
Effect of Particle Heterogeneity in Catalytic Copper-Containing Single-Chain Polymeric Nanoparticles Revealed by Single-Particle Kinetics
 ,
,
Abstract
:1. Introduction
2. Results
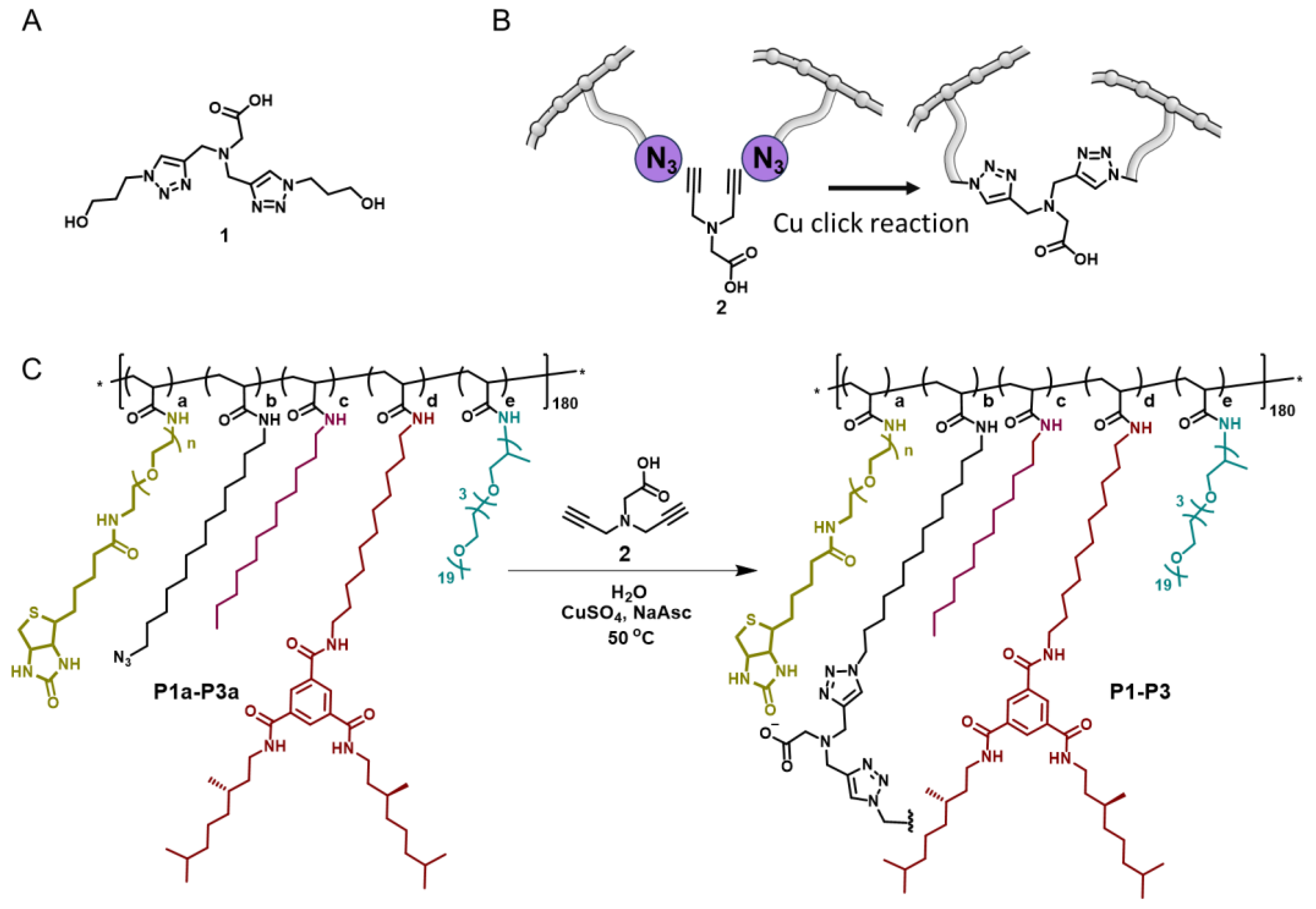
2.1. Design, Synthesis, and Characterization of Cu(I) SCPNs
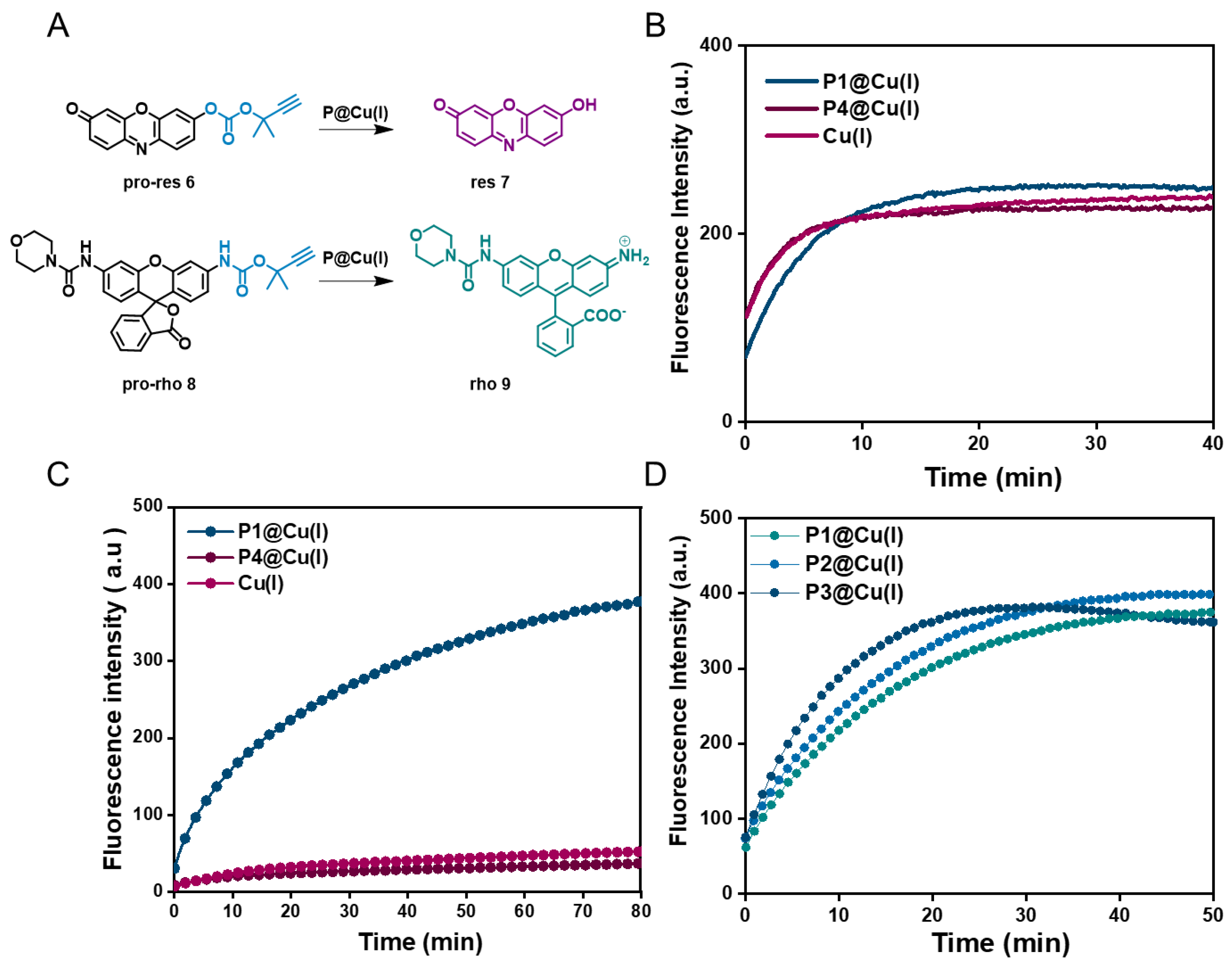
2.2. Catalytic Efficiency of Cu(I) SCPNs—Depropargylation and CuAAC Reactions
2.3. Cu(I) SCPNs Enzyme Kinetics—Michaelis–Menten Model
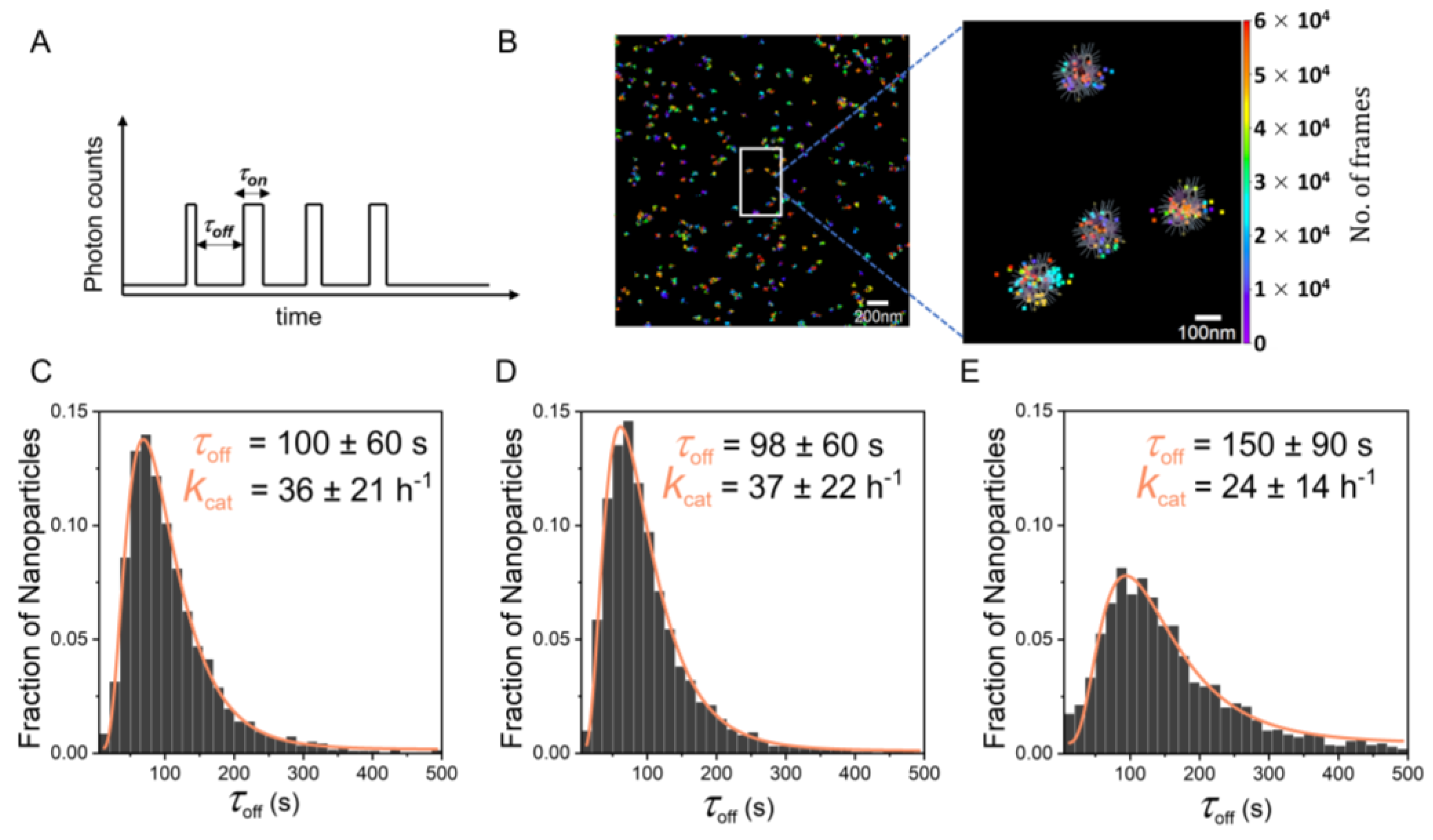
2.4. Single-Particle Kinetics Using Single-Molecule Fluorescence Microscopy
3. Discussion and Conclusions
4. Materials and Methods
4.1. Materials and Instruments
4.2. Pro-Dye and Polymer Synthesis
4.2.1. Pro-Res 6 (2-Methylbut-3-yn-2-yl (3-oxo-3H-Phenoxazin-7-yl) Carbonate)
- Yield = 15 mg, 6%. 1H NMR (400 MHz, CDCl3) δ 7.80 (d, J = 8.7 Hz, 1H), 7.43 (d, J = 9.8 Hz, 1H), 7.29 (s, 1H), 7.23 (d, J = 2.5 Hz, 1H), 6.87 (dd, J = 9.8, 2.0 Hz, 1H), 6.33 (d, J = 2.0 Hz, 1H), 2.65 (s, 1H), 1.82 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 144.34, 135.22, 134.80, 131.15, 118.64, 109.13, 107.30, 83.07, 77.21, 76.05, 73.92, 28.65. FT-IR (ATR): v (cm−1) 3290, 3048, 2994, 1772, 1623, 1608, 1572, 1515, 1246, 1220, 1196, 1129, 885, 848, 772, 667. MALDI-TOF-MS: m/z calc: 323.0; found: 323 [M]−, deprotected product resorufin 212 [M − H]−.
4.2.2. Post-Functionalization of p-PFPA180 to P1a–P3a
- Yield: 380 mg, Mtheoretical = 161 kD, Mn, SEC-DMF = 59.1 kD, Ð = 1.21. 1H NMR (400 MHz, CDCl3) δ 6.92–6.01 (br), 4.15–3.90 (br), 3.84–3.14 (m), 1.90–1.82 (br), 1.72–1.52 (s), 1.41–0.93 (m). FT-IR (ATR): v(cm−1) 3520.56, 2865.86, 2096.31, 1650.15, 1544.45, 1454.07, 1348.63, 1325.4, 1296.42, 1249.42, 1096.62, 947.46, 848.32, 522.91. Mtheoretical = 161 kD, Mn, SEC-DMF = 59.1 kD, Ð = 1.21.
- P2a: p-PFPA180 (180 mg, 1 eq, 0.0042 mmol), 12-azidododecan-1-amine (34 mg, 36 eq, 0,15 mmol), BTA amine (25 mg, 9 eq, 0.037 mmol), biotinylated PEG amine (57 mg, 6 eq, 0.025 mmol), and Jeffamine@1000 (890 mg, 212 eq, 0.8 mmol). The polymer was dried under vacuum at 50 °C to yield a colorless solid and was stored at −19 °C. 1H NMR (400 MHz, CDCl3) δ 8.51–8.39 (br), 6.86–6.32 (br), 4.18–3.13 (m), 1.91–1.51 (m), 1.41–1.04 (m). FT-IR (ATR): v (cm−1) 3519.56, 2866.02, 2096.51, 1650.73, 1543.94, 1454.53, 1348.76, 1325.35, 1294.99, 1249.98, 1094.69, 947.35, 848.13, 523.42. Mtheoretical = 158 kD, Mn, SEC-DMF = 44.3 kD, Ð = 1.20.
- P3a: p-PFPA180 (150 mg, 1 eq, 0.0031 mmol), 12-azidododecan-1-amine (25 mg, 36 eq, 0,11 mmol), dodecyl amine (15.4 mg, 27 eq, 0.083 mmol), biotinylated PEG amine (47 mg, 6 eq, 0.018 mmol), and Jeffamine@1000 (657 mg, 212 eq, 0.65 mmol). The polymer was dried under vacuum at 50 °C to yield a colorless solid and was stored at −19 °C. 1H NMR (400 MHz, CDCl3) δ 6.86–6.13 (br), 4.27–3.95 (br), 3.87–3.13 (m), 1.94–0.82 (m). FT-IR (ATR): v (cm−1): 3301.87, 2863.67, 2096.15, 1649.47, 1540.03, 1454.47, 1346.79, 1324.91, 1295.06, 1249.38, 1199.43, 1098.22, 1039.84, 947.47, 845.32, 522.96. Mtheoretical = 165 kD, Mn,SEC-DMF = 47.5 kD, Ð = 1.25.
4.2.3. Incorporation of the diyne 2 to P1–P3
- P1: 1H NMR (400 MHz, D2O) δ 8.45–8.40 (br), 8.22–8.16 (m), 4.85–4.81 (d), 3.91–3.27 (m), 2.12–1.76 (s), 1.27–0.95 (br). FT-IR (ATR): v (cm−1): 3504.07, 2866.26, 1648.32, 1542.17, 1453.42, 1348.53, 1297.19, 1249.3, 1094.89, 946.76, 845.98. Mtheoretical = 164 kD, Mn, SEC-PBS = 15.5 kD, Ð = 1.38.
- P2: 1H NMR (400 MHz, D2O) δ 8.47–8.43 (br), 8.23–8.14 (m), 4.85–4.81 (s), 3.81–3.29 (m), 1.31–0.99 (br). FT-IR (ATR): v (cm−1): 3437.19, 2867.07, 1647.18, 1544.54, 1452.73, 1348.65, 1294.81, 1249.96, 1094.27, 947.04, 846.59, 521.88. Mtheoretical = 161 kD, Mn, SEC-PBS = 23.4 kD, Ð = 1.35.
- P3: 1H NMR (400 MHz, D2O) δ 8.41–8.37 (br), 8.22–8.16 (m), 4.85–4.81 (s), 4.53–4.35 (s), 3.92–3.27 (m), 1.36–1.12 (br). FT-IR (ATR): v (cm−1): 3436.86, 2869.53, 1646.34, 1548.24, 1454.48, 1348.8, 1296.07, 1250.18, 1091.99, 947.74, 844.89, 805.17, 523.09. Mtheoretical = 168 kD, Mn, SEC-PBS = 16.3 kD, Ð = 1.35.
4.3. Ensemble Catalysis Measurements
4.4. Depropargylation Reactions
4.5. Michaelis–Menten Kinetics
4.6. Single-Particle Kinetic Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Janssen, K.P.F.; De Cremer, G.; Neely, R.K.; Kubarev, A.V.; Van Loon, J.; Martens, J.A.; De Vos, D.E.; Roeffaers, M.B.J.; Hofkens, J. Single Molecule Methods for the Study of Catalysis: From Enzymes to Heterogeneous Catalysts. Chem. Soc. Rev. 2014, 43, 990–1006. [Google Scholar] [CrossRef]
- Kuah, E.; Toh, S.; Yee, J.; Ma, Q.; Gao, Z. Enzyme Mimics: Advances and Applications. Chem. Eur. J. 2016, 22, 8404–8430. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huang, R.; Gopalakrishnan, S.; Cao-Milán, R.; Rotello, V.M. Bioorthogonal Nanozymes: Progress towards Therapeutic Applications. Trends Chem. 2019, 1, 90–98. [Google Scholar] [CrossRef]
- Fedeli, S.; Im, J.; Gopalakrishnan, S.; Elia, J.L.; Gupta, A.; Kim, D.; Rotello, V.M. Nanomaterial-Based Bioorthogonal Nanozymes for Biological Applications. Chem. Soc. Rev. 2021, 50, 13467–13480. [Google Scholar] [CrossRef]
- Garcia, E.S.; Xiong, T.M.; Lifschitz, A.; Zimmerman, S.C. Tandem Catalysis Using an Enzyme and a Polymeric Ruthenium-Based Artificial Metalloenzyme. Polym. Chem. 2021, 12, 6755–6760. [Google Scholar] [CrossRef]
- Chen, J.; Wang, J.; Li, K.; Wang, Y.; Gruebele, M.; Ferguson, A.L.; Zimmerman, S.C. Polymeric “Clickase” Accelerates the Copper Click Reaction of Small Molecules, Proteins, and Cells. J. Am. Chem. Soc. 2019, 141, 9693–9700. [Google Scholar] [CrossRef]
- Chen, J.; Wang, J.; Bai, Y.; Li, K.; Garcia, E.S.; Ferguson, A.L.; Zimmerman, S.C. Enzyme-like Click Catalysis by a Copper-Containing Single-Chain Nanoparticle. J. Am. Chem. Soc. 2018, 140, 13695–13702. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Feng, X.; Xing, H.; Xu, Y.; Kim, B.K.; Baig, N.; Zhou, T.; Gewirth, A.A.; Lu, Y.; Oldfield, E.; et al. A Highly Efficient Single-Chain Metal-Organic Nanoparticle Catalyst for Alkyne-Azide “Click” Reactions in Water and in Cells. J. Am. Chem. Soc. 2016, 138, 11077–11080. [Google Scholar] [CrossRef]
- Liu, Y.; Pujals, S.; Stals, P.J.M.; Paulöhrl, T.; Presolski, S.I.; Meijer, E.W.; Albertazzi, L.; Palmans, A.R.A. Catalytically Active Single-Chain Polymeric Nanoparticles: Exploring Their Functions in Complex Biological Media. J. Am. Chem. Soc. 2018, 140, 3423–3433. [Google Scholar] [CrossRef]
- Cole, J.P.; Hanlon, A.M.; Rodriguez, K.J.; Berda, E.B. Protein-like Structure and Activity in Synthetic Polymers. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 191–206. [Google Scholar] [CrossRef]
- Huerta, E.; Stals, P.J.M.; Meijer, E.W.; Palmans, A.R.A. Consequences of Folding a Water-Soluble Polymer Around an Organocatalyst. Angew. Chem. 2013, 125, 2978–2982. [Google Scholar] [CrossRef]
- Liu, Y.; Turunen, P.; De Waal, B.F.M.; Blank, K.G.; Rowan, A.E.; Palmans, A.R.A.; Meijer, E.W. Catalytic Single-Chain Polymeric Nanoparticles at Work: From Ensemble towards Single-Particle Kinetics. Mol. Syst. Des. Eng. 2018, 3, 609–618. [Google Scholar] [CrossRef]
- Taylor, A.B.; Zijlstra, P. Single-Molecule Plasmon Sensing: Current Status and Future Prospects. ACS Sens. 2017, 2, 1103–1122. [Google Scholar] [CrossRef]
- Elenko, M.P.; Szostak, J.W.; Van Oijen, A.M. Single-Molecule Imaging of an in Vitro-Evolved RNA Aptamer Reveals Homogeneous Ligand Binding Kinetics. J. Am. Chem. Soc. 2009, 131, 9866–9867. [Google Scholar] [CrossRef]
- Leake, M.C. The Physics of Life: One Molecule at a Time. Philos. Trans. R. Soc. B 2013, 368, 20120248. [Google Scholar] [CrossRef]
- Leake, M.C.; Greene, N.P.; Godun, R.M.; Granjon, T.; Buchanan, G.; Chen, S.; Berry, R.M.; Palmer, T.; Berks, B.C. Variable Stoichiometry of the TatA Component of the Twin-Arginine Protein Transport System Observed by in Vivo Single-Molecule Imaging. Proc. Natl. Acad. Sci. USA 2008, 105, 15376–15381. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.P.; Xun, L.; Xie, X.S. Single-Molecule Enzymatic Dynamics. Science 1998, 282, 1877–1882. [Google Scholar] [CrossRef]
- Wang, Y. A Plasmonic Nanotorch: Pushing Plasmon-Enhanced Fluorescence for Applications in Single-Molecule Enzymology. Ph.D. Thesis, Eindhoven University of Technology, Eindhoven, The Netherlands, 2020. [Google Scholar]
- Turunen, P.; Rowan, A.E.; Blank, K. Single-Enzyme Kinetics with Fluorogenic Substrates: Lessons Learnt and Future Directions. FEBS Lett. 2014, 588, 3553–3563. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Kong, J.S.; Yeh, Y.T.E.; Chen, P. Single-Molecule Nanocatalysis Reveals Heterogeneous Reaction Pathways and Catalytic Dynamics. Nat. Mater. 2008, 7, 992–996. [Google Scholar] [CrossRef]
- Zhou, X.; Xu, W.; Liu, G.; Panda, D.; Chen, P. Size-Dependent Catalytic Activity and Dynamics of Gold Nanoparticles at the Single-Molecule Level. J. Am. Chem. Soc. 2010, 132, 138–146. [Google Scholar] [CrossRef]
- Zhou, X.; Andoy, N.M.; Liu, G.; Choudhary, E.; Han, K.S.; Shen, H.; Chen, P. Quantitative Super-Resolution Imaging Uncovers Reactivity Patterns on Single Nanocatalysts. Nat. Nanotechnol. 2012, 7, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Dong, B.; Chen, K.; Zhao, F.; Cheng, X.; Ma, C.; Lee, S.; Zhang, P.; Kang, S.H.; Ha, J.W.; et al. Optical Super-Resolution Imaging of Surface Reactions. Chem. Rev. 2017, 117, 7510–7537. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zhang, Y.; Xu, W. Single-Molecule Nanocatalysis Reveals Catalytic Activation Energy of Single Nanocatalysts. J. Am. Chem. Soc. 2016, 138, 12414–12421. [Google Scholar] [CrossRef] [PubMed]
- Archontakis, E.; Deng, L.; Zijlstra, P.; Palmans, A.R.A.; Albertazzi, L. Spectrally PAINTing a Single Chain Polymeric Nanoparticle at Super-Resolution. J. Am. Chem. Soc. 2022, 144, 23698–23707. [Google Scholar] [CrossRef] [PubMed]
- Sathyan, A.; Croke, S.; Pérez-López, A.M.; de Waal, B.F.M.; Unciti-Broceta, A.; Palmans, A.R.A. Developing Pd(II) Based Amphiphilic Polymeric Nanoparticles for pro-Drug Activation in Complex Media. Mol. Syst. Des. Eng. 2022, 7, 1736–1748. [Google Scholar] [CrossRef]
- Deng, L.; Sathyan, A.; Adam, C.; Unciti-Broceta, A.; Sebastian, V.; Palmans, A.R.A. Enhanced Efficiency of Pd(0)-Based Single Chain Polymeric Nanoparticles for in Vitro Prodrug Activation by Modulating the Polymer’s Microstructure. Nano. Lett. 2024, 14, 3. [Google Scholar] [CrossRef] [PubMed]
- Lozhkin, B.; Ward, T.R. Bioorthogonal Strategies for the in Vivo Synthesis or Release of Drugs. Bioorg. Med. Chem. 2021, 45, 116310. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.Y.; Zhang, B.; Wang, L.; Huang, S.; Wang, S.; You, Y.; Zhu, G.; Zhu, A.; Geng, M.; Li, L. An Efficient and Easily-Accessible Ligand for Cu(I)-Catalyzed Azide–Alkyne Cycloaddition Bioconjugation. Chem. Commun. 2020, 56, 14401–14403. [Google Scholar] [CrossRef] [PubMed]
- Huerta, E.; Van Genabeek, B.; Stals, P.J.M.; Meijer, E.W.; Palmans, A.R.A. A Modular Approach to Introduce Function into Single-Chain Polymeric Nanoparticles. Macromol. Rapid Commun. 2014, 35, 1320–1325. [Google Scholar] [CrossRef]
- ter Huurne, G.M.; de Windt, L.N.J.; Liu, Y.; Meijer, E.W.; Voets, I.K.; Palmans, A.R.A. Improving the Folding of Supramolecular Copolymers by Controlling the Assembly Pathway Complexity. Macromolecules 2017, 50, 8562–8569. [Google Scholar] [CrossRef]
- Wijker, S.; Deng, L.; Eisenreich, F.; Voets, I.K.; Palmans, A.R.A. En Route to Stabilized Compact Conformations of Single-Chain Polymeric Nanoparticles in Complex Media. Macromolecules 2022, 55, 6220–6230. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, V.O.; Presolski, S.I.; Díaz, D.D.; Fokin, V.V.; Finn, M.G. Ligand-Accelerated Cu-Catalyzed Azide-Alkyne Cycloaddition: A Mechanistic Report. J. Am. Chem. Soc. 2007, 129, 12705–12712. [Google Scholar] [CrossRef] [PubMed]
- English, B.P.; Min, W.; Van Oijen, A.M.; Kang, T.L.; Luo, G.; Sun, H.; Cherayil, B.J.; Kou, S.C.; Xie, X.S. Ever-Fluctuating Single Enzyme Molecules: Michaelis-Menten Equation Revisited. Nat. Chem. Biol. 2005, 2, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Kong, J.S.; Chen, P. Single-Molecule Kinetic Theory of Heterogeneous and Enzyme Catalysis. J. Phys. Chem. C 2009, 113, 2393–2404. [Google Scholar] [CrossRef]
- Ye, R.; Mao, X.; Sun, X.; Chen, P. Analogy between Enzyme and Nanoparticle Catalysis: A Single-Molecule Perspective. ACS Catal. 2019, 9, 1985–1992. [Google Scholar] [CrossRef]
- Latocheski, E.; Dal Forno, G.M.; Ferreira, T.M.; Oliveira, B.L.; Bernardes, G.J.L.; Domingos, J.B.; Forno, G.M.D.; Ferreira, T.M.; Oliveira, B.L.; Bernardes, G.J.L.; et al. Mechanistic Insights into Transition Metal-Mediated Bioorthogonal Uncaging Reactions. Chem. Soc. Rev. 2020, 49, 7710–7729. [Google Scholar] [CrossRef]
- Shieh, P.; Dien, V.T.; Beahm, B.J.; Castellano, J.M.; Wyss-Coray, T.; Bertozzi, C.R. CalFluors: A Universal Motif for Fluorogenic Azide Probes across the Visible Spectrum. J. Am. Chem. Soc. 2015, 137, 7145–7151. [Google Scholar] [CrossRef]
- Sathyan, A.; Loman, T.; Deng, L.; Palmans, A.R.A. Amphiphilic Polymeric Nanoparticles Enable Homogenous Rhodium-Catalysed NH Insertion Reactions in Living Cells. Nanoscale 2023, 15, 12710–12717. [Google Scholar] [CrossRef]
- Leenders, C.M.A.; Jansen, G.; Frissen, M.M.M.; Lafleur, R.P.M.; Voets, I.K.; Palmans, A.R.A.; Meijer, E.W. Monosaccharides as Versatile Units for Water-Soluble Supramolecular Polymers. Chem. Eur. J. 2016, 22, 4608–4615. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | a | b | c | d | e | Ð | Mn, SEC (kDa) | Mn, theoretical (kDa) | RH (nm) |
|---|---|---|---|---|---|---|---|---|---|
| pPFPA180 | 1.19 x | 30.2 x | 42 | - | |||||
| P1a | 3 | 18 | - | - | 71 | 1.21 y | 59.1 y | 161 | 4.3 |
| P2a | 3 | 18 | - | 5 | 69 | 1.20 y | 44.3 y | 158 | 5.0 |
| P3a | 3 | 18 | 12 | - | 67 | 1.25 y | 47.5 y | 165 | 5.0 |
| P1 | 3 | 18 | - | - | 71 | 1.38 z | 15.5 z | 164 | 3.4 |
| P2 | 3 | 18 | - | 5 | 69 | 1.35 z | 23.4 z | 161 | 4.9 |
| P3 | 3 | 18 | 12 | - | 67 | 1.35 z | 16.3 z | 168 | 4.8 |
| P4 | - | - | 20 | - | 80 | 1.16 y | 24.4 y | 181.0 | 6.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sathyan, A.; Archontakis, E.; Spiering, A.J.H.; Albertazzi, L.; Palmans, A.R.A. Effect of Particle Heterogeneity in Catalytic Copper-Containing Single-Chain Polymeric Nanoparticles Revealed by Single-Particle Kinetics. Molecules 2024, 29, 1850. https://doi.org/10.3390/molecules29081850
Sathyan A, Archontakis E, Spiering AJH, Albertazzi L, Palmans ARA. Effect of Particle Heterogeneity in Catalytic Copper-Containing Single-Chain Polymeric Nanoparticles Revealed by Single-Particle Kinetics. Molecules. 2024; 29(8):1850. https://doi.org/10.3390/molecules29081850
Chicago/Turabian StyleSathyan, Anjana, Emmanouil Archontakis, A. J. H. Spiering, Lorenzo Albertazzi, and Anja R. A. Palmans. 2024. "Effect of Particle Heterogeneity in Catalytic Copper-Containing Single-Chain Polymeric Nanoparticles Revealed by Single-Particle Kinetics" Molecules 29, no. 8: 1850. https://doi.org/10.3390/molecules29081850