
Investigating Ligand Sphere Perturbations on MnIII–Alkylperoxo Complexes
by
 , , and
, , and
Samuel A. Brunclik
1 ,
,
Elizabeth N. Grotemeyer
1,
Zahra Aghaei
1,
Mohammad Rasel Mian
2 and
Timothy A. Jackson
1,* 1
Department of Chemistry and Center for Environmentally Beneficial Catalysis, University of Kansas, Lawrence, KS 66045, USA
2
Protein Structure and X-ray Crystallography Laboratory, University of Kansas, Lawrence, KS 66045, USA
*
Author to whom correspondence should be addressed.
Molecules 2024, 29(8), 1849; https://doi.org/10.3390/molecules29081849
Submission received: 27 March 2024
/
Revised: 15 April 2024
/
Accepted: 16 April 2024
/
Published: 18 April 2024
(This article belongs to the Special Issue Recent Advances in Coordination Chemistry of Metal Complexes)
Abstract
:Manganese catalysts that activate hydrogen peroxide carry out several different hydrocarbon oxidation reactions with high stereoselectivity. The commonly proposed mechanism for these reactions involves a key manganese(III)-hydroperoxo intermediate, which decays via O–O bond heterolysis to generate a Mn(V)–oxo species that institutes substrate oxidation. Due to the scarcity of characterized MnIII–hydroperoxo complexes, MnIII–alkylperoxo complexes are employed to understand factors that affect the mechanism of the O–O cleavage. Herein, we report a series of novel complexes, including two room-temperature-stable MnIII–alkylperoxo species, supported by a new amide-containing pentadentate ligand (6Medpaq5NO2). We use a combination of spectroscopic methods and density functional theory computations to probe the effects of the electronic changes in the ligand sphere trans to the hydroxo and alkylperoxo units to thermal stability and reactivity. The structural characterizations for both MnII(OTf)(6Medpaq5NO2) and [MnIII(OH)(6Medpaq5NO2)](OTf) were obtained via single-crystal X-ray crystallography. A perturbation to the ligand sphere allowed for a marked increase in reactivity towards an organic substrate, a modest change in the distribution of the O–O cleavage products from homolytic and heterolytic pathways, and little change in thermal stability.
1. Introduction
Manganese-dependent enzymes are vital to a variety of organisms due to their role in carrying out important oxidation-reduction reactions [1,2,3,4,5,6,7,8,9,10,11,12,13]. For example, manganese superoxide dismutase (MnSOD) and manganese lipoxygenase (MnLOX) detoxify free radicals and convert polyunsaturated fatty acids into alkyl hydroperoxides, respectively [1,2,3,4,14,15,16,17]. Inspired by these and other Mn enzymes, a range of synthetic manganese catalysts have been developed to carry out a variety of highly stereo-selective oxidation reactions [18], including asymmetric olefin epoxidation and late-stage C–H bond functionalization [19,20,21,22,23,24,25,26,27]. Due to the abundance and low cost of manganese, as well as the low environmental impact of obtaining manganese, increasing the use of such synthetic catalysts in the industry could reduce the environmental impacts of the current chemical processes [28,29].
At present, the proposed catalytic mechanisms for these synthetic manganese catalysts invoke oxygen–oxygen heterolysis of a MnIII–hydroperoxo intermediate to generate a manganese(V)–oxo species as the active oxidant [27,30]. However, due to the scarcity of MnIII–hydroperoxo complexes that have been trapped and spectroscopically characterized, there is a key gap in our understanding of the mechanism of this O–O cleavage step [31,32,33,34,35]. Nam and co-workers have previously reported MnIII–hydroperoxo complexes supported by tetramethylcyclam (TMC) and a ring-contracted derivative [33,34]. These complexes have shown intense electronic absorption bands in the UV region from ~300–400 nm. Resonance Raman spectra of the [MnIII(OOH)(TMC)]2+ complex revealed a band at 792 cm−1 that was sensitive to 16O/18O labeling [34]. Both of these complexes were shown to oxidize thioanisole, but only one could oxidize hydrocarbons [33,34]. However, the studies of the reactivity of these manganese(III)–hydroperoxo complexes primarily focused on their ability to react directly with various substrates and not on the pathways for O–O activation [33,34].
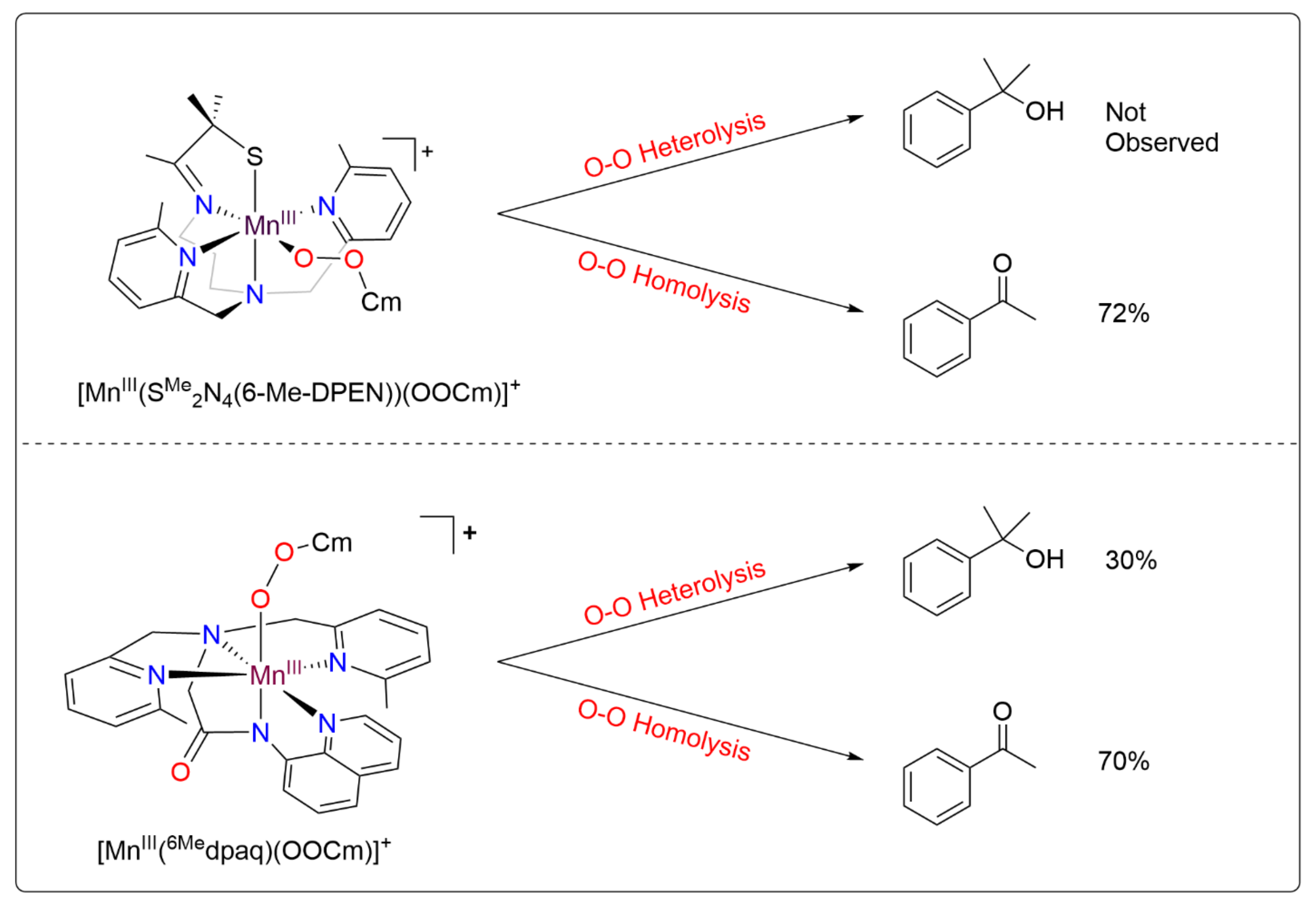
Metal–alkylperoxo complexes, which are typically more stable than their metal–hydroperoxo analogs, have often been used to investigate the factors that affect O–O cleavage pathways [36,37,38,39,40,41]. For manganese, this use of alkylperoxo complexes has been justified by a similar performance by some Mn catalysts using either hydrogen peroxide (H2O2) or cumene hydroperoxide (CmOOH) as an oxidant, suggesting a common O–O activation mechanism between the two [42,43]. Previous studies of MnIII–alkylperoxo complexes supported by pentadentate N4S ligands by Kovacs and co-workers have yielded unique structure-reactivity correlations (the N4S ligand denotes a ligand with four nitrogen donors and one thiolate donor) [44]. For example, complexes with shorter O–O bonds yielded slower thermal decay rates. In addition, the O–O bond lengths were shown to be inversely related to the Mn–N bond lengths of the equatorial 6-Me-pyridyl or quinolyl ligands (see Figure 1 top for a representative structure). Further study of the MnIII–cumylperoxo complexes in this series revealed an exclusive formation of acetophenone as the organic product, which suggests O–O homolysis (as a heterolytic cleavage would produce 2-phenyl-2-propanol; see Figure 1) [44,45]. Taken together, the observations for this series of complexes establish that the local geometry around MnIIIalkyl–peroxo centers controls the rate of decay for O–O homolysis. However, these findings stand in contrast to the proposed heterolytic mechanism for the MnIII–hydroperoxo intermediates formed in the reaction with H2O2. Recently, Kovacs and co-workers expanded this series of complexes by introducing an alkoxide-containing derivative [46], [MnIII(OOtBu)(N4O)]+ (the N4O ligand denotes a ligand with four nitrogen donors and one alkoxide donor). The markedly greater thermal stability of this complex further underscored the relationship between the primary coordination sphere and thermal decay rates.
In our effort to improve our understanding of the role of the ligand environment in controlling the properties of MnIII–alkylperoxo complexes, we produced a series of MnIII–OOR complexes supported by amide-containing N5 ligands (see Figure 1 for a representative example) [47]. The initial complexes in this series were only thermally stable at lower temperatures (−15 °C), and a large excess of tBuOOH was required to form the MnIII–alkylperoxo complexes in high yield [47]. Seeking to improve the thermal stability of these complexes, we followed the work of Kovacs and co-workers and introduced 6-Me-pyridyl donors that enforced longer equatorial Mn–N bonds [44]. Two new complexes were generated using these ligand derivatives, and the increased thermal stability of these complexes at 25 °C allowed us to obtain the X-ray crystallographic structure of [MnIII(OOCm)(6Medpaq)](OTf) [48]. In contrast to that observed for the N4S complexes, these complexes had high thermal stability but showed direct reactivity with triphenylphosphine to yield triphenylphosphine oxide and MnII products [48].
Further investigation into the geometric and electronic differences of these MnIII–OOR complexes, using density functional theory (DFT) compared with the experimental results, showed that the [MnIII(OOtBu)(N4S)]+ complexes decay via a two-state mechanism for the O–O homolysis that is favored by the presence of π-acidic sulfur [49]. This two-state mechanism involves a crossing from the quintet spin state to the triplet spin state as the bond cleavage progresses [49]. In contrast, the [MnIII(OOtBu)(6Medpaq)]+ complexes follow a single-state reaction pathway for homolysis, remaining in the quintet spin state [49]. The lack of a lower-lying triplet pathway for O–O homolysis for [MnIII(OOtBu)(6Medpaq)]+ was consistent with the high thermal stability of this complex. Experimental analysis of the thermal decay of [MnIII(OOCm)(6Medpaq)]+ in several solvents revealed acetophenone as a major product, supporting O–O homolysis as the dominant decay pathway (70% in benzonitrile) [48]. However, 2-phenyl-2-propanol was also observed as a decay product across all the solvents (30% in benzonitrile), showing that O–O heterolysis also occurs (Figure 1) [48]. This result was of particular interest because it differs from the exclusive decay of the analogous [MnIII(OOCm)(N4S)]+ complex by O–O homolysis and shows that changes to the ligand sphere can be used to favor O–O heterolysis [44,45].
On the basis of these results, we are interested in further probing the effect of the ligand sphere on the reactivity and decay pathways of MnIII–alkylperoxo complexes. Previous modifications of the ligand sphere of MnIII–OOR complexes had primarily been made to coordination sites cis to the alkylperoxo unit [48]. However, the site trans to the alkylperoxo unit is often very important when it comes to O–O cleavage (i.e., the so-called “push” effect) [50]. We, therefore, sought to develop a MnIII–alkylperoxo complex that would perturb the electronic properties trans to the alkylperoxo unit in order to further probe the effect of ligand sphere modifications on reactivity and decay pathways.
Herein, we report MnII, MnIII–hydroxo, and MnIII–alkylperoxo complexes, supported by a new 6Medpaq5NO2 ligand (2-(bis((6-methylpyridin-2-yl)methyl)amino)-N-(5-nitroquinolin-8-yl)acetamide; see Figure 2). This new ligand incorporates a strongly electron-withdrawing nitro group at the 5 position of the quinoline, para to its connection with the axially bound amide group. We use a combination of experimental methods and DFT computations to probe the effects of the electronic changes in the ligand sphere trans to the hydroxo and alkylperoxo units on thermal stability and reactivity. The [MnIII(OH)(6Medpaq5NO2)](OTf) (2) complex reacts stoichiometrically with either tBuOOH or CmOOH in CH3CN to form the [MnIII(OOtBu)(6Medpaq5NO2)]+ (3a) or [MnIII(OOCm)(6Medpaq5NO2)]+ (3b) complexes, respectively. These MnIII–alkylperoxo complexes are stable in solution at room temperature with half-lives of ca. 4 days. The MnIII–alkylperoxo complexes follow the previously established trend for dpaq derivatives of high thermal stability while showing evidence for a direct reaction with phosphines in kinetic studies. These results further illuminate the key extent of control that the ligand sphere has over substrate reactivity and O–O cleavage pathways of MnIII–alkyperoxo adducts.
2. Results
2.1. Formation and Characterization of MnII(OTf)(6Medpaq5NO2) (1)
Complex 1 was prepared by the metallation of H6Medpaq5NO2 with MnII(OTf)2 in dichloromethane. The X-ray crystal structure of 1 reveals a monomeric, six-coordinate MnII center coordinated by the pentadentate 6Medpaq5NO2 ligand and a triflate anion (Figure 3). The overall structure is similar to that of the previously reported [MnII(OH2)(6Medpaq)](OTf) complex [48], with the exception that a triflate ion completes that coordination sphere rather than a water molecule. When compared with [MnII(OH2)(6Medpaq)](OTf), the Mn–O1 bond of 1 is notably longer (2.108(3) vs. 2.169(7) Å, respectively; see Table 1), which likely reflects the weaker donating properties of the triflate anion. The Mn–N1 through Mn–N4 distances of 1 are all very similar to those of [MnII(OH2)(6Medpaq)](OTf), indicating that the electron-withdrawing property of the nitro group has little effect on the MnII–N distances. In particular, the Mn–N(amido) distance (Mn–N2) of 1 is identical, within error, to that of [MnII(OH2)(6Medpaq)](OTf). The most notable Mn–N bond difference in 1 is the significant contraction of the Mn–N5 bond, as compared to that of [MnII(H2O)(6Medpaq)](OTf) (2.357(8) vs. 2.417(3) Å, respectively). This change brings the Mn–N4 and Mn–N5 bonds in 1 much closer in length to one another.
The frozen-solution EPR spectrum of 1 at 9 K shows an intense six-line signal near g ≈ 2, consistent with a mononuclear S = 5/2 MnII species (Supplementary Materials Figure S4). In addition, an Evans NMR experiment reveals a magnetic moment of 5.7 µB (Supplementary Materials Figure S5), very close to that expected for a high-spin MnII center (5.9 µB). Thus, the EPR and solution magnetic moment confirm that 1 remains mononuclear in solution.
2.2. Formation and Characterization of [MnIII(OH)(6Medpaq5NO2)](OTf) (2)
Complex 1 reacts rapidly with 0.5 equiv. PhIO, resulting in the formation of a golden amber-colored solution with a single absorption shoulder centered at ca. 550 nm (ε = 226 M−1 cm−1; see Supplementary Materials Figure S6). We refer to this new species as 2. Similar spectral changes were observed previously for the reaction of [MnII(OH2)(6Medpaq)](OTf) with PhIO. The magnetic moment of 2 (4.8 µB) is consistent with a mononuclear S = 2 MnIII center (4.9 µB) (Supplementary Materials Figure S7).
X-ray diffraction studies of single crystals obtained from 2 establish this species as the mononuclear MnIII-hydroxo complex [MnIII(OH)(6Medpaq5NO2)](OTf). In this complex, a six-coordinate MnIII center is in a pseudo-octahedral geometry with the hydroxo ligand trans to the amide functional group (N2) (Figure 4). This coordination matches with that previously reported for the MnIII–hydroxo complexes of dpaq and its derivatives (Table 2). The Mn–O1 bond distance of 1.826(4) Å is just slightly elongated from that observed in [MnIII(OH)(6Medpaq)](OTf) and [MnIII(OH)(dpaq)](OTf) (1.806(6) and 1.806(13) Å, respectively) and is within the range of the Mn–OH bond lengths reported for other MnIII–hydroxo complexes (1.81–1.86 Å). Further bond comparisons with [MnIII(OH)(6Medpaq)](OTf) reveal that the addition of the nitro group has perturbed some of the Mn–N bond lengths more than others. Specifically, Mn–N1 and Mn–N2, involving the quinolyl and amide groups, respectively, remain largely unchanged within error. In contrast, Mn–N3 of 2 has contracted by ca. 0.02 Å relative to [MnIII(OH)(6Medpaq)](OTf), while Mn–N4 has elongated by 0.075 Å and Mn–N5 has contracted by 0.039 Å.
We used 1H NMR spectroscopy to gain further insight into the solution structure of 2 in MeCN. The high-spin MnIII center in 2 caused protons near the metal ion to show broadened and shifted 1H NMR resonances (Figure 5). On the basis of a detailed analysis of the 1H NMR spectrum of the related [MnIII(OH)(dpaq)]+ complex [52], we provide tentative assignments for some of the resonances of 2 in Table 2. The 1H NMR spectrum of 2 in CD3CN shows nine broad peaks (Figure 5) spanning the chemical shift range of 67 to −61 ppm (Table 3). There is a cluster of four downfield peaks from 67 to 45 ppm, two peaks near 10 ppm, and three upfield peaks at −16.8 (shoulder), −19.4, and −60.7 ppm. The spectrum bears a strong resemblance to that of [MnIII(OH)(6Medpaq)](OTf), which is included in Figure 5 (bottom) for comparison. Specifically, the 1H NMR spectrum of [MnIII(OH)(6Medpaq)](OTf) shows three peaks in the downfield region of 67 to 45 ppm, and the chemical shifts for these peaks are nearly identical to three of the downfield peaks of 2 (Table S1). In addition, the upfield region of the 1H NMR spectrum of [MnIII(OH)(6Medpaq)](OTf) shows a broad peak at −9.6 ppm, which corresponds well to the broad peak of 2 at −17.1 ppm, and the spectrum of [MnIII(OH)(6Medpaq)](OTf) has two sharper peaks at −19.3 and −61.6 ppm, which are reproduced in the 1H NMR spectrum of 2 at −19.4 and −60.7 ppm. The [MnIII(OH)(6Medpaq)](OTf) complex has an additional upfield peak at −45.0 ppm that is clearly absent in the 1H NMR spectrum of 2 (Figure 5, top and bottom). We attribute this peak in [MnIII(OH)(6Medpaq)](OTf) to the 5-quinolyl proton, as this proton has been substituted by the nitro group in 2. In support, the 1H NMR spectrum of [MnIII(OH)(dpaq)](OTf) has a proton resonance at −33.7 ppm that was lacking in the spectrum of the nitro-substituted [MnIII(OH)(dpaq5NO2)](OTf) complex [52].
2.3. Formation of [MnIII(OOtBu)(6Medpaq5NO2)]+ (3a) and [MnIII(OOCm)(6Medpaq5NO2)]+ (3b)
The addition of 1.5 equiv. of tBuOOH to the MnII complex 1 in CH3CN at 298 K results in the formation of a green solution (3a) with a prominent electronic absorption feature at 665 nm (ε = 231 M−1 cm−1) (Supplementary Materials Figure S8). (The extinction coefficient was determined by assuming a full conversion took place under these conditions.) A similar reaction occurs upon the addition of CmOOH to 1 in CH3CN, with the feature occurring as a shoulder at ca. 650 nm (ε = 170 M−1 cm−1), resulting in an amber-green solution (3b) (Supplementary Materials Figure S8).
In addition to this method of formation, both 3a and 3b can be formed by first generating 2 from the reaction of 1 and 0.5 equiv. PhIO, followed by the addition of 1.0 equiv. of the alkyl hydrogen peroxide (Figure 6). In this method, the formation of each complex is accompanied by isosbestic behavior, indicating the lack of any accumulating intermediate. Similar behavior was observed in the formation of [MnIII(OOtBu)(6Medpaq)]+ from the reaction of the corresponding MnIII–hydroxo complex with tBuOOH and CmOOH [48]. The solution-phase magnetic moments of 3a and 3b (4.5 and 4.9 µB, respectively) were determined using the Evans NMR method (Supplementary Materials Figure S9). These values are consistent with the S = 2 MnIII centers, confirming both that these complexes are mononuclear in solution and that the reaction of 2 with alkylhydroperoxides does not result in the oxidation of the MnIII center. ESI-MS data, collected for 3a and 3b, reveal prominent peaks with a m/z of 599.18 and 661.20, respectively, which are consistent with the expected m/z of [MnIII(OOtBu)(6Medpaq5NO2)]+ (calculated m/z of 599.18) and [MnIII(OOCm)(6Medpaq5NO2)]+ (calculated m/z of 661.20; see Supplementary Materials Figures S12 and S13).
2.4. Spectroscopic Characterization of 3a and 3b
Because we were unable to obtain crystals suitable for X-ray diffraction for either MnIII–alkylperoxo complex, we used 1H NMR spectroscopy to aid in characterizing the structures of these complexes. The 1H NMR spectra of 3a and 3b show a set of three downfield peaks in the range of 67 to 45 ppm and two upfield peaks at −21 and −59 ppm (Figure 7 and Table 3). The broad peak at 47.0 ppm for 3b has a shoulder at 47.6 ppm, suggesting the presence of two closely spaced resonances. (The 1H NMR spectrum of 3b also shows a set of sharp peaks in the 20—0 ppm region, which we attribute to a small amount of free ligand. The previously reported [MnIII(OH)(dpaq5NO2)]+ complex showed similar peaks in the 1H NMR spectrum in the presence of D2O. We speculate that water, formed as a product of the reaction of [MnIII(OH)(dpaq5NO2)]+ and CmOOH, causes the appearance of a small amount of free ligand in this case.) Although the 1H NMR spectra of both 3a and 3b have fewer peaks than 2, the peaks that are observed are at positions similar to the resonances of 2. Specifically, the upfield peaks of 3a and 3b (at ca. −21 and −59 ppm) have chemical shifts nearly identical to the upfield peaks of 2 (ca. −19 and −60 ppm). In addition, the three downfield peaks of 3a and 3b at ca. 67, 48, and 46 ppm are at similar positions as the corresponding resonances in 2 (67, 55, and 46 ppm). The similarities in the 1H NMR spectra of 3a and 3b with that of 2 are consistent with the 6MedpaqNO2 ligand having a similar coordination mode for the MnIII–alkylperoxo and MnIII–hydroxo complexes.
The 1H NMR spectra of 3a and 3b also bear a strong resemblance to the corresponding spectra of the MnIII–alkylperoxo complexes [MnIII(OOtBu)(6Medpaq)]+ and [MnIII(OOCm)(6Medpaq)]+ (Supplementary Materials Figure S14). The 1H NMR spectra for the [MnIII(OOtBu)(6Medpaq)]+ and [MnIII(OOCm)(6Medpaq)]+ complexes showed three downfield peaks from 67 to 45 ppm and two upfield peaks at −47 and −60 ppm (Supplementary Materials Figure S14). The major difference between these spectra and those of 3a and 3b is the upfield signal at −47 ppm in the spectra of [MnIII(OOtBu)(6Medpaq)]+ and [MnIII(OOCm)(6Medpaq)]+ that is lost in the spectra of 3a and 3b. This difference arises because of the substitution of the 5-quinolyl proton in the former complexes with the nitro group in the latter complexes.
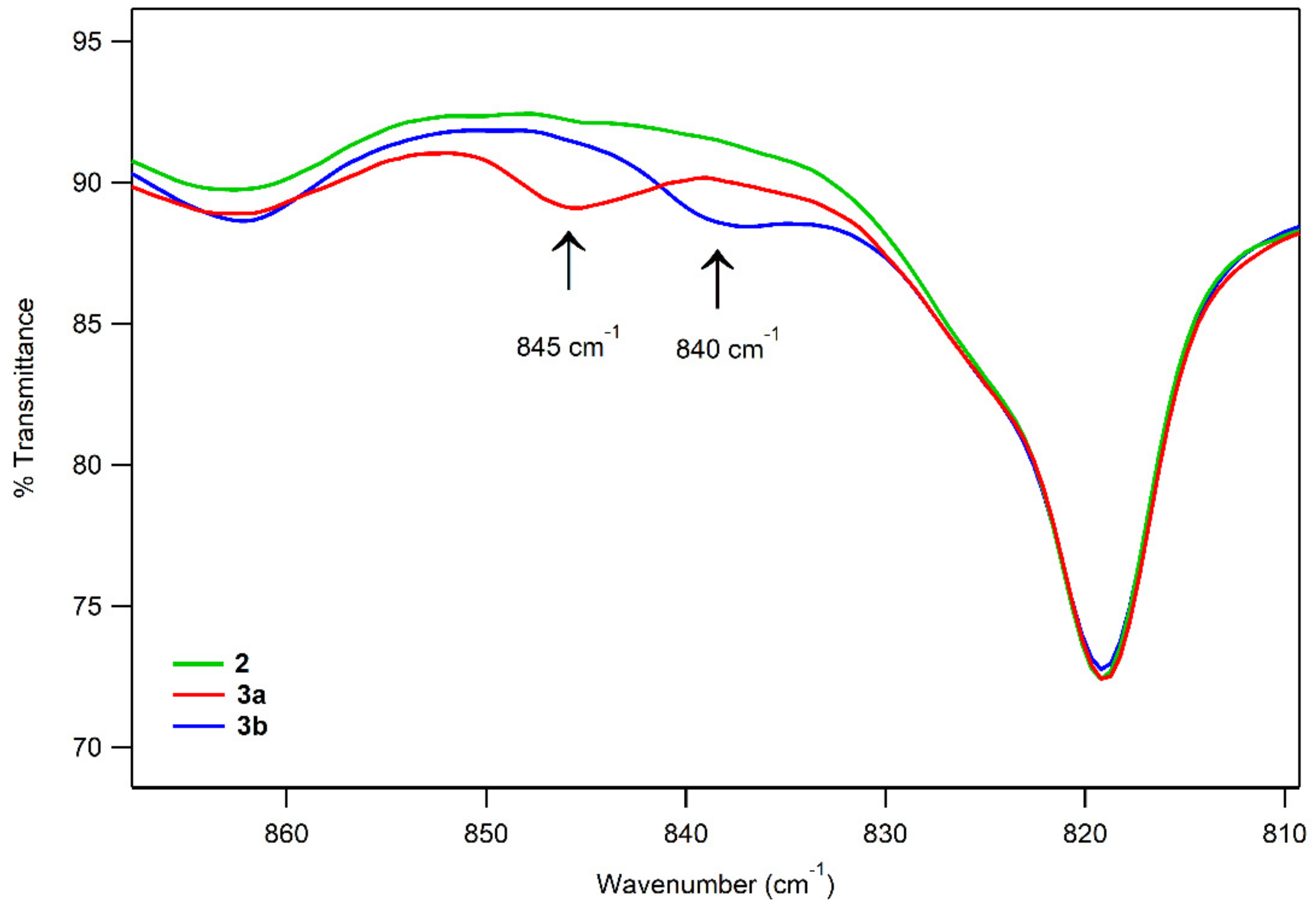
The solution FT-IR spectra of 3a and 3b show features at 845 cm−1 and 840 cm−1, respectively, that are absent in the FT-IR spectrum of 2 (Figure 8). These features have energies similar to those of the O–O vibrations previously reported for MnIII–alkylperoxo complexes (850–895 cm−1) [44,45,47]. The specific O–O vibrational energies for 3a and 3b are ~20–30 cm−1 lower than those of the related complexes [MnIII(OOtBu)(6Medpaq)]+ and [MnIII(OOCm)(6Medpaq)]+ (861 and 875 cm−1) [48]. The vibrational energies are closest to that reported for an alkoxide-ligated MnIII–OOtBu complex (850 cm−1) [46]. As emphasized in that study, coupling of the O–O stretch with other vibrational modes can make it difficult to relate O–O vibrational energies to either the peroxo bond length or thermal stability. Indeed, in the cases of 3a and 3b, we observed a relatively low energy O–O vibration for complexes with high thermal stability.
The EPR analyses of the frozen solutions of 3a and 3b in 1:1 CH3CN:toluene, collected at 10 K, showed only weak signals in the perpendicular mode (near g = 2) and no signals in the parallel mode (Supplementary Materials Figure S15). This is consistent with the lack of X-band signals for many MnIII complexes due to the moderate-to-large zero-field splitting relative to the microwave energy. The weak signals near g = 2 are attributed to a minority of MnII species in the solution. In support, the spin quantification [53] of the frozen solutions of 3a and 3b in CH3CN showed that the g = 2 signals account for only ~15 and 10%, respectively, of the Mn in solution (Supplementary Materials Figures S15 and S16).
2.5. DFT Structures of 3a and 3b
We used DFT computations to predict the structures of 3a and 3b The optimized structures are show in Figure 9 and selected metric parameters are in Table 4. In the DFT-optimized structures of 3a and 3b, the tert-butylperoxo and cumylperoxo ligands are bound trans to the amide nitrogen (N2) at N2–Mn–O1 angles of 176.4° and 173.9°, respectively. The Mn–O1 bond lengths for both 3a and 3b (1.83 Å) are the same as that of the MnIII–hydroxo complex (1.826(4) Å). The average of the Mn–N(pyridine) distances (Mn–NAve:4,5) of 3b (2.33 Å) is essentially the same as that of the previously reported [MnIII(OOCm)(6Medpaq)]+ complex (2.34 Å), while that of 3a is slightly longer (2.35 Å). The oxygen–oxygen bond (O1–O2) for both complexes is 1.45 Å, which is consistent with that determined from the X-ray crystal structure for [MnIII(OOCm)(6Medpaq)]+ (1.47 Å), and is only slight shorter than the range of values determined for the [MnIII(OOCm)(N4S)]+ complexes (1.457(5) to 1.51(2) Å). For the DFT structure, projections of the peroxo O1–O2 bond onto the equatorial plane show the bond bisecting the N1–Mn–N4 bond angle, with the 3b O1–O2 bond being closer to the Mn–N4 bond than in the 3a complex. The Mn–O1–O2 bond angle of 3a (107.3°) is smaller than that of both the [MnIII(OOCm)(6Medpaq)]+ (110.4(2)°) complex and 3b (114.2°).
2.6. Thermal Decay Pathways of 3a and 3b
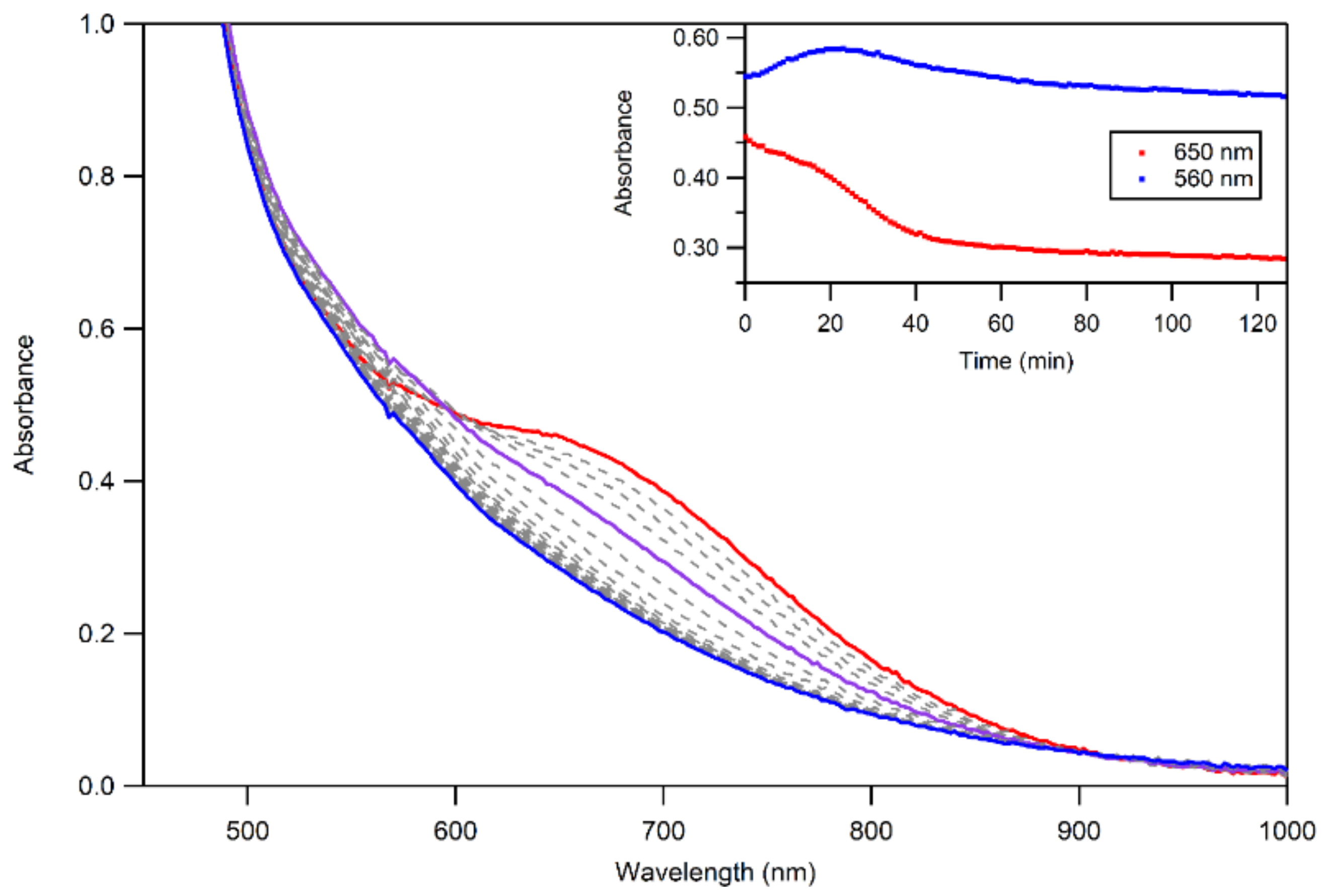
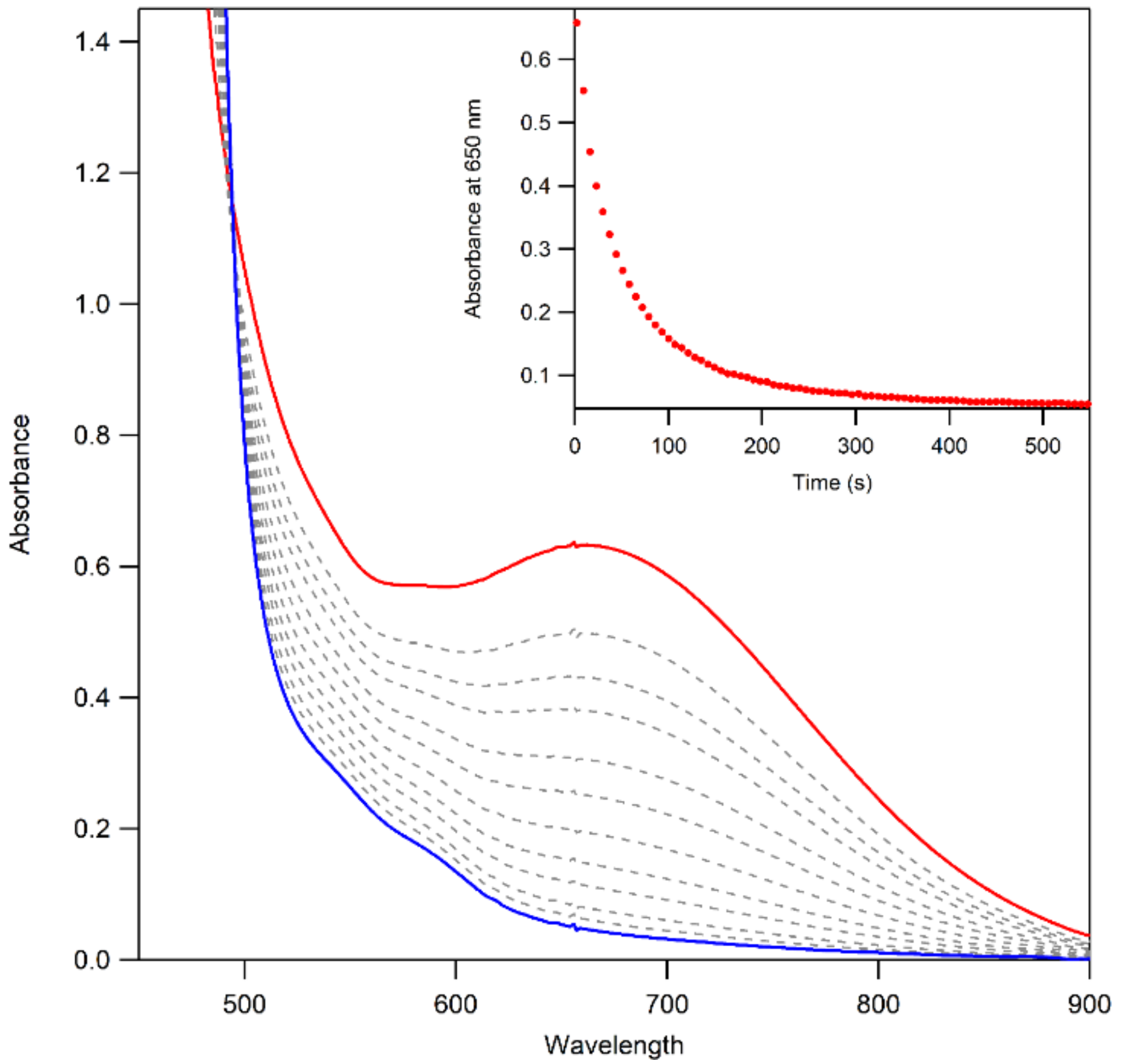
The thermal decay of 3a and 3b in CH3CN resulted in the loss of intensity of the 650 nm feature associated with the MnIII–alkylperoxo complex (Figure 10). The half-lives of the 1.25 mM solutions of 3a and 3b are ca. 4 and 3 days, respectively, in CH3CN at 298 K. Given this robust thermal stability, our analyses of the decay products of 3a and 3b were facilitated by experiments at 323 K. Both 3a and 3b decay to give a final spectrum that shows a steady rise from ~900–400 nm with no clear absorption maxima. The final spectra resemble neither that of 2 nor that expected for the MnII products. This final decay product is thus different from that observed for the [MnIII(OOR)(6Medpaq)]+ complexes. In that case, the MnIII–alkylperoxo complexes underwent thermal decay to re-form the MnIII–hydroxo complexes. A close inspection of the thermal decay profiles of 3a and 3b shows evidence for the formation of the MnIII–hydroxo complex 2, but, in this case, 2 is an intermediate. For example, we observed a rise in absorption at 560 nm (characteristic of 2) for the first ~30 min of decay at 323 K (Figure 10, inset). However, 2 is not stable under these conditions and decays after ~40 min. Further support for the presence of the MnIII–hydroxo complex 2 as an intermediate in this process is provided by the isolation of X-ray diffraction-quality single crystals of 2 from a solution of 3b that was stored at 258 K for over 8 weeks. Due to the complexity of the thermal decay process, we were not able to fit the thermal decay profiles into a standard kinetic model.
We also examined the decay products of 3a and 3b by EPR spectroscopy. A frozen-solution EPR sample of the final thermal decay products of 3 mM 3b was prepared by allowing the complex to decay at 343 K in CH3CN, then drying and redissolving in 1:1 CH3CN:toluene. The EPR spectra, collected at 7 K, reveal an intense signal at g = 2.01, characteristic of a mononuclear MnII species (Supplementary Materials Figure S18). A quantitative analysis of the EPR signal intensity showed that the g = 2.01 signal from a MnII species accounts for ca. 70% of the Mn in solution. Combined with the findings from UV–Vis spectroscopy, this result suggests that there is a partial conversion of 3b to MnII products and a partial generation of another product that gives rise to the broad absorption signal (Figure 10). A similar quantitative EPR analysis of a frozen solution of decay products from the thermal decay of 3a revealed that the EPR-active MnII species accounted for ca. 34% of the Mn in solution (Supplementary Materials Figure S19).
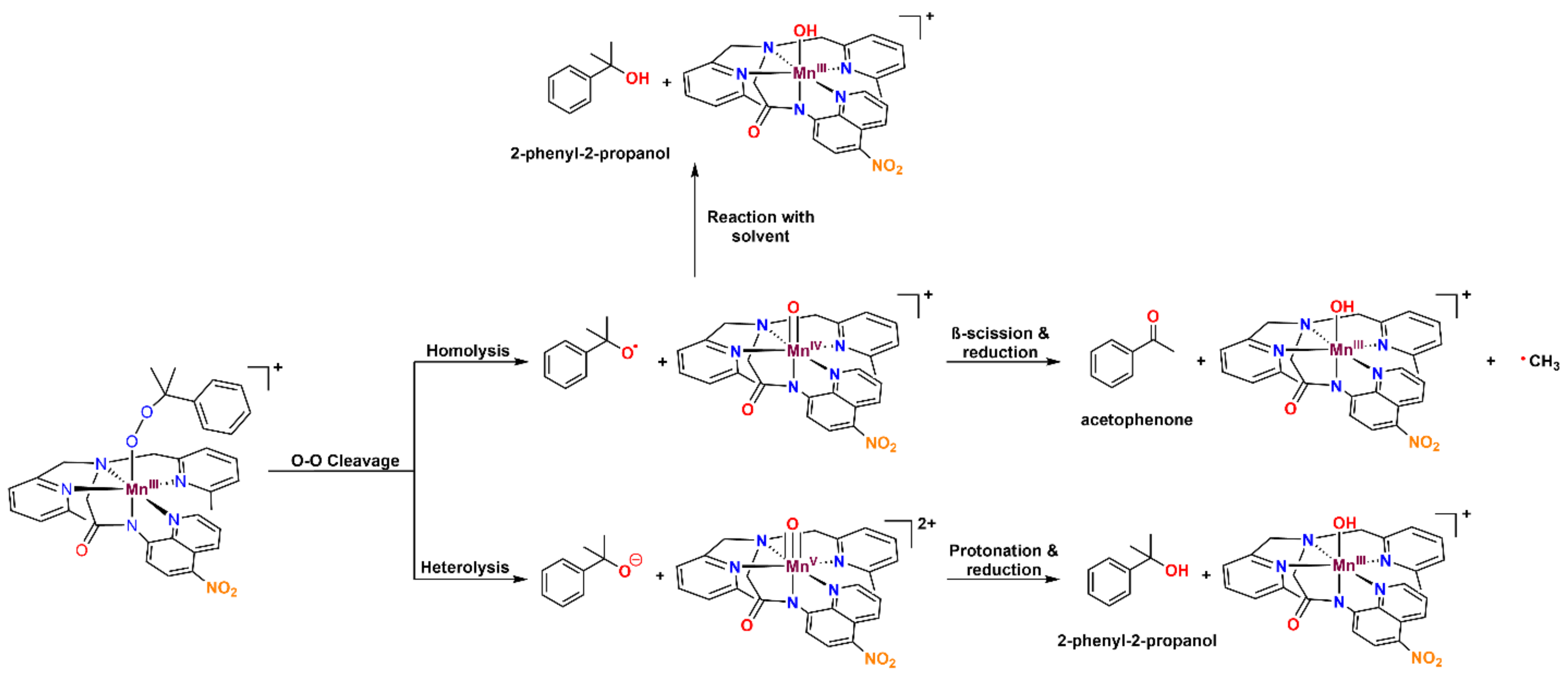
The organic products formed by the decay of metal–cumylperoxo complexes are often used to probe the relative ratios of O–O homolysis and heterolysis. Homolysis yields the cumyloxyl radical, which decays by β-scission to give acetophenone (Figure 11). Heterolysis gives cumyl oxyanion, which can be protonated to form 2-phenyl-2-propanol (cumyl alcohol). The product analysis, following the thermal decay of 3b in CH3CN at 298 K (Figure 11 and Table 5), revealed 49.7 ± 3.5% 2-phenyl-2-propanol and 35.2 ± 2.8% acetophenone relative to the initial concentration of 3b. The recovery of 85% of the cumylperoxo decay products suggests that O–O cleavage is the dominant decay pathway. The high-valent Mn–oxo species generated by the O–O cleavage is likely quite reactive and rapidly decays to give the mixture of Mn products. The organic products of the thermal decay of 3a were not quantified due to the volatility of acetone, one of the potential products, which renders quantification unreliable.
The ~50:35 2-phenyl-2-propanol/acetophenone ratio for 3b marks a ~10% increase in the product ratio compared to [MnIII(OOCm)(6Medpaq)]+ (61:26 2-phenyl-2-propanol: acetophenone; see Table 5). However, the relationship of metal–cumylperoxo decay products to O–O homolysis versus heterolysis in CH3CN is complicated by the reaction of the cumyloxyl radical with solvent. Specifically, the β-scission of the cumyloxyl radical must compete with the cumyloxyl radical abstracting a hydrogen atom from solvent. This latter reaction leads to the formation of 2-phenyl-2-propanol from O–O homolysis (Figure 11). Because the reaction between the cumyloxyl radical and solvent is much slower in a deuterated solvent, we analyzed the decay of 3b in CD3CN. The decay of 3b at 298 K in CD3CN resulted in 24.0 ± 1.6% 2-phenyl-2-propanol and 71.5 ± 4.3% acetophenone relative to the initial 3b concentration (Table 5). This shift in product distribution from 50:35 2-phenyl-2-propanol/acetophenone in CH3CN to 24:72 2-phenyl-2-propanol/acetophenone in CD3CN suggests that a substantial fraction of the 2-phenyl-2-propanol produced by the decay of 3b in CH3CN was generated by the reaction of the cumuloxyl radical with solvent. However, we still observed a different 2-phenyl-2-propanol/acetophenone ratio for 3b than for the previously reported [MnIII(OOCm)(6Medpaq)]+ (71:24 and 40:50, respectively). Thus, the inclusion of the nitro substituent in 3b appears to shift the O–O homolysis/heterolysis ratio. Because the nitro group weakens the donation from the amide ligand trans to the alkylperoxo group, this result illustrates that subtle changes in the donor properties of a trans ligand can have a noticeable effect on the relative energies of the O–O cleavage pathways.
2.7. Oxidation of TEMPOH by the MnII–Hydroxo Complex 2
The addition of 10–50 equiv. TEMPOH to an anerobic solution of 2 (3 mM in CH3CN) at 238 K resulted in the rapid loss of intensity of the broad feature ca. 550 nm (Figure 12, right). The resulting final spectrum resembles that of 1, but the absorption signal is too intense to be from 1 alone. The same final spectrum is observed no matter how much TEMPOH is added. Analysis of the product solution via 1H NMR shows no signals associated with 2, suggesting that the products are a mixture of 1 and other MnII complexes (Supplementary Materials Figure S20). The decay of 2 upon the addition of TEMPOH could be well-fitted using a first-order model (Figure 12, right inset), yielding a pseudo-first-order rate constant (kobs). A linear fit of kobs versus TEMPOH concentration (Figure 12, left) yielded a second-order rate constant for the reaction of TEMPOH with 2 of 6.1 M−1s−1 at 298 K in CH3CN. This rate fits well with those of similar MnIII–hydroxo complexes, including [MnIII(OH)(dpaq5NO2)]+ (7 M−1s−1) [52] and [MnIII(OH)(6Medpaq)]+ (3.4 M−1s−1) [54].
2.8. Oxidation of Triphenylphosphine by MnIII–Alkylperoxo Complexes
The addition of 100 equiv. PPh3 to a solution of 3a (1.00 mM in CH3CN) at 298 K resulted in the loss of intensity of the electronic absorption feature at 660 nm over the course of ca. 6 min (Figure 13). Further analysis of the product solution via 1H NMR showed only a trace amount of signals consistent with those of 2 or 3a, indicating that the reaction yields a mixture of 1 and other MnII products. The decay of the 660 nm absorption signal was unable to be fit with a first-order model, and, as such, we were unable to obtain a pseudo-first-order rate constant or second-order rate constant for the reaction (see Supplementary Materials for more details regarding the complexity of this reaction). While we were unable to obtain a rate constant for the reaction of 3a with PPh3, the t1/2 of the reaction (~40 s) was approximately 16 times faster than that reported for the reaction of [MnIII(OOtBu)(6Medpaq)]+ with PPh3 (~650 s). Thus, we observe a dramatic rate enhancement in the reaction of 3a with PPh3, indicating that 3a is a significantly more electrophilic oxidant. The increased electrophilicity can be rationalized by the nitro substituent in 3a, which can mitigate the charge donation from the amide to the MnIII–OOR unit. A 31P NMR analysis of the organic products revealed the formation of Ph3PO (Supplementary Materials Figure S22). Quantification of the organic products of the reaction of 3b with PPh3 at 298 K in CH3CN via GC-MS revealed a distribution of 67.4 ± 3.7% 2-phenyl-2-propanol and 15.1 ± 2.4% acetophenone based on the initial concentration of the MnIII–alkylperoxo complex. The reaction between [MnIII(OOCm)(6Medpaq)]+ and PPh3 was previously characterized in the same fashion and was found to yield a distribution of 88.5 ± 0.3% 2-phenyl-2-propanol to 1.5 ± 0.3% acetophenone [48]. The increase in acetophenone in 3b compared to the previous complex is consistent with the increased portion of homolytic cleavage observed in the thermal decay.
3. Discussion
In order to further probe the effect of the ligand sphere on the reactivity and decay pathways of MnIII–alkylperoxo complexes, we developed a new ligand (6Medpaq5NO2) that perturbs the electronic properties trans to the alkylperoxo unit. This perturbation only minimally affects the thermal stability of the MnIII–alkylperoxo complex, as complexes 3a and 3b have half-lives in MeCN at room temperature of ~4 days. While MnIII–alkylperoxo complexes that are stable at room temperature are still relatively rare, these complexes all contain two common features [44,46,48]. First, the complexes contain at least two N-donor ligands with elongated Mn–N bonds cis to the alkylperoxo unit. Second, the complexes lack strong π-acidic ligands cis to the alkylperoxo ligand. As we have discussed in a recent computational study, the lack of a π-acidic ligand increases the MnIII quintet-triplet spin gap, which disfavors a two-state mechanism predicted to promote O–O cleavage for both the MnIII–alkylperoxo and MnIII–hydroperoxo complexes [49].
An analysis of the organic decay products of the MnIII–cumylperoxo complex 3b examined in this work illustrates that trans effects also modulate the O–O cleavage pathways of MnIII–alkylperoxo complexes. Specifically, the weaker trans donor in 3b leads to an increased production of homolysis reaction products compared to the [MnIII(OOCm)(6Medpaq)]+ complex (Table 5). While the basis for this change in reaction products is unclear at present, we note that a small change in the relative energies of the reaction barriers for homolysis and heterolysis would account for the observed change in the reaction products.
In contrast to the moderate effect on the O–O cleavage pathways, the new complexes 3a and 3b show much faster rates of reaction with the substrate triphenylphosphine. In fact, the rate of reaction with 3a is sixteen-fold faster than that of the previous [MnIII(OOtBu)(6Medpaq)]+ complex. To date, there are few examples of MnIII–alkylperoxo complexes that react directly with substrates, so it is difficult to make general statements about the factors governing reactivity. However, the increase in the rate of triphenylphosphine oxidation by 3a and 3b suggests that these complexes are more electrophilic than the previously reported [MnIII(OOR)(6Medpaq)]+ complexes. We propose that the weaker donation from the trans amide ligand causes this increase in the electrophilicity of the MnIII–alkylperoxo unit. For comparison, we note that MnIII–hydroperoxo complexes have been shown to attack thioanisole and its derivatives, which is also a showcase of electrophilic reactivity [33,34].
When taken as a whole, data for this new series of complexes suggest that it is possible to make alterations in the ligand sphere of MnIII–alkylperoxo complexes that allow for the separate tuning of reactivity and stability. Herein, a change to the ligand sphere allowed for (1) a marked increase in reactivity towards an organic substrate, (2) a modest change in the distribution of O–O cleavage products from homolytic and heterolytic pathways, and (3) little change in thermal stability. Such an ability to tune these parameters separately is promising for future study, pointing to the possibility of a complex designed for increased reactivity and O–O heterolytic cleavage while maintaining stability under mild conditions.
4. Materials and Methods
4.1. General Methods and Instrumentation
All chemicals were used as obtained from commercial sources unless noted otherwise. 2-(bis((6-methylpyridin-2-yl)methyl)amino)-N-(quinoline-8-yl)acetamide (H6Medpaq) was synthesized according to a reported procedure [48]. The concentration of tert-butyl hydroperoxide (tBuOOH) in the decane stock solution was found to be 4.3 M by iodometric titration. The experiments were performed under standard atmospheric conditions unless otherwise noted.
The electronic absorption experiments were performed using either a Varian Cary 50 Bio UV–Visible spectrophotometer (Varian, Cranford, NJ, USA) or an Agilent 8453 UV–Visible spectroscopy system (Agilent, Santa Clara, CA, USA). The reaction temperature was controlled using either a Unisoku cryostat (Unisoku, Hirakata, Japan) and stirrer or a Quantum Northwest TC 1 temperature controller and stirrer (Quantum Northwest, Liberty Lake, WA, USA). Electrospray ionization mass spectrometry (ESI-MS) experiments were performed using an LCT Premier MircoMass electrospray time-of-flight instrument (MircoMass, Cary, NC, USA). The X-band EPR experiments were conducted using a Bruker EMXplus with Oxford ESR900 continuous-flow liquid helium cryostat and an Oxford ITC503 temperature system (Bruker, Billerica, MA, USA). 1H and 31P NMR spectra were obtained using a Bruker DRX 400 MHz NMR spectrometer (Bruker, Billerica, MA, USA). The hyperfine-shifted 1H NMR spectra were collected within the spectra width of 150 to −100 ppm with 1000 scans to provide a sufficient S/N. Spectra were baseline subtracted with the multipoint fitting procedure using the spline functions in the MestReNova program. GC analysis was performed on the Shimadzu GCMS-QP2010 SE instrument (Shimadzu, Kyoto, Japan). FT-IR spectra were collected using a Shimadzu IRTracer-100 spectrophotometer on a 1 mm ZnSe permanently sealed cell from Buck Scientific (Norwalk, CT, USA).
4.2. Synthesis of (H6Medpaq5NO2)
In a round bottom flask, 2.0011 g (0.004866 mol) (H6Medpaq) was dissolved in 100 mL of H2SO4 and stirred while cooling in an ice bath. A total of 0.5915 g (0.005850 mol) KNO3 was added, and the solution was stirred in an ice bath. After 3 h, the solution was neutralized via the addition of ammonium hydroxide solution until a pH of 7 was reached. The resulting solution was extracted with dichloromethane and water, washed with a sodium carbonate solution, and washed with sodium chloride brine. The solvent was removed under vacuum, and the resulting solid was redissolved into acetonitrile. The solvent was removed, yielding a dry powder, which was characterized by 1H NMR, 13C NMR, and HSQC NMR (Supplementary Materials Figures S1–S3). The final product was obtained as a dark yellow solid with a 96% yield.
4.3. Synthesis of MnII(OTf)(6Medpaq5NO2) (1), [MnIII(OH)(6Medpaq5NO2)]+ (2), and [MnIII(OOR)(6Medpaq5NO2)]+ (3a: R: tBu and 3b: R: Cm)
The reaction of 0.7501 g (0.001648 mol) H6Medpaq5NO2 with 0.789 g (0.001813 mol) MnII(OTf)2∙2CH3CN was performed in 50 mL of dichloromethane under an inert atmosphere in the presence of 0.158 g (0.001648 mol) of NaOtBu as a base. The reaction was stirred for 18 h and yielded a dark orange solution. The solution was passed through a syringe filter, concentrated in vacuo, and layered with diethyl ether. This procedure led to the formation of a yellow-orange precipitate. The solvent was decanted, and the orange was solid dried, redissolved in minimal CH2Cl2, and layered with diethyl ether. The recrystallization procedure was repeated three more times, and an orange solid was obtained with an 89% yield. Elemental analysis: the carbon expected was 47.35%, we found 47.61%; the hydrogen expected was 3.52%, we found 3.63%; the nitrogen expected was 12.74%, we found 12.87%. The powder X-ray diffraction is shown in Supplementary Materials Figure S23 (left).
In a round bottom flask, 0.6013 g (0.0009118 mol) of MnII(OTf)6Medpaq5NO2 was combined with 0.1008 g (0.0004559 mol) of iodosobenzene in 40 mL of CH2Cl2. The solution was stirred at room temperature for 2 h and then layered with diethyl ether. This procedure led to the formation of a dark green-brown precipitate. The solvent was decanted, and the solid was washed with fresh ether. The solid was then dried, redissolved in a mixture of CH2Cl2 and acetonitrile (1:4 v/v), and layered with diethyl ether. This process was repeated one more time, and a green-brown solid was obtained with a 60% yield. The powder X-ray diffraction is shown in Supplementary Materials Figure S23 (right).
The 3a and 3b samples were prepared by dissolving 2 in acetonitrile (typically 1.00–5.00 mM) and reacting with a solution of 1 equivalent of the corresponding alkyl hydrogen peroxide in acetonitrile. The reaction was monitored via UV–Vis spectroscopy (typically at 25–60 °C). The full formation of the MnIII–alkylperoxo complex was determined by monitoring the feature at ca. 650 nm over time for a plateau in absorbance.
4.4. Thermal Decay Reactions of 3a and 3b and Reactivity with PPh3
The sample solutions (1.00–3.00 mM) of 3a and 3b in CH3CN were prepared and transferred to a quartz cuvette. The decay kinetics were monitored on a Varian Cary 50 Bio UV–visible spectrophotometer equipped with a temperature controller and stirrer. Analogous samples were prepared in CH3CN and transferred to quartz cuvettes. The cuvettes were then sealed with a septum and sparged with nitrogen gas before undergoing decay kinetic monitoring.
Samples of 3a and 3b (1.00–3.00 mM) were prepared in CH3CN, transferred into quartz cuvettes, and covered with a rubber septum. A solution of 10–100 equivalents of PPh3 per 50–100 uL was prepared using CH2Cl2. The cuvette was placed in the UV–Vis spectrometer and equilibrated to temperature (25–50 °C) for 5 min before the PPh3 solution was added using a gastight syringe. Analogous experiments were conducted using nitrogen-sparged solutions with a nitrogen headspace in the cuvette.
4.5. Computational Methods
Electronic structure computations were conducted using the ORCA software program (version 5.0.3) [55,56]. Geometry optimizations and frequency calculations used the TPSSh functional [57] with D3 dispersion corrections [58,59] and def2-SVP as the basis set for carbon and hydrogen. The larger basis set def2-TZVP was used for manganese, nitrogen, and oxygen [60,61]. The default integration grids were used with RIJCOSX integral transformation approximations [62]. Auxiliary basis sets were called using the AutoAux command. All open-shell species were considered at the spin-unrestricted level. Frequency calculations were conducted to confirm that all species were optimized to their minima (no imaginary frequencies).
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29081849/s1, Synthesis of H6Medpaq5NO2, Figure S1: 1H NMR spectrum of H6Medpaq5NO2, Figure S2: 13C NMR spectrum of H6Medpaq5NO2, Figure S3: HSQC NMR data for H6Medpaq5NO2, Figure S4: Perpendicular-mode EPR spectrum of 1, Figure S5: Evans NMR spectrum of 1, Figure S6: UV–Vis spectra of 1 with PhIO, Figure S7: Evans NMR spectrum of 2, Figure S8: UV–Vis formation of 3a and 3b, Figure S9: Evans NMR spectra of 3a and 3b, Figure S10: ESI-MS spectrum of 1, Figure S11: ESI-MS spectrum of 2, Figure S12: ESI-MS spectrum of 3a, Figure S13: ESI-MS spectrum of 3b, Figure S14: 1H NMR spectra of 3a, 3b, [MnIII(OOtBu)(6Medpaq)]+, and [MnIII(OOCm)(6Medpaq)]+, Table S1: 1H NMR chemical shifts (ppm) for [MnIII(OH)(6Medpaq)]+, [MnIII(OOtBu)(6Medpaq)]+, and [MnIII(OOCm)(6Medpaq)]+, Figure S15: X-band EPR spectra of 3a and 3b, Figure S16: Perpendicular-mode EPR spectra of 3a, Figure S17: Perpendicular-mode EPR spectra of 3b, Figure S18: Perpendicular-mode EPR spectra of 3b thermal decay products, Figure S19: Perpendicular-mode EPR spectra of 3a thermal decay products, Figure S20: 1H NMR of the products of 2 and TEMPOH, Figure S21: UV–Vis spectra of 2 with PPh3, Reaction of 2 with triphenylphosphine, Figure S22: 31P NMR of the products of 3a and PPh3, X-ray reports for 1 and 2, Table S2: Crystal data and structure refinement for 1 and 2 and powder X-ray diffraction (PXRD) instrumentation and methods, Figure S23: PXRD spectra of 1 and 2, DFT structure coordinates for 3a, DFT structure coordinates for 3b. Refs. [63,64] are cited in Supplementary Materials.
Author Contributions
Conceptualization and Methodology, S.A.B., E.N.G. and T.A.J.; Formal Analysis, S.A.B., E.N.G., Z.A. and M.R.M.; Investigation, S.A.B., E.N.G. and Z.A.; Writing—Original Draft Preparation, S.A.B. and E.N.G.; Writing—Review and Editing, S.A.B. and T.A.J.; Project Administration, T.A.J.; Funding Acquisition, T.A.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the U.S. National Science Foundation (CHE-2154955 to T. A. J.). The calculations were performed at the University of Kansas Center for Research Computing (CRC), including the BigJay Cluster resource, funded through the U.S. NSF Grant MRI-2117449. EPR spectra were acquired on an instrument obtained through the U.S. NSF Grant CHE-0946883. Support for the NMR instrumentation was provided by the NIH Shared Instrumentation Grant #S10OD016360.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The crystallographic data are available through the Cambridge Crystallographic Data Centre (CCDC) at https://www.ccdc.cam.ac.uk/ with structure codes 2339539, and2339541.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Brash, A.R. Lipoxygenases: Occurrence, Functions, Catalysis, and Acquisition of Substrate. J. Biol. Chem. 1999, 274, 23679–23682. [Google Scholar] [PubMed]
- Oliw, E.H.; Jernerén, F.; Hoffmann, I.; Sahlin, M.; Garscha, U. Manganese lipoxygenase oxidizes bis-allylic hydroperoxides and octadecenoic acids by different mechanisms. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2011, 1811, 138–147. [Google Scholar]
- Su, C.; Oliw, E.H. Manganese Lipoxygenase: Purification and characterization. J. Biol. Chem. 1998, 273, 13072–13079. [Google Scholar] [PubMed]
- Su, C.; Sahlin, M.; Oliw, E.H. Kinetics of Manganese Lipoxygenase with a Catalytic Mononuclear Redox Center. J. Biol. Chem. 2000, 275, 18830–18835. [Google Scholar] [PubMed]
- Schramm, V.L.; Wedler, F.C. (Eds.) Manganese in Metabolism and Enzyme Function; Academic Press: Orlando, FL, USA, 1986. [Google Scholar]
- Cotruvo, J.A., Jr.; Stubbe, J. An Active Dimanganese(III)–Tyrosyl Radical Cofactor in Escherichia coli Class Ib Ribonucleotide Reductase. Biochemistry 2010, 49, 1297–1309. [Google Scholar] [PubMed]
- Dismukes, G.C. Manganese Enzymes with Binuclear Active Sites. Chem. Rev. 1996, 96, 2909–2926. [Google Scholar] [PubMed]
- Fraústo da Silva, J.J.R.; Williams, R.J.P. The Biological Chemistry of the Elements, 2nd ed.; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Requena, L.; Bornemann, S. Barley (Hordeum vulgare) oxalate oxidase is a manganese-containing enzyme. Biochem. J. 1999, 343, 185–190. [Google Scholar] [PubMed]
- Vinyard, D.J.; Ananyev, G.M.; Charles Dismukes, G. Photosystem II: The Reaction Center of Oxygenic Photosynthesis. Annu. Rev. Biochem. 2013, 82, 577–606. [Google Scholar] [PubMed]
- Wedler, F.C.; Denman, R.B.; Roby, W.G. Glutamine synthetase from ovine brain is a manganese(II) enzyme. Biochemistry 1982, 21, 6389–6396. [Google Scholar]
- Whiting, A.K.; Boldt, Y.R.; Hendrich, M.P.; Wackett, L.P.; Que, L. Manganese(II)-Dependent Extradiol-Cleaving Catechol Dioxygenase from Arthrobacter globiformis CM-2. Biochemistry 1996, 35, 160–170. [Google Scholar]
- Williams, R.J.P. Free manganese(II) and iron(II) cations can act as intracellular cell controls. FEBS Lett. 1982, 140, 3–10. [Google Scholar] [PubMed]
- Grove, L.E.; Brunold, T.C. Second-sphere tuning of the metal ion reduction potentials in iron and manganese superoxide dismutases. Comment Inorg. Chem. 2008, 29, 134–168. [Google Scholar]
- Miller, A.-F. Superoxide dismutases: Active sites that save, but a protein that kills. Curr. Opin. Chem. Biol. 2004, 8, 162–168. [Google Scholar] [PubMed]
- Sheng, Y.; Abreu, I.A.; Cabelli, D.E.; Maroney, M.J.; Miller, A.-F.; Teixeira, M.; Valentine, J.S. Superoxide Dismutases and Superoxide Reductases. Chem. Rev. 2014, 114, 3854–3918. [Google Scholar] [PubMed]
- Wu, A.J.; Penner-Hahn, J.E.; Pecoraro, V.L. Structural, Spectroscopic, and Reactivity Models for the Manganese Catalases. Chem. Rev. 2004, 104, 903–938. [Google Scholar] [PubMed]
- Call, A.; Capocasa, G.; Palone, A.; Vicens, L.; Aparicio, E.; Choukairi Afailal, N.; Siakavaras, N.; López Saló, M.E.; Bietti, M.; Costas, M. Highly Enantioselective Catalytic Lactonization at Nonactivated Primary and Secondary γ-C–H Bonds. J. Am. Chem. Soc. 2023, 145, 18094–18103. [Google Scholar] [PubMed]
- Aneeja, T.; Neetha, M.; Afsina, C.M.A.; Anilkumar, G. Recent advances and perspectives in manganese-catalyzed C–H activation. Catal. Sci. Technol. 2021, 11, 444–458. [Google Scholar]
- Chen, J.; Jiang, Z.; Fukuzumi, S.; Nam, W.; Wang, B. Artificial nonheme iron and manganese oxygenases for enantioselective olefin epoxidation and alkane hydroxylation reactions. Coord. Chem. Rev. 2020, 421, 213443. [Google Scholar]
- Kasper, J.B.; Vicens, L.; de Roo, C.M.; Hage, R.; Costas, M.; Browne, W.R. Reversible Deactivation of Manganese Catalysts in Alkene Oxidation and H2O2 Disproportionation. ACS Catal. 2023, 13, 6403–6415. [Google Scholar]
- Ottenbacher, R.V.; Talsi, E.P.; Bryliakov, K.P. Chiral Manganese Aminopyridine Complexes: The Versatile Catalysts of Chemo- and Stereoselective Oxidations with H2O2. Chem. Rec. 2018, 18, 78–90. [Google Scholar]
- Ottenbacher, R.V.; Talsi, E.P.; Rybalova, T.V.; Bryliakov, K.P. Enantioselective Benzylic Hydroxylation of Arylalkanes with H2O2 in Fluorinated Alcohols in the Presence of Chiral Mn Aminopyridine Complexes. ChemCatChem 2018, 10, 5323–5330. [Google Scholar]
- Philip, R.M.; Radhika, S.; Abdulla, C.M.A.; Anilkumar, G. Recent Trends and Prospects in Homogeneous Manganese-Catalysed Epoxidation. Adv. Synth. Catal. 2021, 363, 1272–1289. [Google Scholar]
- Sun, W.; Sun, Q. Bioinspired Manganese and Iron Complexes for Enantioselective Oxidation Reactions: Ligand Design, Catalytic Activity, and Beyond. Acc. Chem. Res. 2019, 52, 2370–2381. [Google Scholar] [PubMed]
- Talsi, E.P.; Bryliakov, K.P. Chemo- and stereoselective CH oxidations and epoxidations/cis-dihydroxylations with H2O2, catalyzed by non-heme iron and manganese complexes. Coord. Chem. Rev. 2012, 256, 1418–1434. [Google Scholar]
- Vicens, L.; Olivo, G.; Costas, M. Rational Design of Bioinspired Catalysts for Selective Oxidations. ACS Catal. 2020, 10, 8611–8631. [Google Scholar]
- Bullock, R.M.; Chen, J.G.; Gagliardi, L.; Chirik, P.J.; Farha, O.K.; Hendon, C.H.; Jones, C.W.; Keith, J.A.; Klosin, J.; Minteer, S.D.; et al. Using nature’s blueprint to expand catalysis with Earth-abundant metals. Science 2020, 369, eabc3183. [Google Scholar]
- Nandy, A.; Adamji, H.; Kastner, D.W.; Vennelakanti, V.; Nazemi, A.; Liu, M.; Kulik, H.J. Using Computational Chemistry to Reveal Nature’s Blueprints for Single-Site Catalysis of C–H Activation. ACS Catal. 2022, 12, 9281–9306. [Google Scholar]
- Chen, J.; Song, W.; Yao, J.; Wu, Z.; Lee, Y.-M.; Wang, Y.; Nam, W.; Wang, B. Hydrogen Bonding-Assisted and Nonheme Manganese-Catalyzed Remote Hydroxylation of C–H Bonds in Nitrogen-Containing Molecules. J. Am. Chem. Soc. 2023, 145, 5456–5466. [Google Scholar]
- Leto, D.F.; Jackson, T.A. Peroxomanganese complexes as an aid to understanding redox-active manganese enzymes. J. Biol. Inorg. Chem. 2014, 19, 1–15. [Google Scholar]
- Lin, Y.-H.; Kutin, Y.; van Gastel, M.; Bill, E.; Schnegg, A.; Ye, S.; Lee, W.-Z. A Manganese(IV)-Hydroperoxo Intermediate Generated by Protonation of the Corresponding Manganese(III)-Superoxo Complex. J. Am. Chem. Soc. 2020, 142, 10255–10260. [Google Scholar]
- Sankaralingam, M.; Lee, Y.-M.; Jeon, S.H.; Seo, M.S.; Cho, K.-B.; Nam, W. A mononuclear manganese(iii)–hydroperoxo complex: Synthesis by activating dioxygen and reactivity in electrophilic and nucleophilic reactions. Chem. Comm. 2018, 54, 1209–1212. [Google Scholar] [PubMed]
- So, H.; Park, Y.J.; Cho, K.-B.; Lee, Y.-M.; Seo, M.S.; Cho, J.; Sarangi, R.; Nam, W. Spectroscopic Characterization and Reactivity Studies of a Mononuclear Nonheme Mn(III)–Hydroperoxo Complex. J. Am. Chem. Soc. 2014, 136, 12229–12232. [Google Scholar] [PubMed]
- Tian, Y.-C.; Jiang, Y.; Lin, Y.-H.; Zhang, P.; Wang, C.-C.; Ye, S.; Lee, W.-Z. Hydrogen Atom Transfer Thermodynamics of Homologous Co(III)- and Mn(III)-Superoxo Complexes: The Effect of the Metal Spin State. JACS Au 2022, 2, 1899–1909. [Google Scholar] [PubMed]
- Chavez, F.A.; Rowland, J.M.; Olmstead, M.M.; Mascharak, P.K. Syntheses, Structures, and Reactivities of Cobalt(III)–Alkylperoxo Complexes and Their Role in Stoichiometric and Catalytic Oxidation of Hydrocarbons. J. Am. Chem. Soc. 1998, 120, 9015–9027. [Google Scholar]
- Chen, Y.; Shi, H.; Lee, C.-S.; Yiu, S.-M.; Man, W.-L.; Lau, T.-C. Room Temperature Aerobic Peroxidation of Organic Substrates Catalyzed by Cobalt(III) Alkylperoxo Complexes. J. Am. Chem. Soc. 2021, 143, 14445–14450. [Google Scholar] [PubMed]
- Costas, M.; Mehn, M.P.; Jensen, M.P.; Que, L. Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem. Rev. 2004, 104, 939–986. [Google Scholar]
- Lehnert, N.; Ho, R.Y.N.; Que, L., Jr.; Solomon, E.I. Electronic Structure of High-Spin Iron(III)–Alkylperoxo Complexes and Its Relation to Low-Spin Analogues: Reaction Coordinate of O–O Bond Homolysis. J. Am. Chem. Soc. 2001, 123, 12802–12816. [Google Scholar]
- Lehnert, N.; Ho, R.Y.N.; Que, L.; Solomon, E.I. Spectroscopic Properties and Electronic Structure of Low-Spin Fe(III)–Alkylperoxo Complexes: Homolytic Cleavage of the O–O Bond. J. Am. Chem. Soc. 2001, 123, 8271–8290. [Google Scholar] [PubMed]
- Widger, L.R.; Jiang, Y.; McQuilken, A.C.; Yang, T.; Siegler, M.A.; Matsumura, H.; Moënne-Loccoz, P.; Kumar, D.; de Visser, S.P.; Goldberg, D.P. Thioether-ligated iron(ii) and iron(iii)-hydroperoxo/alkylperoxo complexes with an H-bond donor in the second coordination sphere. Dalton Trans. 2014, 43, 7522–7532. [Google Scholar]
- Du, J.; Miao, C.; Xia, C.; Lee, Y.-M.; Nam, W.; Sun, W. Mechanistic Insights into the Enantioselective Epoxidation of Olefins by Bioinspired Manganese Complexes: Role of Carboxylic Acid and Nature of Active Oxidant. ACS Catal. 2018, 8, 4528–4538. [Google Scholar]
- Miao, C.; Wang, B.; Wang, Y.; Xia, C.; Lee, Y.-M.; Nam, W.; Sun, W. Proton-Promoted and Anion-Enhanced Epoxidation of Olefins by Hydrogen Peroxide in the Presence of Nonheme Manganese Catalysts. J. Am. Chem. Soc. 2016, 138, 936–943. [Google Scholar]
- Coggins, M.K.; Martin-Diaconescu, V.; DeBeer, S.; Kovacs, J.A. Correlation Between Structural, Spectroscopic, and Reactivity Properties Within a Series of Structurally Analogous Metastable Manganese(III)–Alkylperoxo Complexes. J. Am. Chem. Soc. 2013, 135, 4260–4272. [Google Scholar]
- Coggins, M.K.; Kovacs, J.A. Structural and Spectroscopic Characterization of Metastable Thiolate-Ligated Manganese(III)–Alkylperoxo Species. J. Am. Chem. Soc. 2011, 133, 12470–12473. [Google Scholar]
- Downing, A.N.; Coggins, M.K.; Poon, P.C.Y.; Kovacs, J.A. Influence of Thiolate versus Alkoxide Ligands on the Stability of Crystallographically Characterized Mn(III)-Alkylperoxo Complexes. J. Am. Chem. Soc. 2021, 143, 6104–6113. [Google Scholar]
- Parham, J.D.; Wijeratne, G.B.; Rice, D.B.; Jackson, T.A. Spectroscopic and Structural Characterization of Mn(III)-Alkylperoxo Complexes Supported by Pentadentate Amide-Containing Ligands. Inorg. Chem. 2018, 57, 2489–2502. [Google Scholar] [PubMed]
- Opalade, A.A.; Parham, J.D.; Day, V.W.; Jackson, T.A. Characterization and chemical reactivity of room-temperature-stable MnIII–alkylperoxo complexes. Chem. Sci. 2021, 12, 12564–12575. [Google Scholar]
- Brunclik, S.A.; Opalade, A.A.; Jackson, T.A. Electronic structure contributions to O–O bond cleavage reactions for MnIII-alkylperoxo complexes. Dalton Trans. 2023, 52, 13878–13894. [Google Scholar]
- Dawson, J.H.; Holm, R.H.; Trudell, J.R.; Barth, G.; Linder, R.E.; Bunnenberg, E.; Djerassi, C.; Tang, S.C. Magnetic circular dichroism studies. 43. Oxidized cytochrome P-450. Magnetic circular dichroism evidence for thiolate ligation in the substrate-bound form. Implications for the catalytic mechanism. J. Am. Chem. Soc. 1976, 98, 3707–3709. [Google Scholar] [PubMed]
- Wijeratne, G.B.; Corzine, B.; Day, V.W.; Jackson, T.A. Saturation Kinetics in Phenolic O–H Bond Oxidation by a Mononuclear Mn(III)–OH Complex Derived from Dioxygen. Inorg. Chem. 2014, 53, 7622–7634. [Google Scholar]
- Rice, D.B.; Munasinghe, A.; Grotemeyer, E.N.; Burr, A.D.; Day, V.W.; Jackson, T.A. Structure and Reactivity of (μ-Oxo)dimanganese(III,III) and Mononuclear Hydroxomanganese(III) Adducts Supported by Derivatives of an Amide-Containing Pentadentate Ligand. Inorg. Chem. 2019, 58, 622–636. [Google Scholar]
- Eaton, G.R.; Eaton, S.S.; Barr, D.P.; Weber, R.T. Quantitative EPR, 1st ed.; Springer: Vienna, Austria, 2010; p. 185. [Google Scholar]
- Opalade, A.A.; Grotemeyer, E.N.; Jackson, T.A. Mimicking Elementary Reactions of Manganese Lipoxygenase Using Mn-hydroxo and Mn-alkylperoxo Complexes. Molecules 2021, 26, 7151. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar]
- Neese, F. Software update: The ORCA program system—Version 5.0. WIREs Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta—Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [PubMed]
- Neese, F.; Wennmohs, F.; Hansen, A. Efficient and accurate local approximations to coupled-electron pair approaches: An attempt to revive the pair natural orbital method. J. Chem. Phys. 2009, 130, 114108. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Product distribution of the cumylperoxo ligand following the thermal decay of [MnIII(OOCm)(N4S)]+ and [MnIII(OOCm)(6Medpaq)]+.
Figure 1.
Product distribution of the cumylperoxo ligand following the thermal decay of [MnIII(OOCm)(N4S)]+ and [MnIII(OOCm)(6Medpaq)]+.

Figure 2.
Molecular structures of H6Medpaq5NO2 (2-(bis((6-methylpyridin-2-yl)methyl)amino)-N-(5-nitroquinolin-8-yl)acetamide), MnII(OTf)(6Medpaq5NO2) (1), [MnIII(OH)(6Medpaq5NO2)](OTf) (2), [MnIII(OOtBu)(6Medpaq5NO2)](OTf) (3a), and [MnIII(OOCm)(6Medpaq5NO2)](OTf) (3b).
Figure 2.
Molecular structures of H6Medpaq5NO2 (2-(bis((6-methylpyridin-2-yl)methyl)amino)-N-(5-nitroquinolin-8-yl)acetamide), MnII(OTf)(6Medpaq5NO2) (1), [MnIII(OH)(6Medpaq5NO2)](OTf) (2), [MnIII(OOtBu)(6Medpaq5NO2)](OTf) (3a), and [MnIII(OOCm)(6Medpaq5NO2)](OTf) (3b).

Figure 3.
X-ray crystal structure of MnII(OTf)(6Medpaq5NO2) (left) and [MnII(OH2)(6Medpaq)](OTf) (right) [48]. Hydrogen atoms have been omitted for clarity. Ellipsoids, shown at 50% probability; disordered atoms in the nitro moiety omitted for clarity.
Figure 3.
X-ray crystal structure of MnII(OTf)(6Medpaq5NO2) (left) and [MnII(OH2)(6Medpaq)](OTf) (right) [48]. Hydrogen atoms have been omitted for clarity. Ellipsoids, shown at 50% probability; disordered atoms in the nitro moiety omitted for clarity.

Figure 4.
X-ray crystallographic structure of [MnIII(OH)(6Medpaq5NO2)](OTf) (left) and [MnIII(OH)(6Medpaq)](OTf) (right) [48]. The triflate counter anion and hydrogen atoms of the 6Medpaq5NO2 ligand have been removed for clarity.
Figure 4.
X-ray crystallographic structure of [MnIII(OH)(6Medpaq5NO2)](OTf) (left) and [MnIII(OH)(6Medpaq)](OTf) (right) [48]. The triflate counter anion and hydrogen atoms of the 6Medpaq5NO2 ligand have been removed for clarity.

Figure 5.
1H NMR spectra of 2 (top) and [MnIII(OH)(6Medpaq)]+ (bottom) [48] in CD3CN at 298 K.
Figure 5.
1H NMR spectra of 2 (top) and [MnIII(OH)(6Medpaq)]+ (bottom) [48] in CD3CN at 298 K.

Figure 6.
UV–Vis spectra showing the formation of 3a (left) and 3b (right) from the reaction of 3 mM of 2 with 1 equiv. of tBuOOH (left) and CmOOH (right) in CH3CN at 323 K. The absorbance at 650 nm over time for each reaction is shown inset. The down arrows indicate disappearance of 2 (red trace) and the up arrows indicated formation of 3a and 3b (blue traces).
Figure 6.
UV–Vis spectra showing the formation of 3a (left) and 3b (right) from the reaction of 3 mM of 2 with 1 equiv. of tBuOOH (left) and CmOOH (right) in CH3CN at 323 K. The absorbance at 650 nm over time for each reaction is shown inset. The down arrows indicate disappearance of 2 (red trace) and the up arrows indicated formation of 3a and 3b (blue traces).

Figure 7.
1H NMR spectra of 2 (red trace), 3a (green trace), and 3b (blue trace) in CD3CN at 298 K.

Figure 8.
Solution FT-IR spectra of 2 mM solutions of 2 (green), 3a (red), and 3b (blue).

Figure 9.
DFT-optimized structures of 3a (left) and 3b (right).

Figure 10.
UV–Vis spectra of the thermal decay of 3b at 323 K. The initial spectrum is shown in red, and the final spectrum is shown in blue—the formation of 2, as an intermediate, is highlighted in purple.
Figure 10.
UV–Vis spectra of the thermal decay of 3b at 323 K. The initial spectrum is shown in red, and the final spectrum is shown in blue—the formation of 2, as an intermediate, is highlighted in purple.

Figure 11.
Possible decay pathways for 3b.

Figure 12.
Pseudo-first-order rate constants as a function of [TEMPOH] for a solution of 2 at 238 K in CH3CN (left) and UV–Vis spectra of the reaction of 2 (3.00 mM) with 10 equiv. TEMPOH at 238 K in CH3CN (right). Example of pseudo-first-order fit calculation for 1.00 mM solution of 2 with 25 equiv. TEMPOH at 238 K in CH3CN (right inset).
Figure 12.
Pseudo-first-order rate constants as a function of [TEMPOH] for a solution of 2 at 238 K in CH3CN (left) and UV–Vis spectra of the reaction of 2 (3.00 mM) with 10 equiv. TEMPOH at 238 K in CH3CN (right). Example of pseudo-first-order fit calculation for 1.00 mM solution of 2 with 25 equiv. TEMPOH at 238 K in CH3CN (right inset).

Figure 13.
UV–Vis spectra of the reaction of a 3 mM solution of 3a in acetonitrile (red) with 100 equiv. triphenylphosphine at 298 K. The final spectrum is shown in blue. The absorbance at 650 nm over time is shown in the inset.
Figure 13.
UV–Vis spectra of the reaction of a 3 mM solution of 3a in acetonitrile (red) with 100 equiv. triphenylphosphine at 298 K. The final spectrum is shown in blue. The absorbance at 650 nm over time is shown in the inset.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Manganese-ligand bond lengths (Å) and angles from X-ray crystal structures of 1 [MnII(OH2)(6Medpaq)](OTf) and [MnII(dpaq)](OTf) [51]. a From ref. [48]; b from ref. [51].
| Bond | 1 | [MnII(OH2)(L)](OTf) | [MnII(L)](OTf) |
|---|---|---|---|
| a L = 6Medpaq | b L = dpaq | ||
| Mn–O1 | 2.169(7) | 2.108(3) | 2.079(2) |
| Mn–N1 | 2.260(6) | 2.233(3) | 2.214(3) |
| Mn–N2 | 2.150(7) | 2.152(4) | 2.191(3) |
| Mn–N3 | 2.281(6) | 2.280(3) | 2.314(3) |
| Mn–N4 | 2.328(7) | 2.354(4) | 2.244(3) |
| Mn–N5 | 2.357(8) | 2.417(3) | 2.286(3) |
Table 2.
Manganese-ligand bond lengths (Å) and angles from X-ray crystal structures of 2, [MnIII(OH)(6Medpaq)](OTf), [MnIII(OH)(dpaq)](OTf), and [MnIIIMnIII(µ-O)(dpaq5NO2)2](OTf)2. a From ref. [48]. b From ref. [51]; c from ref. [52].
| Bond | [MnIII(OH)(L)](OTf) | [MnIIIMnIII(µ–O)(L)2](OTf)2 | ||
|---|---|---|---|---|
| L = 6Medpaq5NO2 | a L = 6Medpaq | b L = dpaq | c L = dpaq5NO2 | |
| Mn–O1 | 1.826(4) | 1.806(6) | 1.806(13) | 1.7918(4) |
| Mn–N1 | 2.043(4) | 2.041(7) | 2.072(14) | 2.054(2) |
| Mn–N2 | 1.959(5) | 1.962(6) | 1.975(14) | 1.973(2) |
| Mn–N3 | 2.114(4) | 2.130(6) | 2.173(14) | 2.199(2) |
| Mn–N4 | 2.397(4) | 2.322(6) | 2.260(14) | 2.186(2) |
| Mn–N5 | 2.342(5) | 2.381(7) | 2.216(15) | 2.288(3) |
Table 3.
1H NMR chemical shifts (ppm) for 2, 3a, and 3b. Complexes in CD3CN at 298 K.
| 2 | 3a | 3b | |
|---|---|---|---|
| H-quinoline | 67.0 | 67.3 | 68.3 |
| NA a | 58.4 | ||
| H-pyridine | 54.0 | 48.1 | 47.6 |
| H-pyridine | 45.6 | 46.5 | 47.0 |
| NA a | 12.7 | 9.5 | |
| NA a | 9.9 | 8.8 | |
| H-pyridine | −16.8 | ||
| H-quinoline | −19.4 | −21.5 | −21.8 |
| H-quinoline | −60.7 | −58.9 | −59.5 |
a Not assigned.
Table 4.
Manganese-ligand bond lengths (Å) and angles of 3a, 3b, and [MnIII(OOCm)(6Medpaq)](OTf). Bond lengths and angles of 3a and 3b derived from DFT-optimized structures. a From X-ray structure in ref. [48].
Table 4.
Manganese-ligand bond lengths (Å) and angles of 3a, 3b, and [MnIII(OOCm)(6Medpaq)](OTf). Bond lengths and angles of 3a and 3b derived from DFT-optimized structures. a From X-ray structure in ref. [48].
| Bond | 3a | 3b | [MnIII(OOCm)(L)](OTf) |
|---|---|---|---|
| L = 6Medpaq5NO2 | L = 6Medpaq5NO2 | a L = 6Medpaq | |
| Mn–O1 | 1.833 | 1.831 | 1.849(3) |
| Mn–N1 | 2.045 | 2.037 | 2.044(4) |
| Mn–N2 | 1.958 | 1.950 | 1.955(4) |
| Mn–N3 | 2.153 | 2.150 | 2.100(4) |
| Mn–N4 | 2.355 | 2.370 | 2.284(4) |
| Mn–N5 | 2.347 | 2.296 | 2.394(4) |
| O1–O2 | 1.454 | 1.451 | 1.466(4) |
| Mn–O1–O2 | 107.3 | 114.2 | 110.4(2) |
| Source | DFT | DFT | X-Ray |
Table 5.
Product distribution obtained via GC-MS for thermal decay of MnIII–cumylperoxo complexes in acetonitrile and acetonitrile-d3. a From ref. [48].
Table 5.
Product distribution obtained via GC-MS for thermal decay of MnIII–cumylperoxo complexes in acetonitrile and acetonitrile-d3. a From ref. [48].
| Complex | Solvent | 2-Phenyl-2-propanol | Acetophenone |
|---|---|---|---|
| 3b | CH3CN | 49.7 ± 3.5% | 35.2 ± 2.8% |
| a [MnIII(OOCm)(6Medpaq)]+ | CH3CN | 61.3 ± 0.1% | 25.7 ± 0.1% |
| 3b | CD3CN | 24.0 ± 1.6% | 71.5 ± 4.3% |
| a [MnIII(OOCm)(6Medpaq)]+ | CD3CN | 50 ± 0.3% | 40 ± 0.3% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Brunclik, S.A.; Grotemeyer, E.N.; Aghaei, Z.; Mian, M.R.; Jackson, T.A. Investigating Ligand Sphere Perturbations on MnIII–Alkylperoxo Complexes. Molecules 2024, 29, 1849. https://doi.org/10.3390/molecules29081849
AMA Style
Brunclik SA, Grotemeyer EN, Aghaei Z, Mian MR, Jackson TA. Investigating Ligand Sphere Perturbations on MnIII–Alkylperoxo Complexes. Molecules. 2024; 29(8):1849. https://doi.org/10.3390/molecules29081849
Chicago/Turabian StyleBrunclik, Samuel A., Elizabeth N. Grotemeyer, Zahra Aghaei, Mohammad Rasel Mian, and Timothy A. Jackson. 2024. "Investigating Ligand Sphere Perturbations on MnIII–Alkylperoxo Complexes" Molecules 29, no. 8: 1849. https://doi.org/10.3390/molecules29081849