, 7aS)-2,6,7-trihydroxy-2-methyl-4-(4-hydroxyl-methoxylbenzyl)-3a,7a-Dihydrofuro[3,2-b]piperidine (33) (Figures S59 and S60): The synthesized procedure was the same as compound 25. Colorless syrup,yield 46%, [α]25D−30.4 (c 0.08, MeOH). 1H NMR (600 MHz, CD3OD) δ 6.81 (d, J = 1.7 Hz, 1H, HAr), 6.75 (d, J = 8.0 Hz, 1H, HAr), 6.70 (dd, J = 7.9, 1.7 Hz, 1H, HAr), 4.06–4.02 (m, 2H, 2 × O-CH), 3.97 (s, 1H, O-CH), 3.90–3.70 (m, 2H, ArCH2), 3.83 (s, 3H, OCH3), 2.59 (dd, J = 10.8, 4.0 Hz, 1H, NCH), 2.49 (d, J = 13.4 Hz, 1H, NCH2), 2.30 (t, J = 10.9 Hz, 1H, NCH2), 2.16 (d, J = 14.1 Hz, 1H, CH2), 2.03–1.97 (m, 1H, CH2), 1.45 (s, 3H, CH3). 13C NMR (151 MHz, CD3OD) δ 147.7 (CArOCH3), 145.8 (CArOH), 128.4 (CAr), 121.8 (CAr), 114.5 (CAr), 112.2 (CAr), 105.3 (O-C-OH), 82.7 (O-CH), 67.3 (HO-CH), 65.8 (HO-CH), 59.8 (NCH), 58.2 (ArCH2), 55.0 (OCH3), 48.6 (NCH2), 41.5 (CH2), 24.1 (CH3). ESI-HRMS: m/z calcd for C16H24O6N [M + H]+: 326.1598; found: 326.1605.
- (2R, 3aR, 6S, 7S, 7aS)-2,6,7-trihydroxy-2-methyl-4-(4-hydroxyl-3-ethyoxylbenzyl)-3a,7a-Dihydrofuro[3,2-b]piperidine (34) (Figures S61 and S62): The synthesized procedure was the same as compound 25. Colorless syrup, yield 49%, [α]25D−18.5 (c 0.13, MeOH). 1H NMR (600 MHz, CD3OD) δ 6.80 (d, J = 1.7 Hz, 1H, HAr), 6.76 (d, J = 8.0 Hz, 1H, HAr), 6.69 (dd, J = 8.0, 1.8 Hz, 1H, HAr), 4.12–3.99 (m, 4H, OCH2, 2 × O-CH), 3.94 (s, 1H, O-CH), 3.91–3.70 (m, 2H, ArCH2), 2.59 (dd, J = 10.8, 4.0 Hz, 1H, NCH), 2.48 (d, J = 13.5 Hz, 1H, NCH2), 2.28 (t, J = 10.9 Hz, 1H, NCH2), 2.16 (d, J = 12.9 Hz, 1H, CCH2), 2.01 (dd, J = 13.4, 3.8 Hz, 1H, CCH2), 1.45 (s, 3H, CCH3), 1.40 (t, J = 7.0 Hz, 3H, CH2CH3). 13C NMR (151 MHz, CD3OD) δ 146.8 (CArOEt), 146.1 (CArOH), 128.4 (CAr), 121.8 (CAr), 114.6 (CAr), 113.7 (CAr), 105.3 (O-C-OH), 82.6(O-CH), 67.3(HO-CH), 65.8(HO-CH), 64.2 (OCH2), 59.7 (NCH), 58.2 (ArCH2), 48.7 (NCH2), 41.5 (CCH2), 24.1 (CCH3), 13.7 (CH2CH3). ESI-HRMS: m/z calcd for C17H26O6N [M + H]+: 340.1755; found: 340.1760.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shah, N.A.; Levy, C.J. Emerging technologies for the management of type 2 diabetes mellitus. J. Diabetes 2021, 13, 713–724. [Google Scholar] [CrossRef]
- Diabetes. Available online: https://www.who.int/news-room/fact-sheets/detail/diabetes (accessed on 3 March 2023).
- Wu, Z.; Yu, S.; Kang, X.; Liu, Y.; Xu, Z.; Li, Z.; Wang, J.; Miao, X.; Liu, X.; Li, X.; et al. Association of visceral adiposity index with incident nephropathy and retinopathy: A cohort study in the diabetic population. Cardiovasc. Diabetol. 2022, 21, 32. [Google Scholar] [CrossRef] [PubMed]
- Iatcu, C.O.; Steen, A.; Covasa, M. Gut microbiota and complications of type-2 diabetes. Nutrients 2021, 14, 166. [Google Scholar] [CrossRef] [PubMed]
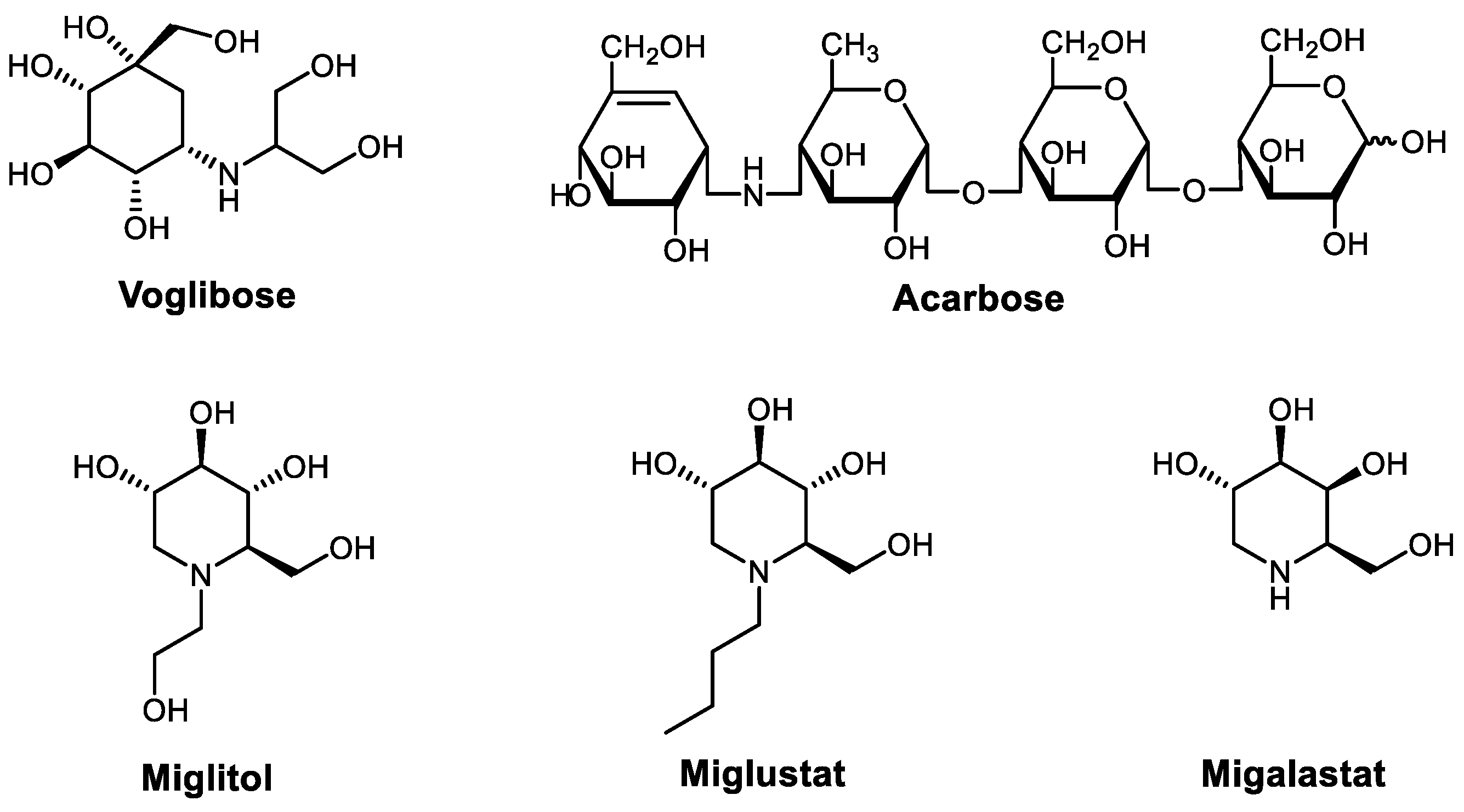
- Usman, B.; Sharma, N.; Satija, S.; Mehta, M.; Vyas, M.; Khatik, G.L.; Khurana, N.; Hansbro, P.M.; Williams, K.; Dua, K. Recent developments in alpha-glucosidase inhibitors for management of type-2 diabetes: An update. Curr. Pharm. Des. 2019, 25, 2510–2525. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.R.; Standl, E.; Tong, N.; Shah, P.; Kalra, S.; Rathod, R. Therapeutic potential of α-glucosidase inhibitors in type 2 diabetes mellitus: An evidence-based review. Expert Opin. Pharmacother. 2015, 16, 1959–1981. [Google Scholar] [CrossRef] [PubMed]
- Hossain, U.; Das, A.K.; Ghosh, S.; Sil, P.C. An overview on the role of bioactive α-glucosidase inhibitors in ameliorating diabetic complications. Food Chem. Toxicol. 2020, 145, 111738. [Google Scholar] [CrossRef] [PubMed]
- Dhameja, M.; Gupta, P. Synthetic heterocyclic candidates as promising α-glucosidase inhibitors: An overview. Eur. J. Med. Chem. 2019, 176, 343–377. [Google Scholar] [CrossRef]
- Borges de Melo, E.; da Silveira Gomes, A.; Carvalho, I. α- and β-Glucosidase inhibitors: Chemical structure and biological activity. Tetrahedron 2006, 62, 10277–10302. [Google Scholar] [CrossRef]
- Matsumura, M.; Monden, T.; Miyashita, Y.; Kawagoe, Y.; Shimizu, H.; Nakatani, Y.; Domeki, N.; Yanagi, K.; Ikeda, S.; Kasai, K. Effects of changeover from voglibose to acarbose on postprandial triglycerides in type 2 diabetes mellitus patients. Adv. Ther. 2009, 26, 660–666. [Google Scholar] [CrossRef]
- Sekar, V.; Chakraborty, S.; Mani, S.; Sali, V.K.; Vasanthi, H.R. Mangiferin from mangifera indica fruits reduces post-prandial glucose level by inhibiting α-glucosidase and α-amylase activity. S. Afr. J. Bot. 2019, 120, 129–134. [Google Scholar] [CrossRef]
- Wang, B.; Zhao, J.; Zhan, Q.; Wang, R.; Liu, B.; Zhou, Y.; Xu, F. Acarbose for postprandial hypotension with glucose metabolism disorders: A systematic review and meta-analysis. Front. Cardiovasc. Med. 2021, 8, 663635. [Google Scholar] [CrossRef]
- Scott, L.J.; Spencer, C.M. Miglitol. Drugs 2000, 59, 521–549. [Google Scholar] [CrossRef]
- Kaku, K. Efficacy of voglibose in type 2 diabetes. Expert Opin. Pharmacother. 2014, 15, 1181–1190. [Google Scholar] [CrossRef] [PubMed]
- Derosa, G.; Maffioli, P. Mini-special issue paper management of diabetic patients with hypoglycemic agents α-glucosidase inhibitors and their use in clinical practice. Arch. Med. Sci. 2012, 5, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ma, S. Recent Advances in Synthetic α-Glucosidase Inhibitors. ChemMedChem 2017, 12, 819–829. [Google Scholar] [CrossRef]
- Paulsen, H. Carbohydrates containing nitrogen or sulfur in the ‘hemiacetal’ ring. Angew. Chem. Int. Ed. Engl. 1966, 5, 495–510. [Google Scholar] [CrossRef]
- Leusmann, S.; Ménová, P.; Shanin, E.; Titz, A.; Rademacher, C. Glycomimetics for the inhibition and modulation of lectins. Chem. Soc. Rev. 2023, 52, 3663–3740. [Google Scholar] [CrossRef]
- Kumar Thakur, A.; Kumar, Y.; K Goyal, K. Pharmacotherapeutics of miglitol: An α-glucosidase inhibitor. J. Anal. Pharm. Res. 2018, 7, 617–619. [Google Scholar] [CrossRef]
- Hollak, C.E.M.; Hughes, D.; van Schaik, I.N.; Schwierin, B.; Bembi, B. Miglustat (Zavesca®) in type 1 Gaucher disease: 5-year results of a post-authorisation safety surveillance programme. Pharmacoepidemiol. Drug Saf. 2009, 18, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Patterson, M.C.; Mengel, E.; Vanier, M.T.; Moneuse, P.; Rosenberg, D.; Pineda, M. Treatment outcomes following continuous miglustat therapy in patients with Niemann-Pick disease Type C: A final report of the NPC Registry. Orphanet J. Rare Dis. 2020, 15, 104. [Google Scholar] [CrossRef]
- Riccio, E.; Zanfardino, M.; Ferreri, L.; Santoro, C.; Cocozza, S.; Capuano, I.; Imbriaco, M.; Feriozzi, S.; Pisani, A. Switch from enzyme replacement therapy to oral chaperone migalastat for treating Fabry disease: Real-life data. Eur. J. Hum. Genet. 2020, 28, 1662–1668. [Google Scholar] [CrossRef] [PubMed]
- Smid, B.E.; Aerts, J.M.F.G.; Boot, R.G.; Linthorst, G.E.; Hollak, C.E.M. Pharmacological small molecules for the treatment of lysosomal storage disorders. Expert Opin. Investig. Drugs 2010, 19, 1367–1379. [Google Scholar] [CrossRef] [PubMed]
- Alonzi, D.S.; Scott, K.A.; Dwek, R.A.; Zitzmann, N. Iminosugar antivirals: The therapeutic sweet spot. Biochem. Soc. Trans. 2017, 45, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tang, S.; Zhang, G.; Pan, Y.; Jiao, W.; Shao, H. Synthesis of N-substituted iminosugar C-glycosides and evaluation as promising α-glucosidase inhibitors. Molecules 2022, 27, 5517. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Luo, H.; Ma, X.; Zou, W.; Shao, H. Stereoselective synthesis of a series of new N-alkyl-3-hydroxypiperidine derivatives containing a hemiketal. Eur. J. Org. Chem. 2011, 4834–4840. [Google Scholar] [CrossRef]
- Winchester, B.G.; Cenci di Bello, I.; Richardson, A.C.; Nash, R.J.; Fellows, L.E.; Ramsden, N.G.; Fleet, G.W.J. The structural basis of the inhibition of human glycosidases by castanospermine analogues. Biochem. J. 1990, 269, 227. [Google Scholar] [CrossRef] [PubMed]
- Heightman, T.D.; Vasella, A.T. Recent insights into inhibition, structure, and mechanism of configuration-retaining glycosidases. Angew. Chem. Int. Ed. 1999, 38, 750–770. [Google Scholar] [CrossRef]
- Jakobsen, P.; Lundbeck, J.M.; Kristiansen, M.; Breinholt, J.; Demuth, H.; Pawlas, J.; Candela, M.P.T.; Andersen, B.; Westergaard, N.; Lundgren, K.; et al. Iminosugars: Potential inhibitors of liver glycogen phosphorylase. Bioorg. Med. Chem. 2001, 9, 733–744. [Google Scholar] [CrossRef]
- De Fenza, M.; Esposito, A.; D’Alonzo, D.; Guaragna, A. synthesis of piperidine nucleosides as conformationally restricted immucillin mimics. Molecules 2021, 26, 1652. [Google Scholar] [CrossRef]
- Choi, Y.S.; George, C.; Comin, M.J.; Barchi, J.J.; Kim, H.S.; Jacobson, K.A.; Balzarini, J.; Mitsuya, H.; Boyer, P.L.; Hughes, S.H.; et al. A conformationally locked analogue of the anti-HIV agent stavudine. An important correlation between pseudorotation and maximum amplitude. J. Med. Chem. 2003, 46, 3292–3299. [Google Scholar] [CrossRef]
- Diot, J.D.; Moreno, I.G.; Twigg, G.; Mellet, C.O.; Haupt, K.; Butters, T.D.; Kovensky, J.; Gouin, S.G. Amphiphilic 1-deoxynojirimycin derivatives through click strategies for chemical chaperoning in N370s Gaucher cells. J. Org. Chem. 2011, 76, 7757–7768. [Google Scholar] [CrossRef] [PubMed]
- Castilla, J.; Rísquez, R.; Cruz, D.; Higaki, K.; Nanba, E.; Ohno, K.; Suzuki, Y.; Díaz, Y.; Mellet, C.O.; Fernández, J.M.G. Conformationally-locked N-glycosides with selective β-glucosidase inhibitory activity: Identification of a new non-iminosugar-type pharmacological chaperone for gaucher disease. J. Med. Chem. 2012, 55, 6857–6865. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Moncayo, M.; García-Moreno, M.I.; Trapero, A.; EgidoGabas, M.; Llebaria, M.; García Fernandez, J.M.; Ortiz Mellet, C. Bicyclic (galacto) nojirimycin analogues as glycosidase inhibitors: Effect of structural modifications in their pharmacological chaperone potential towards β-glucocerebrosidase. Org. Biomol. Chem. 2011, 9, 3698–3713. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Moncayo, M.; García-Moreno, M.I.; Stutz, A.E.; García Fernandez, J.M.; Wrodnigg, T.M.; Ortiz Mellet, C. Fluorescenttagged sp2-iminosugars with potent β-glucosidase inhibitory activity. Bioorg. Med. Chem. 2010, 18, 7439–7445. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Moncayo, M.; Takai, T.; Higaki, K.; Mena-Barragan, T.; Hirano, Y.; Yura, K.; Li, L.; Yu, Y.; Ninomiya, H.; García-Moreno, M.I.; et al. Tuning glycosidase inhibition through aglycone interactions: Pharmacological chaperones for Fabry disease and GM1 gangliosidosis. Chem. Commun. 2012, 48, 6514–6516. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Fernandez, E.M.; Rísquez-Cuadro, R.; Chasseraud, M.; Ahidouch, A.; García-Moreno, M.I.; Ortiz Mellet, C.; OuadidAhidouch, H.; García Fernandez, J.M. Synthesis of N-, S-, and C-glycoside castanospermine analogues with selective neutral α-glucosidase inhibitory activity as antitumour agents. Chem. Commun. 2010, 46, 5328–5330. [Google Scholar] [CrossRef] [PubMed]
- McDevitt, J.P.; Lansbury, P.T. Glycosamino acids: New building blocks for combinatorial synthesis. J. Am. Chem. Soc. 1996, 118, 3818–3828. [Google Scholar] [CrossRef]
- Luo, H.; Zou, W.; Shao, H. Synthesis of N-substituted iminosugars from 2′-carbonyl-C-glycofuranosides. Carbohydr. Res. 2009, 344, 2454–2460. [Google Scholar] [CrossRef]
- Zou, W.; Sandbhor, M.; Bhasin, M. Stereoselective synthesis of polyhydroxylated quinolizidines from C-glycosides by one-pot double-conjugate addition. J. Org. Chem. 2007, 72, 1226–1234. [Google Scholar] [CrossRef]
- Kashtoh, H.; Muhammad, M.T.; Khan, J.J.A.; Rasheed, S.; Khan, A.; Perveen, S.; Javaid, K.; Atia-tul-Wahab; Khan, K.M.; Choudhary, M.I. Dihydropyrano [2,3-c] Pyrazole: Novel in vitro inhibitors of yeast α-Glucosidase. Bioorg. Chem. 2016, 65, 61–72. [Google Scholar] [CrossRef]
- Kashtoh, H.; Baek, K.H. Recent updates on phytoconstituent alpha-glucosidase inhibitors: An approach towards the treatment of type two diabetes. Plants 2022, 11, 2722. [Google Scholar] [CrossRef]
- Ren, L.M.; Qin, X.H.; Cao, X.F.; Wang, L.L.; Bai, F.; Bai, G.; Shen, Y.Q. Structural insight into substrate specificity of human intestinal maltase-glucoamylase. Protein Cell 2011, 2, 827–836. [Google Scholar] [CrossRef]
- Sim, L.; Quezada-Calvillo, R.; Sterchi, E.E.; Nichols, B.L.; Rose, D.R. Human intestinal maltase-glucoamylase: Crystal structure of the N-terminal catalytic subunit and basis of inhibition and substrate specificity. J. Mol. Biol. 2008, 375, 782–792. [Google Scholar] [CrossRef]
- Sim, L.; Willemsma, C.; Mohan, S.; Naim, H.Y.; Pinto, B.M.; Rose, D.R. Structural basis for substrate selectivity in human maltase-glucoamylase and sucrase-isomaltase N-terminal domains. J. Biol. Chem. 2010, 285, 17763–17770. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Dang, P.H.; Vu, N.X.T.; Le, T.H.; Nguyen, T.T. Quinoliniumolate and 2h-1,2,3-triazole derivatives from the stems of Paramignya trimera and their α-glucosidase inhibitory activities: In vitro and in silico studies. J. Nat. Prod. 2017, 80, 2151–2155. [Google Scholar] [CrossRef]
- Zhang, B.W.; Li, X.; Sun, W.L.; Xing, Y.; Xiu, Z.L.; Zhuang, C.L.; Dong, Y.S. Dietary flavonoids and acarbose synergistically inhibit α-glucosidase and lower postprandial blood glucose. J. Agric. Food Chem. 2017, 65, 8319–8330. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. a qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, M.; Yasutake, K.; Hino, M.; Ohwatari, H.; Ohmagari, N.; Takedomi, K.; Tanaka, T.; Nonaka, G. Inhibitory effects of polyphenols from water chestnut (Trapa japonica) husk on glycolytic enzymes and postprandial blood glucose elevation in mice. Food Chem. 2014, 165, 42–49. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μM) | Compound | IC50 (μM) |
|---|---|---|---|
| 6 | >20.0 | 12 | >20.0 |
| 13 | 13.0 ± 0.5 | 14 | 10.0 ± 0.7 |
| 15 | >20.0 | 16 | >20.0 |
| 17 | 4.9 ± 0.2 | 18 | 5.5 ± 0.4 |
| 19 | 5.4 ± 0.3 | 20 | 4.4 ± 0.3 |
| 21 | 0.91 ± 0.1 | 22 | 10.5 ± 0.8 |
| 23 | 9.0 ± 0.6 | 24 | 1.9 ± 0.2 |
| 25 | 8.3 ± 0.4 | 26 | >20.0 |
| 27 | 3.7 ± 0.1 | 28 | 0.5 ± 0.1 |
| 29 | 1.2 ± 0.1 | 30 | 2.6 ± 0.3 |
| 31 | 1.8 ± 0.2 | 32 | 0.07 ± 0.01 |
| 33 | >20.0 | 34 | >20.0 |
| Acarbose | 2.0 ± 0.2 |
| Compound | MW (g/mol) | RB | HBA | HBD | MR | TPSA (Å2) | MlogP |
|---|---|---|---|---|---|---|---|
| 13 | 313.78 | 2 | 5 | 3 | 82.07 | 73.16 | 0.90 |
| 14, 25 | 295.33 | 2 | 6 | 4 | 79.08 | 93.39 | −0.16 |
| 15, 26 | 295.33 | 2 | 6 | 4 | 79.08 | 93.39 | −0.16 |
| 16, 27 | 295.33 | 2 | 6 | 4 | 79.08 | 93.39 | −0.16 |
| 17, 28 | 329.78 | 2 | 6 | 4 | 84.09 | 93.39 | 0.35 |
| 18, 29 | 374.23 | 2 | 6 | 4 | 86.78 | 93.39 | 0.48 |
| 19, 30 | 313.32 | 2 | 7 | 4 | 79.04 | 93.39 | 0.23 |
| 20, 31 | 329.78 | 2 | 6 | 4 | 84.09 | 93.39 | 0.35 |
| 21, 32 | 364.22 | 2 | 6 | 4 | 89.10 | 93.39 | 0.87 |
| 22, 33 | 325.36 | 3 | 7 | 4 | 85.57 | 102.62 | −0.45 |
| 23, 34 | 339.38 | 4 | 7 | 4 | 90.38 | 102.62 | −0.20 |
| 24 | 453.12 | 2 | 6 | 4 | 94.48 | 93.39 | 1.10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Huang, X.; Pan, Y.; Zhang, G.; Tang, S.; Shao, H.; Jiao, W. Synthesis and Biological Evaluation of New Dihydrofuro[3,2-b]piperidine Derivatives as Potent α-Glucosidase Inhibitors. Molecules 2024, 29, 1179. https://doi.org/10.3390/molecules29051179
Wang H, Huang X, Pan Y, Zhang G, Tang S, Shao H, Jiao W. Synthesis and Biological Evaluation of New Dihydrofuro[3,2-b]piperidine Derivatives as Potent α-Glucosidase Inhibitors. Molecules. 2024; 29(5):1179. https://doi.org/10.3390/molecules29051179
Chicago/Turabian StyleWang, Haibo, Xiaojiang Huang, Yang Pan, Guoqing Zhang, Senling Tang, Huawu Shao, and Wei Jiao. 2024. "Synthesis and Biological Evaluation of New Dihydrofuro[3,2-b]piperidine Derivatives as Potent α-Glucosidase Inhibitors" Molecules 29, no. 5: 1179. https://doi.org/10.3390/molecules29051179